

Abstract
During apoptosis, cytochrome c is released into the cytosol as the outer membrane of mitochondria becomes permeable, and this acts to trigger caspase activation. The consequences of this release for mitochondrial metabolism are unclear. Using single-cell analysis, we found that when caspase activity is inhibited, mitochondrial outer membrane permeabilization causes a rapid depolarization of mitochondrial transmembrane potential, which recovers to original levels over the next 30–60 min and is then maintained. After outer membrane permeabilization, mitochondria can use cytoplasmic cytochrome c to maintain mitochondrial transmembrane potential and ATP production. Furthermore, both cytochrome c release and apoptosis proceed normally in cells in which mitochondria have been uncoupled. These studies demonstrate that cytochrome c release does not affect the integrity of the mitochondrial inner membrane and that, in the absence of caspase activation, mitochondrial functions can be maintained after the release of cytochrome c.
Keywords: apoptosis, mitochondria, membrane potential, caspases, ATP
Introduction
Apoptotic cell death is orchestrated by the activation of caspase proteases that cleave key substrates within the cell during the apoptotic process. A major pathway for caspase activation involves a permeabilization of the mitochondrial outer membrane, which releases several proteins, including cytochrome c (Liu et al. 1996; Kluck et al. 1997), Smac/Diablo (Du et al. 2000; Verhagen et al. 2000), and others (Kluck et al. 1999; Kohler et al. 1999). Cytochrome c binds and activates an adapter, the apoptotic protease activating factor (Apaf)-1, to recruit and activate caspase-9 (Li et al. 1997; Srinivasula et al. 1998; Zou et al. 1999; Cain et al. 2000).
In mitochondria, cytochrome c plays an essential role in generation of mitochondrial transmembrane potential (ΔΨm). This potential is essential for various functions including the production of ATP via oxidative phosphorylation. Outer membrane permeability resulting in cytochrome c release should therefore impact on mitochondrial function.
Proapoptotic and antiapoptotic Bcl-2 family proteins regulate the mitochondrial outer membrane permeabilization. However, the exact mechanism by which this event occurs is controversial (for reviews see Gross et al. 1999; Vander Heiden and Thompson 1999; Waterhouse and Green 1999), and the various models that have been suggested impact on mitochondrial function in different ways. Cytochrome c release may proceed through the generation of pores or channels in the outer membrane, composed all or in part of proapoptotic Bcl-2 family proteins (Shimizu et al. 2000). Alternatively, it may occur through disruption of the outer membrane after swelling of the mitochondrial matrix due to opening of the permeability transition pore (Marzo et al. 1998a,Marzo et al. 1998b; Brenner et al. 2000) or as a consequence of a closure of the voltage-dependent anion channels in the mitochondrial outer membrane (Vander Heiden and Thompson 1999; Vander Heiden et al. 2000). In each case, however, the behavior and function of mitochondria should be affected, because the proton gradient generated by the electron transport chain should be impaired. Disruption of the ΔΨm may even kill a cell if downstream apoptotic effects are blocked. Such Bcl-2–regulated, caspase-independent cell death has been described (McCarthy et al. 1997; Martinou et al. 1999; Deshmukh et al. 2000; Haraguchi et al. 2000).
Mitochondrial function may therefore impact on the death of a cell in several ways. Altered ΔΨm may lead to cytochrome c release and activation of caspases or conversely cytochrome c release may alter mitochondrial function, which in the absence of caspase activity may lead to the death of the cell. Here, we take advantage of single-cell analysis to follow changes in the mitochondrial transmembrane potential in relation to mitochondrial outer membrane permeabilization in cells triggered to undergo apoptosis after toxic insults. We observed that a reduction in ΔΨm followed within minutes after the release of cytochrome c. We found, however, that in the absence of caspase activity mitochondria use, cytochrome c at the concentration maintained within the cytoplasm to regenerate ΔΨm and maintain ATP generation.
Materials and Methods
Cell Culture
HeLa cells stably expressing green fluorescent protein (GFP)–tagged cytochrome c (Cc-GFP-HeLa) (Goldstein et al. 2000), Cc-GFP-HeLa cells expressing Bcl-2 and murine embryonic fibroblasts deficient in Apaf-1 (a gift from Dr. F. Cecconi, University of Rome, Rome, Italy) were cultured in DME (GIBCO BRL), and Jurkat cells were grown in RPMI-1640 (GIBCO BRL). All cell lines were maintained at 37°C in a humidified atmosphere of 95% air, 5% CO2, and all media were supplemented with 2 mM glutamine, 200 μg/ml penicillin, 100 μg/ml streptomycin sulphate, and 10% FBS. Adherent cells were subcultured 1:10 by incubating them in 0.25% trypsin (GIBCO BRL) when they were 70% confluent, and resuspending cells were subcultured in growth medium. Suspension cells were subcultured 1:10 when they reached 106 cells/ml.
To induce death, Cc-GFP-HeLa cells were treated with actinomycin D (1 μM), staurosporine (1 μM), or UVC ultraviolet C (180 mJ/cm2). For UV treatment, cells were washed in PBS and irradiated with UV light in PBS at 37°C. The PBS was then aspirated, and the media were replaced. Apaf−/− cells were treated with actinomycin (1 μM), and Jurkat cells were treated with actinomycin D (500 nM), staurosporine (500 nM), or etoposide (40 μM). In experiments where glucose-free medium was used, the cells were washed once with serum-free, glucose-free DME and cultured for 12–15 h in DME containing 10% FBS dialyzed against PBS.
Detection of Apoptotic Events by Flow Cytometry
Cells having undergone specific apoptotic events were detected by flow cytometry using a FACScan® (Becton Dickinson) with a 488-nm laser line and analyzed using Cell Quest software. Phosphatidylserine exposed on the outside of cells was determined by annexin V binding. In brief, cells were pelleted and resuspended in 100 μl of Annexin V-FITC (Calbiochem) diluted 1:100 in annexin buffer (10 mM Hepes, 100 mM NaCl, 10 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2). Cells were incubated for 5 min at 37°C, and 200 μl annexin buffer containing propidium iodide (0.5 μg/ml) was added before FACS® analysis. Annexin V-FITC fluorescence was detected in FL-1, and propidium iodide was detected in FL-2.
Cytochrome c release was detected by incubating 4 × 104 Cc-GFP-HeLa cells in 100 μl of ice-cold cell lysis and mitochondria intact (CLAMI) buffer (250 mM sucrose, 70 mM, KCl 50 μg/ml digitonin in PBS) for 5 min. Aliquots from the suspensions were stained with 0.1% Trypan blue in PBS to ensure that >95% of the cells were lysed. 200 μl of ice-cold digitonin-free CLAMI buffer was added to the cell suspension, and the cells were measured immediately by flow cytometry. Cytochrome c–GFP fluorescence was detected in FL-1. At the concentration used here, digitonin selectively permeabilizes the plasma membrane, allowing any cytochrome c that has been released from the mitochondria to exit the cells. The fluorescence of cells with intact mitochondria was approximately one-third brighter than cells in which the mitochondria had released cytochrome c.
