

Abstract
Proteins with expanded polyglutamine (polyQ) tracts have been linked to neurodegenerative diseases. One common characteristic of expanded-polyQ expression is the formation of intracellular aggregates (IAs). IAs purified from polyQ-expressing cells were dissociated and studied by protein blot assay and mass spectrometry to determine the identity, condition, and relative level of several proteins sequestered within aggregates. Most of the sequestered proteins comigrated with bands from control extracts, indicating that the sequestered proteins were intact and not irreversibly bound to the polyQ polymer. Among the proteins found sequestered at relatively high levels in purified IAs were ubiquitin, the cell cycle–regulating proteins p53 and mdm-2, HSP70, the global transcriptional regulator Tata-binding protein/TFIID, cytoskeleton proteins actin and 68-kD neurofilament, and proteins of the nuclear pore complex. These data reveal that IAs are highly complex structures with a multiplicity of contributing proteins.
Keywords: polyglutamine, Huntington's disease, aggregates, inducible expression, ecdysone receptor
Introduction
CAG repeat expansion and concomitant polyglutamine (polyQ) expression have been linked to a variety of neurodegenerative conditions including Huntington's disease (HD), dentatorubro-pallidoluysian atrophy, spinocerebellar ataxia (including types 1, 2, 3, 6, 7, and 12), and spinobulbar muscular atrophy (Holmes et al. 1999; Zoghbi and Orr 2000). The relationship between the expression, distribution, and aggregation of proteins with expanded polyQ tracts and cell death remains to be understood. To date, most of the progress in determining the molecular events surrounding polyQ toxicity has concentrated on mammalian and invertebrate transgenic models (Reddy et al. 1999; Zoghbi and Orr 2000), viral gene transfer models (Senut et al. 2000), and transfection studies with a variety of polyQ-containing gene products (Reddy et al. 1999; Zoghbi and Orr 2000).
One common hallmark of these diseases and other neurodegenerative conditions, including Alzheimer's disease and Parkinson's disease, is the formation of insoluble protein aggregates and inclusion bodies in disease-specific regions of the human brain (for reviews see Perutz 1999; Wanker 2000). What is not clear in polyQ-mediated pathologies, however, is that, although intracellular aggregates (IAs) may correlate to a greater or lesser extent with disease progression, they have not been causally linked to the death of affected cells (Sisodia 1998). Furthermore, aside from the polyQ-bearing protein, the composition of IAs remains largely unknown.
Some proteins known to be colocalized to polyQ IAs are components of protein-folding and proteolytic pathways. These proteins include ubiquitin, molecular chaperones, and components of the 20S proteasome. Recent studies in transgenic mouse and Drosophila melanogaster models of polyQ-mediated toxicity reveal that inhibition or perturbation of these cellular pathways can significantly suppress cytotoxicity of expressed polyQ proteins in vivo (Sherman and Goldberg 2001).
Other proposed components of IAs are proteins with nonpathogenic length polyQ tracts and caspases. IA recruitment of synthetic reporter proteins with short polyQ tracts, and sequestration of intracellular factors with tracts as short as 19 glutamines, such as CREB-binding protein, have been demonstrated (Kazantzev et al. 1999). A second endogenous protein with a nonpathogenic but longer polyglutamine tract (38 glutamines), TATA-binding protein (TBP) (Kao et al. 1990), has also been colocalized to IAs in tissue samples from HD patients (Huang et al. 1998). The protease caspase-8 has been described as a component of IAs in Hela cells transiently transfected with a 79Q construct (Sanchez et al. 1999).
To date, the majority of studies have assayed recruitment or sequestration of proteins to IAs by immunohistochemical colocalization of antibodies to IAs stained with established IA markers, generally antiubiquitin or an antibody against the polyQ protein itself. The benefit of this type of analysis is that differences in the staining pattern or intensity of staining from one IA to another may provide information on heterogeneity within the IA population, which in turn may yield valuable clues as to the pathogenesis of polyQ-mediated cell death. One drawback of this type of analysis is that many antigens may be masked or conformationally altered by association with the IA and could lead to false negative conclusions or misleading staining patterns. Furthermore, it is difficult to assign a quantitative value to positive structures in immunocytochemical studies.
In this report, we describe the isolation, characterization, and composition of IAs from an inducible in vitro model of polyQ-mediated disease using a human cell line that standardizes several parameters of polyQ-mediated cell death. Through a combination of protein blot analysis, immunohistochemical studies, and proteomics using matrix-assisted laser desorption ionization time of flight mass spectrometry (MALDI) analysis (Yates 2000), the identities and relative levels of aggregate-associated proteins were examined. These studies reveal that IAs are complex structures with a multiplicity of sequestered protein components.
Materials and Methods
Construction of polyQ Reporter Fusion Proteins and Vectors
PolyQ reporter constructs were made by PCR amplification of human Huntingtin (Htt) exon I variants with 13- and 96-CAG tracts, preserving polyproline (polyP) sequences immediately downstream of the polyQ tract (see Fig. 1) and fusing these fragments in frame to the NH2 terminus of EGFP (CLONTECH Laboratories, Inc.) in the cloning vector SKSP. Unique AscI and MluI restriction sites were inserted into the 5′ and 3′ coding regions for insertion of oligonucleotides encoding the SV 40 nuclear localization signal (NLS). For the experiments in this report, only the 3′-localized SV 40 NLS was used. The 96Q tract contains a characterized arginine residue at position 42 of the 96Q tract (Senut et al. 2000). The polyQ–eGFP coding region was excised from SKSP using unique SfiI and PmeI cloning sites for insertion into retroviral vector NIT (sequence data available from GenBank/EMBL/DDBJ under accession number AF311318) for expression in transient transfection studies, or into retroviral vector LPR for use with vector CVBE for ecdysteroid-induced cells. LPR is a Moloney murine leukemia virus–based vector with puromycin resistance and six-tandem ecdysone response elements fused to a minimal cytomegalovirus (CMV) promoter directing transgene expression when combined with CVBE (Suhr et al. 1998). Transient transfection analysis was performed by calcium phosphate precipitation by standard methods. For nuclear visualization, cells were stained with either 100 ng/ml propidium iodide or DAPI.
Figure 1.
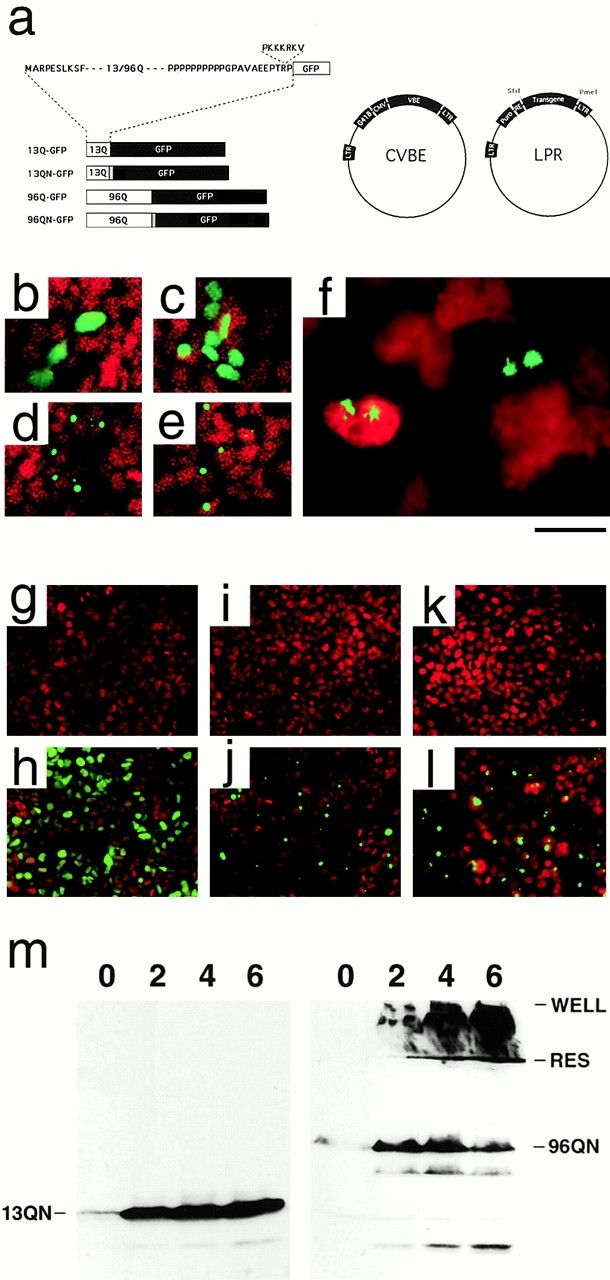
PolyQ–GFP reporter constructs transiently and regulatably expressed in HEK293 cells. (a, left) Schematic of polyQ–GFP fusion proteins indicating Htt exon I–derived sequences (white), SV 40 NLS (gray), and eGFP sequences (black). (a, right) Schematic of CVBE and LPR Moloney murine leukemia virus–based retroviral vectors used in production of inducible polyQ cell lines. CVBE encodes flanking long terminal repeats (LTRs), a G418-resistance gene (G418), the immediate–early CMV promoter (CMV), and the VBE transactivator protein (VBE). LPR encodes long term repeats, a puromycin-resistance gene (Puro), a minimal CMV promoter with six tandem ecdysone response elements (RE), and a SfiI–PmeI polylinker for insertion of the polyQ transgenes. (b–f) Propidium iodide nuclear-stained (red) HEK293 cells transiently transfected with the expression construct 13Q–GFP (b), 13QN–GFP (c), 96Q–GFP (d), and 96QN–GFP (e). GFP produces the bright green fluorescence. (f) IAs in transfected cells revealing stellate fibrous appearance of IAs at high magnification. 72-h vehicle- (g) or ligand-treated (h) 13QN cells stained with the nuclear stain DAPI (red), vehicle- (i) or ligand-treated (j) 96Q cells, and vehicle- (k) or ligand-treated (l) 96QN cells. Bright GFP-positive profiles indicate IA formation. (m) Western blot analysis of induced 13QN (left) or 96QN (right) cells. Numbers at the top of each blot indicate the number of days of induction. WELL, bottom of the well; RES, bottom of the stacking gel and the beginning of the resolving gel. Bar: (b–e) 125 μm; (f) 20 μm; (g–l) 250 μm.
