

Abstract
In this report, we have examined the mechanisms whereby neurotrophins and neural activity coordinately regulate neuronal survival, focussing on sympathetic neurons, which require target-derived NGF and neural activity for survival during development. When sympathetic neurons were maintained in suboptimal concentrations of NGF, coincident depolarization with concentrations of KCl that on their own had no survival effect, synergistically enhanced survival. Biochemical analysis revealed that depolarization was sufficient to activate a Ras-phosphatidylinositol 3-kinase–Akt pathway (Ras–PI3-kinase–Akt), and function-blocking experiments using recombinant adenovirus indicated that this pathway was essential for ∼50% of depolarization-mediated neuronal survival. At concentrations of NGF and KCl that promoted synergistic survival, these two stimuli converged to promote increased PI3-kinase–dependent Akt phosphorylation. This convergent PI3-kinase–Akt pathway was essential for synergistic survival. In contrast, inhibition of calcium/calmodulin-dependent protein kinase II revealed that, while this molecule was essential for depolarization-induced survival, it had no role in KCl- induced Akt phosphorylation, nor was it important for synergistic survival by NGF and KCl. Thus, NGF and depolarization together mediate survival of sympathetic neurons via intracellular convergence on a Ras–PI3-kinase–Akt pathway. This convergent regulation of Akt may provide a general mechanism for coordinating the effects of growth factors and neural activity on neuronal survival throughout the nervous system.
Keywords: nerve growth factor, sympathetic, neurons, ras, mitogen-activated protein kinase, calcium/calmodulin-dependent protein kinase II
The developmental period of naturally occurring neuronal death is critical to the establishment of appropriate neural circuitry. In the mammalian peripheral nervous system, the ultimate survival or death of any given neuron during this period is regulated both by target-derived trophic support and by neuronal activity (Oppenheim 1991). In this regard, the sympathetic nervous system provides one model system in which the role of growth factors and activity has been well-studied. In particular, a large body of work indicates that sympathetic neurons undergo developmental apoptosis during the first three weeks of neonatal life, and that survival during this period is determined largely by competition for limiting concentrations of target-derived NGF acting through the TrkA tyrosine kinase receptor (reviewed in Kaplan and Miller 1997; Francis and Landis 1999). Moreover, recent studies indicate that other members of the same neurotrophin family may actively signal sympathetic neuron apoptosis by binding to the p75 neurotrophin receptor (Aloyz et al. 1998; Bamji et al. 1998). Together, these findings support a model wherein the repertoire of target-derived neurotrophins encountered by a developing sympathetic neuron determines its life versus death by differential signaling through the TrkA versus p75 neurotrophin receptors (Majdan and Miller 1999).
Although these studies have largely emphasized the role of target-derived growth factors in regulating developmental sympathetic neuron apoptosis, a significant number of findings indicate that establishment of appropriate neuronal activity is also essential (Oppenheim 1991; Franklin and Johnson 1992). Of particular relevance is the finding that blockage of either pre- or postganglionic transmission during naturally occurring cell death led to enhanced sympathetic neuron apoptosis (Maderdrut et al. 1988), indicating that, in addition to NGF, endogenous neural activity is essential for survival of these neurons. Similar conclusions derive from studies of other neuronal populations in both the peripheral and central nervous systems (Franklin and Johnson 1992). Moreover, for some neuronal subtypes, the presence of both trophic support and depolarization are required for survival in culture (Ghosh et al. 1994; Meyer-Franke et al. 1995, Meyer-Franke et al. 1998).
Neurotrophins and neural activity have been proposed to interact at a number of levels to promote neuronal survival. In cultured cortical neurons, the survival elicited by KCl acting through voltage-sensitive calcium channels is thought to be due to increased synthesis and secretion of autocrine brain-derived neurotrophic factor (BDNF; Ghosh et al. 1994). Conversely, IGF (insulin-like growth factor)1 treatment leads to potentiation of L-type calcium channels in cerebellar granule cells (Blair and Marshall 1997), an effect that is thought to be largely responsible for IGF-mediated survival of these neurons. As a third example of such interactions, Meyer-Franke et al. 1998 have demonstrated that activity rapidly recruits the TrkB receptor to the membrane of cultured retinal ganglion cells and spinal motor neurons, thereby allowing these neurons to survive in response to BDNF (Meyer-Franke et al. 1998).
