

Abstract
Integrin-mediated adhesion to the extracellular matrix permits efficient growth factor-mediated activation of extracellular signal–regulated kinases (ERKs). Points of regulation have been localized to the level of receptor phosphorylation or to activation of the downstream components, Raf and MEK (mitogen-activated protein kinase/ERK kinase). However, it is also well established that ERK translocation from the cytoplasm to the nucleus is required for G1 phase cell cycle progression. Here we show that phosphorylation of the nuclear ERK substrate, Elk-1 at serine 383, is anchorage dependent in response to growth factor treatment of NIH 3T3 fibroblasts. Furthermore, when we activated ERK in nonadherent cells by expression of active components of the ERK cascade, subsequent phosphorylation of Elk-1 at serine 383 and Elk-1–mediated transactivation were still impaired compared with adherent cells. Elk-1 phosphorylation was dependent on an intact actin cytoskeleton, as discerned by treatment with cytochalasin D (CCD). Finally, expression of active MEK failed to predominantly localize ERK to the nucleus in suspended cells or adherent cells treated with CCD. These data show that integrin-mediated organization of the actin cytoskeleton regulates localization of activated ERK, and in turn the ability of ERK to efficiently phosphorylate nuclear substrates.
Keywords: integrins, extracellular signal–regulated kinase, Elk-1, actin cytoskeleton, translocation
Introduction
Cells utilize several cytoplasmic mitogen-activated protein (MAP) kinase modules to transduce signals from the extracellular environment to the nucleus, ultimately regulating cell proliferation, differentiation, and survival decisions. The extracellular signal–regulated kinase (ERK) family of MAP kinases is activated by several growth factors via a pathway that involves GTP-loading of Ras and sequential phosphorylation and activation of Raf, MEKs (MAP kinase/ERK kinases), and ERK. Recently it has been shown that activation of ERK is also controlled by adhesion via integrins. Fibroblast cell lines or endothelial cells maintained in suspension weakly activate ERK when stimulated with growth factors or G protein receptor agonists compared with cells that are adherent to matrix proteins or anti-integrin antibodies (Miyamoto et al. 1996; Lin et al. 1997; Renshaw et al. 1997; Aplin et al. 1999; Short et al. 2000). Integrins tether the actin cytoskeleton to the membrane via focal complex/adhesion sites (Calderwood et al. 2000), and collaborative signaling is dependent on intact cortical actin structures and/or small focal complexes (Aplin and Juliano 1999). Thus, it is likely that integrins utilize the actin network as a scaffold to enhance growth factor activation of the ERK pathway.
Upon mitogenic stimulation, ERK translocates from the cytoplasm to the nucleus, where it phosphorylates the ternary complex factors Elk-1 and Sap-1a (Chen et al. 1992; Gille et al. 1992; Lenormand et al. 1993). Phosphorylation of Elk-1 increases its affinity for the serum response factor and in concert enhances transcription of growth-related proteins, such as c-Fos (Marais et al. 1993; Whitmarsh et al. 1995). Several sites in the COOH terminus of Elk-1 are phosphorylated by ERK, the most critical of which appears to be serine 383 (Marais et al. 1993). ERK-mediated transcriptional events ultimately impinge on cell cycle elements, such as the induction of cyclin D1 (Albanese et al. 1995; Lavoie et al. 1996), although it is becoming evident that activation of additional pathways, such as the phosphatidyl-inositol-3-kinase pathway, are often required for cell cycle progression (Marshall 1999).
Progression through the G1 phase of the cell cycle is jointly regulated by adhesion to the extracellular matrix and circulating growth factors, and is manifested in alterations in the levels of cyclin D1 and the cyclin-dependent kinase inhibitors, p21cip1 and p27kip1 (Assoian 1997; Bottazzi et al. 1999). Integrin–growth factor collaboration leading to efficient activation of ERK correlates with anchorage-dependent effects on cyclin D1 levels (Roovers et al. 1999). Nonetheless, recent reports provide evidence that simply activating ERK in the absence of integrin engagement is not always sufficient for cyclin D1 expression. Thus, Roovers et al. reported that forced activation of ERK in suspended NIH 3T3 cells substantially overrides the adhesion requirement for expression of cyclin D1 (Roovers et al. 1999). In contrast, Le Gall et al. 1998 found that forced ERK activation is not sufficient to induce cyclin D1 and downstream events, such as hyperphosphorylation of the retinoblastoma protein and S phase entry, in suspended CCL 39 fibroblasts. Together, these data raise the possibility that the regulatory effect of integrins on the ERK pathway may extend beyond the level of ERK activation.
ERK translocation to the nucleus is essential for G1 phase progression (Brunet et al. 1999). In resting conditions, ERK is anchored in the cytoplasm by its association with MEK (Fukuda et al. 1997), the microtubule network (Reszka et al. 1995), and phosphatases, for example MAP kinase phosphatase-3 (MKP-3), and the protein tyrosine phosphatase SL (Camps et al. 1998; Blanco-Aparicio et al. 1999). MEK–ERK association is disrupted upon mitogenic stimulation and a proportion of ERK translocates to the nucleus. The requirement for MAP kinases to move between the cytoplasmic and nuclear compartments to fulfill many of their actions highlights the importance of regulated movement through nuclear pore complexes. Active transport between the cytoplasmic and nuclear compartments is a bidirectional process: specific cytoplasmic proteins are imported into the nucleus, and proteins, tRNA, and mRNAs are exported from the nucleus (Gorlich 1998; Kaffman and O'Shea 1999). Identification of a consensus nuclear localization signal (NLS) characterized by one or more clusters of basic amino acids in numerous proteins has spurred on the study of the mechanism underlying nucleocytoplasmic trafficking. In general, nuclear import of proteins requires the concerted action of importins α and β (also known as karyopherins) acting as carrier molecules, the small GTPase Ran, and pp15 (also known as p10 or NTF2). However, ERKs do not contain a consensus nuclear localization sequence. Rather, translocation is dependent on phosphorylation of ERK at the regulatory threonine and tyrosine residues and also on homodimerization (Fukuda et al. 1997; Khokhlatchev et al. 1998). ERK translocation is likely to be an active mechanism possibly through interactions with the importin proteins that play a key role in protein passage across the nuclear membrane (Gorlich 1998). Once dephosphorylated in the nucleus, ERK is rapidly exported via an active mechanism that is mediated, at least in part, by MEK that has entered the nucleus independently from ERK (Adachi et al. 2000).