Cell Transfection, Retroviral Infection, and Cell Culture
Transient transfection by calcium phosphate precipitation and transfection to produce replication-defective retrovirus was as previously described (Pear et al. 1993; Suhr et al. 1998). All cell cultures were done in DME with 10% FBS in a 10% CO2 environment. For retroviral infection, low titer (multiplicity of infection <0.1) polyQ–green fluorescent protein (GFP) virus variants were used. Infection, selection, and passage of cells were performed as described for other G418-resistant cell types (Suhr et al. 1998). Production of CVBE–LPR coinfected cells was as described for CVBE with MS (Suhr et al. 1998), except that 2 μg/ml puromycin was used for selection of LPR-infected cells. For studies of ligand responsiveness, individual cell populations were passaged to either 24- or 6-well Costar plates. For stimulation of infected cells, 1 μM tebufenozide or vehicle (90% ethanol) was added and replaced only when cells were passaged or the media was changed. For the Western blot time course of induction, 10-cm plates were seeded with 2.5 × 105 13QN or 96QN cells. One set of plates was immediately treated with 1 μM tebufenozide for the 6-d time point, 48 h later for the 4-d time point, and so on. Cells were fed on day 3, and ligand was replaced if necessary. All cells were simultaneously harvested at the end of the experiment, collected by scraping in PBS, pelleted by low-speed centrifugation, and resuspended in 250 μl cold 10 mM Tris-Cl, pH 7.5. The resuspended pellet was then sonicated on ice with 30 1-s bursts at 30% power, the protein concentration was quantified, and aliquots of the preparations were frozen at −70°C until use in protein blot analysis as outlined below.
TUNEL Staining
DNA fragmentation was detected in situ on cultured cells using the immunocytochemical terminal deoxynucleotidyl transferase–mediated dUTP–biotin end labeling (TUNEL) technique (Roche) according to the manufacturer's protocol. Using sheep antifluorescein antibody (Roche), the fluorescein signal was converted into a peroxidase signal for visualization using 0.025% DAB, 0.5% nickel chloride, and 0.018% H2O2 in TBS. Positive and negative controls were included in each experiment.
IA Purification
IAs were purified using a modification of the CsCl gradient procedures described by Scherzinger et al. 1999. PolyQ cells were grown on 10- or 15-cm plates with 1 μM tebufenozide treatment for 5 d, feeding and passaging as necessary, but maintained at >50% confluency. The plates of induced cells were then washed two times with PBS, the cells were collected by scraping in a small volume of PBS, centrifuged to pellet cells, and the pellet was resuspended in 3 ml of cold lysis buffer (50 mM Hepes-KOH, pH 7.4, 150 mM NaCl, 1.5 mM MgCl, 1 mM EGTA, 0.02% sodium azide, 1% NP-40, 10% glycerol, 0.1 mg/ml PMSF). Lysis proceeded on ice for 30 min. The lysate was vortexed vigorously and centrifuged in a microfuge at 1.6 × 104 g for 5 min at 4°C. The insoluble pellet(s) were collectively resuspended in 1 ml of 20 mM Tris, pH 8.0, 15 mM MgCl, and sonicated with 30 1-s bursts at 30% power. 100 μg of DNase was then added, and the mixture was incubated at 37°C for 1 h. After incubation, the lysate was brought up to a final volume of 4 ml with 10 mM Tris HCl, pH 7.5, and dry CsCl was added to a final concentration of 0.16 g/ml. 4 ml of a 0.52 g/ml CsCl Tris solution was placed in an ultracentrifuge tube, overlaid with an equal volume of 0.25 g/ml CsCl solution, and then topped off with the 4 ml of lysate CsCl. This step gradient was centrifuged at 1.1 × 105 g for 24 h in a ultracentrifuge (Beckman Coulter). After centrifugation, thin bands of a greenish hue (that were brightly fluorescent under 495-nm excitation wavelength illumination) were observed ∼1/2–2/3 from the top of the tube. These bands were collected in a volume of 200–300 μl and diluted 50 times (to ∼15 ml) with 10 mM Tris-Cl, pH 7.5. The diluted IAs were centrifuged at 5 × 103 g three times with repeated dilution (sonicating as needed to break up IA clumps). The final pellet was resuspended in 100–300 μl of 10 mM Tris, aliquoted, and frozen at −70°C until use.
Protein Blot Analysis and Quantification
For Western blot analysis, cell lysates and preparations of purified aggregates were heated in loading buffer (50 mM Tris-HCl, pH 6.8, 10% SDS, 0.1% bromophenol blue, 10% glycerol). Equal amounts of protein (∼7.5 μg) were loaded in each lane of a 10% SDS-PAGE gel and electrophoresed. Gels were subsequently transblotted to nitrocellulose membranes using a liquid transfer apparatus (Bio-Rad Laboratories). Membranes were blocked for 1 h in blocking solution (5% nonfat milk in TBS-Tween) and incubated overnight at 4°C with primary antibodies (Table ) diluted in blocking solution. After extensive washes, membranes were incubated for 1 h with peroxidase-conjugated secondary antibodies (1:1,000–1:5,000), and immunoreactive signals were visualized using a chemiluminescence detection kit (Amersham Pharmacia Biotech). Control experiments were carried out by replacing the primary antibody with normal serum or preadsorbing the primary antibody with the corresponding peptide antigen.
Table 1.
Characteristics of the Antisera
| Antigen | Origin | Host | Dilution/Western IFL |
|---|---|---|---|
| Actin | James Lessard | Mouse | 1:1,000/1:3,000 |
| Caspase-3 | Santa Cruz Biotechnology, Inc. | Rabbit | 1:250–500/1:50 |
| Caspase-8 | Santa Cruz Biotechnology, Inc. | Rabbit | 1:250–500/1:50 |
| Caspase-9 | Santa Cruz Biotechnology, Inc. | Rabbit | 1:250–500/1:50 |
| GFP | Santa Cruz Biotechnology, Inc. | Rabbit | 1:3,000/NA |
| GRB-2 | Santa Cruz Biotechnology, Inc. | Rabbit | 1:1,000/1:100–250 |
| HSP70 | Transduction Laboratories | Mouse | NA/1:100 |
| HSP70 | Santa Cruz Biotechnology, Inc. | Goat | 1:500/1:100 |
| Huntingtin | Chemicon | Rat | 1:500/1:50 |
| Lamin B | Santa Cruz Biotechnology, Inc. | Goat | 1:500/1:500 |
| mdm-2 Ab-1 (N) | Santa Cruz Biotechnology, Inc. | Rabbit | 1:100/1:50 |
| mdm-2 Ab-2 (C) | Santa Cruz Biotechnology, Inc. | Rabbit | 1:100/1:50 |
| MEF-2a | Santa Cruz Biotechnology, Inc. | Rabbit | 1:500/1:100 |
| Nck | Santa Cruz Biotechnology, Inc. | Rabbit | 1:500/1:250 |
| Neurofilament 68 kD | Chemicon | Rabbit | 1:1,000/1:500 |
| NPCP/mAb414 | Babco | Mouse | 1:1,000/1:3,000 |
| p53 | Santa Cruz Biotechnology, Inc. | Rabbit | 1:500/1:100 |
| TBP | Santa Cruz Biotechnology, Inc. | Rabbit | 1:1,000/1:100–200 |
| Ubiquitin | Dako | Rabbit | 1:1,000/1:100 |
For quantification of sequestered proteins, each antibody used to detect a sequestered protein species in the Western blot analyses (see Fig. 3) was titrated against increasing concentrations of induced 96QN cell whole cell extracts or purified IA samples to determine the linear range of detection (data not shown). Each antibody was reassessed against the same samples as those described in the legend to Fig. 3 using an amount of protein and dilution of antibody determined to be within the linear range of detection. From these protein blots, the integrated densities of bands within purified IA lanes and control lanes were determined and used to calculate the percentage of protein for an individual protein species sequestered into IAs relative to the amount of the same protein in whole cell extracts. Since no recruited protein species displayed different band intensities in the 5-d induced 13QN and 96QN uninduced lanes, these two values were averaged to provide the basal protein expression level.
Figure 3.
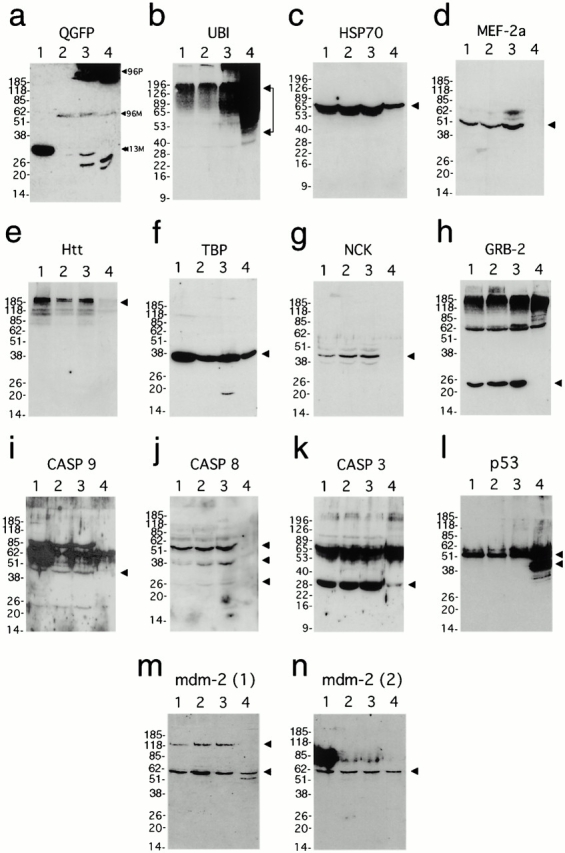
Western blot analysis of protein sequestration in purified IAs and control whole cell extracts. For all protein blots: lane 1, whole cell extracts from 5-d induced 13QN cells; lane 2, uninduced 96QN cells; lane 3, 5-d induced 96QN cells; and lane 4, isolated concentrated aggregates. Arrowheads at the right of autoradiograms indicate predicted molecular weights of individual protein species unless otherwise specified. (a) Processing for polyQ–GFP immunoreactivity. 96-P indicates the position of polyQN–GFP polymers within the stacking gel and near the top of the resolving gel (lanes 3 and 4), 96-M indicates a low level of 96QN monomers (lanes 2–4), and 13-M indicates the 13QN monomer band (lane 1). This autoradiogram was overexposed to reveal the 96QN monomer bands in lanes 3 and 4. (b) Ubiquitin; (c) HSP70; (d) MEF-2a; (e) Htt; (f) TBP; (g) Nck; (h) GRB-2; (i) caspase-9; (j) caspase-8 (putative procaspase-8, upper arrowhead; putative caspase-8 cleavage products, lower arrowheads); (k) caspase-3; (l) p53 and p50 immunoreactive species (upper and lower arrowheads, respectively); (m) mdm-2 antibody-1 (120-kD variant, upper arrowhead; p60 variant, lower arrowhead); and (n) mdm-2 antibody-2 immunoreactivity.
