

Abstract
Agrin released from motor nerve terminals activates a muscle-specific receptor tyrosine kinase (MuSK) in muscle cells to trigger formation of the skeletal neuromuscular junction. A key step in synaptogenesis is the aggregation of acetylcholine receptors (AChRs) in the postsynaptic membrane, a process that requires the AChR-associated protein, rapsyn. Here, we mapped domains on MuSK necessary for its interactions with agrin and rapsyn. Myotubes from MuSK −/− mutant mice form no AChR clusters in response to agrin, but agrin-responsiveness is restored by the introduction of rat MuSK or a Torpedo orthologue. Thus, MuSK −/− myotubes provide an assay system for the structure–function analysis of MuSK. Using this system, we found that sequences in or near the first of four extracellular immunoglobulin-like domains in MuSK are required for agrin responsiveness, whereas sequences in or near the fourth immunoglobulin-like domain are required for interaction with rapsyn. Analysis of the cytoplasmic domain revealed that a recognition site for the phosphotyrosine binding domain–containing proteins is essential for MuSK activity, whereas consensus binding sites for the PSD-95/Dlg/ZO-1-like domain–containing proteins and phosphatidylinositol-3-kinase are dispensable. Together, our results indicate that the ectodomain of MuSK mediates both agrin- dependent activation of a complex signal transduction pathway and agrin-independent association of the kinase with other postsynaptic components. These interactions allow MuSK not only to induce a multimolecular AChR-containing complex, but also to localize that complex to a primary scaffold in the postsynaptic membrane.
Keywords: MuSK, acetylcholine receptors, neuromuscular junction, synapse, agrin
During formation of the vertebrate neuromuscular junction, motor axons release the proteoglycan agrin to trigger differentiation of the underlying postsynaptic membrane (McMahan 1990; Gautam et al. 1996; Cohen et al. 1997; Jones et al. 1997; Burgess et al. 1999; for review see Burden 1998; Sanes and Lichtman 1999). The best studied step in this program of differentiation is the aggregation of acetylcholine receptors (AChRs)1 directly beneath the nerve terminal. An effector of the aggregation process is a cytoplasmic AChR-associated protein called rapsyn; rapsyn induces aggregation of AChRs when both are expressed in heterologous cells, and no AChR clusters form in mutant mice that lack rapsyn (Froehner et al. 1990; Phillips et al. 1991a,Phillips et al. 1991b; Apel et al. 1995; Gautam et al. 1995). Thus, agrin may act in part by promoting interactions of AChRs with rapsyn.
A critical component in the signal transduction pathway that leads from agrin to rapsyn is a muscle-specific receptor tyrosine kinase called MuSK (Valenzuela et al. 1995). MuSK is concentrated, along with AChRs and rapsyn, in the postsynaptic apparatus. Although agrin can interact with numerous components on the myotube surface (for review see Sanes et al. 1998), MuSK became the leading candidate agrin receptor with the observation that MuSK −/− mice, like agrin −/− and rapsyn −/− mice, fail to form neuromuscular junctions (deChiara et al. 1996). Subsequent studies in vitro supported MuSK's candidacy: agrin rapidly stimulated MuSK phosphorylation in cultured myotubes; the introduction of a dominant negative mutant MuSK inhibited agrin's ability to form AChR clusters; only isoforms or fragments of agrin that activated MuSK induced AChR clustering; and chemical cross-linking showed specific interaction of agrin with MuSK (Glass et al. 1996, Glass et al. 1997; Meier et al. 1996; Fuhrer et al. 1997; Hopf and Hoch 1998a,Hopf and Hoch 1998b).
Although genetic and biochemical evidence has established MuSK as a component of the agrin receptor, it remains unclear how agrin interacts with MuSK and how MuSK interacts with rapsyn. Agrin can be cross-linked to MuSK on the surface of myotubes, but direct binding of agrin to purified MuSK has not been demonstrable, suggesting that an accessory component (called MASC, for muscle-associated specificity component; Glass et al. 1996) is required. Moreover, MuSK is unusual among receptor tyrosine kinases in that ligand-dependent activation is insufficient to mediate its effects: a chimera composed of the neurotrophin receptor (trkC kinase) ectodomain fused to the MuSK cytoplasmic domain is activated by neurotrophin, but does not induce AChR clustering (Glass et al. 1997; Jones et al. 1999). Thus, the MuSK ectodomain appears to play roles in addition to that of ligand binding. One possible role is the interaction of MuSK with rapsyn. Indeed, although MuSK and rapsyn cocluster in heterologous cells (Gillespie et al. 1996), this association requires extracellular MuSK sequences, suggesting that the ectodomain interacts with a rapsyn-associated transmembrane linking molecule (RATL; Apel et al. 1997). By virtue of this linkage, AChR–rapsyn complexes could be induced by and become associated with agrin/MASC/MuSK complexes, thereby forming a postsynaptic apparatus.
To begin to understand how MuSK functions, we wanted to define critical regions of its extracellular and cytoplasmic domains. To this end, we devised an assay system using a myogenic cell line derived from MuSK −/− mice. Agrin did not induce AChR clustering in MuSK −/− myotubes, but agrin sensitivity was restored by introduction of wild-type MuSK. Therefore, we were able to test the ability of a series of mutant MuSK constructs to induce AChR clustering and to coaggregate with rapsyn and AChRs. We show that distinct portions of the MuSK ectodomain are necessary for interactions with agrin/MASC and rapsyn/RATL. Within the cytoplasmic domain, a recognition site for phosphotyrosine binding (PTB) domain–containing proteins is essential for activity, whereas a recognition site for PSD-95/Dlg/ZO-1-like (PDZ) domain–containing proteins is dispensable. Based on these and previous results, we propose a model in which the ectodomain of MuSK not only mediates ligand-dependent activation of a complex signal transduction pathway, but also directs ligand-independent localization of a multimolecular AChR-containing complex to the postsynaptic membrane.
Materials and Methods
Mutagenesis of MuSK
Expression vectors encoding rat MuSK, rat trkC, and a rat MuSK-rat trkC chimera were described previously (Apel et al. 1997). In all three vectors, the cDNA is expressed under the control of the Rous Sarcoma virus long terminal repeat. In the chimera, the extracellular domain of MuSK except for the last three amino acids before the predicted transmembrane domain (amino acids 1–492) is followed by 32 amino acids of the extracellular domain plus the complete transmembrane and cytoplasmic domains of rat trkC. In the trkC and MuSK-trkC constructs, a myc tag consisting of three tandem copies of a 12–amino acid myc epitope was fused in-frame at the carboxy-terminal of the coding region.
MuSK and the MuSK-trkC chimera are called construct 1 and construct 1T, respectively. Constructs 2–6 are identical to construct 1, and constructs 2T, 5T, 6T, 14T, and 15T are identical to construct 1T except that in each case a fragment of MuSK was deleted. Deletions, constructed by PCR, were as follows: 2 and 2T, amino acids 25–112, inclusive; 3, amino acids 115–204; 4, amino acids 205–299; 5 and 5T, amino acids 299–396; 6 and 6T, amino acids 397–484; 14T, 25–204; and 15T, amino acids 25–299.
Constructs 7–13 were derived from a vector in which the rat MuSK cDNA was expressed under the control of late adenovirus promoter, and a Srf I site was engineered in-frame just upstream of the transmembrane domain. Deletions were constructed by PCR, removing successively larger fragments of the ectodomain as follows: 7, amino acids 447–492; 8, amino acids 398–492; 9, amino acids 283–492; 10, 233–492; 11, amino acids 191–492; 12, amino acids 142–492; and 13, amino acids 99–492.
In constructs 16–25, the ectodomain of MuSK was intact, but portions of the cytoplasmic domain were mutated or deleted. The mutations, generated by mutagenesis, were as follows: 16, Lys608 was replaced by Ala; 17, Tyr553 was replaced by Phe; 18, Asp545-Arg546-Leu547-His548 and Pro549 were replaced by Leu-Tyr-Leu-Ser-Ser; 19, amino acids 545–549 were replaced by Ile-Pro-Ile-Leu-Glu; 24, Tyr831 was replaced by Phe; and 25, Val866 Gly867 and Val868 were deleted. The deletions, constructed by PCR, were as follows: construct 20, amino acids 515–594, inclusive; 21, amino acids 655–868; 22, amino acids 721–868; and construct 23, amino acids 799–868.
The coding sequence of a tyrosine kinase from Torpedo electric organ (Jennings et al. 1993) was inserted in place of rat MuSK in construct 7. In all constructs, ligation junctions were sequenced to confirm correct insertion and maintenance of the reading frame.
Cell Culture
MuSK +/− mice were bred with transgenic mice bearing a temperature-sensitive SV40 T antigen under the control of an interferon-inducible promoter (Jat et al. 1991). MuSK +/−, transgene-positive offspring were backcrossed to MuSK +/− mice and a myogenic line was established from hindlimb muscles of a MuSK −/− transgene-positive embryonic day 18 pup (Sugiyama et al. 1997) using the methods detailed by Donoghue et al. 1992. Cells were cultured as myoblasts at 33°C in DME containing 10% heat-inactivated FCS, glutamine, penicillin, streptomycin, and 20 U/ml γ-interferon (R&D Systems, Inc.). To induce formation of myotubes, cultures were transferred to 37°C and the medium was replaced with DME containing 2% horse serum, glutamine, and antibiotics, but no FCS or interferon. Cells were transfected as myoblasts with Fugene6 reagent (Roche Molecular Biochemical), used according to the manufacturer's instructions. Fusion was initiated 1 d after transfection and cultures were harvested 5–7 d later. Between 5 and 20% of myotubes expressed the exogenous protein, as assessed by immunostaining.
For immunoblotting analysis, the cells were cultured on 10-cm plates. In some experiments, a recombinant carboxy-terminal fragment of agrin (∼100 nM; y = 4, z = 8 form; Ferns et al. 1993) was added to the cultures for 10 min before harvesting. For immunohistochemistry, cells were cultured on 13-mm-diam glass coverslips. Where specified, the recombinant agrin fragment was added to the cultures for 18 h before harvesting.