ΔΨm was measured primarily using tetramethylrhodamine ethyl ester (TMRE). Cells were incubated at 37°C for 20 min in media containing TMRE (50 nM). TMRE fluorescence was detected by flow cytometry using FL-2. ΔΨm was also measured using DiOC(6)3 (40 nM), and CMTM-Ros (150 nM) also diluted in media.
Western Blotting
To determine the extent of cytochrome c release in cells by western blotting, 1.5 × 106 Jurkat cells were incubated on ice for 5 min in 100 μl of ice-cold CLAMI buffer containing 200 μg/ml digitonin. The lysis of >95% of the cells was confirmed by Trypan blue exclusion. The cells were pelleted (1,000 g for 5 min at 4°C), and the supernatant containing cytosolic protein was stored at −80°C. The pellets were incubated at 4°C for 10 min in universal immunoprecipitation buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 0.2% Triton X-100, 0.3% NP-40 1× complete™ protease inhibitor (Roche). The samples were centrifuged (10,000 g for 10 min at 4°C), and the supernatant containing mitochondrial protein was stored at −80°C. Protein from each sample were boiled for 5 min in 5× sample loading buffer and electrophoresed in individual lanes of 15% SDS-PAGE gels. The proteins were transferred to supported Hybond C nitrocellulose (Amersham Pharmacia Biotech) and Western blotted using anti–cytochrome c (7H8.2C12; PharMingen) and antiactin (C4; ICN Biomedicals) diluted 1:1,000. The immobilized proteins were incubated with horseradish peroxidase secondary antibody, and the signal was detected using Dura Signal chemiluminescence reagent (Pierce Chemical Co.).
Confocal Microscopy
Confocal microscopy was performed using a Nikon Eclipse TE 300 microscope and a MRC 1024 confocal microscope (Bio-Rad Laboratories) using an Ar/Kr laser. For immunocytochemistry, Cc-GFP-HeLa cells were grown on LabTek four-well chamber slides (Nalge Nunc International). Cells were fixed with 4% paraformaldehyde in PBS for 15 min. The cells were washed in blocking buffer (0.05% saponin, 3% BSA, in PBS) and incubated overnight at 4°C with anti-Bax antibody (PharMingen) diluted 1:200 in blocking buffer. The cells were washed in blocking buffer and incubated for 1 h at room temperature with rabbit Ig conjugated to Texas red (Amersham Pharmacia Biotech) diluted 1:200 in blocking buffer. The fluorescence of GFP and Texas red were detected by confocal microscopy using excitation wavelengths of 488 or 522 nm and detection wavelengths of 568 or 605 nm, respectively. Images were Kalman averaged.
For time-lapse analysis, Cc-GFP-HeLa cells were grown on glass-bottom microwell dishes (MatTek). Cells were treated with apoptotic stimuli in phenol red–free DME, supplemented with 10% FBS, 20 mM Hepes, pH 7.2, 2 mM l-glutamine, 200 μg/ml penicillin, 100 μg/ml streptomycin sulphate, and TMRE (50 nM), and returned to an incubator for 2–12 h. The media were overlaid with mineral oil (Sav-On), and the dish was placed on the confocal microscope. The temperature was maintained at 37°C using an MS-C Temp Controller (Narishige). Cells were excited using a 488-nm laser line attenuated at 96%. Cytochrome c–GFP and TMRE fluorescence were detected using 568 or 605 nm, respectively. Images were Kalman averaged three times each at 2-min intervals. Untreated cells followed under these conditions for 400 frames were undamaged, to the extent that mitosis was observed in many cells during this period.
Images were analyzed with Metamorph v4.0 (Universal Imaging Corp.) by drawing regions around individual cells and then computing standard deviation (punctate/diffuse) and integrated brightness (total brightness). For ΔΨm, the total brightness of TMRE was divided by the total brightness of the cytochrome c–GFP to account for any movement of the cell, except in Apaf-deficient murine embryonic fibroblasts, which contained no cytochrome c–GFP. The punctate–diffuse index and the relative TMRE fluorescence index were calculated by dividing each value by the average of the first six values. In Fig. 3 C, the relative TMRE fluorescence of each cell was calculated by dividing each value by the average of the first 119 values. Quicktime movies were processed using NIH Image J software (National Institutes of Health).
Figure 3.

Time course analysis of ΔΨm during apoptosis. Cc-GFP-HeLa cells stained with TMRE and treated with actinomycin D (1 μM) in the presence (+zVAD) or absence (−zVAD) of 100 μM zVADfmk as indicated, and were followed by time-lapse confocal microscopy. Images were taken every 2 min and the relative brightness of TMRE fluorescence and punctate–diffuse index of cytochrome c–GFP was calculated and plotted over time. (A) Two cells, which commenced cytochrome c–GFP release at 534 and 474 min show a subsequent drop in ΔΨm, commencing at 536 and 478 min, respectively. (B) Similar cells in the absence or presence of zVADfmk (100 μM) were followed by time-lapse confocal microscopy, and the relative brightness of TMRE fluorescence was calculated and plotted over time. For comparative purposes, cytochrome c–GFP release (indicated by arrow) was set at 4 h in each case. (C) The mean brightness of TMRE in individual cells aligned for cytochrome c release at 4 h. n represents the number of cells averaged. Error bars indicate SD.
Measurement of Cellular ATP
ATP assays were performed using the ATP bioluminescence assay kit HSII (Roche) following the manufacturer's instructions. In brief, 105 cells were resuspended in 100 μl lysis buffer and stored at −80°C. Aliquots from each sample were diluted to 100 μl in dilution buffer. 100 μl of luciferase reagent was added to each sample and after a delay of 1 s, the luminescence was integrated over 10 s using a monolight 2010 luminescence recorder (Analytical Luminescence Laboratory). The ATP concentration of the samples was determined by comparing values to a standard curve for ATP performed at the same time. The ATP concentration was standardized using the total cellular protein estimated by the micro BCA assay (Pierce Chemical Co.).
Permeabilized Cell Assays for ΔΨm
Cc-GFP-HeLa cells (2 × 106) were incubated for 20 min at 37°C in media containing TMRE (50 nM). Buffers in all subsequent steps contained TMRE (50 nM). The cells were trypsinized and incubated for 5 min on ice in 1 ml of ice-cold mitochondria isolation buffer (MIB) (200 mM mannitol, 50 mM sucrose, 10 mM Hepes, 10 mM succinate, 70 mM KCl, 1 mM DTT) containing 50 μg/ml digitonin. When >95% of cells were permeable to Trypan blue, the cells were washed twice in ice-cold MIB containing 0.1% BSA. For Fig. 2 A, the cells were incubated for 30 min at 37°C in 1 ml of MIB containing truncated Bid (tBid) (20 μg/ml) and diluted in 1:10 in MIB containing BSA (0.1%) ATP (1 mM), creatin-phosphate (5 mM), creatin kinase (0.1 mg/ml), oligomycin (10 μg/ml), and the concentrations of cytochrome c indicated. Carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP) (10 μM) and KCN (1 mM) were added as indicated. The cells were analyzed by flow cytometry after 20 min, measuring cytochrome c–GFP fluorescence in FL-1 and TMRE fluorescence in FL-2. Analysis confirmed that >95% of cells treated with tBid had released cytochrome c–GFP. For Fig. 2 B, the permeabilized apoptotic cells were directly resuspended in the MIB containing cytochrome c, and the cells were analyzed by flow cytometry after 20 min.