As ERK translocation is a critical determinant in the transcriptional and biological responses to activation of this pathway, we addressed whether ERK activity, in the absence of integrin engagement, was able to impinge on nuclear events. We demonstrate that under conditions of equivalent ERK activity, ERK-mediated phosphorylation of the transcription factor Elk-1 is diminished in the absence of integrin engagement or upon disruption of the actin cytoskeleton. Additionally, Elk-1–driven gene transcription is low in nonadherent cells despite ERK being activated. Both during nonadherent conditions and in the absence of an intact cytoskeleton in adherent cells, ERK predominantly accumulated in the cytoplasm rather than translocating to the nucleus. Thus, integrin-mediated adhesion permits ERK to efficiently localize in the nucleus and phosphorylate a key downstream nuclear substrate.
Materials and Methods
Plasmids
FLAG epitope–tagged Elk-1 in pCMV5 (Yang et al. 1998) was provided by Dr. A. Sharrocks (University of Manchester, Manchester, England). The 22W Raf cDNA, a gift from Dr. C. Der (University of North Carolina at Chapel Hill), was subcloned into pcDNA3.1 (Invitrogen). pMCL-MEK1-ΔED, encoding an active version of MEK1 with an NH2-terminal deletion of residues 32–51 and serine residues within its activation loop replaced with acidic amino acids, was from Dr. N. Ahn (University of Colorado, Boulder, CO) (Mansour et al. 1994).
Cell Culture and Transfection
NIH 3T3 and Tet-off NIH 3T3 cells were maintained in DMEM containing 10% bovine calf serum. Additionally, 2 μg/ml tetracycline was included in the medium for the Tet-off cell lines as described previously (Roovers et al. 1999; Zhu et al. 2000). Tet-off NIH 3T3 cells harboring either a construct encoding a constitutively active form of MEK1, MEK1-S218D/S222D, [tet-MEK*-3T3], or cyclin D1 [tet-cyclin D1-3T3] were used. To retain high percentages of cells expressing active MEK, clones were used at low (<20) passage numbers. The tet-MEK*-3T3 cell clone 7B was used in these experiments. Transient transfections were performed with SuperFect (QIAGEN) according to the manufacturer's instructions.
Cell Adhesion and Preparation of Cell Lysate
Confluent cells were serum starved and processed as described previously (Aplin and Juliano 1999). In brief, cells were detached with trypsin, which was subsequently quenched with soybean trypsin inhibitor (GIBCO BRL). Cells were resuspended in DMEM with 1% BSA and incubated nonadherently at 37°C for 45 min in a rotator. Cells were then plated onto dishes coated with 10 μg/ml human fibronectin (Collaborative Biomedical Product) for a further 3 h. As indicated, some cells were treated stimulated with 10–20 ng/ml EGF; no significant differences were seen between concentrations within this range. Cells were lysed in a modified RIPA buffer and cell lysates were cleared by centrifugation at 16,000 g.
Immunofluorescence Microscopy
Cells replated on glass coverslides were prepared for immunofluorescence microscopy as described previously (Aplin and Juliano 1999). Elk-1 was detected with anti–Elk-1 polyclonal antibody (New England Biolabs, Inc.). Slides were viewed on a ZEISS Axioskop microscope equipped for epifluorescence and images were captured using MetaMorph imaging software. For confocal microscopy experiments, tet-MEK*-3T3 and tet-cyclin D1-3T3 cells were incubated in serum-free DMEM for 24 h and subsequently reseeded onto coverslips for the control monolayer and cytochalasin D (CCD; 2 μg/ml final concentration)-treated cultures, or into petri dishes coated with 1% agarose for the suspension cultures. The cells were maintained in 10% FCS in the absence of tetracycline for either 6 or 9 h to allow for efficient expression of ectopic active MEK and cyclin D1. At selected times spanning G1 phase, control monolayer, CCD, and suspension cultures were fixed and stained as described previously (Zhu et al. 1999). MEK1 was detected with an anti-MEK1 monoclonal antibody (Transduction Laboratories), and ERK was detected with an anti-ERK1 polyclonal antibody (clone K-23; Santa Cruz Biotechnology, Inc.). The immunofluorescence analysis for cyclin D1 was performed using the ammonium sulfate fraction of a polyclonal cyclin D1 antibody prepared against recombinant murine cyclin D1. Slides were visualized using a Leica TCS 4D confocal immunofluorescence microscope and 1-μm sections were captured at 40× magnification using an Image Graphics image recorder.
Immunoprecipitation, Western Blotting, and Kinase Reactions
Immunoprecipitations were performed either overnight or for 2 h at 4°C from precleared lysates followed by a further incubation with protein G-Sepharose for 2 h at 4°C. FLAG–Elk-1, endogenous focal adhesion kinase (FAK), and hemagglutinin (HA)-ERK1 were immunoprecipitated with anti-FLAG antibody (Sigma-Aldrich), clone 2A7 (Upstate Biotechnology), and anti-HA antibody, 12CA5 (Babco), respectively. Precipitates were washed three times with cold RIPA buffer, and boiled with SDS-PAGE sample buffer to dissociate the proteins. For analysis by Western blotting, samples were separated by SDS-PAGE under reducing conditions. Phosphoserine 383 and total levels of Elk-1 were detected using antibodies from New England Biolabs, Inc. and Santa Cruz Biotechnology, Inc. Antibodies to the Raf COOH terminus (clone C-12; Santa Cruz Biotechnology, Inc.), FAK (clone 77, Transduction Laboratories), MEK (Transduction Laboratories), dually phosphorylated ERK (New England Biolabs, Inc.), total ERK (Santa Cruz Biotechnology, Inc.), and phosphotyrosine (clone 4G10; Upstate Biotechnology) were also used. Immunoreactivity was detected using horseradish peroxidase–conjugated secondary antibodies and enhanced chemiluminescence. HA-ERK1 in vitro kinase assays using myelin basic protein as substrate have also been described previously (Aplin and Juliano 1999).
Luciferase Reporter Assays
Elk-1 transcriptional activity was determined using a construct encoding a fusion between the GAL4 DNA binding domain and the transactivation domain of Elk-1 (GAL4–Elk-1). NIH 3T3 cells were transfected with 500 ng GAL4–Elk-1, 500 ng of a reporter plasmid controlling the transcription of firefly luciferase (pFR-luc), 10 ng of pRL-CMV-luc (Renilla luciferase under the control of the CMV promoter), and 1 μg of either pcDNA3-22W Raf or empty vector. Cells were transfected for 4 h, serum starved for 8 h, and then detached from the dish and rolled for 45 min in DMEM/BSA. The subsequent increase in luciferase activity was determined in cells either maintained in suspension or replated on fibronectin for a further 4 h.