The quail fibroblast cell line QT-6 (Moscovici et al. 1977) was obtained from the American Type Culture Collection and cultured in DME containing 10% FCS, 1% DMSO, penicillin, and streptomycin. Cells were cultured on glass coverslips and transfected using the calcium phosphate method described by Phillips et al. 1991b.
Immunoblotting
Cultured cells were lysed and solubilized in NP-40, the extracts were subjected to immunoprecipitation with antibodies to MuSK and the resulting precipitates were immunoblotted with antibodies to phosphotyrosine (4G10; Upstate Biotechnology Inc.). After detection of the phosphorylated species, the immunoblots were stripped and reprobed with antiserum to the MuSK cytoplasmic domain (see below) to ascertain total MuSK levels.
Immunohistochemistry
MuSK −/− myotubes were incubated live for 1 h with rhodamine-α-bungarotoxin (rBTX; Molecular Probes, Inc.), washed, and fixed for 20 min at room temperature in 2% paraformaldehyde in PBS. In some experiments, cells were permeabilized by incubation for 10 min in 1% Triton X-100. Nonspecific binding sites were blocked by overnight incubation with 10% FCS at 4°C and then cultures were incubated sequentially with antibodies to MuSK (for 2 h) and fluorescein-conjugated second antibody (for 1 h). AChR clusters were counted with rhodamine illumination using a 40× oil objective. 10 fields were chosen at random on each coverslip, all clusters >3 μm in diameter were counted, and this number was divided by the number of myotubes that crossed the field.
QT-6 cells were rinsed in PBS, fixed in 1% paraformaldehyde, 100 mM l-lysine, and 10 mM sodium metaperiodate, rinsed again, and permeabilized with 1% Triton X-100. They were incubated with a mixture of mouse anti-rapsyn (Peng and Froehner 1985) and rabbit anti-MuSK, washed, and reincubated with a mixture of fluorescein- and rhodamine-conjugated second antibodies.
Rabbit antisera were generated to the cytoplasmic domain (serum 41101K) and to the ectodomain of rat MuSK, using standard techniques (Glass et al. 1996). Antiserum to a soluble fusion between the ectodomain of mouse MuSK and immunoglobulin Fc segment (Meier et al. 1997) was a gift of M. Ruegg (Basel, Switzerland).
Results
An Assay for MuSK Function
When myoblasts fuse to form myotubes, AChR subunit genes are activated and AChRs synthesized. Most of the receptors are diffusely distributed at low density on the myotube surface, but some form high density aggregates, which are readily visualized with the selective ligand rBTX (Fig. 1 a). Addition of a recombinant carboxy-terminal fragment of agrin increased the number of such AChR clusters ∼10-fold (Fig. 1 b). In contrast, myoblasts derived from MuSK −/− mutant mice form myotubes that are unresponsive to agrin, even though they bear normal levels of diffuse AChRs (Glass et al. 1996). Likewise, myotubes of an immortalized myogenic cell line derived from MuSK −/− muscle did not form AChR clusters, either spontaneously or in response to agrin (Fig. 1c and Fig. d). These results are consistent with the notion that MuSK is a critical component of the agrin receptor and that even spontaneous AChR clusters are dependent on MuSK.
Figure 1.
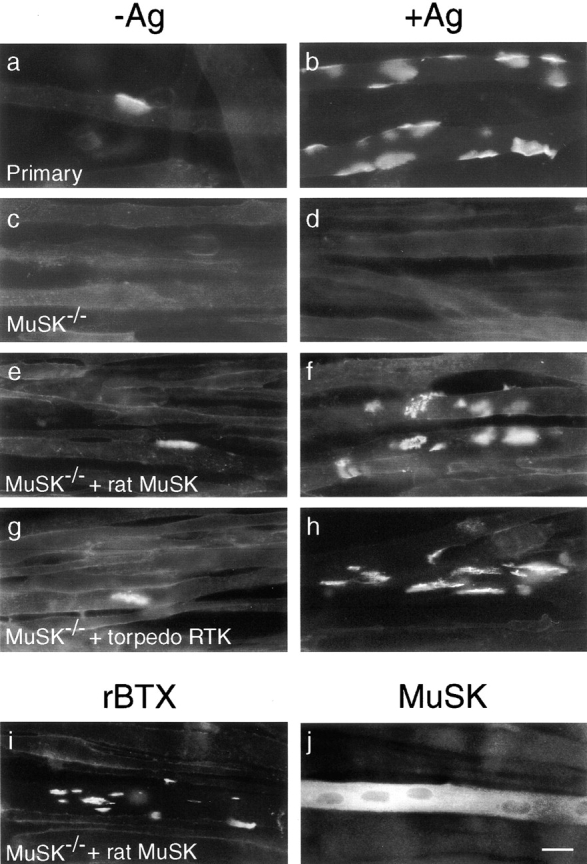
MuSK restores the ability of MuSK−/− myotubes to cluster AChRs spontaneously and in response to agrin. Myoblasts from wild-type mice (a and b) or from a MuSK −/− myogenic cell line (c–j) were fused to form myotubes, treated with agrin (b, d, f, and h–j) or left untreated (a, c, e, and g) for 18 h, and then stained with rBTX (a–i) or anti-MuSK (j). (a and b) Wild-type myotubes formed small numbers of AChR clusters spontaneously and many more after treatment with recombinant agrin. (c and d) MuSK −/− myotubes formed neither spontaneous nor agrin-induced AChR clusters. (e and f) Transfection of MuSK −/− cells with rat MuSK restores their ability to form small numbers of spontaneous and large numbers of agrin-induced AChR clusters. (g and h) Transfection with a Torpedo receptor tyrosine kinase also rescues spontaneous and agrin-induced clustering, suggesting that this MuSK homologue is also a MuSK orthologue. (i and j) After transfection with rat MuSK and agrin treatment, this culture was doubly stained with rBTX and anti-MuSK. AChR clusters formed on the MuSK-positive myotube, but remained diffusely distributed on adjacent, MuSK-negative myotubes. Bar, 20 μm.
To establish an assay for MuSK function, we transfected MuSK −/− myoblasts with an expression vector encoding rat MuSK, induced fusion, incubated the myotubes with or without agrin, then stained them with rBTX. MuSK-transfected myotubes formed a small number of clusters in the absence of exogenous agrin, and approximately eightfold more clusters in the presence of agrin (Fig. 1e and Fig. f). AChR clusters were present only on those myotubes that had been transfected and were expressing MuSK (Fig. 1i and Fig. j), and over 80% of MuSK-positive myotubes in the agrin-treated cultures bore AChR clusters (data not shown). Thus, spontaneous as well as agrin-induced AChR clustering requires MuSK. Because MuSK −/− myotubes form AChR clusters in response to other, possibly nonphysiological, clustering agents (Sugiyama et al. 1997; Gautam et al. 1999), we believe that MuSK is not required as a structural component of AChR clusters. Instead, spontaneous clustering is likely to result from a low level of MuSK activation, either by an endogenous ligand or in the absence of ligand. Data that distinguish these alternatives are presented below.
To test the specificity of the response to agrin, we introduced two other kinases into MuSK −/− myotubes: rat trkC (Lamballe et al. 1991) and a receptor tyrosine kinase isolated from Torpedo electric organ (Jennings et al. 1993). TrkC, the receptor for neurotrophin 3 is homologous to MuSK in its cytoplasmic domain, but unrelated in its extracellular domain. In contrast, the Torpedo kinase is homologous to MuSK throughout its length and is selectively expressed in muscle; therefore, it is a plausible MuSK orthologue. Myotubes transfected with trkC formed no AChR clusters either spontaneously or in response to neurotrophin 3, whereas the Torpedo kinase endowed MuSK −/− myotubes with the ability to form AChR clusters spontaneously and in response to agrin (Fig. 1g and Fig. h, and data not shown). This result, combined with the inactivity of trkC in this assay, supports the presumption that the Torpedo kinase is orthologous to MuSK; we call it Torpedo MuSK hereafter. Together, these results confirm a specific requirement of MuSK for agrin signaling. More important for the present study, the ability of MuSK to restore agrin sensitivity to MuSK −/− cells provides an assay system to determine the relationship between MuSK's structure and its function.
A Domain Required for Agrin to Activate MuSK
The extracellular segment of MuSK contains five distinct domains: four immunoglobulin-like domains and a cysteine rich region called a C6 box. All five domains are conserved in sequence and arrangement in rat, mouse, human, Xenopus, chicken, and Torpedo MuSK (Jennings et al. 1993; Ganju et al. 1995; Valenzuela et al. 1995; Fu et al. 1999; D.J. Glass, and G.D. Yancopoulos, unpublished). To determine which portions of the MuSK ectodomain are required for its activation, we generated the mutants diagrammed in Fig. 2 a. In a first series (constructs 2–6), sequences were deleted that encoded either the first, second, or third immunoglobulin-like domain, the C6 box, or the fourth immunoglobulin-like domain. In a second series (constructs 7–13), nested deletions extended variable distances toward the amino terminus from a common site just upstream of the predicted transmembrane domain. Each mutant, as well as wild-type MuSK (construct 1), was transfected into MuSK −/− myoblasts, which were fused, treated, and stained as described above.
Figure 2.