Figure 2.

Cytochrome c concentration limits respiration in mitochondria that have undergone outer membrane permeabilization. (A) Permeabilized cells stained with TMRE (50 nM), either untreated or treated with tBid, were incubated at 37°C for 20 min with the concentrations of horse heart cytochrome c indicated. ΔΨm was measured by flow cytometry. FCCP (10 μM) was used as a control for depolarized mitochondria. KCN (1 mM) was used to block the involvement of cytochrome c in the electron transport chain. (B) Cc-GFP-HeLa cells were treated for 12 h with actinomycin D (1 μM) and stained with TMRE (50 nM). The cells were permeabilized with digitonin and incubated for 20 min with horse heart cytochrome c. ΔΨm and cytochrome c–GFP were measured by flow cytometry. The cells were gated for cytochrome c–GFP release, and the relative fluorescence of TMRE was compared with that of cells that had not released cytochrome c–GFP. Error bars indicate SD.
Online Supplemental Material
Supplemental video of Fig. 4 shows loss and regeneration of ΔΨm after cytochrome c release. Cc-GFP-HeLa cells were treated with actinomycin D (1 μM) in the presence of N-benzoylcarbonyl-Val-Ala-Asp-fluoromethylketone (zVADfmk) (100 μM), and confocal images were taken every 2 min. The cytochrome c–GFP (green, left) shows the coordinate release of cytochrome c in the individual cells (the staining goes from punctate to diffuse upon release). TMRE fluorescence in the same cells (red, right) shows the loss and recovery of ΔΨm. The red and green images are of the same cells taken at the same time. The frames are separate rather than overlaid for clarity, and a mathematical representation of loss and regeneration of ΔΨm in a similarly treated cell is shown in Fig. 4 A. Video is available at http://www.jcb.org/cgi/content/full/153/2/319/DC1.
Figure 4.
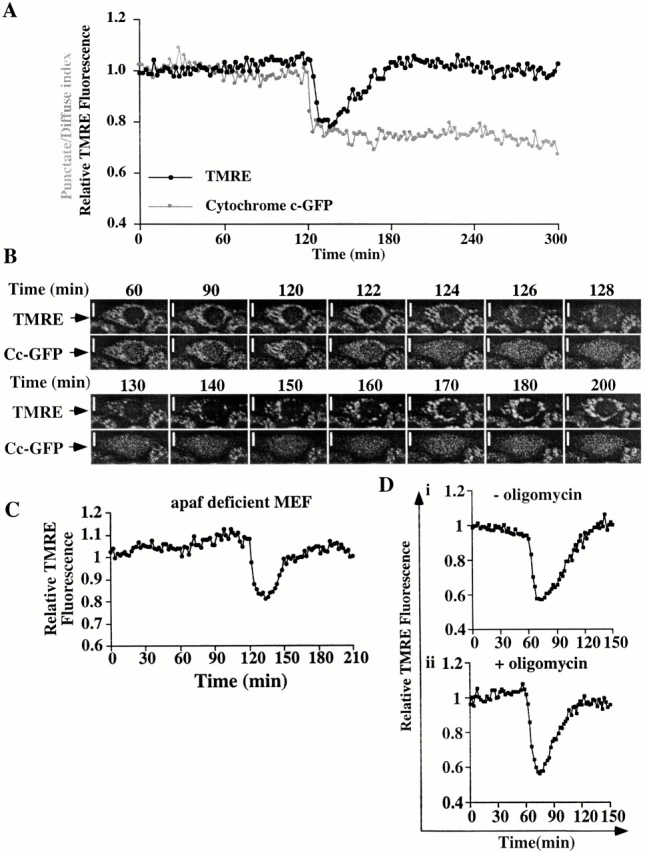
Dissipation and regeneration of ΔΨm after cytochrome c release in the absence of caspases. (A) Time lapse of the relative brightness of TMRE and the punctate–diffuse index of cytochrome c–GFP in one Cc-GFP-HeLa cell treated with actinomycin D (1 μM) in the presence of zVADfmk showing a drop in ΔΨm, followed by a slow regeneration of ΔΨm after cytochrome c–GFP release. Cytochrome c–GFP release was set at 120 min. (B) Pictographic representation of ΔΨm and cytochrome c–GFP in the cell depicted in A shows that ΔΨm is regenerated, whereas cytochrome c–GFP remains diffuse throughout the cell. More cells can be seen in Quicktime movie format at http://www.jcb.org/cgi/content/full/153/2/319/DC1. In the movie, green (left) indicates cytochrome c–GFP, whereas red (right) indicates TMRE staining. (C and D) Time-lapse of the relative brightness of TMRE fluorescence of one Apaf-1–deficient murine embryonic fibroblast (apaf-deficient MEF) treated with 1 μM actinomycin D (C), one Cc-GFP-HeLa cell treated with 1 μM staurosporine in the presence of 100 μM zVADfmk (Di), or one Cc-GFP-HeLa cell treated with 1 μM staurosporine in the presence of 100 μM zVADfmk and 10 μg/ml of oligomycin (Dii). Bars, 10 μm.
Results
Cytochrome c Maintains ΔΨm after Mitochondrial Outer Membrane Permeabilization
To examine the relationship between ΔΨm and the permeabilization of the outer mitochondrial membrane, we took advantage of a recently described HeLa cell line (Cc-GFP-HeLa) stably expressing cytochrome c–GFP in the mitochondrial intermembrane space (Goldstein et al. 2000). In these cells, the release of cytochrome c–GFP faithfully mirrors the release of cytochrome c, and this occurs in a kinetically rapid all-or-nothing manner during apoptosis. We monitored ΔΨm using TMRE, which incorporates into the mitochondria in a nernstian manner in relation to ΔΨm (Farkas et al. 1989; Fink et al. 1998).
Previously, we have found that in CEM and HeLa cells treated with UV radiation in the presence of the caspase inhibitor zVADfmk, cytochrome c can be released without significantly affecting ΔΨm (Bossy-Wetzel et al. 1998). To confirm this finding in the cell lines used in the this study, we treated Jurkat or Cc-GFP-HeLa cells with actinomycin D, staurosporine, UV, or etoposide and examined ΔΨm by TMRE staining. As shown in Fig. 1, loss of ΔΨm in Jurkat cells treated with etoposide or Cc-GFP-HeLa cells treated with actinomycin D was prevented by addition of the caspase inhibitor. Similar results were obtained in both cell lines treated with staurosporine and in Cc-GFP-HeLa cells treated with UV (data not shown), and when other dyes including CMTM-ROS and DiOC(6)3 were used to measure ΔΨm (not shown). In every case, zVADfmk did not affect cytochrome c release in Cc-GFP-HeLa cells (Goldstein et al. 2000).