Luciferase activities were determined using the dual luciferase assay kit (Promega). Cells were extracted and assayed sequentially for firefly and Renilla luciferase activities. Cell lysate (20 μl) was incubated with 100 μl of luciferin reagent and luminescence recorded for 10 s in an Analytical Luminescence Laboratory Monolight 2010 luminometer. Subsequently, Stop and Glo® reagent (100 μl) was added and the specific luminescence from the Renilla luciferase was recorded for an additional 10 s. Firefly activities were normalized to Renilla luciferase activity.
Results
Growth Factor–mediated Phosphorylation of Elk-1 at Serine 383 Is Adhesion Dependent
Activation of ERK by growth factors in human and mouse fibroblasts is dependent on the state of adhesion (Miyamoto et al. 1996; Lin et al. 1997; Renshaw et al. 1997; Aplin and Juliano 1999). The transcription factor Elk-1 is a substrate for ERK, and phosphorylation at several COOH-terminal sites, including serine 383, is critical for its transcriptional potential (Marais et al. 1993; Whitmarsh et al. 1995). We initially determined whether Elk-1 displays anchorage-dependent phosphorylation in response to growth factors. NIH 3T3 fibroblasts express low levels of endogenous Elk-1, hence we used transient transfection of a FLAG-tagged version of Elk-1 which localized to the nucleus as determined by immunofluorescence. Identical results were obtained with antibodies to Elk-1 (Fig. 1 A) and the FLAG epitope (data not shown). The phosphorylation status of Elk-1 was monitored by immunoprecipitation followed by Western blotting with a serine 383 phosphorylation state–dependent antibody. When transfected cells adherent to fibronectin were stimulated with EGF, Elk-1 was robustly phosphorylated at serine 383 (Fig. 1 B). In contrast, Elk-1 was weakly phosphorylated upon EGF treatment in nonadherent cells. Thus, Elk-1 phosphorylation is adhesion dependent in response to growth factors in a manner that closely correlates with adhesion effects on ERK activation (Lin et al. 1997; Aplin and Juliano 1999).
Figure 1.


Adhesion to fibronectin and EGF collaborate to provide efficient phosphorylation of the Elk-1 transcription factor. NIH 3T3 cells were transfected with either pCMV5 (Vector) or pCMV5-FLAG-Elk-1. In A, 1 μg of green fluorescent protein (GFP) was included in the transfections to identify transfected cells. After 48 h, transfected cells were serum starved before being replated in DMEM/BSA on fibronectin-coated coverslips (A) or maintained in suspension (Sus) or replated on fibronectin (Fn)-coated dishes for a further 3 h (B). (A) Localization of Elk-1 was determined by immunofluorescence with an Elk-1 antibody and TRITC-conjugated anti–rabbit secondary antibody. The scale bar depicts a 10 micron distance. (B) After the 3-h incubation, cells were treated with 20 ng/ml EGF for 15 min as indicated. Ectopically expressed Elk-1 was immunoprecipitated (IP) from cell lysates from each condition with an M2 FLAG epitope antibody. Immunoprecipitates were analyzed by Western blotting (WB) with antibodies to determine phosphorylated and total Elk-1 levels.
Expression of Active Raf or MEK1 Renders ERK Activity Adhesion Independent
Next we sought to determine whether restoring the ability of cells to activate ERK, while maintained in suspension, was able to impinge on nuclear events by phosphorylating Elk-1. To this end we used an active mutant of Raf, known as 22W Raf, that has an NH2-terminal deletion of 305 amino acids and high transforming potential (Stanton and Cooper 1987). 22W Raf was expressed as a 36-kD protein in NIH 3T3 cells that was recognized by a COOH-terminal Raf antibody (Fig. 2 A). Whereas activation of an epitope-tagged version of ERK1 (HA-ERK1) was anchorage dependent in response to growth factor, expression of 22W Raf led to a robust activation of HA-ERK1 in cells either maintained in suspension or adherent to fibronectin (Fig. 2 B). Importantly, the activation of HA-ERK1 was equivalent in cells under these conditions at this 3-h time point (Fig. 2 B) and throughout a 2–6-h time period (Fig. 2 C). Additionally, we used a constitutively active version of MEK1, referred to as MEK1-ΔED. Akin to 22WRaf, expression of MEK1-ΔED efficiently activated ERK1 regardless of the cellular state of adhesion (Fig. 2 D). Activation of the c-Jun NH2-terminal kinase (JNK) pathway also results in phosphorylation of Elk-1 (Whitmarsh et al. 1995). As expected, expression of 22W Raf or MEK1-ΔED did not result in activation of an epitope-tagged version of JNK1 (data not shown). Together, these data show that the adhesion-dependent requirement for the activation of ERK is bypassed by expression of active versions of Raf and MEK.
Figure 2.

Expression of active Raf or MEK in suspended cells is sufficient to activate ERK activity. (A) NIH 3T3 cells transfected either with pcDNA3 vector (Vec) or pcDNA3-22W Raf were analyzed by Western blotting (WB) with the anti-Raf COOH-terminal (C-term) antibody, C12, 48 h after transfection. (B) Cells transfected with HA-ERK1 and either vector (Vec) or 22W Raf were replated onto fibronectin (Fn) or maintained in suspension (Sus) for 3 h, as above. Some cells were then treated with 10 ng/ml EGF for 5 min as indicated. HA-ERK was immunoprecipitated (IP) from cell lysates and assayed for activity by in vitro kinase assay using myelin basic protein (MBP) as a substrate. (C) HA-ERK activity was measured in vector and 22W Raf–expressing cells either under nonadherent or adherent conditions at several time points. Values were normalized to the activity of HA-ERK at the 2 h time point on Fn. Shown is the mean and standard deviation of three independent experiments. (D) Cells were cotransfected with equivalent levels of HA-ERK1 and MEK1-ΔED. HA-ERK activity was analyzed, as above, in cells maintained in DMEM/BSA in suspension and adherent conditions for 3 h.
Active ERK Requires Adhesion-dependent Signals to Efficiently Phosphorylate Elk-1
To examine whether active ERK was able to phosphorylate Elk-1 at serine 383 in the absence of integrin engagement, we cotransfected cells with Elk-1 and either 22W Raf or empty vector. As before, control transfected cells exhibited little phosphorylation of Elk-1 when either held in suspension or readhered to fibronectin for 3 h in the absence of growth factors (Fig. 3 A). Expression of 22W Raf stimulated ERK phosphorylation of Elk-1 at serine 383 in adherent cells, but this effect was markedly reduced under suspension conditions (Fig. 3 A). The levels of Elk-1 and 22W Raf were comparable between the suspension and fibronectin conditions (Fig. 3 A, and latter data not shown). Similarly, expression of MEK1-ΔED resulted in efficient phosphorylation of serine 383 in Elk-1 when cells were adherent to fibronectin but not under suspension conditions (Fig. 3 B). In both the 22W Raf and MEK1-ΔED experiments, there remained a small but noticeable increase in the phosphorylation of Elk-1 above control conditions in suspended cells. Thus, the adhesion effect on ERK-mediated phosphorylation of Elk-1 is potent but not complete.