Amino-terminal regions in the MuSK ectodomain are required to mediate agrin-dependent AChR clustering. (a) Mutant constructs. The top line shows motifs in the MuSK ectodomain, including four immunoglobulin-like domains (Ig I–IV) and a region containing six phylogenetically conserved cysteine residues (C6). Subsequent lines show structures of wild-type and mutant constructs tested by expression in MuSK −/− myotubes. To the right of each construct is indicated whether or not it rescued the ability of MuSK −/− myotubes to form AChR clusters in the absence of agrin (−Ag) or to form additional clusters in the presence of agrin (+Ag). All mutants permitted spontaneous clustering (+), but mutants lacking amino-terminal regions (Ig-I and -II) were unable to respond to agrin (=, spontaneous level of clustering; ↑, reduced relative to wild-type; ↑↑, wild-type level of clustering). (b–g) Examples of AChR clusters on MuSK−/−myotubes that had been transfected with mutant constructs, treated, and stained as in Fig. 1. (b and c) Construct 2. (d and e) Construct 3. (f and g) Construct 10. (h) Quantitation of the extent to which MuSK constructs 1, 2, and 3 rescued the ability of MuSK −/− myotubes to form AChR clusters in the absence or presence of agrin. Cultures were doubly stained with rBTX and anti-MuSK as in Fig. 1 (i and j), and only MuSK-positive (i.e., successfully transfected) myotubes were scored. Bar is 20 μm.
Four principal results were obtained from this series of experiments. First, all of the constructs restored the ability of MuSK −/− myotubes to form spontaneous AChR clusters (Fig. 2b, Fig. d, and Fig. f). Because the deletions span the entire ectodomain, we conclude that spontaneous clustering reflects ligand-independent activation of MuSK rather than activation by an endogenous ligand.
Second, all of the mutants with deletions confined to the carboxy-terminal three fifths of the ectodomain were able to mediate agrin-induced AChR clustering (Fig. 2f and Fig. g, constructs 4–11). We did not rigorously control for differences in transfection efficiency, but conclude that differences between any of these mutants and wild-type MuSK were less than twofold. Thus, the third and fourth immunoglobulin domains and the C6 box are dispensable for agrin-mediated AChR clustering. Interestingly, Hesser et al. 1999 recently reported on the occurrence of a naturally occurring splice variant of MuSK that lacks the third immunoglobulin-like repeat, yet retains the ability to induce AChR clustering.
Third, mutants with deletions in the amino-terminal two fifths of the ectodomain were markedly impaired in their ability to mediate agrin-induced AChR clustering (Fig. 2, b–e, constructs 2, 3, 12, and 13). This inability did not result from failure of these mutants to reach the cell surface, because staining of nonpermeabilized cells with antibodies to MuSK showed clear immunoreactivity (see below). Thus, sequences in or near the first or second immunoglobulin-like domain interact, directly or indirectly, with agrin.
Fourth, whereas mutants lacking sequences within the first immunoglobulin-like domain (constructs 2 and 13) were completely agrin-insensitive, agrin had limited ability to stimulate AChR clustering (∼2.5-fold above spontaneous levels) in myotubes transfected with mutants lacking the second immunoglobulin-like domain (Fig. 2 h, constructs 3 and 12, and data not shown; only transfected [MuSK-positive] myotubes were scored). The difference between constructs 2 and 13 on the one hand and constructs 3 and 12 on the other indicates that sequences in or near immunoglobulin-like domain 1 are sufficient to endow MuSK with agrin responsiveness.
Agrin-dependent Tyrosine Phosphorylation of MuSK Ectodomain Mutants
Previous studies have shown that agrin causes rapid phosphorylation of MuSK, and that this phosphorylation is required for MuSK to mediate agrin's effects (Glass et al. 1996, Glass et al. 1997; Meier et al. 1996). If this is so, MuSK ectodomain mutants able to mediate agrin-induced AChR clustering should exhibit agrin-dependent phosphorylation. To test this prediction, we assayed agrin-dependent phosphorylation of MuSK. Wild-type or mutant MuSK was precipitated from lysates of transfected MuSK −/− cells with an antibody to an intracellular epitope, which was present in all mutants. Precipitated material was separated by gel electrophoresis, transferred to nitrocellulose, and probed with antiphosphotyrosine antibody. The blots were stripped and reprobed with anti-MuSK, to permit normalization to MuSK protein levels and to confirm the electrophoretic mobility of the mutants.
As shown in Fig. 3, MuSK was undetectable in untransfected MuSK −/− cells, but readily detectable in transfected cells. Wild-type MuSK was phosphorylated at a low level in the absence of added agrin, and its phosphorylation was increased >10-fold 10 min after the addition of agrin to the cells (lanes 3 and 4). Tyrosine phosphorylation of construct 11 (which lacks immunoglobulin-like domains 3 and 4 and the C6 box yet remains responsive to agrin) was also greatly stimulated by agrin (lanes 5 and 6). In contrast, construct 13, which lacks most of the ectodomain and is agrin unresponsive, was highly phosphorylated in the absence of agrin and showed only slightly increased phosphorylation after treatment with agrin (lanes 7 and 8). This result raises the possibility that the ectodomain of MuSK keeps the cytoplasmic domain inactive in the absence of agrin; ligand binding would alter the conformation of the ectodomain to relieve the inhibition.
Figure 3.
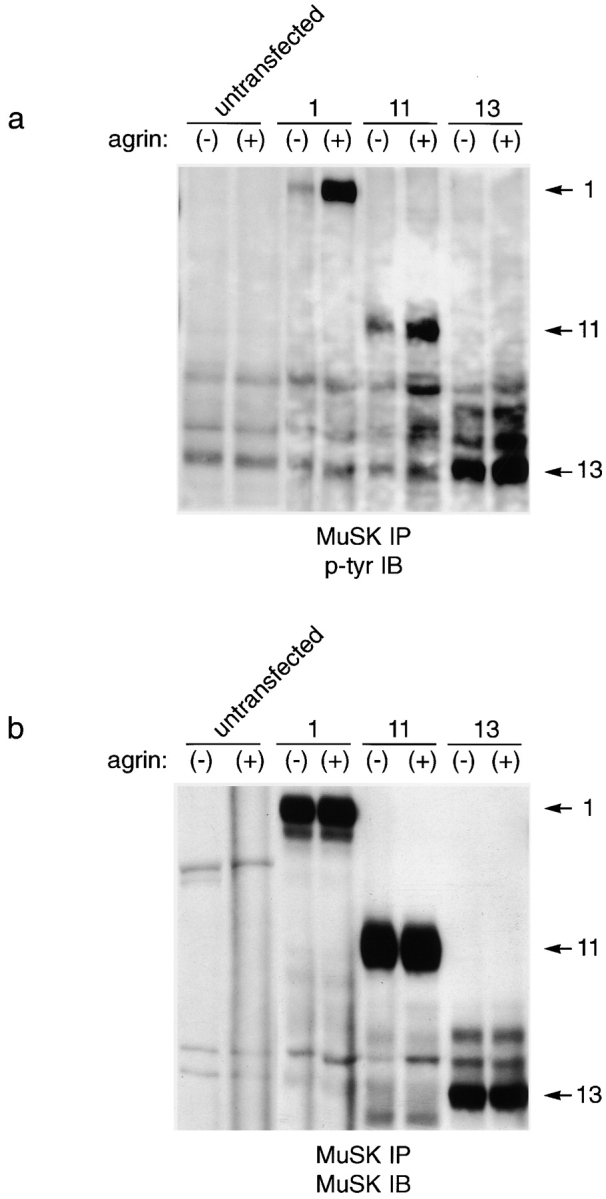
Tyrosine phosphorylation of MuSK ectodomain mutants. MuSK −/− myotubes were transfected with the indicated constructs (Fig. 2 a), treated with agrin for 10 min (+) or left untreated (−), and then subjected to immunoprecipitation with anti-MuSK. Immunoblots were probed with antiphosphotyrosine (a), stripped, and reprobed with anti-MuSK (b). (lanes 1 and 2) Untransfected myoblasts; (lanes 3 and 4) myoblasts transfected with wild-type MuSK (construct 1); (lanes 5 and 6) myoblasts transfected with construct 11; and (lanes 7 and 8) myoblasts transfected with construct 13. Wild-type MuSK and construct 11, both of which mediate agrin-induced AChR clustering, also show agrin-dependent tyrosine phosphorylation. Mutant 13, which does not mediate agrin-dependent tyrosine phosphorylation, shows a high level of agrin-independent phosphorylation but only slight agrin-dependent phosphorylation.
A Domain Required for MuSK to Cocluster with Rapsyn
MuSK and rapsyn cocluster when coexpressed in QT6 fibroblasts (Gillespie et al. 1996; Apel et al. 1997). Surprisingly, even though rapsyn is a cytoplasmic protein, it is the ectodomain of MuSK that is essential for this association: chimeras composed of the ectodomain of MuSK and the cytoplasmic domain of trkC cocluster with rapsyn in QT6 cells, whereas chimeras containing the ectodomain of trkC and the cytoplasmic domain of MuSK do not (Apel et al. 1997). Therefore, we hypothesized that RATL mediates the association.
We asked whether the portions of MuSK required for association with rapsyn were distinguishable from those required for activation by agrin. For this purpose, we modified construct 2, which lacks the first immunoglobulin-like domain and is agrin unresponsive. In construct 2T (Fig. 4 a), the ectodomain from construct 2 was fused to the transmembrane and cytoplasmic domains of trkC, to exclude any contribution of cytoplasmic MuSK sequences to its localization. We used trkC as a negative control and the chimera between wild-type MuSK and trkC (construct 1T) as a positive control. These constructs were transfected into QT-6 cells either alone or with an expression vector encoding rapsyn. 2 d later, cells were fixed, permeabilized, and doubly stained with antibodies specific for rapsyn and MuSK or trkC. Rapsyn formed small aggregates when transfected by itself, whereas construct 1T, construct 2T, and trkC were all diffusely distributed when introduced alone (Fig. 4b and Fig. c, and data not shown). Cotransfection of rapsyn and trkC had no effect on the distribution of either component, whereas cotransfection of rapsyn with constructs 1T or 2T led to nearly perfect colocalization of the two components in most cells (Fig. 4, d–g, and data not shown). No differences were detected between constructs 1T and 2T in this assay. Thus, sequences required for activation by agrin are not required for colocalization with rapsyn.
Figure 4.