Figure 1.

Loss of ΔΨm is caspase dependent during apoptosis. (A) Jurkat cells treated with etoposide (40 μM) or Cc-GFP-HeLa cells were treated with actinomycin D (1 μM) in the presence or absence of zVADfmk (100 μM), harvested at the times indicated, stained with TMRE (50 nM), and analyzed by flow cytometry. Low fluorescence indicates a loss of ΔΨm. (B) A representation of A, in which untreated cells (thin lines) are overlaid directly on cells treated with the apoptosis inducer in the presence of zVADfmk (zVAD) (100 μM). In the Cc-GFP-Hela cells, 57% of cells had released cytochrome c–GFP by this point.
The maintenance of ΔΨm may be through either the electron transport chain or through ATP-dependent reversal of ATP synthase (Simbula et al. 1997). We have previously shown that oligomycin, an ATP synthase inhibitor, does not dissipate the ΔΨm maintained after cytochrome c release, whereas inhibitors of complex III (which donates electrons to cytochrome c) and complex IV (which accepts electrons from cytochrome c) do (Goldstein et al. 2000). This suggested that cytochrome c that is still present in the cell after its release from mitochondria is sufficient to maintain electron transport and ΔΨm.
To test this idea, we induced the release of intermembrane proteins from mitochondria in digitonin-permeabilized cells, using recombinant tBid to mimic the apoptotic process (von Ahsen et al. 2000). As shown in Fig. 2 A, treatment with tBid caused a rapid loss of ΔΨm, to the same extent produced by the protonophore FCCP. Upon addition of cytochrome c, however, ΔΨm was maintained, despite the presence of oligomycin to block the generation of ΔΨm by hydrolysis of ATP. ΔΨm was not maintained in the presence of the complex IV inhibitor KCN. Thus, the loss of ΔΨm was due to the release of cytochrome c and not other mitochondrial changes.
Then, we examined the role of cytochrome c in maintaining ΔΨm in cells induced to undergo apoptosis in the presence of caspase inhibitors. Cc-GFP-HeLa cells were treated with actinomycin D plus zVADfmk, and cytochrome c–GFP release was monitored (not shown). At a time point when the majority of cells exhibited cytochrome c–GFP release, the cells were treated with digitonin to permeabilize the plasma membrane. This resulted in a complete loss of ΔΨm (Fig. 2 B) as all available cytochrome c was washed free. Addition of exogenous cytochrome c significantly restored ΔΨm in these cells. Therefore, the outer membranes of mitochondria in permeabilized tBid-treated or apoptotic cells are permeable to cytochrome c, which can restore electron transport in these mitochondria. It follows that the maintenance of ΔΨm after mitochondrial outer membrane permeabilization is through the use of the low levels of cytosolic cytochrome c. By comparing the total amount of cytochrome c in these cells with standard concentrations of horse heart cytochrome c in immunoblots, we estimated that if cytochrome c were evenly dispersed throughout the cell, it would be ∼10 μM (not shown). Addition of 10 μM cytochrome c in our permeabilized cell assays was sufficient to maintain the ΔΨm (Fig. 2 A).
Analysis of Single Cells Undergoing Apoptosis Shows a Drop and Recovery of ΔΨm after Cytochrome c–GFP Release
Competing models that explain the permeabilization of the mitochondrial outer membrane make different predictions regarding changes in ΔΨm before the release of cytochrome c. Outer membrane rupture after permeability transition (Marzo et al. 1998b) or closure of the voltage-dependent anion channels (Vander Heiden and Thompson 1999) involve dissipation or increase in ΔΨm, respectively.
In time-lapse confocal microscopy experiments, we consistently observed that, upon addition of the protonophore FCCP, TMRE staining was lost in <2 min. At the concentration used, TMRE did not suffer from self-quenching associated with other dyes used to track ΔΨm and did not affect mitochondrial function or cell viability (data not shown). We therefore used TMRE fluorescence to track ΔΨm in individual Cc-GFP-HeLa cells after treatment with actinomycin D, UV, or staurosporine. As shown in several examples depicted in Fig. 3A and Fig. B, ΔΨm was never seen to dissipate before cytochrome c–GFP release, although in some cases an increase was observed (Fig. 3 B). When individual cells treated with actinomycin D in the presence or absence of zVADfmk were standardized for cytochrome c–GFP release at 4 h, the mean ΔΨm showed that there were no significant or consistent changes before mitochondrial outer membrane permeabilization (Fig. 3 C).
Dissipation of ΔΨm commenced shortly after cytochrome c release (Fig. 3 A). Previously, we observed that reducing the temperature by 13°C did not influence the duration of cytochrome c release (Goldstein et al. 2000). Using the relative TMRE fluorescence in individual cells treated with actinomycin D plus zVADfmk, we observed that the duration of ΔΨm loss (the time taken to reduce ΔΨm from its value at the time cytochrome c–GFP was released until ΔΨm was <10% of its lowest value before cytochrome c–GFP release) increased from 11.9 ± 2.7 min at 37°C (n = 43) to 33.2 ± 7.3 min at 24°C (n = 16). This was not due to the time required for diffusion of TMRE from the depolarized mitochondria, as FCCP caused a complete loss of staining in <2 min at either temperature (data not shown).
Strikingly, however, upon the release of cytochrome c, ΔΨm rapidly decreased in all cases, whether or not zVADfmk was present. Therefore, the single-cell analysis appeared to directly contradict our bulk level observations in Fig. 1. Careful observations over time, though, resolved this apparent paradox. As shown in Fig. 4 A, after the drop in ΔΨm upon cytochrome c release in cells treated with actinomycin D plus zVADfmk, ΔΨm recovered over the next 30–60 min and remained high (until accumulated visual light damage due to repeated assessments eventually reduced ΔΨm).
The images in Fig. 4 B show the fluorescence in this cell over time, clearly demonstrating the loss and recovery of TMRE staining after cytochrome c release. Therefore, the observations on cell populations in Fig. 1 represent cells in which the large majority of those that have released cytochrome c have already recovered ΔΨm.
The loss and recovery of ΔΨm was not due to the presence of Cc-GFP, since a similar effect was seen in HeLa cells that did not express Cc-GFP (not shown), nor was it dependent on the use of caspase inhibitors, as a similar loss and recovery in ΔΨm was observed in a transformed cell line from Apaf-1–deficient murine embryonic fibroblasts treated with actinomycin D (Fig. 4 C). Furthermore, this effect was not restricted to apoptosis induced by actinomycin D and was not dependent on the function of complex V (which, as we noted, could maintain transmembrane potential by hydrolyzing ATP), since both drop and recovery of ΔΨm occurred in cells treated with staurosporine plus zVADfmk in the presence or absence of oligomycin (Fig. 4 D). Similarly, cyclosporin A, which interferes with the mitochondrial permeability transition, had no effect at doses up to 400 μM on either the drop or recovery phases after cytochrome c release (not shown). Therefore, this is not an effect of a reversible permeability transition observed as a flicker, i.e., a rapid decrease and increase of fluorescence of dyes used to measure ΔΨm (Huser and Blatter 1999; Ichas et al. 1997).