Figure 3.



Phosphorylation and transcriptional activity of Elk-1 mediated by activated ERK are impaired in nonadherent cells. NIH 3T3 cells were transfected with FLAG–Elk-1 and either vector (Vec), 22W Raf (A), or MEK1-ΔED (B). Serum-starved cells were either replated on fibronectin-coated plates (Fn) or maintained in suspension (Sus) for 3 h. FLAG-Elk-1 immunoprecipitates (IP) were analyzed by Western blotting (WB) for levels of serine 383 phosphorylated and total Elk-1. Shown are representatives of at least three independent experiments with equivalent results. In C, cells were transfected with GAL4–Elk-1, pFR-luc reporter, and either vector or 22W Raf. After a brief serum starvation, cells were replated as above on fibronectin-coated plates (Fn) or maintained in suspension (Sus). The increase in GAL4-Elk-1 transactivation of pFR-luc during a 4-h time period was determined by assaying for firefly luciferase activity. For each experiment, three separate samples were assayed for each condition and all readings were normalized to the activity of Renilla luciferase under the control of a constitutively active CMV promoter (pRL-CMV-luc). The enhanced GAL4–Elk-1–driven luciferase activity in 22W Raf–expressing cells in adherent compared with suspended cells is statistically significant (*P < 0.05).
To address the effect on activation of a reporter driven by Elk-1, we cotransfected cells with either empty vector or 22W Raf cDNA, in addition to the GAL4–Elk-1 fusion and firefly luciferase reporter constructs. GAL4–Elk-1 activity was low in vector-transfected cells either maintained in suspension or replated onto fibronectin during a 4-h time period (Fig. 3 C). Expression of 22W Raf enhanced GAL4–Elk-1 activity. However, 22W Raf–expressing cells maintained in suspension showed only a 2-fold increase over vector-transfected cells compared with a 4.5-fold increase under adherent conditions (Fig. 3 C). Similarly, GAL4–Elk-1 activity was impaired in 22W Raf–expressing cells treated with CCD (data not shown). These findings demonstrate that, under conditions whereby ERK activation is equivalent, Elk-1 transactivation is strongly enhanced under adherent conditions.
Disruption of the Actin Cytoskeleton Inhibits ERK Phosphorylation of Elk-1
Integrin signaling events are typically dependent on an intact actin cytoskeleton. We used the actin depolymerizing agent, CCD, that caps the ends of growing actin fibers and inhibits integrin-mediated tyrosine phosphorylation of focal adhesion proteins (Burridge et al. 1992). Treatment of 22W Raf–expressing cells with CCD significantly reduced ERK phosphorylation of Elk-1 in adherent cells (Fig. 4 A, top). Under these conditions, cells remained round but firmly attached when viewed by microscopy, and FAK phosphotyrosine levels were dramatically reduced (Fig. 4 A, bottom). Overall, in these transient transfection experiments, the CCD effect was not quite as dramatic as the inhibition of Elk-1 phosphorylation in suspension. In vitro kinase assays demonstrated that ERK activation by 22W Raf was not inhibited by treatment with CCD (Fig. 4 B). In contrast, treatment of 22W Raf–expressing cells with the microtubule-disrupting agent, colchicine, did not inhibit Elk-1 phosphorylation at serine 383 (Fig. 4 C). Thus, the ability of ERK, once activated, to phosphorylate Elk-1 is dependent on an intact actin cytoskeleton, but not the microtubule network.
Figure 4.
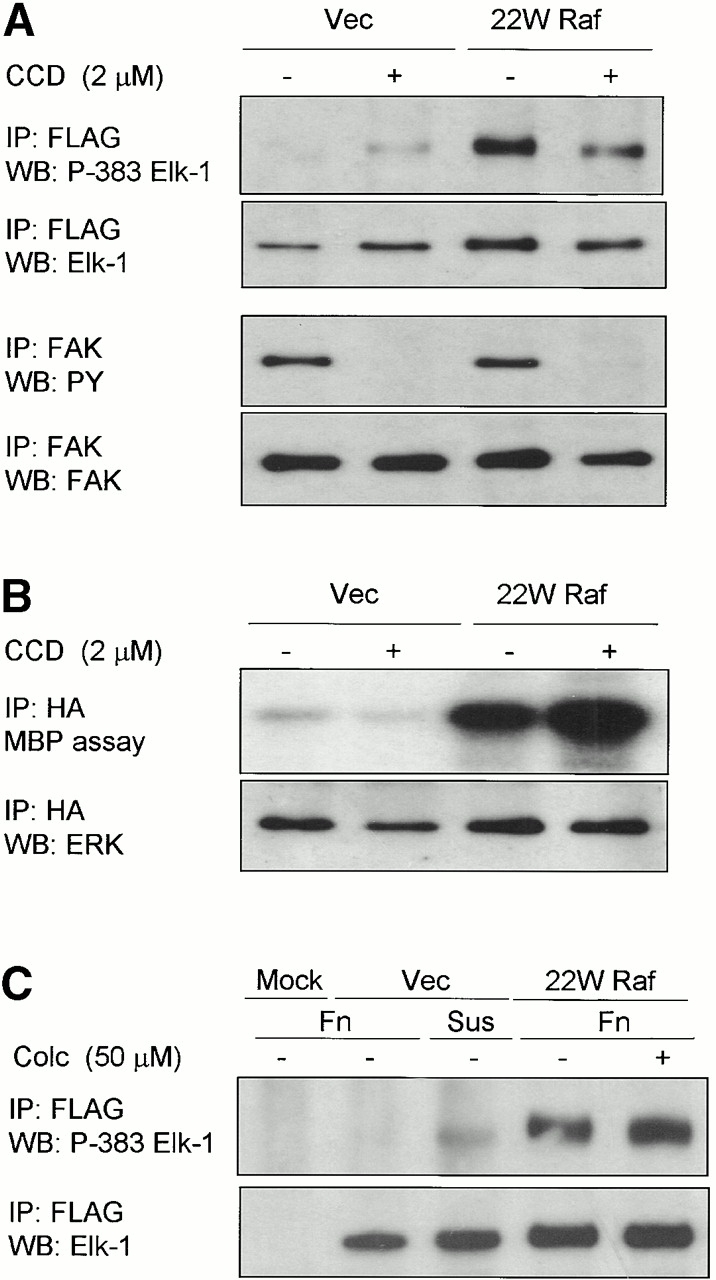
Disruption of the actin cytoskeleton, but not the microtubule network, inhibits the ability of activated ERK to phosphorylate Elk-1. NIH 3T3 cells were transfected either with vector (Vec) or 22W Raf and either FLAG-Elk-1 (A and C) or HA-ERK1 (B). In A and B, cells were treated accordingly with 2 μM CCD throughout adhesion to fibronectin-coated plates (Fn). In C, serum-starved cells were treated with 50 μM colchicine (Colc), as indicated, before replating either on fibronectin-coated plates (Fn) or maintained in suspension (Sus) for 3 h. FLAG-Elk-1 immunoprecipitates (IP) were analyzed by Western blotting (WB) for levels of serine 383 phosphorylated and total Elk-1 (A and C). Additionally, in A (bottom) endogenous FAK was immunoprecipitated from cell lysates and blotted for tyrosine phosphorylation (PY) and total levels of FAK. In B, HA-ERK was immunoprecipitated and activity measured by in vitro kinase assay. Shown are representatives of at least three independent experiments with equivalent results.