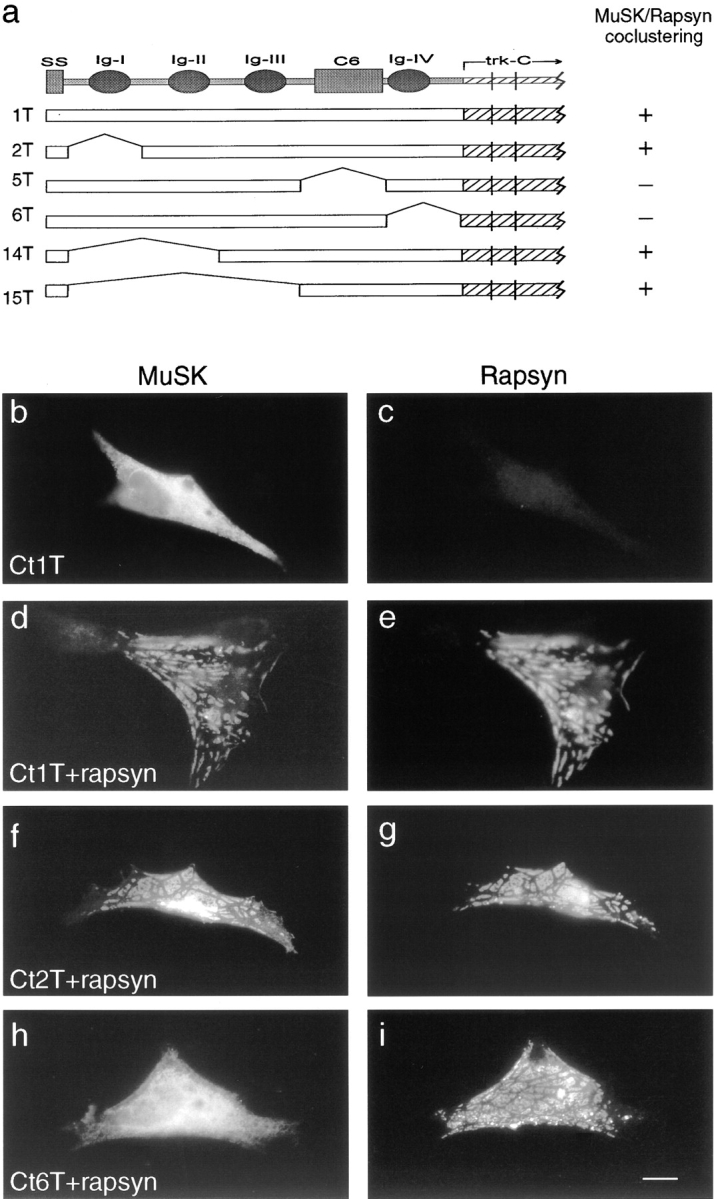
Carboxy-terminal regions of the MuSK ectodomain are required for association with rapsyn in heterologous cells. (a) MuSK constructs tested for their ability to cocluster with rapsyn in QT-6 cells. In all constructs, the ectodomain was derived from MuSK and the transmembrane and cytoplasmic domains were derived from TrkC. Abbreviations are as in Fig. 2, and areas of the MuSK ectodomain included in constructs 1T, 2T, 5T, and 6T are identical to those in 1, 2, 5, and 6, respectively, in Fig. 2. To the right of each construct is indicated whether it coclustered with rapsyn in QT-6 cells. (b–i) Localization of MuSK and rapsyn in cells transfected with expression vectors encoding MuSK construct 1T alone, (b and c) rapsyn plus MuSK constructs that cocluster well (d and e, 1T; f and g, 2T), or rapsyn plus a MuSK construct that does not cocluster (h and i, 6T). Cells were doubly stained with antibodies specific for MuSK (b, d, f, and h) and rapsyn (c, e, g, and i). Bar, 10 μm.
Based on these results, we generated and tested constructs in which additional sequences were deleted. Constructs 14T and 15T, in which immunoglobulin-like domains 1 and 2 or 1–3, respectively, were deleted behaved like constructs 1T and 2T in this assay: they were diffusely distributed when introduced on their own, but coclustered with rapsyn when both components were expressed (Fig. 4 a). In contrast, constructs 5T and 6T, which lacked carboxy-terminal portions of the ectodomain (the C6 box and the fourth immunoglobulin-like domain, respectively), were diffusely distributed both in the absence and in the presence of rapsyn (Fig. 4h and Fig. i, and data not shown). For each construct, similar results were obtained when AChR subunits were cotransfected along with rapsyn. That is, constructs 1T, 2T, 14T, and 15T aggregated with rapsyn and AChRs, whereas 5T and 6T did not (data not shown). Thus, juxtamembranous portions of the MuSK ectodomain are required for its association with rapsyn, presumably via RATL.
Together, the results in Fig. 2 and Fig. 4 show that sequences in the MuSK ectodomain required for colocalization with rapsyn are dispensable for activation by agrin. What role might this juxtamembranous region play in muscle cells? One obvious possibility is that it is required for the association of MuSK with AChR rich aggregates. To test this hypothesis, we transfected MuSK −/− cells with MuSK mutants, and then stained nonpermeabilized cells with anti-MuSK and rBTX to determine whether the MuSK constructs reached the cell surface and whether they colocalized with AChRs. As shown in Fig. 5, constructs that contained the fourth immunoglobulin-like domain of the MuSK ectodomain (constructs 1, 2, 4, and 5) were concentrated at AChR clusters. In contrast, the two constructs that lacked sequences within the fourth immunoglobulin-like domain (constructs 6 and 7) were present on the myotube surface, but not concentrated at significantly higher levels in AChR rich than in AChR poor areas. Thus, the domain required for MuSK to associate with rapsyn in QT-6 cells is required for association with rapsyn–AChR clusters in myotubes.
Figure 5.
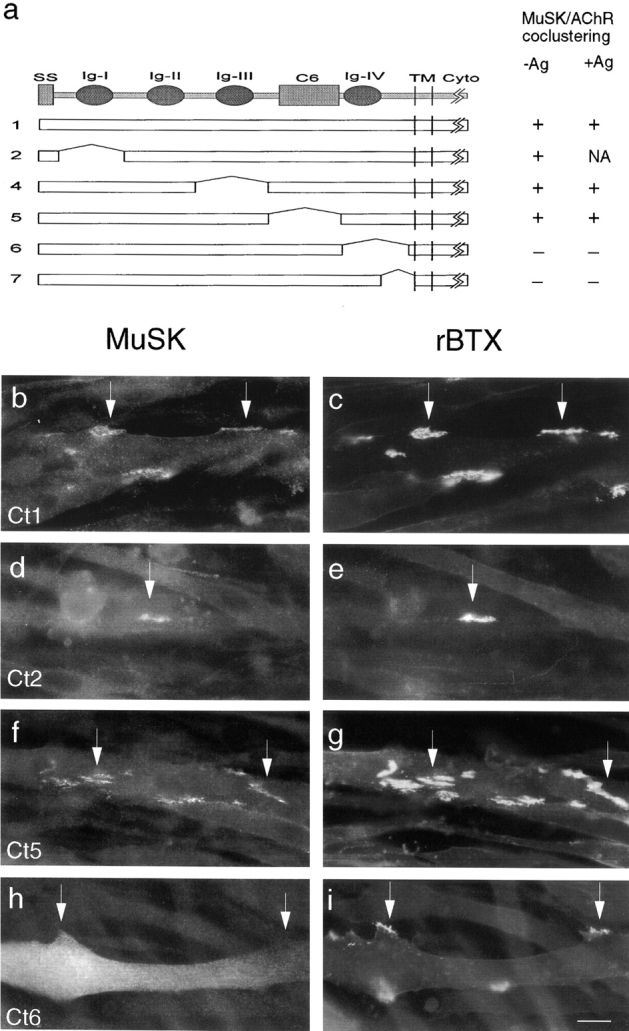
Carboxy-terminal regions of the MuSK ectodomain are required for association with AChR clusters in myotubes. (a) MuSK constructs, numbered as in Fig. 2, which were tested for their ability to associate with AChR clusters in MuSK −/− muscle cells. To the right of each construct is indicated whether it was concentrated at spontaneous or agrin-induced clusters. NA, not applicable, because agrin did not induce AChR clusters in these cells. (b–i) MuSK and AChR localization in cells transfected with expression vectors encoding MuSK constructs that colocalize (b and c, construct 1; d and e, construct 2; f and g, construct 5) or do not colocalize (h and i, construct 6) with AChRs. Cells were treated with agrin for 18 h, and then doubly stained with anti-mouse MuSK antibodies (b, d, f, and h) and rBTX (c, e, g, and i). Arrows indicate corresponding points in the two images of each field. Bar, 20 μm.
Two results from this series deserve further comment. First, cells transfected with construct 2 were unresponsive to agrin, but MuSK was concentrated at spontaneous clusters in these cells; likewise MuSK was present at both spontaneous and agrin-induced AChR clusters in cells transfected with constructs 1, 4, and 5. Thus, the association of MuSK with rapsyn in muscle cells does not require that MuSK be activated by agrin. Second, construct 5T, which lacked the C6 box, failed to colocalize with rapsyn and AChRs in QT-6 cells but construct 5 did do so in myotubes. We speculate that deletion of sequences adjacent to the fourth immunoglobulin-like domain prevented that domain from assuming an appropriate conformation in heterologous cells but not in muscle cells.
Essential Binding Sites for ATP and for PTB Domain-containing Proteins
How does agrin-induced phosphorylation of MuSK lead to postsynaptic differentiation? According to a widely accepted model (for review see Schlessinger and Ullrich 1992), phosphorylation activates multiple signal transduction pathways that eventually combine to generate the biological response. The signal transduction components are recruited to the kinase by adaptor proteins that bind to conserved sequences. Therefore, one strategy for defining the pathways through which MuSK signals, is to identify critical sites in its cytoplasmic domain.