An alternative mechanism, whereby neurotrophins and neural activity could interact to promote neuronal survival, derives from the fact that these two different stimuli activate at least some of the same intracellular signaling pathways. Neurotrophin binding to TrkA leads to activation of a number of signaling pathways, including the phosphatidylinositol 3-kinase (PI3-kinase)–Akt and Ras–mitogen-activated protein kinase kinase (MEK)–mitogen-activated protein (MAP) kinase pathways (reviewed in Kaplan and Stephens 1994; Kaplan and Miller 1997). Of these signaling proteins, Ras, PI3-kinase, and Akt have all been shown to be essential for NGF-mediated survival of sympathetic neurons (Bartlett et al. 1997; Markus et al. 1997; Crowder and Freeman 1998). In contrast, depolarization-mediated survival of sympathetic neurons requires calcium influx via L-type calcium channels (Franklin et al. 1995). This calcium influx can result in activation of the calmodulin/calmodulin-dependent protein kinase pathway (Hanson and Schulman 1992) and in increased Ras activity (Farnsworth et al. 1995). The resultant activation of the Ras–MEK–MAP kinase pathway (Rosen et al. 1994), therefore, provides at least one point of convergence between neurotrophin and depolarization-induced signaling.
In this report, we have chosen to utilize neonatal sympathetic neurons, which require both NGF and neural activity for survival during naturally occurring cell death in vivo, to examine the cellular interactions between these two extrinsic cues. Our data demonstrate that NGF and depolarization synergize to regulate neuronal survival, and that this synergy is mediated intracellularly by convergence on the Ras–PI3-kinase–Akt pathway. Such convergence may play an essential role in regulating neuronal survival in both the peripheral and central nervous systems.
Materials and Methods
Primary Neuronal Cultures
Mass cultures of sympathetic neurons were derived from the superior cervical ganglia of postnatal day 1 Sprague-Dawley rats, as previously described (Ma et al. 1992), except that neurons were dissected into plating Ultraculture medium (serum free) containing 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (all from Biowhittaker). Neurons were plated in the medium supplemented with 3% rat serum (Harlan Sprague Dawley Inc.), 50 ng/ml mouse 2.5 S NGF prepared from mouse salivary gland (Cedarlane Labs, Ltd.), and 0.5% cytosine arabinoside (Sigma Chemical Co.). For 3[4,5-dimethylthio-zol-2-yl]2,5-diphenyltetrazolium bromide (MTT; Sigma Chemical Co.) assays and biochemistry, neurons were plated on tissue culture dishes (Falcon Plastics) coated with rat-tail collagen. For microscopy, cells were plated on 16-well glass chamber slides (Nunc Inc.) coated first with collagen/poly-d-lysine, followed by a second coating of collagen. Neurons were plated at a density of 2,500–3,000/well of a 96-well plate for the MTT assays and 40,000 cells/well of a 6-well plate for biochemistry. In all cases, neurons were cultured for 5 d in the above-mentioned medium. After 5 d of culture, cells were washed three times for 1 h in Ultraculture supplemented with 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. After the washes, cells were induced in the same media used for the washes, supplemented with NGF or KCl in the presence or absence of kinase inhibitors. MTT and TdT-mediated dUTP nick end labeling (TUNEL) assays were performed 48 h after induction, and cell extracts for Western blotting were taken 15 min after induction. The kinase inhibitors KN-62, K-252a, LY294002, and PD98059 were obtained from Calbiochem-Novabiochem Corp. Nifedipine was obtained from BIOMOL.
Adenoviral Infection
For adenoviral infection, cells were grown for 3 d in plating medium as described earlier and then switched into similar media containing the desired MOI (multiplicity of infection = pfu/cell) of adenovirus and no cytosine arabinoside. Infection was allowed to proceed for 24 h, after which the cells were switched back to plating media containing cytosine arabinoside without virus for an additional 24 h before the washout and induction with NGF or KCl.
Recombinant adenoviruses expressing dominant-negative Ras (N17-Ras), dominant-negative Akt (dnAkt; Boudreau, M., C. Tudan, and D.R. Kaplan, manuscript submitted for publication), and dnTrkA (Ferrari et al. 1995) were amplified, purified, and titered as previously described (Slack et al. 1996; Aloyz et al. 1998).
Survival Assays and TUNEL
Survival assays were performed 48 h after washout and induction of neurons as previously described (Aloyz et al. 1998; Bamji et al. 1998). In brief, 20 μl of MTT reagent was added to the medium in each well of a 96-well plate containing the cultured neurons. After a 2.5 h incubation at 37°C, the medium/MTT mixture was removed and the cells were lysed with 100 μl of isopropanol containing 2 μl/ml of concentrated HCl. The absorbance of the lysate at 570 and 630 nm was determined using a Biotek model ELX-800 UV plate reader (Mandel Scientific Inc.).