Nuclear Accumulation of ERK Is Impaired by Disruption of the Actin Cytoskeleton
The above findings suggested that ERK might be unable to redistribute properly to the nucleus upon activation in nonadherent conditions. To investigate this possibility, we used an NIH 3T3 cell line that expresses active MEK1 (tet-MEK*-3T3) under inducible control. Upon induction of active MEK in these cells, ERK is activated similarly under adherent and suspension conditions (Roovers et al. 1999). We used this system to analyze the role of adhesion upon the localization of ERK, after induction of active MEK. By confocal analysis, we determined that in adherent tet-MEK*-3T3 cells, ERK was localized primarily in the nuclear compartment, whereas MEK was present in the cytoplasm (Fig. 5 A, left). In contrast, in nonadherent cells, ERK extensively colocalized with MEK in the cytoplasmic compartment (Fig. 5 A, right). Higher magnification images confirmed that ERK was inefficiently translocated to the nucleus in nonadherent conditions (Fig. 5 B). Consistent with a role for integrin-associated actin structures playing an important role, ERK was poorly localized to the nucleus in adherent tet-MEK*-3T3 cells treated with CCD (Fig. 5 B). Quantification of >50 MEK-positive cells revealed that nuclear ERK staining was predominant in 75% of adherent cells, but only in 20 and 35% of cells treated with CCD and incubated in suspension, respectively. Moreover, some suspended and CCD-treated cells showed diffuse, rather than cytoplasm-specific, ERK staining. Similar observations on the immunolocalization of ERK were found in cells transiently expressing 22W Raf, in that ERK poorly distributed to the nucleus in nonadherent conditions (data not shown). The confocal analysis agrees well with our Elk-1 results which indicate that adhesion-dependent translocation of ERK in NIH 3T3 cells is potent but not complete.
Figure 5.


Nuclear accumulation of ERK is impaired in nonadherent cells and by disruption of the actin cytoskeleton. Tet-Mek*-3T3 cells were serum starved and stimulated with 10% FCS in the absence of tetracycline for 6–9 h. The localization of active MEK and ERK was compared in adherent (Adh) and nonadherent (Sus) cells or adherent untreated (Adh) vs. CCD-treated (Adh/CCD) monolayers via confocal microscopy. Bottom panels are of (A) overlays of the MEK and ERK images or (B) images showing DAPI staining of nuclei. Scale bars represent either a 10 or 5 micron distance, as indicated. (C and D) tet-MEK*-3T3 cells were transfected with FLAG–Elk-1 as before. Cells were serum starved overnight, after which in some populations tetracycline was removed from the media to induce expression of active MEK. Cells were detached and either maintained in suspension (Sus) or replated onto fibronectin (Fn) and lysed 6 h after induction. (C) Levels of MEK expression and activation of ERK determined by Western blotting (WB) of whole cell lysates. (D) FLAG–Elk-1 was immunoprecipitated (IP) and analyzed by Western blotting for phosphorylated and total levels of Elk-1.
We next examined whether the inability of ERK to accumulate in the nucleus of nonadherent tet-MEK*-3T3 cells, induced to express active MEK, corresponded to inefficient phosphorylation of a nuclear target. To this end we transfected these cells with FLAG–Elk-1 and induced expression of active MEK in suspension or adherent conditions. Consistent with earlier published findings (Roovers et al. 1999), induction of active MEK (by removal of tetracycline from the culture media) was independent of anchorage (Fig. 5 C, top). Western blotting with an antibody that recognizes dually phosphorylated ERK demonstrated that ERK was phosphorylated to comparable levels upon removal of tetracycline in both suspended and adherent cells (Fig. 5 C, middle and bottom). The level of Elk-1 phosphorylation at serine 383 in uninduced cells was higher than observed in the vector controls in the transient transfection experiments. This is likely due to the slightly elevated levels of active ERK in the noninduced cells. Nevertheless, Elk-1 phosphorylation was noticeably and consistently increased in adherent, but not suspension, cells that were induced to express active MEK (Fig. 5 D). Taken together, data from these confocal immunolocalization and Elk-1 phosphorylation experiments indicate that despite being active, ERK requires a state of cellular adhesion for efficient nuclear translocation, and that integrin–cytoskeletal connections are important for this process.
Disruption of the Actin Cytoskeleton Does Not Prevent Nuclear Accumulation of Cyclin D1
Similar to ERK, cyclin D1 does not contain a consensus nuclear localization sequence; rather, it is localized to the nucleus via such sequences in the cyclin-dependent kinase inhibitors, p21cip1 and p27kip1 (LaBaer et al. 1997; Cheng et al. 1999). To examine the possibility that disruption of the actin cytoskeleton has a global effect on nucleocytoplasmic transport, we analyzed nuclear localization of cyclin D1 in CCD-treated cells. Confocal immunolocalization experiments showed that nuclear accumulation of ectopically expressed cyclin D1 in tet-cyclin D1-3T3 cells, was not altered upon disruption of the actin cytoskeleton as indicated by its colocalization with DAPI-stained nuclei (Fig. 6). Thus, nuclear accumulation of cyclin D1 protein via a consensus nuclear signal import mechanism is not dependent on an intact actin cytoskeleton.
Figure 6.

Targeting of cyclin D1 to the nucleus in not affected by disruption of the actin cytoskeleton. Tet-cyclin D1-3T3 cells were G0-synchronized and stimulated with 10% FCS in the absence of tetracycline in monolayer in the absence and presence of CCD. The cells were fixed 6 h after stimulation, stained with anti-cyclin D1 antibody and DAPI nuclear stain, and analyzed via confocal microscopy. An overlay of the cyclin D1 and DAPI images is shown. The scale bars show a 10 micron distance. Adh, adherent.