As a first step, we tested a construct in which a critical lysine residue in the consensus ATP binding site was replaced with alanine (Fig. 6 a, construct 16). Such mutations in other receptor tyrosine kinases abolish tyrosine kinase activity (Chou et al. 1987; Honegger et al. 1987), and we showed previously that this MuSK mutant inhibits AChR clustering when expressed in wild-type myotubes (Glass et al. 1997), suggesting that it had reduced activity. Therefore, if MuSK is acting as a receptor tyrosine kinase, construct 16 should be unable to mediate AChR clustering. Alternatively, if MuSK were only a substrate for other kinases, this mutation might not abolish activity. Here, we transfected construct 16 into MuSK −/− cells, and then assayed for tyrosine phosphorylation of the mutant by immunoblotting and AChR clustering by staining with rBTX. Construct 16 was devoid of detectable phosphotyrosine (Fig. 7, lanes 1 and 2) and was completely unable to induce formation of AChR clusters, either spontaneously or in response to agrin. Inactivity did not reflect poor expression or misrouting because construct 16 was expressed at levels similar to those of wild-type MuSK and reached the cell-surface without impediment (data not shown). These results indicate that the kinase activity of MuSK is essential for its biological activity.
Figure 6.

Roles of binding sites for ATP and for PTB domain proteins in the MuSK cytoplasmic domain. (a) Mutant constructs. To the right of each construct is indicated whether or not it rescued the ability of MuSK −/− myotubes to form AChR clusters in the absence of agrin (−Ag) or to form additional clusters in the presence of agrin (+Ag). Plus sign, spontaneous clustering; −, no clustering; ↑↑, wild-type level of agrin sensitivity; and ↑, reduced clustering relative to wild-type. In constructs 16 and 17, a conserved ATP binding site and a binding site for PTB domain proteins, respectively, were mutated. In constructs 18 and 19, the specificity of the PTB site was altered, as detailed in Results. (b–g) Distribution of AChRs on MuSK −/− myotubes that had been transfected with mutant constructs, treated, and stained as in Fig. 1. (b and c) Construct 1; (d and e) construct 17; and (f and g) construct 19. (h) Quantitation of the extent to which MuSK constructs 17, 18, and 19 rescued the ability of MuSK −/− myotubes to form AChR clusters in the absence or presence of agrin. Constructs 16 and 17 were inactive, whereas constructs 18 and 19 showed reduced activity relative to wild-type (compare with Fig. 2 h and 8 h). Cultures were doubly stained with rBTX and anti-MuSK, and only MuSK-positive (i.e., successfully transfected) myotubes were scored. Bar, 20 μm.
Figure 7.

Tyrosine phosphorylation of MuSK cytoplasmic mutants. MuSK −/− myotubes were transfected with indicated constructs (Fig. 6 a), treated with agrin for 10 min (+), or left untreated (−), and then subjected to immunoprecipitation with anti-MuSK. Immunoblots were probed with antiphosphotyrosine (a), and then stripped and reprobed with anti-MuSK (b). Constructs 16 and 17, which do not mediate agrin-dependent tyrosine phosphorylation, are not detectably phosphorylated. Constructs 18 and 19 mediate lower levels of agrin-induced AChR clustering than wild-type MuSK; agrin-dependent tyrosine phosphorylation is greatly reduced for construct 18 but not construct 19.
Next, we tested the function of the cytoplasmic sequence NPMY, which is perfectly conserved among rat, mouse, human, Xenopus, and Torpedo MuSKs (Jennings et al. 1993; Valenzuala et al., 1995; Ganju et al. 1995; Fu et al. 1999), and which corresponds to a consensus binding site (NPXY) for signaling proteins that contain a PTB domain (for review see van der Geer and Pawson 1995; Borg and Margolis 1998). We changed the critical tyrosine residue to phenylalanine (construct 17), a mutation known to abolish binding of most PTB domain proteins. This mutation had no detectable effect on the expression level or the ability of MuSK to reach the cell surface (Fig. 8 d). However, myotubes transfected with construct 17 formed neither spontaneous nor agrin-induced AChR clusters (Fig. 6d, Fig. e, and Fig. h). Moreover, construct 17 was not detectably phosphorylated either spontaneously or after addition of agrin (Fig. 7, lanes 3 and 4). These results suggest that PTB domain–containing proteins are required for activation of MuSK's kinase activity.
Figure 8.

Binding sites predicted to recognize p85 and PDZ domain-containing proteins are inessential for MuSK to cluster AChRs or to associate with AChR clusters. (a) Mutant constructs. To the right of each construct is indicated whether or not it rescued the ability of MuSK −/−myotubes to form AChR clusters in the absence of agrin (−Ag) or to form additional clusters in the presence of agrin (+Ag). +, spontaneous clustering; −, no clustering; ↑↑, wild-type level of agrin sensitivity; and ↑, reduced clustering relative to wild-type. In constructs 20–23, large segments of the cytoplasmic domain were deleted. In constructs 24 and 25, putative binding sites for p85 and PDZ domains were mutated. Constructs 20–23 were completely inactive, whereas constructs 24 and 25 showed activity equivalent to that of wild-type MuSK. (b and c) Cells that had been transfected with construct 25, incubated with or without agrin, and then stained with rBTX. (d–g) Cells that had been transfected with construct 17 (d and e) or 25 (f and g), treated with agrin, and then doubly stained with rBTX (d and f) and anti-MuSK antibodies (e and g). The MuSK mutant with a three residue carboxy-terminal truncation (construct 25) is unable to bind PDZ domains in PICK (data not shown) but can induce and associate with AChR clusters. MuSK and AChRs are both diffusely distributed in myotubes transfected with construct 17. (h) Quantitation of the extent to which MuSK constructs 24 and 25 rescued the ability of MuSK−/− myotubes to form AChR clusters in the absence or presence of agrin. Cultures were doubly stained with rBTX and anti-MuSK, and only MuSK-positive (i.e., successfully transfected) myotubes were scored. Bar, 20 μm.
Muscle fibers express at least two PTB domain proteins, insulin receptor substrate 1 (IRS-1) and shc (Araki et al. 1993; Giorgino et al., 1995; Altiok et al. 1997). To determine whether either of these was activated by MuSK, we incubated wild-type myotubes with or without agrin, immunoprecipitated IRS-1 or shc, and probed immunoblots with antiphosphotyrosine. Neither protein was detectably phosphorylated in response to agrin treatment (data not shown). These results raised the possibility that MuSK associates with PTB proteins other than IRS-1 or shc. In support of this possibility, MuSK lacks residues upstream of NPXY that favor interaction with IRS-1 or shc. Although all PTB domains recognize the NPXY motif, residues amino-terminal of this tetrapeptide dictate binding specificity. A hydrophobic residue such as I or L is present five residues upstream of Y in sites that bind shc, and L is present eight residues upstream of Y in sites that bind IRS-1 (Gustafson et al. 1995; Eck et al. 1996; Kuriyan and Cowburn 1997; Borg and Margolis 1998; Cattaneo and Pelicci 1998). In rat, mouse, human, Xenopus, and Torpedo MuSKs, the fifth and eighth residues are H and D, respectively, so the extended sequence (DRLHPNPMY) would not be expected to bind either shc or IRS-1.
To assess the importance of the sequence upstream of NPMY in MuSK (DRLHP), we altered it either to LYLSS (construct 18), which is found upstream of NPXY in the IRS-1 binding site of the insulin receptor, or to IPILE (construct 19), which is upstream of NPXY in the shc binding site of trkB and trkC. Both of these constructs mediated agrin-dependent AChR clustering, but at lower levels than wild-type MuSK (Fig. 6, a and f–h). In addition, construct 18 was poorly phosphorylated in response to agrin (Fig. 7, lanes 5 and 6). The fact that residues adjacent to the NPXY motif modulate MuSK activity supports the idea that PTB domain proteins are involved in MuSK signaling. However, the finding that optimizing the MuSK cytoplasmic domain to bind shc or IRS-1 decreased the ability of MuSK to induce AChR clustering suggests that wild-type MuSK interacts with a PTB domain protein that is neither shc nor IRS1.
Inessential Binding Sites for Phosphatidylinositol-3-kinase and PDZ Domain Proteins
The essential ATP- and PTB domain–binding sites of MuSK are within the amino-terminal third of its cytoplasmic domain. To ask whether the carboxy-terminal portion of the cytoplasmic domain also contained essential sites, we deleted the last 214, 148, or 70 amino acids (Fig. 8 a, constructs 21–23). All three of these constructs were completely inactive in the presence or absence of agrin (data not shown), suggesting that sites in the carboxy-terminal fifth of the cytoplasmic domain were essential for activity. Examination of the sequence revealed two consensus binding sites in this region. The first was YXXM, which is the consensus binding site for p85, the regulatory subunit of phosphatidylinositol-3-kinase. This enzyme mediates signal transduction by several tyrosine kinases, including the trk kinases which, as noted above, are related to MuSK (Songyang et al. 1993; for review see Fruman et al. 1998). The second was VXV at the carboxy terminus, which resembles the consensus binding site for PDZ domain proteins. Proteins in this family that have been implicated in the aggregation of numerous membrane proteins include components of the postsynaptic membrane such as glutamate receptors and ephrins (Fanning and Anderson 1998; Hata et al. 1998; O'Brien et al. 1998; Torres et al. 1998). Moreover, using the yeast two-hybrid system, we have identified PDZ domain proteins that bind to MuSK, and shown that deletion of its carboxy-terminal three residues abolishes the ability of MuSK to bind these proteins (Torres, R., E.D. Apel, H. Zhou, D.J. Glass, J.R. Sanes, and G.D. Yancopoulos, unpublished observations).
We changed the essential tyrosine of the putative p85 binding site to phenylalanine in construct 24, and deleted the carboxy-terminal three resides (VGV) in construct 25. Surprisingly, both constructs were equivalent to wild-type MuSK in their abilities to mediate spontaneous and agrin-induced AChR clustering (Fig. 8b, Fig. c, and Fig. h). Thus, both the p85- and the PDZ domain–binding sites are dispensable for MuSK function in cultured myotubes. In view of the similarity of MuSK to trk, it is interesting that the p85-binding domain in trk is dispensable for at least some aspects of neurotrophin signaling (Hallberg et al. 1998). In light of the known role of PDZ domain proteins in aggregation of postsynaptic components, we also asked whether construct 25 was impaired in its ability to associate with AChR–rapsyn clusters. The association of this mutant with AChRs and rapsyn in myotubes was indistinguishable from that of wild-type MuSK (Fig. 8f and Fig. g).