To calculate degree of sequestration, two parameters were established. The first parameter established the number of whole cell extract equivalents of IAs per unit volume of purified IAs relative to a unit weight of 96QN–GFP whole cell extracts. By quantifying the intensity of the polymerized 96QN–GFP band for 5 μl of purified IAs sample relative to the polymerized 96QN–GFP band for 7.5 μg 96QN–GFP whole cell extracts, it was determined that this value averaged 16.3, indicating that the amount of IA protein in 5 μl of purified IAs represents the amount of IA protein in 7.5 μg × 16.3, or ∼122 μg of 5-d 96QN–GFP whole cell extract. For blots in which the amount of purified IAs or whole cell extract was reduced or increased to bring detection within the linear range, the correction factor was proportionally adjusted.
Since only a subpopulation of cells at the time of harvest contained large IAs likely to equilibrate within the CsCl gradient band taken during purification, a second parameter was determined that corrected for IA heterogeneity within the 96QN–GFP cell population. 18% of cells displayed large IAs at the 5-d time point, so, to normalize to whole cell extracts, the protein adjusted value was divided by 0.18 to produce the final value reported.
Protein Isolation and MALDI Analysis
For isolation of individual protein bands from IA preparations, 40 IA equivalents (relative to the amount used in protein blot analyses above) in a volume of 80 μl (a 2.5× relative concentration) prepared for gel loading as above, were subjected to 10% SDS-PAGE on a 1-mm slab gel and run at low constant voltage (90 V) for 8 h. The gel was then stained overnight with 40% methanol, 10% acetic acid, 0.25% Coomassie brilliant blue and destained for 8 h the next day in 40% methanol, 10% acetic acid solution. Four prominent stained bands that ranged in size from ∼70 to 30 kD were excised from the gel with a scalpel, transferred to a microfuge tube, treated with TFA–acetonitrile for 1 h at 37°C, and vacuum lyophilized. The lyophilized samples were treated with trypsin for 16 h at 37°C, and peptides were extracted according to Bienvenut et al. 1999. Extracted peptides were dried by vacuum centrifugation and applied to the MALDI sample plate via use of C18 Ziptips (Millipore). The sample was dissolved in 10 μl 0.1% TFA and 5% acetonitrile, applied to a Ziptip, and equilibrated in the same buffer (see manufacturer's instructions). The bound peptides were washed with the same buffer (4 × 10 μl) and then eluted with 0.1% TFA, 80% acetonitrile, 10 mg/ml α-cyano-4-hydroxy-trans-cinnamic acid (∼1 μl) directly onto the MALDI plate. MALDI analysis was accomplished using a Voyager RP (PerSeptive Biosystems) with internal calibration using insulin (5,734 Da) and a synthetic peptide LAP 821 (1,740 Da). Between 10 and 23 peaks with closely matching peaks on samples run without internal calibration were used to challenge public protein databases using Protein Prospector (http://prospector.ucsf.edu) and ProFound (http://prowl.rockefeller.edu) (Zhang and Chait 2000). Values were compared with both mammalian and human protein databases at lower (±200–500 ppm) and higher (±1,000 ppm) mass tolerances.
Immunohistochemistry
Antisera used in these studies are listed in Table .
Cell Culture.
Cells were fixed for 20 min in phosphate-buffered 4% paraformaldehyde and rinsed twice in 0.1 M TBS. Primary antibodies were diluted in 0.1 M TBS containing 5% normal donkey serum and 0.3% Triton X-100 (NDST). Cells were preincubated in 5% NDST for 1 h at room temperature and then incubated in the primary antibodies overnight at 4°C. Cells were rinsed three times in 5% NDST and incubated for 2 h at room temperature with donkey secondary antibodies (Jackson ImmunoResearch Laboratories) conjugated to FITC, cyanin-3, or cyanin-5, and diluted 1:250 in 5% NDST. Cells were then washed five times in 0.1 M TBS, with the third wash containing 10 ng/ml 4, 6-diamidino-2-phenylindole (Sigma-Aldrich) for nuclear staining. TRITC–phalloidin staining was performed by incubating fixed cultured cells in 30 nM TRITC–phalloidin for 30 min followed by extensive rinsing with PBS.
Human Tissue.
Postmortem caudate–putamen tissues from control (n = 3) and age matched HD grade 3 (n = 3) patients (mean age: 70.8 yr; range: 60–78 yr) were obtained from the Harvard Brain Tissue Resource Center, Cambridge, MA. Tissue blocks were cryoprotected at 4°C in increasing concentrations of phosphate-buffered sucrose (10, 20, and 30%). Coronal serial 40-μm-thick sections were cut on a freezing microtome, collected either in 0.1 M TBS for immediate immunostaining, or in cryoprotectant for further storage at −20°C. Free-floating sections were placed in 0.3% hydrogen peroxide for 20 min and then preincubated in 0.1 M TBS containing 5% normal donkey serum, 5% normal AB human serum, and 0.3% Triton X-100 (5% NHDST) for 2–4 h at room temperature. Sections were then incubated in the primary antibodies (Table ) diluted in 5% NDHST for 72 h at 4°C. Sections were rinsed three times in 5% NDHST, incubated for 1 h at room temperature with the adequate biotinylated donkey secondary antibodies (Jackson ImmunoResearch Laboratories) diluted 1:200 in 5% NDHST, rinsed twice in 0.1 M TBS, and incubated with an avidin–biotin peroxidase complex (1:120) for 1 h at room temperature. After intense rinses in 0.1 M TBS, tissue-bound peroxidase was visualized using 0.025% DAB, 0.5% nickel chloride, and 0.018% H2O2 in TBS. Sections were rinsed, mounted on gelatin-coated slides, dried, and coverslipped.
Results
Expression and Aggregation of polyQ–GFP Fusion Proteins
Long (96Q) and short (13Q) polyQ tracts derived from human Htt exon I (Huntington Disease Collaborative Research Group 1993) were fused to a GFP reporter protein to produce 96Q–GFP and 13Q–GFP constructs (Senut et al. 2000), as shown in Fig. 1 a. Unique sequences were designed into the regions flanking the polyQ tracts to allow insertion of SV 40 NLS and other sequences either COOH- or NH2-terminal to the polyQ tract. A long polyproline tract was located immediately COOH-terminal to the polyQ tract as in native Htt (Fig. 1 a). PolyQ–GFP constructs were inserted either into a constitutive expression vector for pilot studies and testing by transient transfection or into the regulated reporter retroviral vector system CVBE–LPR shown in Fig. 1 a. Fluorescent microscopy of cells transiently transduced with 13Q–GFP expression plasmids revealed that transfected cells expressed GFP at high levels throughout the cytoplasm and nucleus (Fig. 1 b). Addition of an SV 40 NLS resulted in efficient localization of the 13QN–GFP reporter protein to the nucleus, as shown in Fig. 1 c. Transfected 96Q–GFP and 96QN–GFP expression constructs, on the other hand, initially displayed uniform GFP-positive fluorescence throughout the cytoplasm or nucleus but, within 24–72 h of transfection, condensed into the bright granules indicative of IAs (Fig. 1d and Fig. e). Most cells displayed one or two IAs, and IAs were found within the cytoplasm, surrounding the nuclear envelope, and within the nucleus itself. At higher magnification, IAs were observed to have a stellate appearance with fibrous projections emanating from a central core (Fig. 1 f). In the absence of the NLS, IAs were approximately evenly divided between the cytoplasm and nucleus. The addition of the NLS resulted in >90% perinuclear/nuclear localization of IAs (data not shown).
To circumvent the toxic consequences of chronic constitutive polyQ expression, the polyQ–GFP fusion constructs were used in a positively regulated retroviral vector system (Suhr et al. 1998). Transactivator and reporter retroviruses bearing the polyQ–GFP constructs were introduced into HEK293 cells as described in Materials and Methods. Three inducible lines were produced: CVBE–LPR–96Q–GFPQ–GFP (96Q), CVBE–96QN–GFP (96QN), and CVBE–LPR13QN–GFP (13QN). CVBE–LPR13Q–GFP cells were not included in the study since preliminary experiments revealed that they were indistinguishable in all respects from the 13QN cell population, except for localization of the polyQ–GFP protein. In Fig. 1g–l, inducible 13QN, 96Q, and 96QN cell lines were stimulated with 1 μM tebufenozide and allowed to grow for ≤1 wk. In the absence of ligand, only widely scattered cells (<0.1%) displayed even low green fluorescence (Fig. 1g, Fig. i, and Fig. k). After ligand stimulation, green fluorescence was rapidly observed and, as predicted from transient transfection studies, displayed diffuse nuclear fluorescence in 13Q/N–GFP cells (Fig. 1 h) and punctate intense positivity indicative of IAs by 72 h in 96Q- and 96QN-induced cells (Fig. 1j and Fig. l).
Western blot analysis of the time course of polyQ induction revealed a low level of 13QN expression in the absence of ligand that is increased ∼50-fold to a steady state level of expression within 48 h of ligand addition (Fig. 1 m, left). A similar pattern is observed in 96QN-induced cells (Fig. 1 m, right); however, at times >24 h after induction, IA formation was observed with increasing frequency, as evidenced by the increasing accumulation of insoluble material within the stacking gel at the top of the blot. By day 6, there is an apparent loss of monomeric 96QN protein as increasing free protein is trapped within the growing aggregates.
Morphologic Changes and Increased Toxicity Induced by 96QN Expression
Study of induced cells revealed no obvious time-dependent morphological changes in any of the induced lines with the exception of 96QN, which at time points >4 d exhibited irregularities in nuclear morphology, including nuclear hypertrophy, the formation of small satellite structures of nuclear material, and an increase in the formation of multinucleated syncitia. Fig. 2, a–d, shows examples of the occasional multinucleated syncytia that form at 5 d after induction. One hallmark of these syncytia is the frequent presence of large cytoplasmic IAs at the center of a ring of nuclei (Fig. 2, a and c). The number of syncytia within representative cell populations at different times after induction is shown in Fig. 2 e. At the 5-d time point, 96QN cells develop over fivefold more multinucleated syncytia than 5-d 13QN cells, 96Q cells, or 96QN cells at the earlier time points.
Figure 2.