Discussion
Cellular adhesion via integrin receptors is intimately involved with regulation of signaling cascades. Phosphorylation of the Elk-1 transcription factor in response to growth factor treatment is impaired in nonadherent conditions. Under these conditions, growth factor activation of ERK is impaired. However, in these studies we bypassed this regulatory step by expression of active forms of Raf and MEK. Despite being able to render ERK activity anchorage independent, Elk-1 phosphorylation continued to display an adhesion requirement. Furthermore, we showed by confocal microscopy that when the ERK pathway is activated, nuclear translocation of ERK is hindered in suspended cells. Both localization of ERK from the cytoplasm into the nucleus and phosphorylation of Elk-1 were inhibited on CCD treatment, highlighting the importance of the integrin–actin cytoskeletal connection. By contrast, treatment with colchicine failed to abrogate 22W Raf–mediated phosphorylation of Elk-1, arguing against a requirement for an intact microtubule network in ERK translocation. Nuclear localization of ectopically expressed cyclin D1 was unaltered by disruption of the actin cytoskeleton, indicating that interference with nuclear localization is not a generalized effect. Thus, in addition to integrins being able to regulate growth factor activation of ERK, we suggest the presence of a second integrin-regulated checkpoint in the ERK cascade. This checkpoint is downstream of activation, but occurs at the level of active ERK accumulation in the nucleus and phosphorylation of its nuclear substrates.
The observation that integrity of the actin cytoskeleton is necessary for trafficking of ERK to the nucleus is a novel and interesting finding. Although it is well established that activation of the ERK pathway contributes to induction of cyclin D1, previous studies have yielded inconsistent results with regard to the role of ERK signaling in the adhesion-dependent expression of cyclin D1 expression. Roovers et al. 1999 reported that forced activation of the MEK/ERK pathway leads to the expression of cyclin D1 in suspended 3T3 cells, whereas Le Gall et al. 1998, using a similar approach, failed to see cyclin D1 expression when the MEK/ERK pathway was activated in suspended CCL39 cells. As relatively low levels of ERK signaling are sufficient to induce cyclin D1 (for a review see Roovers and Assoian 2000), our results may provide an explanation for these discrepant observations. Perhaps the different results obtained by Roovers et al. 1999 and Le Gall et al. 1998 reflects the fact that ERK translocation to the nucleus is strongly dependent on integrin signaling in some cell lines, whereas it is less strictly dependent on integrin signaling in others. Indeed, we do see low levels of nuclear ERK staining when induced tet-MEK*-3T3 cells used in Roovers et al. 1999 are cultured in suspension or treated with CCD.
We favor the explanation that integrins support efficient ERK translocation to the nucleus, as we find that in cells expressing active MEK, ERK preferentially colocalizes with MEK in the cytoplasm of nonadherent but not adherent cells. Other recent findings have pointed towards an adhesion dependence of ERK translocation to the nucleus (Danilkovitch et al. 2000). In these studies, macrophage-stimulating protein showed reduced ERK activation and a further lack of detectable ERK translocation to the nucleus in suspended RE7 epithelial cells; however, this study did not examine ERK translocation under conditions where high ERK activity is maintained in suspended cells. ERK is deactivated by the activity of a variety of cellular phosphatases, including MKPs, and dephosphorylation of nuclear ERK leads to its rapid export (Khokhlatchev et al. 1998), thus possibly presenting an alternative explanation of our results. However, in our system it is unlikely that dephosphorylation of ERK is upregulated in suspended cells, as ERK activation mediated by active versions of either Raf or MEK was unaltered in suspension versus adherent conditions. Additionally, under serum-free conditions levels of the nuclear-localized MKP-2 are unaltered in suspended versus adherent cells (data not shown). The activities controlling Elk-1 dephosphorylation are not well characterized, although recent studies in COS cells have implicated a role for the calcium-dependent protein phosphatase 2B (calcineurin) (Sugimoto et al. 1997; Tian and Karin 1999).
Integrin-mediated adhesion has been shown to recruit a variety of structural and signaling molecules into specialized sites and to cause the membrane localization of the GTPase, Rac (Burridge et al. 1992; del Pozo et al. 2000). However, the mechanism underlying effects of adhesion on ERK trafficking to the nucleus is as yet undetermined. ERK is sequestered in the cytoplasm through its interaction with binding partners, such as its upstream activator MEK, and efficient ERK-mediated activation of gene transcription is enhanced through the binding of the scaffolding protein, MEK partner 1 (MP-1) (Schaeffer et al. 1998). Thus, the balance of ERK interactions between its upstream activators and scaffolding proteins may be altered by the state of cellular adhesion. Nuclear translocation of ERK is dependent on its ability to homodimerize; ERK mutants defective in this ability poorly translocate to the nucleus when microinjected into fibroblasts (Khokhlatchev et al. 1998). An intriguing notion is that integrins, via the formation of an actin-based platform, enhance the ability of ERK monomers to homodimerize. Consistent with this idea, recent evidence suggests, at least under in vitro conditions, that ERK can directly bind to actin and actin-binding proteins, such as α-actinin (Leinweber et al. 1999). Furthermore, active ERK molecules can be detected at sites of integrin-mediated adhesion (Fincham et al. 2000). Future research will be directed at understanding the mechanism underlying the adhesion regulation of ERK nucleocytoplasmic trafficking.
Our findings add credence to the emerging theme that cell adhesion molecules regulate nuclear signaling events. Ingber and colleagues have shown that integrin “hard-wiring” is able to impact on nuclear structure (Maniotis et al. 1997). Further, certain integrins may provide direct modulation of nuclear events. For example, engagement of the leukocyte integrin LFA-1/αLβ2 has been shown to initially bind and subsequently promote the nuclear localization of the c-Jun coactivator, JAB1, leading to enhanced activating protein 1 (AP-1) transcriptional activity (Bianchi et al. 2000). High expression levels of the cell–cell adhesion molecules, E- and N-cadherin, reduce the nuclear localization and transcription potential of β-catenin, by recruiting it to sites of cell–cell contact (Sadot et al. 1998; Orsulic et al. 1999). In conjunction with our current findings on integrin regulation of ERK localization, these other reports highlight an important role for cell adhesion molecules and the actin cytoskeleton in the nuclear trafficking of signaling molecules.
Acknowledgments
We thank members of the Juliano lab for helpful discussions, Brian Hogan for technical support, and Xiaoyun Zhu for assistance with the early confocal experiments. We are grateful to Channing Der, Andrew Sharrocks, and Natalie Ahn for plasmid gifts.