Discussion
Numerous polypeptides promote growth or differentiation by binding to the ectodomain of receptor tyrosine kinases. This binding induces dimerization or conformational changes that lead to autophosphorylation. Once phosphorylated, the cytoplasmic domain recruits and/or phosphorylates additional components that transduce the biological signal (Schlessinger and Ullrich 1992). In keeping with this model, agrin interacts with and activates MuSK, and MuSK phosphorylation is required for AChR clustering. However, agrin and MuSK are unusual among growth factor/receptor tyrosine kinase pairs in at least two respects. First, agrin does not bind MuSK directly, but requires an accessory component, MASC (Glass et al. 1996). Second, the ectodomain serves not only to activate MuSK but also to recruit AChR–rapsyn aggregates, via another accessory component, RATL (Apel et al. 1997). Here, we have mapped the domains of the MuSK ectodomain that mediate interactions with agrin–MASC and rapsyn–RATL. In addition, we have taken the first steps toward mapping the cytoplasmic domains through which MuSK transduces its signals.
In initial studies, we characterized a MuSK −/− myogenic cell line and showed that introduction of rat MuSK or a related Torpedo kinase rescued the ability of MuSK-deficient myotubes to form AChR clusters, both spontaneously and in response to agrin. Although these experiments were designed to establish an assay for structure–function analysis, they led to three novel conclusions. First, the Torpedo receptor kinase first described by Jennings et al. 1993 and later shown to be homologous to MuSK (Valenzuela et al. 1995) is likely to be a true orthologue of MuSK. Second, the requirement for MuSK is limited to muscle cells. Results in vivo had not excluded the possibility that expression of MuSK was also required at earlier stages of development or in other tissues to generate agrin-sensitive myotubes, but analysis of rescued MuSK −/− cells demonstrates that this is not the case. Third, formation of spontaneous as well as agrin-induced AChR clusters requires MuSK. In principle, MuSK might be activated at low level in a ligand-independent fashion, or muscles might produce an endogenous ligand. We favor the hypothesis that activation is ligand-independent, based on our inability to block spontaneous clustering by deletion of any portion of the ectodomain.
Using MuSK −/− cells as an assay system, we found that the amino-terminal two fifths of the ectodomain is both necessary and sufficient to confer agrin sensitivity on MuSK: deletions within this region compromised agrin sensitivity, whereas constructs lacking the remainder of the ectodomain were fully agrin-sensitive. The possibility that these sequences are required for basal MuSK function was excluded by the observation that a construct lacking them restored the ability of MuSK −/− cells to form spontaneous AChR clusters. The critical region includes two immunoglobulin-like domains, which are known to mediate ligand binding in numerous cell surface receptors. Thus, these domains may interact with agrin, MASC, or both.
Further analysis suggests that the region of the first immunoglobulin-like domain is more critical than that of the second immunoglobulin-like domain for interactions with agrin. Deletion of sequences within the first immunoglobulin-like domain completely abolished agrin-sensitivity (constructs 2 and 13), whereas deletion of the second immunoglobulin-like domain left MuSK with significant albeit impaired agrin sensitivity (constructs 3 and 12). One interpretation of these results is that only sequences in or near the first immunoglobulin-like domain is required for agrin responsiveness, but its conformation is altered by mutations in neighboring areas. Alternatively, the second immunoglobulin-like domain may contain sites that stabilize or enhance interactions of MuSK with agrin–MASC. We cannot yet distinguish between these alternatives.
Next, we attempted to determine which portions of MuSK are necessary for it to associate with rapsyn. Although rapsyn is entirely intracellular, we showed previously that MuSK–rapsyn interactions require the MuSK ectodomain, implying the existence of a transmembrane linker, RATL (Apel et al. 1997). Studies in both fibroblasts and MuSK −/− myotubes suggest that the juxtamembranous portion of the ectodomain is both necessary and sufficient for the association of MuSK and rapsyn–RATL: MuSK fails to coaggregate with rapsyn in its absence, whereas deletion of more amino-terminal sequences does not impair MuSK–rapsyn coaggregation. The critical region includes the fourth immunoglobulin-like domain and a unique juxtamembranous segment, either or both of which might be involved in MuSK–rapsyn interactions; additional constructs will be needed to distinguish these possibilities.
The observation that agrin induced AChR clustering in constructs lacking the juxtamembranous domain showed that MuSK–rapsyn/RATL interactions are dispensable for at least some aspects of MuSK function. What roles might such interactions play? We showed previously that MuSK-dependent phosphorylation of AChRs is reduced in rapsyn-deficient cells, which is consistent with the idea that rapsyn and RATL bring AChRs into proximity with MuSK (Apel et al. 1997). One possibility is that this interaction is more important in vivo than in the transfected MuSK −/− cells, which express MuSK at higher than normal levels. Alternatively, the requirement of rapsyn for AChR phosphorylation may reflect a role of rapsyn (which is present in MuSK −/− cells) but not of MuSK–rapsyn interactions. In either case, it is important to note that AChR phosphorylation may not be required for AChR clustering (Meyer and Wallace 1998). Another possibility, which we favor, is that MuSK–rapsyn interactions (and possibly AChR phosphorylation) may be important for the localization of AChR clusters to synaptic sites but not for their formation per se. Although it is natural to interpret our results on colocalization as reflecting recruitment of MuSK to an AChR–rapsyn cluster, the relevant interaction in vivo may be one that recruits AChR and rapsyn to a subsynaptic concentration of MuSK. This point is crucial in evaluating the model proposed below.
In a final series of studies, we initiated an analysis of the MuSK cytoplasmic domain. Two observations from this series were surprising. First, a binding site (NPXY) for PTB domains in adaptor proteins (van der Geer and Pawson 1995; Borg and Margolis 1998) is required for MuSK activity. This result was surprising because similar mutations of other kinases such as trkB (Minichiello et al. 1998) or erbB3 (Vijapurkar et al. 1998) impair but do not abolish activity. Therefore, it will be important to learn which PTB protein(s) bind(s) to MuSK, and what signaling components they recruit to the complex. Second, a carboxy-terminal binding site (VXV) for PDZ domains in scaffolding proteins (Fanning and Anderson 1998) is dispensable both for MuSK-activated AChR clustering and for association of MuSK with AChR–rapsyn clusters. This result was also unexpected in view of our evidence that MuSK can bind PDZ proteins through its carboxy-terminal VXV motif (Torres, R., E.D. Apel, H. Zhou, D.J. Glass, J.R. Sanes, and G.D. Yancopoulos, unpublished observations) and that PDZ proteins play multiple roles in assembly of the postsynaptic apparatus at neuron–neuron synapses (Butz et al. 1998; Hata et al. 1998; O'Brien et al. 1998; Torres et al. 1998). We favor the possibility that PDZ domain proteins do play roles in MuSK signaling, but that these roles are not readily detected in the assays we have used to date.
Our new results, along with those presented previously (Glass et al. 1996, Glass et al. 1997; Apel et al. 1997; Jones et al. 1999), suggest a model in which MuSK plays critical roles in three distinct steps that together lead to the formation of the postsynaptic membrane (Fig. 9). First, a signal from the nerve leads to formation of a primary synaptic scaffold of which MuSK is a component, and for which MuSK is required (Gautam et al. 1995; Moscoso et al. 1995; deChiara et al. 1996; Apel et al. 1997). The observation that MuSK clusters beneath nerve terminals in rapsyn-deficient mutant mice, whereas other components of the synaptic membrane do not, demonstrates that the primary scaffold is assembled by mechanisms distinct from those responsible for AChR clustering. The neural signal that induces formation of the primary scaffold is unknown; it may be agrin, but no published data bear directly on this point. Second, agrin released from the nerve terminal interacts with MuSK, presumably via MASC, to activate MuSK kinase. This interaction requires sequences in or near the first immunoglobulin-like domain of MuSK, and may also depend on the second immunoglobulin-like domain. Once phosphorylated, MuSK recruits PTB domain–containing proteins to initiate a signaling process that leads to formation of aggregates that contain AChRs, rapsyn, and other components of the postsynaptic membrane and cytoskeleton. Third, AChR–rapsyn clusters are recruited to the primary scaffold. This step is mediated by RATL, and requires juxtamembranous sequences of the MuSK ectodomain. It is agrin-independent in that it is mediated by constructs unable to interact with agrin. Although shown as a late step in the model, it probably occurs in parallel with formation of the primary scaffold in vivo (Noakes et al. 1993; Apel et al. 1997; Bowen et al. 1998).
Figure 9.

Model of MuSK-mediated postsynaptic differentiation (see Discussion). In a first step, the nerve induces subterminal clustering of MuSK, forming a primary synaptic scaffold. In a second step, nerve-derived Z+ agrin activates MuSK, inducing coclustering of rapsyn and AChRs via intracellular pathways that involve PTB domain–containing proteins but not PDZ domain proteins. In a third step, MuSK uses RATL to recruit rapsyn–AChR clusters to the primary scaffold. Sequences in or near immunoglobulin-like domains 1 are required to mediate associations with agrin, presumably via MASC, whereas juxtamembranous sequences in or near immunoglobulin-like domain 4 are required to mediate associations with rapsyn, presumably via RATL.
We believe this model is attractive because it accounts for a cardinal difference between synaptogenesis and other biological responses mediated by receptor tyrosine kinases. MuSK-dependent aggregation of postsynaptic specializations is not fundamentally different from other kinase-dependent developmental programs. What is different is that MuSK localizes the specialized domain with a submicron level of precision. By using separate accessory proteins to catalyze formation of the postsynaptic apparatus and to recruit that apparatus to subsynaptic sites, MuSK can independently control the composition and the location of the synapse, both of which are critical for synaptic function.