For the TUNEL experiments, cells were briefly rinsed in PBS, pH 7.2, and fixed for 15 min in 4% paraformaldehyde (Sigma Chemical Co.), 0.25% glutaraldehyde (Fluka AG), and 0.2% Triton X-100 (Sigma Chemical Co.) in PBS, pH 7.2. Cells were then permeabilized with 0.5% Triton X-100 for 5 min and washed three times with PBS. TUNEL reaction was performed for 1 h at 37°C. Each 100 μl of TUNEL reaction mixture contained 20 μl of TdT buffer, 1.5 μl of TdT enzyme (both from Promega Corp.), and 1 μl of biotin-16-dUTP (Boehringer Mannheim Corp.). After the TUNEL reaction, cells were rinsed three times in PBS and incubated for 45 min at room temperature with Cy3-conjugated streptavidin (Jackson ImmunoResearch Laboratories) diluted 1:500 in PBS. Cells were then counterstained for 1 min with Hoechst 33258 (Sigma Chemical Co.) and diluted 1:1,000 in PBS. Cells were washed three times with PBS after each of these incubations and then mounted. For each treatment, random images were captured and processed. Digital image acquisition and analysis was performed with the Northern Eclipse software (Empix Inc.) using a Sony XC-75CE CCD video camera.
Western Blot Analysis and Immunoprecipitation
Sympathetic neurons were rinsed briefly in cold TBS and then lysed in TBS lysis buffer (137 mM NaCl, 20 mM Tris, pH 8.0, 1% vol/vol NP-40, and 10% vol/vol glycerol; Knusel et al. 1994) supplemented with Mini Complete protease inhibitor cocktail (Boehringer Mannheim Corp.) and 1.5 mM sodium vanadate. Lysates were scraped into Eppendorf tubes and rocked for 10 min at 4°C. Samples were then cleared by centrifugation. Protein concentration was determined by the BCA assay (Pierce Chemical Co.) using BSA as a standard.
For immunoprecipitation, samples were diluted into immunoprecipitation buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1% vol/vol NP-40, 0.5% wt/wt sodium deoxycholate, 0.1% wt/vol SDS, 5% vol/vol glycerol, 10 mM sodium fluoride, 5 mM EGTA, 1 mM EDTA, and 30 mM glycerolphosphate) containing the primary antibody, and incubated with gentle agitation for 4 h at 4°C. 30 μl of protein A–Sepharose beads (Pharmacia Biotech, Inc.), which were preincubated 1 h in cold immunoprecipitation buffer, were added and the samples were further incubated overnight at 4°C. After immunoprecipitation, beads were washed four times in 3% NETF buffer (50 mM Tris, pH 7.5, 100 mM NaCl, 5 mM EDTA, 50 mM NaF, 3% vol/vol NP-40). The NETF buffer was then replaced with 2× sample buffer and placed in a boiling water bath for 10 min. Immunoprecipitated samples were then separated by SDS-PAGE.
For SDS-PAGE, samples were diluted in sample buffer (Laemmli 1974) and placed in a boiling water bath for 5 min. Equal amounts of protein were separated on 7.5–15% gradient gels and transferred onto nitrocellulose overnight at 100 mA. For all antibodies, except antiphosphotyrosine (which was blocked with 3% BSA in TBS), blots were blocked in 5% skim milk (Carnation) in TBS overnight at 4°C. Primary antibodies used included antiphosphotyrosine (mAb 4G10; Upstate Biotechnology Inc.), anti-panTrk 203 (Hempstead et al. 1992), antiextracellular signal-regulated kinase (anti-ERK) 1 and 2 (pAb C-18; Santa Cruz), anti-tyr/thr phosphorylated ERK1 and 2 (Promega Corp.), anti-Akt and antiphosphoserine 473 Akt (New England Biolabs). For Western blots, secondary antibodies used were HRP-conjugated anti-mouse (1:10,000) and anti-rabbit (1:10,000) pAbs (Boehringer Mannhiem Corp.). All incubations were performed in 2.5% skim milk in TBS + 0.1% Tween-20 (Sigma Chemical Co.). For detection, blots were washed with TBS and antibody localization visualized using the ECL chemiluminescence kit (Nycomed Amersham Inc.).