This work was supported by National Institutes of Health grants GM26165 (R.L. Juliano) and CA72639 (R.K. Assoian). Sheryl Stewart is supported by a predoctoral fellowship from the American Heart Association.
Footnotes
Abbreviations used in this paper: CCD, cytochalasin D; ERK, extracellular signal–regulated kinase; FAK, focal adhesion kinase; HA, hemagglutinin; MAP, mitogen-activated protein; MEK, MAP kinase/ERK kinase; MKP, MAP kinase phosphatase.
References
- Adachi M., Fukuda M., Nishida E. Nuclear export of MAP kinase (ERK) involves a MAP kinase kinase (MEK)-dependent active transport mechanism. J. Cell Biol. 2000;148:849–856. doi: 10.1083/jcb.148.5.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albanese C., Johnson J., Watanabe G., Eklund N., Vu D., Arnold A., Pestell R.G. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J. Biol. Chem. 1995;270:23589–23597. doi: 10.1074/jbc.270.40.23589. [DOI] [PubMed] [Google Scholar]
- Aplin A.E., Juliano R.L. Integrin and cytoskeletal regulation of growth factor signaling to the MAP kinase pathway. J. Cell Sci. 1999;112:695–706. doi: 10.1242/jcs.112.5.695. [DOI] [PubMed] [Google Scholar]
- Aplin A.E., Short S.M., Juliano R.L. Anchorage-dependent regulation of the mitogen-activated protein kinase cascade by growth factors is supported by a variety of integrin alpha chains. J. Biol. Chem. 1999;274:31223–31228. doi: 10.1074/jbc.274.44.31223. [DOI] [PubMed] [Google Scholar]
- Assoian R.K. Anchorage-dependent cell cycle progression. J. Cell Biol. 1997;136:1–4. doi: 10.1083/jcb.136.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi E., Denti S., Granata A., Bossi G., Geginat J., Villa A., Rogge L., Pardi R. Integrin LFA-1 interacts with the transcriptional co-activator JAB1 to modulate AP-1 activity. Nature. 2000;404:617–621. doi: 10.1038/35007098. [DOI] [PubMed] [Google Scholar]
- Blanco-Aparicio C., Torres J., Pulido R. A novel regulatory mechanism of MAP kinases activation and nuclear translocation mediated by PKA and the PTP-SL tyrosine phosphatase. J. Cell Biol. 1999;147:1129–1136. doi: 10.1083/jcb.147.6.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottazzi M.E., Zhu X., Bohmer R.M., Assoian R.K. Regulation of p21cip1 expression by growth factors and the extracellular matrix reveals a role for transient ERK activity in G1 phase. J. Cell Biol. 1999;146:1255–1264. doi: 10.1083/jcb.146.6.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A., Roux D., Lenormand P., Dowd S., Keyse S., Pouyssegur J. Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:664–674. doi: 10.1093/emboj/18.3.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge K., Turner C.E., Romer L.H. Tyrosine phosphorylation of paxillin and pp125FAK accompanies cell adhesion to extracellular matrixa role in cytoskeletal assembly. J. Cell Biol. 1992;119:893–903. doi: 10.1083/jcb.119.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood D.A., Shattil S.J., Ginsberg M.H. Integrins and actin filamentsreciprocal regulation of cell adhesion and signaling. J. Biol. Chem. 2000;275:22607–22610. doi: 10.1074/jbc.R900037199. [DOI] [PubMed] [Google Scholar]
- Camps M., Nichols A., Gillieron C., Antonsson B., Muda M., Chabert C., Boschert U., Arkinstall S. Catalytic activation of the phosphatase MKP-3 by ERK2 mitogen-activated protein kinase. Science. 1998;280:1262–1265. doi: 10.1126/science.280.5367.1262. [DOI] [PubMed] [Google Scholar]
- Chen R.H., Sarnecki C., Blenis J. Nuclear localization and regulation of ERK- and RSK-encoded protein kinases. Mol. Cell. Biol. 1992;12:915–927. doi: 10.1128/mcb.12.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M., Olivier P., Diehl J.A., Fero M., Roussel M.F., Roberts J.M., Sherr C.J. The p21cip1 and p27kip1 CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:1571–1583. doi: 10.1093/emboj/18.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danilkovitch A., Donley S., Skeel A., Leonard E.J. Two independent signaling pathways mediate the antiapoptotic action of macrophage-stimulating protein on epithelial cells. Mol. Cell. Biol. 2000;20:2218–2227. doi: 10.1128/mcb.20.6.2218-2227.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Pozo M.A., Price L.S., Alderson N.B., Ren X.D., Schwartz M.A. Adhesion to the extracellular matrix regulates the coupling of the small GTPase Rac to its effector PAK. EMBO (Eur. Mol. Biol. Organ.) J. 2000;19:2008–2014. doi: 10.1093/emboj/19.9.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fincham V.J., James M., Frame M.C., Winder S.J. Active ERK/MAP kinase is targeted to newly forming cell-matrix adhesions by integrin engagement and v-Src. EMBO (Eur. Mol. Biol. Organ.) J. 2000;19:2911–2923. doi: 10.1093/emboj/19.12.2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M., Gotoh Y., Nishida E. Interaction of MAP kinase with MAP kinase kinaseits possible role in the control of nucleocytoplasmic transport of MAP kinase. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:1901–1908. doi: 10.1093/emboj/16.8.1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gille H., Sharrocks A.D., Shaw P.E. Phosphorylation of transcription factor p62TCF by MAP kinase stimulates ternary complex formation at c-fos promoter. Nature. 1992;358:414–417. doi: 10.1038/358414a0. [DOI] [PubMed] [Google Scholar]
- Gorlich D. Transport into and out of the cell nucleus. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:2721–2727. doi: 10.1093/emboj/17.10.2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaffman A., O'Shea E.K. Regulation of nuclear localizationa key to a door. Annu. Rev. Cell Dev. Biol. 1999;15:291–339. doi: 10.1146/annurev.cellbio.15.1.291. [DOI] [PubMed] [Google Scholar]
- Khokhlatchev A.V., Canagarajah B., Wilsbacher J., Robinson M., Atkinson M., Goldsmith E., Cobb M.H. Phosphorylation of the MAP kinase ERK2 promotes its homodimerization and nuclear translocation. Cell. 1998;93:605–615. doi: 10.1016/s0092-8674(00)81189-7. [DOI] [PubMed] [Google Scholar]
- LaBaer J., Garrett M.D., Stevenson L.F., Slingerland J.M., Sandhu C., Chou H.S., Fattaey A., Harlow E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997;11:847–862. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- Lavoie J.N., L'Allemain G., Brunet A., Muller R., Pouyssegur J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J. Biol. Chem. 1996;271:20608–20616. doi: 10.1074/jbc.271.34.20608. [DOI] [PubMed] [Google Scholar]