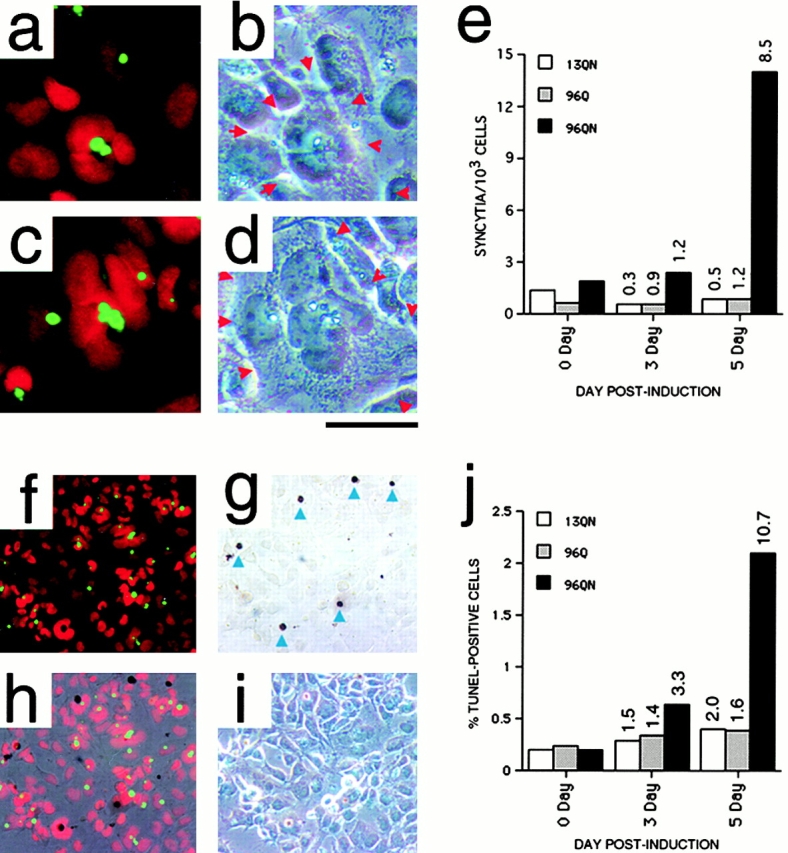
Morphological changes and cell death in induced polyQ-expressing cells. (a–d) 5-d induced 96QN cells matched fluorescence (a and c) and phase–contrast images (b and d), revealing multinucleated syncitia-like cell structures. In a and c, nuclei are red, and polyQ–GFP IAs are green. In b and d, red arrowheads indicate the syncitia boundaries. (e) Quantification of the number of syncitia-like structures/103 cells in a typical experiment in each induced cell population at 0, 3, and 5 d. Numbers indicate fold change relative to day 0. (f–i) TUNEL positivity in induced 96QN cells. (f) Fluorescent view; (g) bright-field view reveling DAB-stained TUNEL-positive profiles; (h) overlay of both views; and (i) corresponding phase–contrast view. Arrows in panel g indicate TUNEL profiles that were scored as positive for quantification. Overlay and phase–contrast views reveal that the TUNEL positive cells are often rounded up and resting just above the monolayer of surviving cells below. (j) Quantification of the number of TUNEL-positive profiles in a typical experiment in each induced cell population at 0, 3, and 5 d. Numbers indicate fold change relative to day 0. The slight increase in apoptotic cells at 3 and 5 d for 13QN and 96Q cells is probably the consequence of high cell density near the end of the culture period. Bar: (a–d) 50 μm; (f–i) 250 μm.
Changes in cellular/nuclear morphology paralleled a low but detectable increase in cell death within the stimulated polyQ–GFP populations. Fig. 2f–i, shows TUNEL staining of a representative 96QN population stimulated for 5 d with tebufenozide. A comparison of 13QN-, 96Q-, and 96QN-stimulated cells reveals that only nuclear-localized long polyQ tract-producing cells displayed increased TUNEL positivity during the 5-d time course of the experiment (Fig. 2 j). At 3 d after induction, 97QN cells displayed over twofold more TUNEL-positive profiles than other cell types and over fivefold more TUNEL-positive bodies by day 5, representing ∼2% of the total cell population.
Purification of IAs from Day 5 Induced 96QN Cells
IAs were purified from induced polyQ cells at the 5-d time point (corresponding to the maximum change in cellular morphology and cytotoxicity) using a variation of IA isolation procedures described by Scherzinger et al. 1999.
Western blot analyses comparing either whole cell extracts or purified aggregates of different cell populations are shown in Fig. 3, a–n. Fig. 3 a shows a blot of 5-d induced whole cell extract from 13QN cells (lane 1), uninduced 96QN cells (lane 2), 5-d induced 96QN cells (lane 3), and purified IAs (lane 4) processed for detection of GFP. In Fig. 3 a, monomeric 13QN is observed migrating near an apparent molecular mass of 35 kD and monomeric 96QN near 52–53 kD as in Fig. 1 m. IAs are observed throughout the stacking gel and accumulate at the extreme top of the resolving gel. Anti-GFP staining localizes the purified IAs (lane 4) almost exclusively at the top of the resolving gel with only a weak band corresponding to monomeric 96QN. The low level of monomeric 96QN protein indicates that, although the purified IAs are denatured, they are not resolubilized into free monomers to a significant degree. The possibility that 96QN polymers may be covalently linked together by the action of transglutaminase is further discussed below.
To begin specific identification and characterization of IA components, an identical blot to Fig. 3 a was probed with antiubiquitin primary antibody to examine ubiquitin sequestration within the purified IAs. Western blot for ubiquitin (Fig. 3 b) revealed a positive signal in all four lanes and substantially increased positivity in induced 96QN whole cell extracts, with most immunoreactive species extending from ∼50–60 kD up and into the stacking gel (Fig. 3 b, lane 3). Ubiquitin staining in the purified IA lane (Fig. 3 b, lane 4) reveals a significant intensification of the pattern observed in lane 3, indicating a dramatic concentration of ubiquitin in the purified IA sample. The smear of signal from 50 kD up likely represents ligated ubiquitin polymers and other ubiquitinated species, including 96QN polymers and other sequestered proteins.
After published reports of chaperone protein recruitment described in the Introduction, we also examined sequestration of the 70-kD heat shock protein HSP70 (Wu et al. 1985). Western blot analysis of HSP70 also revealed the presence of a significant amount of 70-kD HSP70 sequestered within the purified IAs from the 5-d 96QN cells (Fig. 3 c, lane 4). Unlike ubiquitin, which forms polymers and covalent linkages to target proteins (Ciechanover 1994), HSP70 is liberated from the IA in monomeric form and displays an identical migration pattern to the three control lanes. No HSP70 is detected at higher molecular weights or within the stacking gel, suggesting that essentially all of the sequestered HSP70 protein is released during the boiling/SDS/β-mercaptoethanol treatment of the IAs for loading.
Another class of protein either predicted or demonstrated to be sequestered within IAs is proteins with nonpathological length polyQ tracts, such as native Htt, TBP, or myocyte-specific enhancer factor (MEF-2a) (Suzuki et al. 1996). MEF-2a was detected at approximately equivalent levels in each of the three control lanes, but could not be detected in the purified IA lane even after prolonged exposure (Fig. 3 d). Htt staining revealed a clear doublet of high molecular weight bands of the correct size in the control lanes but not in lane 4 (Fig. 3 e). Fig. 3 f reveals the presence of significant sequestered TBP in the purified IAs. No significant signal is detected at higher molecular weights, and all signal is concentrated within the ∼38-kD band, suggesting that TBP is not cross-linked to the 96QN polymer at the 5-d time of harvest.
A second class of protein potentially sequestered within IAs are SH2/SH3 proteins capable of binding extended polyP tracts (Sittler et al. 1998). We examined two polyP binding proteins determined in pilot experiments to be detected in HEK293 cells—Grb-2 (Lowenstein et al. 1992) and a related protein, Nck (Lehmann et al. 1990). As shown in Fig. 3g and Fig. h, although both proteins were detected at relatively high and equivalent levels in control lanes, neither protein was detected in purified aggregates.
Members of the caspase family of cysteine proteases were also examined because they are integral to the progression of apoptosis (Thornberry and Lazebnik 1998) and have been implicated in polyQ-mediated cell death via possible recruitment to IAs (Sanchez et al. 1999). We examined caspase-9 (Duan et al. 1996), caspase-8 (Muzio et al. 1996), and caspase-3 (Fernandes-Alnemri et al. 1994) (Fig. 3, i–k), which in pilot studies of HEK293 cells, demonstrated detectable levels in whole cell extracts. Of the three, only caspase-3 (Fig. 3 k) was clearly detected in lane 4. There is also intensification of a high molecular weight band up at the border of the resolving gel, which may indicate that some level of caspase-3 is still trapped within the 96QN polymer. Caspase-9 identification was complicated by several nonspecific bands and an apparent low level of expression and, though not observed to be sequestered within IAs in these experiments, may be observed in ongoing experiments in other cell types.
The final group of proteins examined for sequestration were the tumor suppressor protein p53 (Lane and Crawford 1979) and the p53-regulating protein mdm-2 (Momand et al. 1992). Western blot analysis revealed that p53 was sequestered at high relative levels (Fig. 3 l). In addition, a tight triplet of additional p53-positive bands was observed within the IAs at ∼48–50 kD that was not observed in the control lanes. Mdm-2 was also observed in purified IAs at high levels (Fig. 3m and Fig. n). Mdm-2 modulates p53 by binding and promoting the ubiquitination and subsequent proteolysis of p53 (Fuchs et al. 1998). An ∼58–60-kD form of mdm-2 described in previous reports (Pochampally et al. 1998) was the predominant mdm-2 species observed, although a high molecular weight 120-kD band was also detected within the control lanes (Fig. 3 m). Only the 58–60-kD band was found to sequester within the IAs and, like p53, displayed a second band of immunoreactivity at a slightly lower molecular mass (56/57 kD), only within the concentrated IAs in lane 4 (Fig. 3 m). Use of an mdm-2 antibody located further toward the COOH terminus in the mdm-2 protein revealed only the 58–60-kD band and did not detect the 56/57-kD band or the high molecular weight band observed with the first antibody (Fig. 3 n). Immunodetection of the 56/57-, 58–60-, and 120-kD bands was blocked by use of manufacturer-supplied blocking peptides as described in Materials and Methods (data not shown).
MALDI Analysis of Predominant Protein Species
Fig. 4 a shows Coomassie-stained protein species in a concentrated IA sample on a 1-mm 10% SDS–polyacrylamide gel. Four bands at ∼68, 54, 50, and 41 kD stood out as much more prominent than the ∼30 other bands of lesser intensity within the sharply resolved region of the gel from 100 kD to the dye front. These four bands were excised from the gel and subjected to MALDI analysis to provide tentative identification of each protein species.