Kinase Assays
100 μg of total protein was immunoprecipitated, except that after immunoprecipitation, beads were washed twice in 3% NETF buffer, followed by two washes in NETF buffer without NP-40, and a single wash in reaction buffer (20 mM MOPS, pH 7.2, 25 mM β-glycerolphosphate, 5 mM EGTA, 2 mM EDTA, 20 mM MgCl2, 2 mM sodium orthovanadate, 1 mM dithiothreitol, PKA inhibitor peptide [Upstate Biotechnology, Inc.], and 5% glycerol). The beads were resuspended in 20 μl of reaction buffer and 10 μl of myelin basic protein cocktail (2 mg/ml MBP in reaction buffer) for ERK assays, or histone H2B cocktail (1.6 mg/ml histone H2B in reaction buffer) for Akt assays. The reactions were initiated with 10 μl of 50 μM γ[32P]ATP (∼3,000 cpm/pmol) in a final volume of 40 μl and incubated for 20 min at 30°C. The reactions were terminated with the addition of 5× SDS sample buffer for 5 min and loaded onto an SDS-PAGE gel. After transfer of protein, the membrane was immunoblotted for the appropriate protein, exposed to film, and the bands excised and subjected to Cerenkov counting.
Akt peptide assays were conducted similarly, except an Akt specific substrate peptide was used in place of histone H2B. In brief, the beads were suspended in 20 μl reaction buffer with 10 μl Akt-1 substrate peptide (RPRAATF; Santa Cruz) dissolved as 1 mg/ml in assay buffer, and the reaction was started with the addition of 10 μl of 250 μM γ[32P]ATP (3,000 Ci/mmol prepared in assay dilution buffer). The reaction proceeded for 20 min at 30°C and was stopped by spotting 25 μl on 2.5-cm P81 filter paper (Whatman Inc.) and the remaining protein was charged with loading buffer for Western analysis. The P81 filter papers were washed 10 times in 0.75% phosphoric acid and counted in the scintillation counter to monitor incorporation of γ[32P]ATP.
Results
NGF and Depolarization Synergistically Promote Sympathetic Neuron Survival
To test the hypothesis that neurotrophins and neuronal activity together regulate neuronal survival in a fashion distinct from either alone, we focussed on postnatal day 1 sympathetic neurons of the neonatal rat superior cervical ganglion (SCG). Survival of these neurons, which are cultured at the time when they undergo naturally occurring cell death in vivo, can be maintained with either NGF (Belliveau et al. 1997) or depolarization as induced by chronic KCl treatment (Franklin et al. 1995).
To perform these experiments, sympathetic neurons were selected for five days in 50 ng/ml NGF, were switched to varying concentrations of NGF and/or KCl for two days, and survival was then measured using MTT assays, which measure mitochondrial function. As previously reported, maximal sympathetic neuron survival was obtained with either 10 ng/ml NGF or 50 mM KCl (Fig. 1 a; Franklin et al. 1995; Belliveau et al. 1997). Interestingly, when suboptimal concentrations of NGF and KCl were added together, these two stimuli acted synergistically to promote neuronal survival (Fig. 1 a). Specifically, while 2.5 ng/ml NGF supported only 29% survival and 5 or 10 mM KCl supported no survival, the combination of 2.5 ng/ml NGF plus 5 or 10 mM KCl supported 46 and 59% neuronal survival, respectively. Similarly, 5 ng/ml NGF caused 53% survival, but the addition of 10 mM KCl, which on its own has no survival effect, supported ∼100% neuronal survival (Fig. 1 a).
Figure 1.

NGF and KCL synergistically regulate sympathetic neuron survival. a, Sympathetic neuron survival in response to various combinations of NGF and KCl, as monitored using MTT assays. Neonatal sympathetic neurons were cultured in 50 ng/ml NGF for five days, washed free of neurotrophin-containing medium, and then incubated for two days in various concentrations of NGF (ng/ml) and/or KCl (mM), as indicated on the x-axis. Data is derived from one representative experiment performed in triplicate. Similar results were obtained in three separate experiments. In these assays, absolute values are normalized so that the value obtained with 20 ng/ml NGF is considered 100% survival. Error bars represent SD. *Indicate those NGF plus KCl values that are significantly different (P < 0.01, t test) from 2.5 (*) or 5 ng/ml NGF (**). b, Quantitative analysis of sympathetic neuron apoptosis in response to various combinations of NGF and KCl, as monitored by TUNEL. NGF-selected neurons were incubated for two days in different concentrations of NGF (ng/ml) and/or KCl (mM), as indicated on the x-axis. Each point represents combined data from three separate experiments, each of which was performed in triplicate. The data are expressed as percentage of TUNEL-negative cells/total Hoechst-labeled nuclei. Error bars and significance are as in a.