Acknowledgments
We thank E.D. Apel (Washington University) for valuable advice and suggestions, Markus Ruegg (University of Basel) for antisera, and Richard Scheller (Stanford University) for agrin-producing cells.
This work was supported by grants to J.R. Sanes from the National Institutes of Health and the Muscular Dystrophy Association.
Footnotes
1.used in this paper: AChR, acetylcholine receptor; MASC, muscle-associated specificity component; MuSK, muscle-specific receptor tyrosine kinase; PDZ, PSD-95/Dlg/ZO-1-like; PTB, phosphotyrosine-binding; RATL, rapsyn-associated transmembrane linking molecule; rBTX, rhodamine-α-bungarotoxin
References
- Altiok N., Altiok S., Changeux J.P. Heregulin-stimulated acetylcholine receptor gene expression in musclerequirement for MAP kinase and evidence for a parallel inhibitory pathway independent of electrical activity. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:717–725. doi: 10.1093/emboj/16.4.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apel E.D., Roberds S.L., Campbell K.P., Merlie J.P. Rapsyn may function as a link between the acetylcholine receptor and the agrin-binding dystrophin-associated glycoprotein complex. Neuron. 1995;15:115–126. doi: 10.1016/0896-6273(95)90069-1. [DOI] [PubMed] [Google Scholar]
- Apel E.D., Glass D.J., Moscoso L.M., Yancopoulos G.D., Sanes J.R. Rapsyn is required for MuSK signaling and recruits synaptic components to a MuSK-containing scaffold. Neuron. 1997;18:623–635. doi: 10.1016/s0896-6273(00)80303-7. [DOI] [PubMed] [Google Scholar]
- Araki E., Sun X.J., Haag B.L., Chuang L.M., Zhang Y., Yang-Feng T.L., White M.F., Kahn C.R. Human skeletal muscle insulin receptor substrate-1. Characterization of the cDNA, gene, and chromosomal localization. Diabetes. 1993;42:1041–1054. doi: 10.2337/diab.42.7.1041. [DOI] [PubMed] [Google Scholar]
- Borg J.P., Margolis B. Function of PTB domains. Curr. Top. Microbiol. Immunol. 1998;228:23–38. doi: 10.1007/978-3-642-80481-6_2. [DOI] [PubMed] [Google Scholar]
- Bowen D.C., Park J.S., Bodine S., Stark J.L., Valenzuela D.M., Stitt T.N., Yancopoulos G.D., Lindsay R.M., Glass D.J., DiStefano P.S. Localization and regulation of MuSK at the neuromuscular junction. Dev. Biol. 1998;199:309–319. doi: 10.1006/dbio.1998.8936. [DOI] [PubMed] [Google Scholar]
- Burden S.J. The formation of neuromuscular synapses. Genes Dev. 1998;12:133–148. doi: 10.1101/gad.12.2.133. [DOI] [PubMed] [Google Scholar]
- Burgess R.W., Nguyen Q.T., Son Y.-J., Lichtman J.W., Sanes J.R. Alternatively spliced isoforms of nerve- and muscle-derived agrintheir roles at the neuromuscular junction. Neuron. 1999;23:33–44. doi: 10.1016/s0896-6273(00)80751-5. [DOI] [PubMed] [Google Scholar]
- Butz S., Okamoto M., Sudhof T.C. A tripartite protein complex with the potential to couple synaptic vesicle exocytosis to cell adhesion in brain. Cell. 1998;18:773–782. doi: 10.1016/s0092-8674(00)81736-5. [DOI] [PubMed] [Google Scholar]
- Cattaneo E., Pelicci P.G. Emerging roles for SH2/PTB-containing Shc adaptor proteins in the developing mammalian brain. Trends Neurosci. 1998;21:476–481. doi: 10.1016/s0166-2236(98)01282-x. [DOI] [PubMed] [Google Scholar]
- Chou C.K., Dull T.J., Russell D.S., Gherzi R., Lebwohl D., Ullrich A., Rosen O.M. Human insulin receptors mutated at the ATP-binding site lack protein tyrosine kinase activity and fail to mediate postreceptor effects of insulin. J. Biol. Chem. 1987;262:1842–1847. [PubMed] [Google Scholar]
- Cohen I., Rimer M., Lomo T., McMahan U.J. Agrin-induced postsynaptic-like apparatus in skeletal muscle fibers in vivo. Mol. Cell. Neurosci. 1997;9:237–253. doi: 10.1006/mcne.1997.0623. [DOI] [PubMed] [Google Scholar]
- deChiara T.M., Bowen D.C., Valenzuela D.M., Simmons M.V., Poueymirou W.T. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell. 1996;85:501–512. doi: 10.1016/s0092-8674(00)81251-9. [DOI] [PubMed] [Google Scholar]
- Donoghue M.J., Morris-Valero R., Johnson Y.R., Merlie J.P., Sanes J.R. Mammalian muscle cells bear a cell-autonomous, heritable memory of their rostrocaudal position. Cell. 1992;69:67–77. doi: 10.1016/0092-8674(92)90119-w. [DOI] [PubMed] [Google Scholar]
- Eck M.J., Dhe-Paganon S., Trub T., Nolte R.T., Shoelson S.E. Structure of the IRS-1 PTB domain bound to the juxtamembrane region of the insulin receptor. Cell. 1996;85:695–705. doi: 10.1016/s0092-8674(00)81236-2. [DOI] [PubMed] [Google Scholar]
- Fanning A.S., Anderson J.M. PDZ domains and the formation of protein networks at the plasma membrane. Curr. Top. Microbiol. Immunol. 1998;228:209–233. doi: 10.1007/978-3-642-80481-6_9. [DOI] [PubMed] [Google Scholar]
- Ferns M., Campanelli J.T., Hoch W., Scheller R.H., Hall Z.M. The ability of agrin to cluster AChRs depends on alternative splicing and on cell surface proteoglycans. Neuron. 1993;11:491–502. doi: 10.1016/0896-6273(93)90153-i. [DOI] [PubMed] [Google Scholar]
- Froehner S.C., Luetje C.W., Scotland P.B., Patrick J. The postsynaptic 43K protein clusters muscle nicotinic acetylcholine receptors in Xenopus oocytes. Neuron. 1990;5:403–410. doi: 10.1016/0896-6273(90)90079-u. [DOI] [PubMed] [Google Scholar]
- Fruman D.A., Meyers R.E., Cantley L.C. Phosphoinositide kinases. Annu. Rev. Biochem. 1998;67:481–507. doi: 10.1146/annurev.biochem.67.1.481. [DOI] [PubMed] [Google Scholar]
- Fu A.K.Y., Smith F.D., Zhou H., Chu A.H., Tsim K.W.K., Peng B.H., Ip N.Y. Xenopus muscle-specific kinasemolecular cloning and prominent expression in neural tissues during early embryonic development. Eur. J. Neurosci. 1999;11:373–382. doi: 10.1046/j.1460-9568.1999.00443.x. [DOI] [PubMed] [Google Scholar]
- Fuhrer C., Sugiyama J.E., Taylor R.G., Hall Z.W. Association of muscle-specific kinase MuSK with the acetylcholine receptor in mammalian muscle. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:4951–4960. doi: 10.1093/emboj/16.16.4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganju P., Walls E., Brennan J., Reith A.D. Cloning and developmental expression of Nsk2, a novel receptor tyrosine kinase implicated in skeletal myogenesis. Oncogene. 1995;11:281–290. [PubMed] [Google Scholar]
- Gautam M., Noakes P.G., Mudd J., Nichol M., Chu G.C., Sanes J.R., Merlie J.P. Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn-deficient mice. Nature. 1995;377:232–236. doi: 10.1038/377232a0. [DOI] [PubMed] [Google Scholar]
- Gautam M., Noakes P.G., Moscoso L., Rupp F., Scheller R.H., Merlie J.P., Sanes J.R. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 1996;85:525–535. doi: 10.1016/s0092-8674(00)81253-2. [DOI] [PubMed] [Google Scholar]
- Gautam M., DeChiara T.M., Glass D.J., Yancopoulos G.D., Sanes J.R. Distinct phenotypes of mutant mice lacking agrin, MuSK, or rapsyn. Dev. Brain Res. 1999;114:171–178. doi: 10.1016/s0165-3806(99)00013-9. [DOI] [PubMed] [Google Scholar]
- Gillespie S.K., Balasubramanian S., Fung E.T., Huganir R.L. Rapsyn clusters and activates the synapse-specific receptor tyrosine kinase MuSK. Neuron. 1996;16:953–962. doi: 10.1016/s0896-6273(00)80118-x. [DOI] [PubMed] [Google Scholar]
- Giorgino F., Smith R.J. Dexamethasone enhances insulin-like growth factor-I effects on skeletal muscle cell proliferation. Role of specific intracellular signaling pathways. J. Clin. Invest. 1995;96:1473–1483. doi: 10.1172/JCI118184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass D.J., Bowen D.C., Stitt T.N., Radziejewski C., Bruno J., Ryan T.E., Gies D.R., Shah S., Mattsson K., Burden et al S.J. Agrin acts via a MuSK receptor complex. Cell. 1996;85:513–523. doi: 10.1016/s0092-8674(00)81252-0. [DOI] [PubMed] [Google Scholar]
- Glass D.J., Apel E.D., Shah S., Bowen D.C., deChiara T.M., Stitt T.N., Sanes J.R., Yancopoulos G.D. Kinase domain of the muscle-specific receptor tyrosine kinase (MuSK) is sufficient for phosphorylation but not clustering of acetylcholine receptorsrequired role for the MuSK ectodomain? Proc. Natl. Acad. Sci. USA. 1997;94:8848–8853. doi: 10.1073/pnas.94.16.8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson T.A., He W., Craparo A., Schaub C.D., O'Neill T.J. Phosphotyrosine-dependent interaction of SHC and insulin receptor substrate 1 with the NPEY motif of the insulin receptor via a novel non-SH2 domain. Mol. Cell. Biol. 1995;15:2500–2508. doi: 10.1128/mcb.15.5.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata Y., Nakanishi H., Takai Y. Synaptic PDZ domain-containing proteins. Neurosci. Res. 1998;32:1–7. doi: 10.1016/s0168-0102(98)00069-8. [DOI] [PubMed] [Google Scholar]