- Le Gall M., Grall D., Chambard J.C., Pouyssegur J., Van Obberghen-Schilling E. An anchorage-dependent signal distinct from p42/44 MAP kinase activation is required for cell cycle progression. Oncogene. 1998;17:1271–1277. doi: 10.1038/sj.onc.1202057. [DOI] [PubMed] [Google Scholar]
- Leinweber B.D., Leavis P.C., Grabarek Z., Wang C.L., Morgan K.G. Extracellular regulated kinase (ERK) interaction with actin and the calponin homology (CH) domain of actin-binding proteins. Biochem. J. 1999;344:117–123. [PMC free article] [PubMed] [Google Scholar]
- Lenormand P., Sardet C., Pages G., L'Allemain G., Brunet A., Pouyssegur J. Growth factors induce nuclear translocation of MAP kinases (p42mapk and p44mapk) but not of their activator MAP kinase kinase (p45mapkk) in fibroblasts. J. Cell Biol. 1993;122:1079–1088. doi: 10.1083/jcb.122.5.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T.H., Chen Q., Howe A., Juliano R.L. Cell anchorage permits efficient signal transduction between Ras and its downstream kinases. J. Biol. Chem. 1997;272:8849–8852. [PubMed] [Google Scholar]
- Maniotis A.J., Chen C.S., Ingber D.E. Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc. Natl. Acad. Sci. USA. 1997;94:849–854. doi: 10.1073/pnas.94.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour S.J., Matten W.T., Hermann A.S., Candia J.M., Rong S., Fukasawa K., Vande Woude G.F., Ahn N.G. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. 1994;265:966–970. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- Marais R., Wynne J., Treisman R. The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain. Cell. 1993;73:381–393. doi: 10.1016/0092-8674(93)90237-k. [DOI] [PubMed] [Google Scholar]
- Marshall C. How do small GTPase signal transduction pathways regulate cell cycle entry? Curr. Opin. Cell Biol. 1999;11:732–736. doi: 10.1016/s0955-0674(99)00044-7. [DOI] [PubMed] [Google Scholar]
- Miyamoto S., Teramoto H., Gutkind J.S., Yamada K.M. Integrins can collaborate with growth factors for phosphorylation of receptor tyrosine kinases and MAP kinase activationroles of integrin aggregation and occupancy of receptors. J. Cell Biol. 1996;135:1633–1642. doi: 10.1083/jcb.135.6.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsulic S., Huber O., Aberle H., Arnold S., Kemler R. E-cadherin binding prevents beta-catenin nuclear localization and beta-catenin/LEF-1-mediated transactivation. J. Cell Sci. 1999;112:1237–1245. doi: 10.1242/jcs.112.8.1237. [DOI] [PubMed] [Google Scholar]
- Renshaw M.W., Ren X.D., Schwartz M.A. Growth factor activation of MAP kinase requires cell adhesion. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:5592–5599. doi: 10.1093/emboj/16.18.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reszka A.A., Seger R., Diltz C.D., Krebs E.G., Fischer E.H. Association of mitogen-activated protein kinase with the microtubule cytoskeleton. Proc. Natl. Acad. Sci. USA. 1995;92:8881–8885. doi: 10.1073/pnas.92.19.8881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roovers K., Davey G., Zhu X., Bottazzi M.E., Assoian R.K. α5β1 integrin controls cyclin D1 expression by sustaining mitogen-activated protein kinase activity in growth factor-treated cells. Mol. Biol. Cell. 1999;10:3197–3204. doi: 10.1091/mbc.10.10.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roovers K., Assoian R.K. Integrating the MAP kinase signal into the G1 phase cell cycle machinery. BioEssays. 2000;22:818–826. doi: 10.1002/1521-1878(200009)22:9<818::AID-BIES7>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Sadot E., Simcha I., Shtutman M., Ben-Ze'ev A., Geiger B. Inhibition of β-catenin-mediated transactivation by cadherin derivatives. Proc. Natl. Acad. Sci. USA. 1998;95:15339–15344. doi: 10.1073/pnas.95.26.15339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer H.J., Catling A.D., Eblen S.T., Collier L.S., Krauss A., Weber M.J. MP1a MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science. 1998;281:1668–1671. doi: 10.1126/science.281.5383.1668. [DOI] [PubMed] [Google Scholar]
- Short S.M., Boyer J.L., Juliano R.L. Integrins regulate the linkage between upstream and downstream events in G-protein-coupled receptor signaling to mitogen-activated protein kinase. J. Biol. Chem. 2000;275:12970–12977. doi: 10.1074/jbc.275.17.12970. [DOI] [PubMed] [Google Scholar]
- Stanton V.P., Jr., Cooper G.M. Activation of human raf transforming genes by deletion of normal amino-terminal coding sequences. Mol. Cell. Biol. 1987;7:1171–1179. doi: 10.1128/mcb.7.3.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto T., Stewart S., Guan K.L. The calcium/calmodulin-dependent protein phosphatase calcineurin is the major Elk-1 phosphatase. J. Biol. Chem. 1997;272:29415–29418. doi: 10.1074/jbc.272.47.29415. [DOI] [PubMed] [Google Scholar]
- Tian J., Karin M. Stimulation of Elk1 transcriptional activity by mitogen-activated protein kinases is negatively regulated by protein phosphatase 2B (calcineurin) J. Biol. Chem. 1999;274:15173–15180. doi: 10.1074/jbc.274.21.15173. [DOI] [PubMed] [Google Scholar]
- Whitmarsh A.J., Shore P., Sharrocks A.D., Davis R.J. Integration of MAP kinase signal transduction pathways at the serum response element. Science. 1995;269:403–407. doi: 10.1126/science.7618106. [DOI] [PubMed] [Google Scholar]
- Yang S.H., Whitmarsh A.J., Davis R.J., Sharrocks A.D. Differential targeting of MAP kinases to the ETS-domain transcription factor Elk-1. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:1740–1749. doi: 10.1093/emboj/17.6.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X., Roovers K., Davey G., Assoian R.K. Methods for analysis of adhesion-dependent cell cycle progression. In: Guan J.-L., editor. Signaling Through Cell Adhesion Molecules. CRC Press; Boca Raton, FL: 1999. pp. 129–140. [Google Scholar]
- Zhu X., Scharf E., Assoian R.K. Induction of anchorage-independent growth by transforming growth factor-beta linked to anchorage-independent expression of cyclin D1. J. Biol. Chem. 2000;275:6703–6706. doi: 10.1074/jbc.275.10.6703. [DOI] [PubMed] [Google Scholar]