One possible explanation for this synergy is that KCl causes enhanced synthesis and/or secretion of a nonneurotrophin growth factor, thereby conditioning the media and causing enhanced survival. If this hypothesis were true, the synergistic survival should be density-dependent. To test this possibility, sympathetic neurons were plated in 50 ng/ml NGF at densities of 350 and 1,500 cells/well for five days (Fig. 2). Neurons were then switched to varying concentrations of NGF and/or KCl for an additional two days. The survival of these cultures was measured by MTT assay. This analysis (Fig. 2) demonstrated that the cell density had no effect on synergistic survival mediated by NGF and KCl.
Figure 2.

Sympathetic neuron survival in response to NGF and KCl is similar at different cell densities. Sympathetic neuron survival at different densities, as monitored by MTT assays. Neonatal sympathetic neurons were cultured in 50 ng/ml NGF for five days at 350 or 1,500 cells/well, washed free of neurotrophin-containing medium, and then incubated for two days in various concentrations of NGF (ng/ml) or KCl (mM), as indicated on the x-axis. Data is derived from one representative experiment performed in triplicate. In these assays, absolute values are normalized so that the value obtained with 10 ng/ml NGF is considered 100% survival. Error bars represent SD. *Indicate the NGF plus KCl values that are significantly different (P < 0.01, t test) from 2.5 (*) or 5 ng/ml NGF (**). Insets are phase-contrast photographs of representative fields of neurons at the different cell densities.
To confirm that the MTT assays were accurately measuring increases in neuronal survival, we performed similar experiments, but measured the extent of neuronal apoptosis rather than mitochondrial function. 48 h after being switched into various concentrations of NGF and/or KCl, neurons were stained for apoptotic nuclei using TUNEL-labeling and stained for all nuclei using Hoechst 33258 (Fig. 1 b and Fig. 3). We then determined the percentage of TUNEL-negative cells in representative fields from each survival condition. This analysis confirmed the results obtained with the MTT assays. Specifically, 57% of cells treated with 5 ng/ml NGF were TUNEL-negative (Fig. 1 b and 3, a and b) and 17% of cells treated with 10 mM KCl were TUNEL-negative (Fig. 1 b and 3, e and f). In contrast, when neurons were cultured in 5 ng/ml NGF plus 10 mM KCl, the number of TUNEL-negative neurons was increased to the same level as observed with 10 ng/ml NGF or 50 mM KCl (5 ng/ml NGF + 10 mM KCl = 78%; 10 ng/ml NGF = 79%; 50 mM KCl = 77%; Fig. 1 b and 3, i and j). Thus, suboptimal amounts of NGF and KCl synergistically support sympathetic neuron survival in a density-independent fashion.
Figure 3.
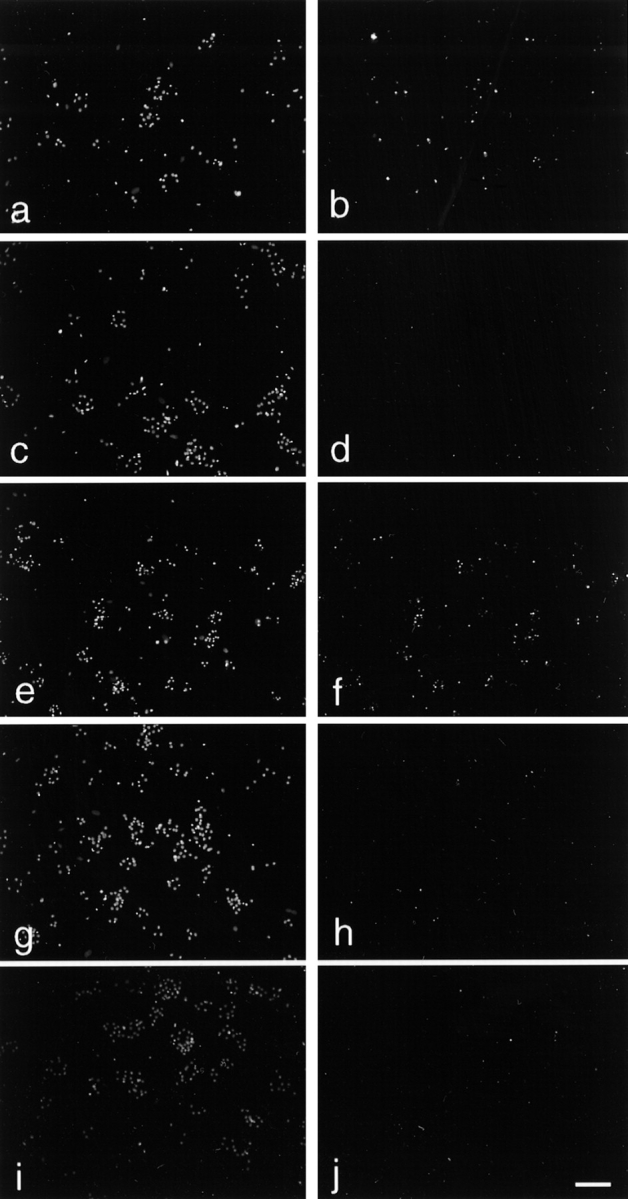
Quantitation of sympathetic neuron apoptosis in response to NGF and KCl. Immunofluorescence microscopy showing Hoechst 33258 (left) and TUNEL (right) of the same field of sympathetic neurons treated with NGF and/or KCL. Neurons were cultured five days in 50 ng/ml NGF, and then switched into the following concentrations of NGF and/or KCl for an additional two days before analysis: 5 ng/ml NGF (a and b); 10 ng/ml NGF (c and d); 10 mM KCl (e and f); 50 mM KCl (g and h); and 5 ng/ml NGF + 10 mM KCl (i and j). TUNEL labeling is markedly reduced in j, compared with that seen in b or f. Bar, 100μm.