- Hesser B.A., Sander A., Witzemann V. Identification and characterization of a novel splice variant of MuSK. FEBS (Fed. Eur. Biochem. Soc.) Lett. 1999;442:133–137. doi: 10.1016/s0014-5793(98)01641-x. [DOI] [PubMed] [Google Scholar]
- Hallberg B., Ashcroft M., Loeb D.M., Kaplan D.R., Downward J. Nerve growth factor induced stimulation of Ras requires Trk interaction with Shc but does not involve phosphoinositide 3-OH kinase. Oncogene. 1998;17:691–697. doi: 10.1038/sj.onc.1201980. [DOI] [PubMed] [Google Scholar]
- Honegger A.M., Dull T.J., Felder S., Van Obberghen E., Bellot F., Szapary D., Schmidt A., Ullrich A., Schlessinger J. Ponit mutation at the ATP binding site of EGF receptor abolishes protein-tyrosine kinase activity and alters cellular routing. Cell. 1987;51:199–209. doi: 10.1016/0092-8674(87)90147-4. [DOI] [PubMed] [Google Scholar]
- Hopf C., Hoch W. Dimerization of the muscle-specific kinase induces tyrosine phosphorylation of acetylcholine receptors and their aggregation on the surface of myotubes J. Biol. Chem. 273 1998. 6467 6473a [DOI] [PubMed] [Google Scholar]
- Hopf C., Hoch W. Tyrosine phosphorylation of the muscle-specific kinase is exclusively induced by acetylcholine receptor-aggregating agrin fragments Eur. J. Biochem 253 1998. 382 389b [DOI] [PubMed] [Google Scholar]
- Jat P.S., Noble M.D., Ataliotis P., Tanaka Y., Yannoutsos N., Larsen L., Kioussis D. Direct derivation of conditionally immortal cell lines from an H-2Kb-tsA58 transgenic mouse. Proc. Natl. Acad. Sci. USA. 1991;88:5096–5100. doi: 10.1073/pnas.88.12.5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings C.G.B., Dyer S.M., Burden S.J. Muscle-specific trk-related receptor with a kringle domain defines a distinct class of receptor tyrosine kinases. Proc. Natl. Acad. Sci. USA. 1993;90:2895–2899. doi: 10.1073/pnas.90.7.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones G., Meier T., Lichtsteiner M., Witzemann V., Sakmann B., Brenner H.R. Induction by agrin of ectopic and functional postsynaptic-like membrane in innervated muscle. Proc. Natl. Acad. Sci. USA. 1997;94:2654–2659. doi: 10.1073/pnas.94.6.2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones G., Moore C., Hashemolhosseini S., Brenner H.R. Constitutively active MuSK is clustered in the absence of agrin and induces ectopic postsynaptic-like membranes in skeletal muscle fibers. J. Neurosci. 1999;19:3376–3383. doi: 10.1523/JNEUROSCI.19-09-03376.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriyan J., Cowburn D. Modular peptide recognition domains in eukaryotic signaling. Annu. Rev. Biophys. Biomed. Struct. 1997;26:259–288. doi: 10.1146/annurev.biophys.26.1.259. [DOI] [PubMed] [Google Scholar]
- Lamballe F., Klein R., Barbacid M. TrkC, a new member of the trk family of tyrosine protein kinases, is a receptor for neurotrophin-3. Cell. 1991;66:967–979. doi: 10.1016/0092-8674(91)90442-2. [DOI] [PubMed] [Google Scholar]
- McMahan U.J. The agrin hypothesis. Cold Spring Harbor Symp. 1990;60:407–418. doi: 10.1101/sqb.1990.055.01.041. [DOI] [PubMed] [Google Scholar]
- Meier T., Gesemann M., Cavalli V., Ruegg M.A., Wallace B.G. AChR phosphorylation and aggregation induced by an agrin fragment that lacks the binding domain for α-dystroglycan. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:2625–2631. [PMC free article] [PubMed] [Google Scholar]
- Meier T., Hauser D.M., Chiquet M., Landmann L., Ruegg M.A., Brenner H.R. Neural agrin induces ectopic postsynaptic specializations in innervated muscle fibers. J. Neurosci. 1997;17:6534–6544. doi: 10.1523/JNEUROSCI.17-17-06534.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer G., Wallace B.G. Recruitment of a nicotinic acetylcholine receptor mutant lacking cytoplasmic tyrosine residues in its beta subunit into agrin-induced aggregates. Mol. Cell. Neurosci. 1998;11:324–333. doi: 10.1006/mcne.1998.0689. [DOI] [PubMed] [Google Scholar]
- Minichiello L., Casagranda F., Tatche R.S., Stucky C.L., Postigo A., Lewin G.R., Davies A.M., Klein R. Point mutation in trkB causes loss of NT4-dependent neurons without major effects on diverse BDNF responses. Neuron. 1998;21:335–345. doi: 10.1016/s0896-6273(00)80543-7. [DOI] [PubMed] [Google Scholar]
- Moscoso L.M., Chu G.C., Gautam M., Noakes P.G., Merlie J.P., Sanes J.R. Synapse-associated expression of an acetylcholine receptor-inducing protein, ARIA/heregulin, and its putative receptors, ErbB2 and ErbB3, in developing mammalian muscle. Dev. Biol. 1995;172:158–169. doi: 10.1006/dbio.1995.0012. [DOI] [PubMed] [Google Scholar]
- Moscovici C., Moscovici G., Jimenez H., Lai M.M.C., Hayman M.J., Vogt P.K. Continuous tissue culture cell lines derived from chemically induced tumors of Japanese quail. Cell. 1977;11:95–103. doi: 10.1016/0092-8674(77)90320-8. [DOI] [PubMed] [Google Scholar]
- Noakes P.G., Phillips W.D., Hanley T.A., Sanes J.R., Merlie J.P. 43K protein and acetylcholine receptors colocalize during the initial stages of neuromuscular synapse formation in vivo. Dev. Biol. 1993;155:275–280. doi: 10.1006/dbio.1993.1025. [DOI] [PubMed] [Google Scholar]
- O'Brien R.J., Lau L.F., Huganir R.L. Molecular mechanisms of glutamate receptor clustering at excitatory synapses. Curr. Opin. Neurobiol. 1998;8:364–369. doi: 10.1016/s0959-4388(98)80062-7. [DOI] [PubMed] [Google Scholar]
- Peng H.B., Froehner S.C. Association of the postsynaptic 43K protein with newly formed acetylcholine receptor clusters in cultured muscle cells. J. Cell Biol. 1985;100:1698–1705. doi: 10.1083/jcb.100.5.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips W.D., Kopta C., Blount P., Gardner P.D., Steinbach J.H., Merlie J.P. ACh receptor-rich membrane domains organized in fibroblasts by recombinant 43-kilodalton protein Science 251 1991. 568 570a [DOI] [PubMed] [Google Scholar]
- Phillips W.D., Maimone M.M., Merlie J.P. Mutagenesis of the 43-kD postsynaptic protein defines domains involved in plasma membrane targeting and AChR clustering J. Cell Biol. 115 1991. 1713 1723b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes J.R., Apel E.D., Gautam M., Glass D., Grady R.M., Martin P.T., Yancopoulos G.D. Agrin receptors at the skeletal neuromuscular junction. Ann. NY Acad. Sci. 1998;841:1–13. doi: 10.1111/j.1749-6632.1998.tb10905.x. [DOI] [PubMed] [Google Scholar]
- Sanes J.R., Lichtman J.W. Development of the vertebrate neuromuscular junction. Annu. Rev. Neurosci. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- Schlessinger J., Ullrich A. Growth factor signaling by receptor tyrosine kinases. Neuron. 1992;9:383–391. doi: 10.1016/0896-6273(92)90177-f. [DOI] [PubMed] [Google Scholar]
- Songyang Z., Shoelson S.E., Chaudhuri M., Gish G., Pawson T., Haser W.G., King F., Roberts T., Ratnofsky S., Lechleider R.J. SH2 domains recognize specific phosphopeptide sequences. Cell. 1993;72:767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- Sugiyama J.E., Glass D.J., Yancopoulos G.D., Hall Z.W. Laminin-induced acetylcholine receptor clusteringan alternative pathway. J. Cell Biol. 1997;139:181–191. doi: 10.1083/jcb.139.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres R., Firestein B.L., Dong H., Staudinger J., Olson E.N., Huganir R.L., Bredt D.S., Gale N.W., Yancopoulos G.D. PDZ proteins bind, cluster, and synaptically colocalize with Eph receptors and their ephrin ligands. Neuron. 1998;21:1453–1463. doi: 10.1016/s0896-6273(00)80663-7. [DOI] [PubMed] [Google Scholar]
- Valenzuela D.M., Stitt T.N., DiStefano P.S., Rojas E., Mattsson K., Compton D.L., Nunez L., Park J.S., Start J.L., Gies D.R. Receptor tyrosine kinase specific for the skeletal muscle lineageexpression in embryonic muscle, at the neuromuscular junction, and after injury. Neuron. 1995;15:573–584. doi: 10.1016/0896-6273(95)90146-9. [DOI] [PubMed] [Google Scholar]
- Van der Geer P., Pawson T. The PTB domaina new protein module implicated in signal transduction. Trends Biochem. Sci. 1995;20:277–280. doi: 10.1016/s0968-0004(00)89043-x. [DOI] [PubMed] [Google Scholar]
- Vijapurkar U., Cheng K., Koland J.G. Mutation of a Shc binding site tyrosine residue in ErbB3/HER3 blocks heregulin-dependent activation of mitogen-activated protein kinase. J. Biol. Chem. 1998;273:20996–21002. doi: 10.1074/jbc.273.33.20996. [DOI] [PubMed] [Google Scholar]