One of the major functions of the electron transport chain is to generate ATP via complex V. If, after the permeabilization of the outer membrane, the mitochondria can use the decreased amounts of cytochrome c to sustain electron transport, then ATP levels should be maintained. We therefore examined ATP levels in Cc-GFP-HeLa cells deprived of glucose and provided with pyruvate to ensure that the majority of ATP generation was dependent on mitochondrial electron transport and the function of complex V. Consistent with our hypothesis, we noted a decrease in ATP levels in the glucose-deprived cells upon treatment with oligomycin for 1 h (indicated by the dashed lines in Fig. 5 Aiii), and the majority of cells died within 12 h (not shown). In the presence of glucose, Cc-GFP-HeLa cells were resistant to oligomycin-induced death for >3 d (not shown).
Figure 5.

Mitochondria that maintain ΔΨm after cytochrome c release also produce ATP. (A) Cc-GFP-HeLa cells cultured in the absence of glucose for 12–15 h were treated with actinomycin D (1 μM) in the presence or absence of zVADfmk (100 μM). (Ai) After 12 h, the cells were stained with TMRE (50 nM) and analyzed by flow cytometry. Cells treated in the presence of zVADfmk, maintained ΔΨm. (Aii) Similarly treated cells were harvested at the times indicated and percentage of cells with polarized mitochondria, and the percentage of cells that had not released cytochrome c were determined by flow cytometry. (Aiii) Aliquots of cells in Aii were analyzed for total cellular ATP. (B) Cc-GFP-HeLa cells cultured in the absence of glucose for 12–15 h were treated with actinomycin D (1 μM) in the presence or absence of zVADfmk (100 μM). After 12 h, when ∼80% of the cells had released cytochrome c, oligomycin (10 μg/ml) was added to the sample indicated. All cells were harvested 1 h later, and total cellular ATP was measured. Error bars indicate SEM.
When the glucose-deprived cells were induced to undergo mitochondrial outer membrane permeabilization by treatment with actinomycin D (±zVADfmk), ∼80% of the cells released cytochrome c–GFP after 12 h (Fig. 5 Aii). ATP levels were reduced in the absence of zVADfmk, however, in the presence of zVADfmk, ΔΨm (Fig. 5 , Ai and Aii) and ATP levels were maintained (Fig. 5, Aiii and B). Therefore, the maintenance of ΔΨm provided enough electromotive force to maintain ATP generation, despite the low levels of cytochrome c available. This effect was only temporary, however, since ATP levels dropped over the after 12 h, and extensive nonapoptotic death was observed after 24 h under these conditions (not shown). We further confirmed the ability of mitochondria to generate ATP under these conditions by treating Cc-GFP-HeLa cells with actinomycin D in the presence or absence of zVADfmk. After 12 h, when permeabilization of the outer mitochondrial membrane had occurred in ∼80% of the cells, oligomycin was added to cells that were treated in the presence of zVADfmk. After 1 h, the cells were harvested, and total cellular ATP levels were measured. ATP levels were significantly lower in the treated cells compared with those in control cells, unless caspase activation was blocked by zVADfmk. (Fig. 5 B). The addition of oligomycin for 1 h caused a loss of ATP even in the presence of zVADfmk. Therefore, ATP levels were maintained by mitochondrial complex V function in these cells.
These data strongly support the conclusion that most mitochondria remain functional after the permeabilization of the outer mitochondrial membrane using the reduced levels of available cytochrome c now diffuse through the cytosol. Upon caspase activation, this effect is lost, and ATP levels then decrease dramatically. The loss of mitochondrial function and drop in ATP correspond to caspase-independent death of the cells.
Mitochondrial Transmembrane Potential Is Not Required for Apoptosis, Mitochondrial Outer Membrane Permeabilization, or the Antiapoptotic Effects of Bcl-2
The mitochondrial transmembrane potential, maintained by the low level of cytochrome c in apoptotic cells before caspase activation, is required for several mitochondrial functions, including the maintenance of ATP generation, as noted above. Therefore, we determined whether this was also required for the mitochondrial changes that function to promote or prevent apoptosis.
Uncouplers such as FCCP, carbonyl cyanide m-chlorophenylhydrazone (CCCP), and 2,4, dinitrophenol (DNP) dissipate the transmembrane potential by preventing the generation of a proton gradient. We examined the effects of FCCP on cytochrome c release and apoptosis in Jurkat cells and Cc-GFP-HeLa cells treated with various inducers of apoptosis. In no case did dissipation of the transmembrane potential block, diminish, or delay cytochrome c release or apoptosis and often slightly increased sensitivity to proapoptotic agents (Fig. 6). Similar results were observed when CCCP or DNP were used in place of FCCP (data not shown).
Figure 6.

Apoptosis and cytochrome c release proceed in the absence of hyperpolarization. (A) Jurkat cells were treated with etoposide (40 μM) or actinomycin D (500 nM), and Cc-GFP-HeLa cells were treated with UV (180 mJ/cm2) or actinomycin D (1 μM) in the presence or absence of FCCP (5 μM) for the times indicated. The cells were analyzed for phosphatidylserine exposure as a measure of apoptosis. (B) Similar cells to those assayed in A were analyzed for cytochrome c release (Bi) by western blotting (Jurkat) or cytochrome c–GFP release (Bii) by flow cytometry (Cc-GFP-HeLa). (C) Untreated Cc-GFP-HeLa cells or Cc-GFP-HeLa cells treated with UV (180 mJ/cm2) in the presence or absence of the concentrations of FCCP indicated were harvested at 6 h and assayed for phosphatidylserine exposure (annexin V-FITC binding) or cytochrome c–GFP release (CLAMI assay) by flow cytometry.
The proapoptotic Bcl-2 family members tBid and Bax can translocate to the mitochondria during apoptosis and effectively and rapidly trigger mitochondrial outer membrane permeabilization (von Ahsen et al. 2000; Wei et al. 2000). Immunocytochemistry of Cc-GFP-HeLa cells treated with UV (Fig. 7 A) or actinomycin D (not shown) revealed that Bax had translocated to the mitochondria of all cells in which cytochrome c–GFP was in the cytoplasm. Furthermore, the addition of recombinant tBid or Bax to digitonin-permeabilized Cc-GFP-HeLa cells resulted in loss of green fluorescence, indicative of the release of cytochrome c–GFP from mitochondria (Fig. 7 B). Dissipation of the mitochondrial transmembrane potential with FCCP had no effect on the ability of these proapoptotic Bcl-2 family proteins to permeabilize the mitochondrial outer membrane (Fig. 7 B). Furthermore, Bax overexpressed in Cc-GFP-HeLa treated with FCCP induced cytochrome c–GFP release in a similar number of cells as compared with control cells (not shown).