Figure 4.

Protein and Western blot analyses of sequestered protein in concentrated IAs. (a) Coomassie-stained protein gel of isolated IAs revealing multiple bands from ∼10 kD (dye) up to the boundary of the stacking/resolving gel (stack). The four predominant bands used for MALDI analysis are labeled at the right. Lane organization is as described in the legend to Fig. 3. (b) Protein blot analysis of 68-kD neurofilament light polypeptide; (c) protein blot analysis of actin; (d) protein blot analysis with monoclonal antibody 414–detecting putative NPCP proteins Nup62, Nup153, Nup214, and Nup358 as labeled; (e) protein blot analysis of lamin revealing no detectable sequestration in purified IAs.
12 peptide masses were used to query the peptide database for the ∼68–70-kD protein band. 7 of the top 10 matches with 4/12 to 7/12 matched masses covering 10–19% of the peptide were from the 68-kD light neurofilament protein (NFL) from a variety of species including human (Julien et al. 1987). Subsequent protein blot analysis shown in Fig. 4 b reveals not only that NFL is expressed at readily detectable levels in the HEK293 cells but that it is also recruited to IAs at high levels within the purified IA fraction.
Submission of 10 peptide masses for the 40–41-kD band at lower mass tolerances returned no strong matches; however, resubmission of the same data set increasing the mass tolerance returned all 10 top matches as various species of the microfilament protein actin (Gunning et al. 1983) with molecular mass ranges from 37.8 to 42.1 kD—all in close agreement with the molecular weight of the excised band. Matched masses ranged from 6/10 to 8/10 and covered 28–36% of the protein. Subsequent protein blot analysis using an antiactin monoclonal antibody (Lessard 1988), shown in Fig. 4 c, confirms detection in both control lanes and high levels in purified IA preparations.
Identification of the 54-kD band with 10 submitted weights returned no strong matches at low weight tolerance allowances; however, at higher error tolerance, two proteins of the target molecular weight were returned with four to five matches. The first was the DEAD box protein Dbp-5 (5/10 matches) recently described by Schmitt et al. 1999, with a predicted molecular mass of 53.9 kD, and the second was RanBP3-a (4/10 matches), with a predicted molecular mass of 53.2 kD (Mueller et al. 1998). RanBP3, a splice variant of RanBP3-a, was also returned but has a slightly higher molecular mass of 59.6 kD. Human Dbp-5 is an RNA-dependent ATPase and localizes within the cytoplasm and at the nuclear rim, interacting with the cytoplasmic fibrils of the nuclear pore complex (NPC) via the NH2-terminal region of the nucleoporin CAN/Nup159p. Dbp-5 is also a DEAD box protein, a family of RNA-binding proteins involved in a variety of mRNA processing and import/export functions (Schmitt et al. 1999). RanBP3-a is one member of a family of Ran-GTPase binding proteins (Dasso and Pu 1998) involved in the regulation of nuclear transport that are also characteristically associated with the cytoplasmic side of the NPC. Although we could not directly examine Dbp-5 or RanBP3-a protein expression by immunochemical methods, we could readily examine recruitment of known associated NPC components. Using mAb414 (Davis and Blobel 1986), a well-characterized monoclonal antibody against FXFG nucleoporins collectively referred to in this report as NPC proteins (NPCPs), we performed protein blot analysis on the control extracts and concentrated IA preparations and found a high level of association of NPCP components with the purified IA fraction. The 62-kD size of the most intense band (Fig. 4 d) suggests that this band represents Nup62, a protein of the central gated channel of the nuclear pore involved in the trafficking of protein and mRNA to and from the nucleus. At higher molecular weights, three bands that correspond to the approximate molecular weights of three other NPCPs known to cross-react with mAb414—Nup153, Nup214/CAN, and Nup358/RanBP2 (Ryan and Wente 2000)—are also tentatively identified. The recruitment of this multiplicity of NPC proteins suggests that the recruitment of both RanBP3-a and Dbp-5 may likely stem from broad interaction with most, if not all, of the NPC and associated components. To test whether or not the identification of nucleoporins associated with the purified IAs represented IA association with the nuclear envelope as a structure (i.e., via fortuitous copurification of nuclear membranes), we probed an identical blot with the ubiquitous nuclear structural protein lamin B. Fig. 4 e shows that, although lamins are readily detected in the whole cell extracts in Fig. 4, lanes 1–3, no lamin protein is detectable in the purified IA sample either comigrating with native lamins or in the insoluble polyQ protein higher on the gel.
The closest return for the remaining band at 50 kD approximating this molecular weight is Tat-binding protein-1 (TatBP-1; 49.1 kD) from human and mouse (8/23 matches, 21% of the peptide). TatBP-1 was characterized early on for its interaction with the HIV Tat protein (Nelbock et al. 1990) and has since been recognized as a component of the 19S regulatory subunit of the 26S proteasome (Dubiel et al. 1993). We were unable to confirm the identity of the 50-kD band as TatBP-1 by immunochemical methods using available antibodies, which may be due to technical limitations or may indicate that the band is not TatBP-1.
Semiquantitative Analysis of Protein Sequestration into polyQ IAs
Our attempts to accurately quantify the purified IA samples by standard methods were unsuccessful due to the lack of solubility of the sample. However, by titrating different sample volumes, taking multiple exposures of the Western chemiluminescent assay, and performing scanning densitometry on the resulting GFP positivity, we determined that 5 μl of purified IAs contained 16.3-fold more polymerized insoluble 96QN–GFP immunoreactivity than 7.5 μg of 5-d 96QN cell whole extract. Using this value and semiquantitative protein blot analysis techniques described in Materials and Methods, we determined the relative levels of individual protein species for each cell type and treatment described in Fig. 3 and Fig. 4.
Comparison of individual protein species in the three control lanes revealed that only ubiquitin displayed a difference greater than twofold between lanes. Ubiquitin was increased ∼20-fold in 5-d 96QN whole cell extracts (Fig. 3 b, lane 3), compared with 13QN and uninduced 96QN cells (Fig. 3 b, lanes 1 and 2). This increase likely represents an accumulation of ubiquitin protein on IAs as opposed to a dramatic increase in ubiquitin expression.
Relative sequestration of proteins to IAs is summarized in Table . Of the proteins tested (and excepting 96QN–GFP itself and ubiquitin), p53 and Nup-p62 are the most sequestered as a function of total cellular protein, with a level equivalent to or exceeding the entire intracellular pool sequestered in IAs. Other protein species are recruited at levels of 13–58% of cellular stores with caspase-3 sequestered at the lowest level, <1% of cellular stores. For all of these proteins, but in particular for those that appear sequestered at lower levels, it must be kept in mind that, if there is significant heterogeneity in IA sequestration, then the relative percentage of recruitment of an individual protein species may be dramatically increased in a subpopulation of IA-containing cells.
Table 2.
Protein Sequestration to IAs
| Class | Protein | Seq to IA | Polymer | Relative seq |
|---|---|---|---|---|
| PolyQ | 96QN-GFP | + | + | ≈2,000% |
| PolyQ | TBP | + | − | 13.4% |
| PolyQ | MEF-2a | − | NA | NA |
| PolyQ | Htt | − | NA | NA |
| Proteolysis | Ubiquitin | + | + | 282.7% |
| Proteolysis | HSP70 | + | − | 14.9% |
| PolyP | Grb-2 | − | NA | NA |
| PolyP | Nck | − | NA | NA |
| Protease | Caspase-3 | + | −/+ | 0.4% |
| Protease | Caspase-8 | − | NA | NA |
| Protease | Caspase-9 | − | NA | NA |
| Cell cycle | p53:53 kD | + | − | 255.7% |
| Cell cycle | p53:50 kD | + | − | 191.4% |
| Cell cycle | mdm-2 Ab 1:60 kD | + | − | 32.2% |
| Cell cycle | mdm-2 Ab 1:57 kD | + | − | 24.5% |
| Cell cycle | mdm-2 Ab 2:60 kD | + | − | 31.9% |
| Structural | NFL | + | − | 57.7% |
| Structural | Actin | + | − | 26.6% |
| Structural nuclear | Lamin B | − | NA | NA |
| Structural nuclear | NPCP-Nup62 | + | − | 124% |
| Structural nuclear | NPCP-NupHMW | + | − | NQ |
Class, assigned group or class of protein. Protein, protein common name and molecular mass of individual species (where applicable). Seq to IA, sequestration of protein to IAs (+ indicates strong evidence of sequestration; − indicates no measurable sequestration). Polymer, indicates whether protein blot analysis supports the presence of higher molecular weight species consistent with isopeptide bond formation for individual proteins (+ indicates strong evidence of polymerization; −/+ indicates potential polymerization; − indicates no evidence of polymerization; NA indicates protein is not sequestered. Relative Seq, relative amount of sequestered protein to total protein for each individual protein species. Where more than one protein band is observed, the molecular weights indicate the band identity (NQ indicates that the value was not quantified due to lack of corresponding signal detected in the control samples).
Immunohistochemical Analysis of Sequestration into IAs In Vitro and In Vivo
Ubiquitin, HSP70, TBP, actin, and NPCPs could be localized to IAs in 5-d 96QN cells or human postmortem tissue from HD individuals. Ubiquitin (Fig. 5 a) and HSP70 (Fig. 5 b) were detected in vitro colocalizing with IAs of 96QN-expressing cells, whereas HSP70 visualization required the use of an alternate antibody from the antibody used for protein blot analysis. Although not directly detected within 96QN cell IAs in situ, an intensification of TBP positivity was observed in induced 96QN cells relative to uninduced cells (Fig. 5c and Fig. d). This intensification was determined to localize with cells in later stages of apoptotic cell death (Suhr, S., manuscript in preparation). Parallel immunohistochemical localization experiments were performed with postmortem striatal tissue samples from HD individuals (n = 3) and non-HD age-matched controls (n = 3). Striatal tissue samples were examined for altered intracellular distribution of each of the putative sequestered proteins. Punctate ubiquitin-positive profiles were seldom observed throughout the striatum of all three control cases (Fig. 5 e) but were frequently observed in HD striatal samples (Fig. 5 f). Although HSP70 and caspase-3 displayed a weak intensity of immunopositive staining in all tested tissues, no obvious differences in the distribution pattern of staining were observed between HD or control samples (data not shown). Immunohistochemistry for TBP, on the other hand, revealed the presence of scattered darkly positive profiles exclusively within the striatal samples of the three HD patients that were not observed in the age-matched controls or after preabsorption with antigen (Fig. 5, g–i). Higher magnification of these inclusions revealed two distinct types of structure: diffusely stained spheres of various size and labeling intensity (Fig. 5j and Fig. k) and intensely stained inclusion-like objects with a halo of lighter immunostaining (Fig. 5l and Fig. m). These positive profiles might correspond to a combination of the intensification of antigen observed in the in vitro studies and localization of TBP to polyQ IAs in a subset of these cells.