Figure 7.



Bcl-2 family members maintain their proapoptotic and antiapoptotic functions in the presence of uncouplers of ΔΨm. (A) Confocal micrographs of immunocytochemistry of Bax (left) in Cc-GFP-HeLa cells treated with UV (180 mJ/cm2) for 6 h in the absence (top) or presence (bottom) of FCCP (5 μM). Cytochrome c–GFP fluorescence (right) in the same cells is also shown. (B) Permeabilized HeLa cells were treated with tBid (20 μg/ml) or Bax (10 μg/ml) in the absence (left) or presence (right) of FCCP (5 μM). Cytochrome c–GFP fluorescence was detected by flow cytometry in FL-1. Release of cytochrome c–GFP was observed as a drop in overall fluorescence in the cells. (C) Cc-GFP-HeLa cells, which overexpress Bcl-2, were treated with UV (180 mJ/cm2) in the presence or absence of FCCP (5 μM), CCCP (10 μM), or DNP (800 μM). Cells were harvested after 6 h and assayed for phosphatidylserine exposure or cytochrome c–GFP release. The extent of annexin V binding and cytochrome c–GFP release was compared with cells that did not express Bcl-2, which were treated with UV at the same time. Bars, 20 μm.
Bcl-2 functions to inhibit or delay apoptosis, at least in part, by preventing mitochondrial outer membrane permeabilization (Kluck et al. 1997; Yang et al. 1997). Cytochrome c release and apoptosis are effectively blocked in Cc-GFP-HeLa cells that stably express Bcl-2 (Fig. 7 C). Dissipation of the mitochondrial transmembrane potential by FCCP, CCCP, or DNP did not alter this protective mechanism of Bcl-2. Therefore, the maintenance of ΔΨm is not required for either the proapoptotic or antiapoptotic effects of Bcl-2 family members and is not required for either mitochondrial outer membrane permeabilization or apoptosis after cellular stress.
We conclude that the most important functions of the maintenance of ΔΨm after mitochondrial outer membrane permeabilization are to provide a continued source of energy to the cell and to maintain mitochondrial functions if the apoptotic pathway is blocked downstream of this event. This raises the possibility that mitochondrial repair and cell survival might be possible after this potential point-of-no-return in the apoptotic process.
Discussion
Permeabilization of the mitochondrial outer mitochondrial membrane, regulated by Bcl-2 family members, is an integral event during apoptosis. Upon permeabilization, cytochrome c is released to facilitate formation of the apoptosome and caspase activation. The mechanism of outer membrane permeabilization remains controversial, and the consequences of this event on mitochondrial and cellular metabolism—other than activation of caspases—are not well understood. In this paper, we have shown that after the permeabilization of the mitochondrial outer membrane, ΔΨm transiently decreases. When caspase activation is blocked, ΔΨm then recovers and is maintained by the low levels of cytochrome c diffuse throughout the cell. In cells that have released cytochrome c in the absence of caspase activation, the maintenance of ΔΨm is sufficient for the mitochondria to generate ATP.
Several hypotheses for the mechanism of mitochondrial outer membrane permeabilization predict an increase (hyperpolarization) or decrease (permeability transition) in ΔΨm before the permeabilization event. However, in single-cell analysis, using cytochrome c–GFP as an indicator of mitochondrial outer membrane permeabilization and TMRE to follow ΔΨm, we never observed a loss of ΔΨm before cytochrome c release (Fig. 3). Similarly, hyperpolarization did not occur uniformly before cytochrome c release and was only observed in a small subset of cells. In support of these observations, neither cytochrome c release nor the apoptotic response was affected by the addition of uncouplers that dissipate ΔΨm and thereby prevent any hyperpolarization (Fig. 6). Indeed, the presence of these uncouplers in no way affected the function of proapoptotic or antiapoptotic Bcl-2 family members (Fig. 7). These data preclude a requirement for large-scale changes (either an increase or decrease) in ΔΨm as a general mechanism for mitochondrial outer membrane permeabilization during apoptosis.
Although there were no significant changes in ΔΨm before cytochrome c release, in our single-cell experiments we observed a caspase-independent loss of ΔΨm shortly after cytochrome c release (Fig. 3). The loss of ΔΨm after cytochrome c release that we observed in single-cell analysis appeared to contrast with our results obtained by bulk cell analysis, which showed that ΔΨm was maintained after cytochrome c release (Fig. 1; Bossy-Wetzel et al. 1998). This paradox was resolved by the observation that in the presence of a caspase inhibitor or in the absence of Apaf-1 (which prevents caspase activation through the apoptosome), ΔΨm regenerated during the 60 min after cytochrome c release and sustained subsequently (Fig. 4). Since permeabilization of the mitochondrial outer membrane occurs at different times in different cells, this loss and recovery of ΔΨm appears as an overall maintenance of ΔΨm at the bulk cell level.
We found that after cytochrome c release, the ΔΨm was maintained by the electron transport chain rather than by reversal of the ATP synthase (Fig. 4). In digitonin-treated cells, 10 μM cytochrome c was sufficient to maintain electron transport after permeabilization of the mitochondrial outer membrane during apoptosis (Fig. 2). We calculated that, if cytochrome c was evenly dispersed throughout the Cc-GFP-HeLa cells, it would result in a concentration of ∼10 μM. This is consistent with previous estimations that the concentration of cytochrome c in the intermembrane space is between 0.5 and 5 mM (Forman and Azzi 1997). Since the intermembrane space represents ∼10% of the mitochondria and mitochondria can occupy 10–30% of the total cell volume, we can estimate that the final concentration of cytochrome c throughout a cell would range between 5 and 150 μM.
Although there is sufficient cytochrome c to maintain ΔΨm after permeabilization of the mitochondrial outer membrane, we observe a transient depolarization that then recovers in the absence of caspase activity. The mechanism of this depolarization is unknown. One possibility is based on changes in respiration in response to ADP. At high ADP concentrations, mitochondria increase respiration (state 3), resulting in a decrease in ΔΨm. As the ADP is converted to ATP, the ratio of ADP/ATP decreases, and mitochondria enter state 4, with decreased respiration and increased ΔΨm.
Therefore, the drop in ΔΨm we observe after cytochrome c release may represent a shift from state 4 to 3 respiration, induced by a sudden increase in ADP/ATP. This would be followed by an increase in ΔΨm as the ADP is converted to ATP. This is consistent with a model suggesting that the voltage-dependent anion channel closes during apoptosis, and therefore ATP accumulates in the mitochondria, whereas ADP accumulates in the cytosol (Vander Heiden et al. 1999, Vander Heiden et al. 2000). Upon outer membrane permeabilization, ADP levels at the mitochondria would therefore suddenly increase. However, for these transitions to state 3 and 4 to occur, both electron transport and the function of the F0/F1 ATPase are required. Since oligomycin, which inhibits the F0/F1 ATPase, had no effect on either the drop or recovery of ΔΨm after cytochrome c release (Fig. 4), this explanation is unlikely. Another possibility is a transient opening of the adenosine nucleotide translocator, which temporarily dissipates ΔΨm. However, cyclosporin A (≤400 μM), which delays adenosine nucleotide translocator opening (Crompton et al. 1998), had no effect on the kinetics or extent of the depolarization or the recovery of ΔΨm after cytochrome c release (data not shown). A third possibility is that the decrease in cytochrome c concentration results in a transient production of reactive oxygen species that temporarily disrupt electron transport. Cytochrome c, at the high levels found in mitochondria, is a potent antioxidant (Forman and Azzi 1997), and this effect may be compromised as the levels drop upon outer membrane permeabilization.
Although we do not know the underlying mechanism of this ΔΨm depolarization, the effects of temperature on this phenomenon are potentially informative. We have previously reported that the release of cytochrome c from the mitochondria is rapid and temperature independent. That is, reduction of the temperature to 24°C did not affect the interval between initial and complete release of cytochrome c (Goldstein et al. 2000). However, we found that dissipation of ΔΨm to 10% of initial levels in cells incubated at 24°C took more than twice as long as cells incubated at 37°C. This prolonged kinetics suggests that temperature-dependent enzymatic effects are involved in the process by which ΔΨm is lost but do not play a role in cytochrome c release.
In addition to maintenance of ΔΨm, we found that in the absence of caspase activation ATP synthesis by the mitochondria (i.e., oligomycin-sensitive maintenance of ATP levels) continues in cells despite mitochondrial outer membrane permeabilization (Fig. 5). These data show that even though the outer mitochondrial membrane has been permeabilized, the mitochondrial inner membrane remains intact, the electron transport chain and ATP synthase remain functional, and the cytochrome c within the cytosol is sufficient to drive oxidative phosphorylation. These conclusions are supported by previous studies that show that isolated mitochondria treated with tBid maintain their ability to import protein after cytochrome c release (von Ahsen et al. 2000).
Various aspects of mitochondrial metabolism have been reported to impact on the apoptotic process. Apoptosis is an active (ATP-requiring) process and, if sufficient ATP is not present, the cell death deviates from an apoptotic to a necrotic phenotype (Eguchi et al. 1997; Nicotera et al. 1998). The proton gradient across the mitochondrial inner membrane is essential for the production of ATP via oxidative phosphorylation, and a reduction in this gradient will impact on the potential of the cell to produce ATP and survive. Recent studies have reported that sympathetic neurons can be rescued from caspase-independent cell death, even after mitochondrial outer membrane permeabilization and cytochrome c release, however, recovery was not possible after ΔΨm had dissipated (Deshmukh et al. 2000). Similarly, we observe an eventual loss of ΔΨm even in the presence of caspase inhibitors, and this precedes a drop in ATP and death of the cell (Fig. 5; data not shown). Before this drop, however, it remains possible that during the period over which ΔΨm is maintained, nonneuronal cells may have the potential to recover after cytochrome c release. This is consistent with the recent observations that defects in Apaf-1 or caspase-9, which should act downstream of mitochondrial outer membrane permeabilization, can contribute to oncogenic transformation (Soengas et al. 1999).
In conclusion, our results suggest that before or in the absence of caspase activation, mitochondria can maintain several functions, including the generation of ATP, and may contribute to survival of the cells for prolonged periods after cytochrome c release.
Supplemental Material
Acknowledgments
We thank Ruth Kluck and Mauro Degli Esposti for invaluable discussions and an anonymous reviewer for comments.
This work was supported by grants CA69381 and AI40646 from the National Institutes of Health.
Footnotes
The online version of this article contains supplemental material.
Oliver von Ahsen's current address is ZMBH, University of Heidelberg, Im Neuenheimer Feld 282, 69120 Heidelberg, Germany.
Abbreviations used in this paper: Apaf, apoptotic protease activating factor; CCCP, carbonyl cyanide m-chlorophenylhydrazone; Cc-GFP-HeLa, HeLa cells stably expressing GFP-tagged cytochrome c; CLAMI, cell lysis and mitochondria intact; DNP, dinitrophenol, ΔΨm, mitochondrial transmembrane potential; FCCP, carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone; GFP, green fluorescent protein; MIB, mitochondria isolation buffer; tBid, truncated Bid; TMRE, tetramethylrhodamine ethyl ester; zVADfmk, N-benzoylcarbonyl-Val-Ala-Asp-fluoromethylketone.
References
- Bossy-Wetzel E., Newmeyer D.D., Green D.R. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:37–49. doi: 10.1093/emboj/17.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner C., Cadiou H., Vieira H.L., Zamzami N., Marzo I., Xie Z., Leber B., Andrews D., Duclohier H., Reed J.C., Kroemer G. Bcl-2 and Bax regulate the channel activity of the mitochondrial adenine nucleotide translocator. Oncogene. 2000;19:329–336. doi: 10.1038/sj.onc.1203298. [DOI] [PubMed] [Google Scholar]
- Cain K., Bratton S.B., Langlais C., Walker G., Brown D.G., Sun X.M., Cohen G.M. Apaf-1 oligomerizes into biologically active approximately 700-kDa and inactive approximately 1.4-MDa apoptosome complexes. J. Biol. Chem. 2000;275:6067–6070. doi: 10.1074/jbc.275.9.6067. [DOI] [PubMed] [Google Scholar]
- Crompton M., Virji S., Ward J.M. Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur. J. Biochem. 1998;258:729–735. doi: 10.1046/j.1432-1327.1998.2580729.x. [DOI] [PubMed] [Google Scholar]
- Deshmukh M., Kuida K., Johnson E.M., Jr. Caspase inhibition extends the commitment to neuronal death beyond cytochrome c release to the point of mitochondrial depolarization. J. Cell Biol. 2000;150:131–143. doi: 10.1083/jcb.150.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C., Fang M., Li Y., Li L., Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Eguchi Y., Shimizu S., Tsujimoto Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997;57:1835–1840. [PubMed] [Google Scholar]