Figure 5.
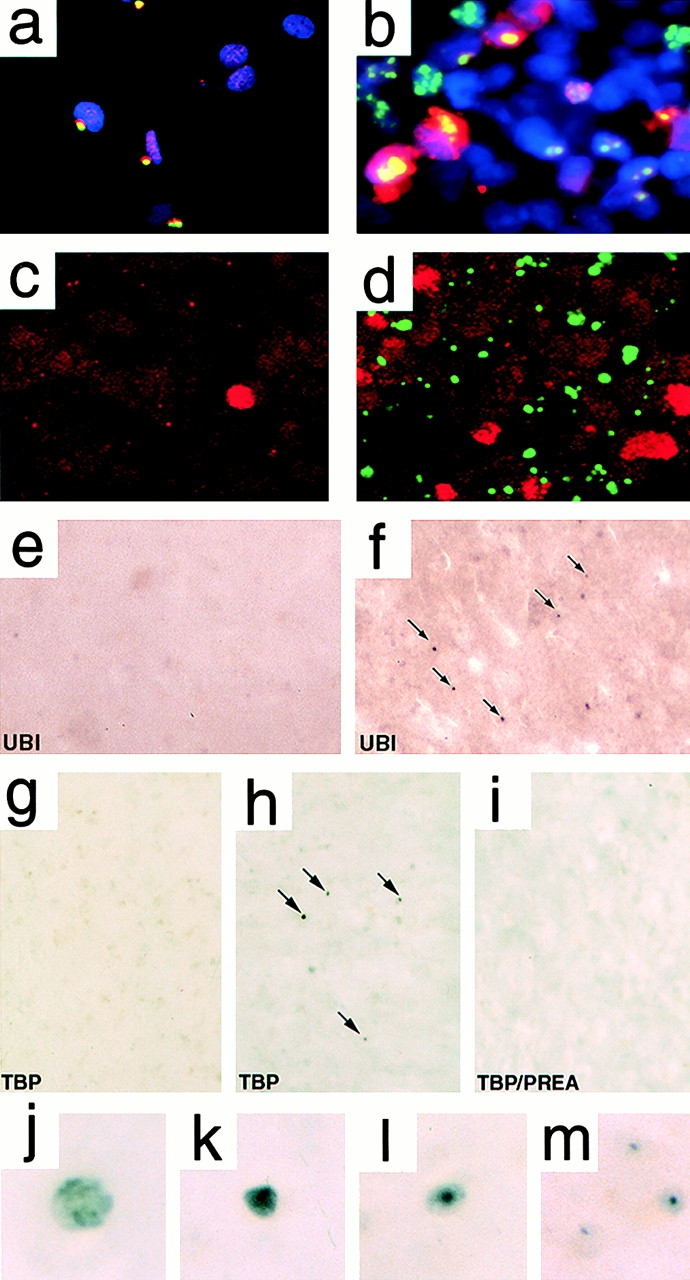
Immunohistochemical analyses of sequestered proteins in 96QN cells and human HD striatum. (a) Colocalization of ubiquitin (red) with IAs (green) resulting in uniform yellow color of all IAs in transfected 96Q cells. Nuclei are in blue. Arrows indicate colocalizing IAs. (b) Colocalization of HSP70 with IAs. Arrows indicate some colocalizing IAs. (c) Uninduced 96QN cells with infrequent intense TBP positivity (red) compared with 5-d induced 96QN cells (d) with frequent intensely TBP-positive cells. 96QN–GFP IAs are green. (e) Ubiquitin-positive profiles in the non-HD control striatum and in the HD striatum (f). Arrows indicate punctate ubiquitin-positive profiles. (g–i) Localization of TBP within punctate profiles in control striatum (g), HD striatum (h), and HD striatum after preabsorbtion with peptide (i). Arrows indicate TBP-positive profiles in HD tissue. (j–m) High magnification of TBP-positive profiles in HD striatum revealing immunoreactivity in a diffuse sphere (j), a dark intense sphere (k), or punctate profiles (l and m) with lighter surrounding immunopositivity.
Although NFL colocalization to polyQ IAs has been described in an earlier report (Nagai et al. 1999), we could see no indication of IA colocalization or altered distribution of either NFL or actin in either HD tissues or induced 96QN cells by immunocytochemistry (Fig. 6, a and b). We were, however, able to colocalize actin with IAs in induced 96QN cells using TRITC–phalloidin staining (Fig. 6, c–e). This discrepancy supports the proposition that some IA-sequestered proteins are not amenable to detection by immunological reagents.
Figure 6.
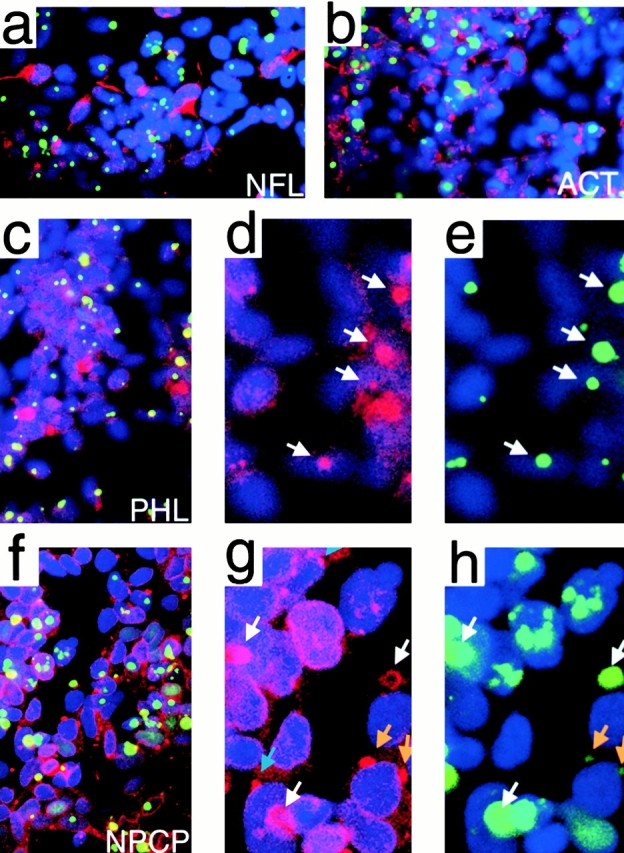
Immunohistochemical fluorescent confocal microscopy of protein species predicted by MALDI analysis to associate with IAs from 96QN cells. Green fluorescence indicates the 96QN–GFP proteins and IAs, blue indicates the DNA stain DAPI, and red CY-3 fluorophore is used for each individual protein tested for localization. Yellow color indicates colocalization of IAs with the test protein antibody. (a) Immunostaining for NFL on 5-d induced 96QN cells. (b) Immunostaining for actin on 5-d induced 96QN cells. (c) Staining of 96QN cells with TRITC–phalloidin. (d and e) Magnification of a region from panel c with separation of the color channels to highlight colocalization by comparison of the red actin channel in panel d with matching signal in of 96QN–GFP green fluorescence in panel e. Arrows indicate some of the colocalizing IAs. (f) Staining of 96QN cells with monoclonal antibody 414 against NPCPs. Bright yellow signal indicates colocalization of NPCP signal with a subset of IAs. (g and h) Individual color channels to highlight colocalization of NPCPs with IAs. White arrows indicate colocalization of spherical monoclonal antibody 414 staining with a subset of IAs, blue arrows indicate areas of NPCP density on the nuclear envelope, and orange arrows indicate colocalization of 96QN–GFP with these perinuclear densities.
NPCP immunohistochemistry revealed several distinctive characteristics of IA colocalization not observed with other antigens. First, it is apparent from the examples shown in Fig. 6f–h, that NPCP only colocalizes strongly with a subset of IAs. Using a multiplicity of fluorescent labels, two distinct populations of IAs are observed in Fig. 6 f: green fluorescent IAs, with little or no detectable contribution of NPCP to the IA, and yellow IAs, indicating a combination of the GFP fluorescence and CY3 red-labeled NPCP. Secondly, in Fig. 6g and Fig. h, a magnification of this field indicates that some IAs are labeled around the edges by NPCP antibody resulting in a halo pattern sometimes appearing associated with nuclei and, in other cases, appearing separate from DAPI-stained nuclei. Third, in addition to these halo structures, there are also examples of smaller IAs in which NPCP positivity colocalizes with the extent of the IA (Fig. 6g and Fig. h, orange arrows). These fainter IAs may not yet be true IAs but, instead, areas of polyQ–GFP association with high density NPCP islands on the nuclear lamina. These areas of NPCP density at the nuclear periphery are not dependent on the presence of polyQ–GFP accumulation, since numerous NPCP-dense regions are observed with no apparent polyQ–GFP colocalization (Fig. 6 g, blue arrows).
Discussion
The observation that cell death increases shortly after the widespread formation of IAs does not necessarily imply a causal link between IAs and cell death; however, it also does not rule out the possibility (discussed in Wanker 2000). Many of the sequestered proteins are presumably recruited through direct interaction with the polyQ-harboring protein, and this interaction may initiate with monomeric soluble polyQ proteins long before IA formation itself takes place. The association of NPCP islands with polyQ–GFP protein or weak IAs may represent this type of early association. If IAs are, in fact, protective, a concept that has been proposed in recent studies (i.e., Saudou et al. 1998), knowledge of IA composition may lead to strategies for accelerating specific sequestration of individual proteins through pharmacological or other means.
It is clear from these experiments that many different proteins, in addition to the 96Q– and 96QN–GFP fusion peptide, are sequestered within IAs. In addition, after denaturation of IAs, and with the exception of ubiquitin and the 96Q peptides themselves, the proteins that are sequestered present at least one band comigrating with proteins from whole cell extracts, suggesting that at least some of the sequestered proteins are likely to be full length and intact. It can be further inferred that these proteins have not been targeted by ubiquitin, have not been cross-linked to 96QN molecules (i.e., via transglutaminase; Cooper et al. 1997; Kahlem et al. 1998), or have not undergone irreversible polymerization, since higher molecular weight immunoreactive species are not observed at levels differing from control cell extracts.