- Farkas D.L., Wei M.D., Febbroriello P., Carson J.H., Loew L.M. Simultaneous imaging of cell and mitochondrial membrane potentials Biophys. J 56 1989. 1053 1069 (erratum published in 57:684) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink C., Morgan F., Loew L.M. Intracellular fluorescent probe concentrations by confocal microscopy. Biophys. J. 1998;75:1648–1658. doi: 10.1016/S0006-3495(98)77607-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman H.J., Azzi A. On the virtual existence of superoxide anions in mitochondriathoughts regarding its role in pathophysiology. FASEB J. 1997;11:374–375. doi: 10.1096/fasebj.11.5.9141504. [DOI] [PubMed] [Google Scholar]
- Goldstein J.C., Waterhouse N.J., Juin P., Evan G.I., Green D.R. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat. Cell Biol. 2000;2:156–162. doi: 10.1038/35004029. [DOI] [PubMed] [Google Scholar]
- Gross A., McDonnell J.M., Korsmeyer S.J. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- Haraguchi M., Torii S., Matsuzawa S., Xie Z., Kitada S., Krajewski S., Yoshida H., Mak T.W., Reed J.C. Apoptotic protease activating factor 1 (Apaf-1)–independent cell death suppression by Bcl-2. J. Exp. Med. 2000;191:1709–1720. doi: 10.1084/jem.191.10.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huser J., Blatter L.A. Fluctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore Biochem. J 343Pt 21999. 311 317 [PMC free article] [PubMed] [Google Scholar]
- Ichas F., Jouaville L.S., Mazat J.P. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell. 1997;89:1145–1153. doi: 10.1016/s0092-8674(00)80301-3. [DOI] [PubMed] [Google Scholar]
- Kluck R.M., Bossy-Wetzel E., Green D.R., Newmeyer D.D. The release of cytochrome c from mitochondriaa primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Kluck R.M., Esposti M.D., Perkins G., Renken C., Kuwana T., Bossy-Wetzel E., Goldberg M., Allen T., Barber M.J., Green D.R., Newmeyer D.D. The pro-apoptotic proteins, Bid and Bax, cause a limited permeabilization of the mitochondrial outer membrane that is enhanced by cytosol. J. Cell Biol. 1999;147:809–822. doi: 10.1083/jcb.147.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler C., Gahm A., Noma T., Nakazawa A., Orrenius S., Zhivotovsky B. Release of adenylate kinase 2 from the mitochondrial intermembrane space during apoptosis. FEBS Lett. 1999;447:10–12. doi: 10.1016/s0014-5793(99)00251-3. [DOI] [PubMed] [Google Scholar]
- Li P., Nijhawan D., Budihardjo I., Srinivasula S.M., Ahmad M., Alnemri E.S., Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Liu X., Kim C.N., Yang J., Jemmerson R., Wang X. Induction of apoptotic program in cell-free extractsrequirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Martinou I., Desagher S., Eskes R., Antonsson B., Andre E., Fakan S., Martinou J.C. The release of cytochrome c from mitochondria during apoptosis of NGF-deprived sympathetic neurons is a reversible event. J. Cell Biol. 1999;144:883–889. doi: 10.1083/jcb.144.5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzo I., Brenner C., Zamzami N., Jurgensmeier J.M., Susin S.A., Vieira H.L., Prevost M.C., Xie Z., Matsuyama S., Reed J.C., Kroemer G. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis Science 281 1998. 2027 2031a [DOI] [PubMed] [Google Scholar]
- Marzo I., Brenner C., Zamzami N., Susin S.A., Beutner G., Brdiczka D., Remy R., Xie Z.H., Reed J.C., Kroemer G. The permeability transition pore complexa target for apoptosis regulation by caspases and bcl-2-related proteins J. Exp. Med 187 1998. 1261 1271b [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy N.J., Whyte M.K., Gilbert C.S., Evan G.I. Inhibition of Ced-3/ICE–related proteases does not prevent cell death induced by oncogenes, DNA damage, or the Bcl-2 homologue Bak. J. Cell Biol. 1997;136:215–227. doi: 10.1083/jcb.136.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicotera P., Leist M., Ferrando-May E. Intracellular ATP, a switch in the decision between apoptosis and necrosis. Toxicol. Lett. 1998;102–103:139–142. doi: 10.1016/s0378-4274(98)00298-7. [DOI] [PubMed] [Google Scholar]
- Shimizu S., Ide T., Yanagida T., Tsujimoto Y. Electrophysiological study of a novel large pore formed by Bax and the voltage-dependent anion channel that is permeable to cytochrome c. J. Biol. Chem. 2000;275:12321–12325. doi: 10.1074/jbc.275.16.12321. [DOI] [PubMed] [Google Scholar]
- Simbula G., Glascott P.A., Jr., Akita S., Hoek J.B., Farber J.L. Two mechanisms by which ATP depletion potentiates induction of the mitochondrial permeability transition. Am. J. Physiol. 1997;273:C479–C488. doi: 10.1152/ajpcell.1997.273.2.C479. [DOI] [PubMed] [Google Scholar]
- Soengas M.S., Alarcon R.M., Yoshida H., Giaccia A.J., Hakem R., Mak T.W., Lowe S.W. Apaf-1 and caspase-9 in p53-dependent apoptosis and tumor inhibition. Science. 1999;284:156–159. doi: 10.1126/science.284.5411.156. [DOI] [PubMed] [Google Scholar]
- Srinivasula S.M., Ahmad M., Fernandes-Alnemri T., Alnemri E.S. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol. Cell. 1998;1:949–957. doi: 10.1016/s1097-2765(00)80095-7. [DOI] [PubMed] [Google Scholar]
- Vander Heiden M.G., Thompson C.B. Bcl-2 proteinsregulators of apoptosis or of mitochondrial homeostasis? Nat. Cell Biol. 1999;1:E209–E216. doi: 10.1038/70237. [DOI] [PubMed] [Google Scholar]
- Vander Heiden M.G., Chandel N.S., Schumacker P.T., Thompson C.B. Bcl-xL prevents cell death following growth factor withdrawal by facilitating mitochondrial ATP/ADP exchange. Mol. Cell. 1999;3:159–167. doi: 10.1016/s1097-2765(00)80307-x. [DOI] [PubMed] [Google Scholar]
- Vander Heiden M.G., Chandel N.S., Li X.X., Schumacker P.T., Colombini M., Thompson C.B. Outer mitochondrial membrane permeability can regulate coupled respiration and cell survival. Proc. Natl. Acad. Sci. USA. 2000;97:4666–4671. doi: 10.1073/pnas.090082297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhagen A.M., Ekert P.G., Pakusch M., Silke J., Connolly L.M., Reid G.E., Moritz R.L., Simpson R.J., Vaux D.L. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- von Ahsen O., Renken C., Perkins G., Kluck R.M., Bossy-Wetzel E., Newmeyer D.D. Preservation of mitochondrial structure and function after Bid- or Bax-mediated cytochrome c release. J. Cell Biol. 2000;150:1027–1036. doi: 10.1083/jcb.150.5.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse N.J., Green D.R. Mitochondria and apoptosisHQ or high-security prison? J. Clin. Immunol. 1999;19:378–387. doi: 10.1023/a:1020550716138. [DOI] [PubMed] [Google Scholar]
- Wei M.C., Lindsten T., Mootha V.K., Weiler S., Gross A., Ashiya M., Thompson C.B., Korsmeyer S.J. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000;14:2060–2071. [PMC free article] [PubMed] [Google Scholar]
- Yang J., Liu X., Bhalla K., Kim C.N., Ibrado A.M., Cai J., Peng T.I., Jones D.P., Wang X. Prevention of apoptosis by Bcl-2release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- Zou H., Li Y., Liu X., Wang X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 1999;274:11549–11556. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.