MALDI Identification of NFL and Actin
Identification of NFL as a component of the IAs was unexpected, given the nonneuronal nature of the HEK293 cell; however, it is tempting to speculate on the existence of a pathological polyQ–NFL interaction that might explain limitation of polyQ-mediated disease manifestation to neural tissues. The connection between polyQ, NFL, and toxicity in HD remains to be established, but it is unlikely that polyQ interaction with NFL is the sole mechanism of cytotoxicity, since polyQ expression is toxic to essentially every cultured cell type tested, and it is unlikely that all cultured cells express NFL.
Actin, on the other hand is much more attractive as a universal pathogenic component of polyQ-mediated toxicity since it is expressed in every cell type and across phyla. In addition, we have observed aberrant staining of 96QN–GFP-expressing cells with an antimyosin monoclonal antibody, suggesting possible effects on the actin/myosin cytoskeleton (Suhr, S., manuscript in preparation). Actin and myosin are also involved in mitosis and cytokinesis, raising the possibility that the low-level syncitia formation we observed arises from a cytokinesis defect due to changes in actin dynamics. We have not observed a clear difference in nonsequestered actin expression, distribution between control and 96QN-expressing cells, or brain sections from human HD tissue, however, precluding us from definitively identifying actin as an important component of polyQ-mediated cytotoxicity.
Sequestration of p53 and mdm-2
The recruitment of three of the identified proteins (p53, mdm-2, and NPCP/nucleoporins) may have particular relevance to potential mechanisms of polyQ-mediated toxicity. With regard to p53 and mdm-2, it is unknown at this time whether their sequestration or interaction with polyQ proteins is directly mediated through binding with the polyQ protein, or if they represent proteins that are brought into IAs through association with other highly recruited proteins. Four highly recruited proteins found or confirmed in this study to be sequestered within IAs—ubiquitin, HSP70, TBP, and mdm-2—are also known to bind p53 (Hughes et al. 1997). Recently, Steffan et al. 2000 showed that p53 coaggregates with the NH2-terminal region of Htt exon I, a region encompassing both the polyQ and polyP tracts, in cultured cells and cell extracts. In addition to cytotoxicity due to polyQ interaction with intact p53, the indication of lower molecular weight p53 immunoreactive species (p48–p50) observed at high levels within the purified IA lane suggest another pathological mechanism. There are several known cleaved forms of p53. A 50-kD form results from the loss of the NH2-terminal 23 amino acid and is postulated to have intact transactivation and apoptosis-inducing determinants, but to have lost the domain responsible for protein–protein interaction with mdm-2 (Okorokov et al. 1997). It is tempting to speculate that the accumulation of the mdm-2–independent p50 accounts for increased p53 activity.
A second potential mediator of net increased p53 activity could arise from the sequestered mdm-2 protein species. Sequestered 58–60-kD mdm-2 and a lower molecular mass 55–56-kD form are observed within IAs. One 58–60-kD isoform of mdm-2 has been found to be formed by caspase cleavage in nonapoptotic cells (Olson et al. 1993). Caspase cleavage to generate the 58–60-kD mdm-2 removes the COOH-terminal domain of mdm-2 involved in mediating ubiquitination and proteolysis of p53, but not p53 binding, and predicts that 58–60-kD mdm-2 could function as a dominant negative factor p53 stabilizer through competition with full-length mdm-2 (Olson et al. 1993). The lower molecular weight mdm-2 band observed only in the IAs at 56/57 kD may represent a novel mdm-2 cleavage product produced by combinations of factors specific to the environment within the IAs. The accumulation of a putative p53 50-kD form and cleaved mdm-2 within the IAs both provide potential means of altering p53 activity.
Interaction of Polyglutamine with NPCPs
The possibility of polyQ interaction with proteins of the nuclear pore and matrix could answer many questions with regard to localization and toxicity of long-tract polyQ both in vivo and in cultured cell models. Three lines of evidence link the nucleus to polyQ-mediated toxicity: (a) the overwhelming majority of reports examining the mechanism of polyQ-mediated toxicity in HD, either through study of human brain tissue, cultured cells, and animal models, agree that the onset of symptoms and cytotoxicity is concurrent with cleavage of the polyQ tract from the large cytoplasmic Htt molecule and subsequent translocation of this NH2-terminal fragment into the nucleus; (b) addition of an NLS accelerates polyQ-mediated toxicity in cultured cell models; and (c) even though lacking evident NLSs within the Htt NH2-terminal fragment (or any other region of Htt), the NH2-terminal polyQ fragment of Htt (or synthetic polyQ reporters) tends to accumulate within or surrounding the nucleus, often producing an inpocket when located adjacent to the nuclear envelope. Indentation of the nuclear envelope has also been found at high levels in ultrastructural studies of HD brain (Roos et al. 1985). Toxic pathways that could result from changes in the NPC, matrix, or envelope, include alteration or inhibition of mRNA and protein trafficking between the nucleus and cytoplasm.
It is our hope that deciphering the contents of IAs in a cellular model of HD will provide new insight into the population of proteins that interact with polyQ-bearing proteins, irrespective of whether sequestration of these proteins within the IA directly contributes to cell death. Much of the speculation about potential mechanisms of polyQ-mediated cell death in this report has focused on p53 and NPCP because they copurify with IAs at higher relative levels than most of the other proteins tested; however, it is equally possible that proteins found at lower levels interact preferentially with soluble or monomeric polyQ protein and are consequently underrepresented in IAs. For this reason, even the most weakly recruited factors should not be dismissed as insignificant to polyQ-mediated pathology until thoroughly studied.
Acknowledgments
The authors wish to thank Scott Zeitlin (Columbia University, New York, NY) for polyQ cDNAs and useful advice. We also wish to thank Ethan Signer, Brian Kaspar and Andrew Willhoite in our laboratory, Harry Higgs and Tom Pollard, and Leslie Thompson for helpful discussions. Thanks also to James Lessard (Children's Hospital Medical Center, Cincinnati, OH) for the actin monoclonal antibody. Thanks also to Mary Lynn Gage for help with the manuscript.
Human tissues were provided by the Harvard Brain Tissue Resource Center (Boston, MA), which is supported, in part, by Public Health Services grant number MH/NS 31862. This work was supported by grants from the Hereditary Disease Foundation and the National Institute of Aging. Purchase of the MALDI instrument at University of California at Los Angeles, Los Angeles, CA, was possible through partial support by National Cancer Institute (National Institutes of Health) Cancer Center Support grant CA 16042-20 to the Jonsson Comprehensive Cancer Center.
Footnotes
Steven T. Suhr and Marie-Claude Senut contributed equally to this work.
Abbreviations used in this paper: CMV, cytomegalovirus; GFP, green fluorescent protein; HD, Huntington's disease; Htt, Huntingtin; IA, intracellular aggregate; MALDI, matrix-assisted laser desorption ionization time of flight mass spectrometry; MEF-2a, myocyte-specific enhancer factor; NDST, normal donkey serum and Triton X-100; NFL, light neurofilament protein; NLS, nuclear localization signal; NPC, nuclear pore complex; NPCP, NPC protein; polyP, polyproline; polyQ, polyglutamine; TatBP-1, Tat-binding protein-1; TBP, TATA-binding protein; TUNEL, terminal deoxynucleotidyl transferase–mediated dUTP–biotin end labeling.
References
- Bienvenut W.V., Sanchez J.-C., Karmine A., Rouge V., Rose K., Binz P.-A., Hochstrasser D.F. Toward a clinical molecular scanner for proteome researchparallel protein chemical processing before and during Western blot. Anal. Chem. 1999;71:4800–4807. doi: 10.1021/ac990448m. [DOI] [PubMed] [Google Scholar]
- Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell. 1994;79:13–21. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- Cooper A.J., Sheu K.F., Burke J.R., Onodera O., Strittmatter W.J., Roses A.D., Blass J.P. Polyglutamine domains are substrates of tissue transglutaminasedoes transglutaminase play a role in expanded CAG/poly-Q neurodegenerative diseases? J. Neurochem. 1997;69:431–434. doi: 10.1046/j.1471-4159.1997.69010431.x. [DOI] [PubMed] [Google Scholar]
- Dasso M., Pu R.T. Nuclear transportRun by ran? Am. J. Hum. Genet. 1998;63:311–316. doi: 10.1086/301990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis L.I., Blobel G. Identification and characterization of a nuclear pore complex protein. Cell. 1986;45:699–709. doi: 10.1016/0092-8674(86)90784-1. [DOI] [PubMed] [Google Scholar]
- Duan H., Orth K., Chinnaiyan A.M., Poirier G.G., Froelich C.J., He W.W., Dixit V.M. ICE-LAP6, a novel member of the ICE/Ced-3 family, is activated by the cytotoxic T-cell granzume B. J. Biol. Chem. 1996;271:16720–16724. doi: 10.1074/jbc.271.28.16720. [DOI] [PubMed] [Google Scholar]
- Dubiel W., Ferrell K., Rechsteiner M. Peptide sequencing identifies MSS1, a modulator of HIV Tat-mediated transactivation, as subunit 7 of the 26S protease. FEBS Lett. 1993;323:276–278. doi: 10.1016/0014-5793(93)81356-5. [DOI] [PubMed] [Google Scholar]
- Fernandes-Alnemri T., Litwack G., Alnemri E.S. CPP32, a novel human apoptotic protien with homology to C. elegans cell death protein Ced-3 and mammalian interleukin-1 β-converting enzyme. J. Biol. Chem. 1994;269:30761–30764. [PubMed] [Google Scholar]
- Fuchs S.Y., Adler Y., Buschmann T., Wu X., Ronai Z. Mdm2 association with p53 targets its ubiquitination. Oncogene. 1998;17:2543–2547. doi: 10.1038/sj.onc.1202200. [DOI] [PubMed] [Google Scholar]
- Holmes S.E., O'Hearn E.E., McInnis M.G., Gorelick-Feldman D.A., Kleiderlein J.J., Callahan C., Kwak N.G., Ingersoll-Ashworth R.G., Sherr M., Sumner A.J. Expansion of a novel CAG trinucleotide repeat in the 5′ region of PPP2R2B is associated with SCA12. Nat. Genet. 1999;23:391–392. doi: 10.1038/70493. [DOI] [PubMed] [Google Scholar]
- Gunning P., Ponte P., Okayama H., Engel J., Blau H., Kedes L. Isolation and characterization of full-length cDNA clones for human α-, β-, and γ-actin mRNAsskeletal but not cytoplasmic actins have an amino-terminal cysteine that is subsequently removed. Mol. Cell. Biol. 1983;3:787–795. doi: 10.1128/mcb.3.5.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.C., Faber P.W., Persichetti F., Mittal V., Vonsattel J.-P., McDonald M.E., Gusella J.F. Amyloid formation by mutant huntingtinthreshold, progressivity and recruitment of normal polyglutamine proteins. Somat. Cell. Mol. Genet. 1998;24:217–223. doi: 10.1023/b:scam.0000007124.19463.e5. [DOI] [PubMed] [Google Scholar]
- Hughes P.E., Alexi T., Schreiber S.S. A role for the tumour suppressor gene p53 in regulating neuronal apoptosis. Neuroreport. 1997;8:v–xii. [PubMed] [Google Scholar]
- Huntington's Disease Collaborative Research Group A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Julien J.P., Grosveld F., Yazdanbaksh K., Flavell D., Meijer D., Mushynski W. The structure of the human neurofilament gene (NF-L)a unique exon-intron organization in the intermediate filament gene family. Biochim. Biophys. Acta. 1987;909:10–20. doi: 10.1016/0167-4781(87)90041-8. [DOI] [PubMed] [Google Scholar]
- Kahlem P., Green H., Djian P. Transglutaminase action imitates Huntington's diseaseselective polymerization of Huntingtin containing expanded glutamine. Mol. Cell. 1998;1:595–601. doi: 10.1016/s1097-2765(00)80059-3. [DOI] [PubMed] [Google Scholar]
- Kao C.C., Lieberman P.M., Schmidt M.C., Zhou Q., Pei R., Berk A.J. Cloning of a transcriptionally active human TATA binding factor. Science. 1990;248:1646–1650. doi: 10.1126/science.2194289. [DOI] [PubMed] [Google Scholar]
- Kazantzev A., Preisinger E., Dranovski A., Goldgaber D., Housman D. Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc. Natl. Acad. Sci. USA. 1999;96:11404–11409. doi: 10.1073/pnas.96.20.11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane D.P., Crawford L.V. T antigen is bound to a host protein in SV40-transformed cells. Nature. 1979;278:261–263. doi: 10.1038/278261a0. [DOI] [PubMed] [Google Scholar]
- Lehmann J.M., Riethmuller G., Johnson J.P. Nck, a melanoma cDNA encoding a cytoplasmic protein consisting of the src homology units SH2 and SH3. Nucleic Acids Res. 1990;18:1048–1054. doi: 10.1093/nar/18.4.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard J.L. Two monoclonal antibodies to actinone muscle selective and one generally reactive. Cell Motil. Cytoskeleton. 1988;10:349–362. doi: 10.1002/cm.970100302. [DOI] [PubMed] [Google Scholar]
- Lowenstein E.J., Daly R.J., Batzer A.G., Li W., Margolis B., Lammers R., Ullrich A., Skolnik E.Y., Bar-Sagi D., Schlessinger J. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell. 1992;70:431–442. doi: 10.1016/0092-8674(92)90167-b. [DOI] [PubMed] [Google Scholar]
- Momand J., Zambetti G.P., Olson D.C., George D., Levine A.J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–1245. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- Mueller L., Cordes V.C., Bischoff F.R., Ponstingl H. Humna RanBP3, a group of nuclear RanGTP binding proteins. FEBS Lett. 1998;427:330–336. doi: 10.1016/s0014-5793(98)00459-1. [DOI] [PubMed] [Google Scholar]
- Muzio M., Chinnaiyan A.M., Kischkel F.C., O'Rourke K., Shevchenko A., Ni J., Scaffidi C., Bretz J.D., Zhang M., Gentz R. FLICE, a novel FADD-homologous ICE/CED-3 like protease is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- Nagai Y., Onodera O., Chun J., Strittmatter W.J., Burke J.R. Expanded polyglutamine domain proteins bind neurofilament and alter the neurofilament network. Exp. Neurol. 1999;155:195–203. doi: 10.1006/exnr.1998.6991. [DOI] [PubMed] [Google Scholar]
- Nelbock P., Dillon P.J., Perkins A., Rosen C.A. A cDNA for a protein that interacts with the human immunodeficiency virus Tat transactivator. Science. 1990;248:1650–1653. doi: 10.1126/science.2194290. [DOI] [PubMed] [Google Scholar]
- Okorokov A.L., Ponchel F., Milner J. Induced N- and C-terminal cleavage of p53a core fragment of p53, generated by interaction with damaged DNA, promotes cleavage of the N-terminus of full-length p53, whereas ssDNA induces C-terminal cleavage of p53. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:6008–6017. doi: 10.1093/emboj/16.19.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson D.C., Marechal V., Momand J., Chen J., Romocki C., Levine A.J. Identification and characterization of multiple mdm-2 proteins and mdm-2-p53 protein complexes. Oncogene. 1993;8:2353–2360. [PubMed] [Google Scholar]
- Pear W.S., Nolan G.P., Scott M.L., Baltimore D. Production of high titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perutz M.F. Glutamine repeats and neurodegenerative diseasesmolecular aspects. Trends Biochem. Sci. 1999;24:58–63. doi: 10.1016/s0968-0004(98)01350-4. [DOI] [PubMed] [Google Scholar]
- Pochampally R., Fodera B., Chen L., Shao W., Levine E.A., Chen J. A 60 kD MDM2 isoform is produced by caspase cleavage in non-apoptotic tumor cells. Oncogene. 1998;17:2629–2636. doi: 10.1038/sj.onc.1202206. [DOI] [PubMed] [Google Scholar]
- Reddy P.H., Williams M., Tagle D.A. Recent advances in understanding the pathogenesis of Huntington's disease. Trends Neurosci. 1999;22:248–255. doi: 10.1016/s0166-2236(99)01415-0. [DOI] [PubMed] [Google Scholar]
- Roos R.A., Bots G.T., Hermans J. Neuronal nuclear membrane indentation and astrocyte/neuron ratio in Huntington's disease. A quantitative electron microscopic study. J. Hirnforsch. 1985;26:689–693. [PubMed] [Google Scholar]
- Ryan K.J., Wente S.R. The nuclear pore complexa protein machine bridging the nucleus and cytoplasm. Curr. Opin. Cell. Biol. 2000;12:361–371. doi: 10.1016/s0955-0674(00)00101-0. [DOI] [PubMed] [Google Scholar]
- Sanchez I., Xu C.-J., Juo P., Kakizaka A., Blenis J., Yuan J. Caspase-8 is required for cell death induced by expanded polyglutamine repeats. Neuron. 1999;22:623–633. doi: 10.1016/s0896-6273(00)80716-3. [DOI] [PubMed] [Google Scholar]
- Saudou F., Finkbeiner S., Devys D., Greenberg M.E. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- Scherzinger E., Sittler A., Schweiger K., Heiser V., Lurz R., Hasenbank R., Bates G.P., Lehrach H., Wanker E.E. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrilsimplications for Huntington's disease pathology. Proc. Natl. Acad. Sci. USA. 1999;96:4604–4609. doi: 10.1073/pnas.96.8.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt C., von Kobbe C., Bachi A., Pante N., Rodrigues J.P., Boscheron C., Rigaut G., Wilm M., Seraphin B., Carmo-Fonseca M., Izaurralde E. Dbp5, a DEAD-box protein required for mRNA export, is recruited to the cytoplasmic fibrils of nuclear pore complex via a conserved interaction with CAN/Nup159p. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:4332–4347. doi: 10.1093/emboj/18.15.4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senut M.-C., Suhr S.T., Kaspar B., Gage F.H. Intraneuronal aggregate formation and cell death after viral expression of expanded polyglutamine tracts in the adult rat brain. J. Neurosci. 2000;20:219–229. doi: 10.1523/JNEUROSCI.20-01-00219.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman M.Y., Goldberg A.L. Cellular defences against unfolded proteinsa cell biologist thinks about neurodegenerative diseases. Neuron. 2001;29:15–32. doi: 10.1016/s0896-6273(01)00177-5. [DOI] [PubMed] [Google Scholar]
- Sisodia S.S. Nuclear inclusions in glutamine repeat disordersare they pernicious, coincidental, or beneficial? Cell. 1998;95:1–4. doi: 10.1016/s0092-8674(00)81743-2. [DOI] [PubMed] [Google Scholar]
- Sittler A., Walter S., Wedemeyer N., Hasenbank R., Scherzinger E., Eickhoff H., Bates G.P., Lehrach H., Wanker E.E. SH3GL3 associates with the Huntingtin exon 1 protein and promotes the formation of polygln-containing protein aggregates. Molec. Cell. 1998;2:427–436. doi: 10.1016/s1097-2765(00)80142-2. [DOI] [PubMed] [Google Scholar]
- Steffan J.S., Kazantsev A., Spasic-Boskovic O., Greenwald M., Zhu Y.-Z., Gohler H., Wanker E.E., Bates G.P., Housman D.E., Thompson L.M. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA. 2000;97:6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhr S.T., Gil E.B., Senut M.-C., Gage F.H. High level transactivation by a modified Bombyx ecdysone receptor in mammalian cells without exogenous retinoid x receptor. Proc. Natl. Acad. Sci. USA. 1998;95:7999–8004. doi: 10.1073/pnas.95.14.7999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki E., Lowry J., Sonoda G., Testa J.R., Walsh K. Structures and chromosome locations of the human MEF2A gene and a pseudogene MEF2AP. Cytogenet. Cell Genet. 1996;73:244–249. doi: 10.1159/000134348. [DOI] [PubMed] [Google Scholar]
- Thornberry N.A., Lazebnik Y. Caspasesenemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- Wanker E.E. Protein aggregation in Huntington's and Parkinson's diseaseimplications for therapy. Mol. Med. Today. 2000;6:387–391. doi: 10.1016/s1357-4310(00)01761-5. [DOI] [PubMed] [Google Scholar]
- Wu B., Hunt C., Morimoto R. Structure and expression of the human gene encoding major heat shock protein HSP70. Mol. Cell. Biol. 1985;5:330–341. doi: 10.1128/mcb.5.2.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates J.R., III. Mass Spectrometry from genomics to proteomics. Trends Genet. 2000;16:5–8. doi: 10.1016/s0168-9525(99)01879-x. [DOI] [PubMed] [Google Scholar]
- Zhang W., Chait B.T. ProFound—An expert system for protein identification using mass spectrometric peptide mapping information. Anal. Chem. 2000;72:2482–2489. doi: 10.1021/ac991363o. [DOI] [PubMed] [Google Scholar]
- Zoghbi H.Y., Orr H.T. Glutamine repeats and neurodegeneration. Annu. Rev. Neurosci. 2000;23:217–247. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]