Depolarization Signals Survival via a TrkA-independent Mechanism
Previous work with cortical neurons indicates that depolarization-induced survival requires an autocrine neurotrophin loop (Ghosh et al. 1994). To determine if a similar mechanism could explain the functional synergy observed here, we asked whether KCl-mediated survival requires Trk receptor activation. Lysates of neurons maintained in varying concentrations of NGF or KCl were immunoprecipitated with anti-panTrk, and the level of Trk receptor activation monitored by Western blot analysis with antiphosphotyrosine (Fig. 4 a). Both 5 and 10 ng/ml NGF led to a robust increase in Trk autophosphorylation, but Trk autophosphorylation was undetectable in the presence of KCl, as previously reported by ourselves and others (Franklin et al. 1995; Bamji et al. 1998).
Figure 4.

KCl signals via a TrkA-independent mechanism. a, KCl does not cause Trk autophosphorylation. NGF-selected sympathetic neurons were washed free of neurotrophin, and then exposed to various concentrations of NGF (ng/ml) or KCl (mM) for 15 min. Cellular lysates were immunoprecipitated with anti-panTrk and probed with antiphosphotyrosine (p-tyr) to visualize Trk autophosphorylation. All samples were normalized for equal amounts of protein. b, Dominant-negative TrkA inhibits NGF-mediated, but not KCl-mediated sympathetic neuron survival. NGF-selected neurons were infected with recombinant adenovirus expressing dnTrkA at titers ranging from 10 to 150 MOI, or with 200 MOI of a control adenovirus expressing the TTA transactivator, and were then switched to 10 ng/ml NGF (10N) or 50 mM KCl (50K) one day later. After 48 h, cell survival was measured by MTT assays. As controls, uninfected sister cultures were either withdrawn from NGF (0N), or maintained in 10 ng/ml NGF or 50 mM KCl. Results are those obtained in one representative experiment performed in triplicate, and represent the mean ± SD. Similar results were obtained in three separate experiments. *Represents those values that are significantly different from 10 ng/ml NGF alone (P < 0.01). c, K252a blocks Trk activation in response to NGF, whereas nifedipine has no effect. NGF-selected neurons were treated with various concentrations of NGF (ng/ml) or KCl (mM) for 15 min in the presence of 200 nm K-252a or 1 μM nifedipine (NIF), and then probed for phosphotyrosine (p-tyr). All samples were normalized for equal amounts of protein. d and e, K252a selectively blocks NGF-mediated sympathetic neuron survival, whereas nifedipine selectively blocks KCl-mediated survival. d, NGF-selected neurons were treated for two days with 200 nm K-252a in the presence of 10 ng/ml NGF (10N) or 50 mM KCl (50K) and survival was measured by MTT assays. Results derive from one representative experiment performed in triplicate and each point is the mean ± SD. Similar results were obtained in three separate experiments. *Indicates those values that are significantly different from 10 ng/ml NGF (P < 0.01). e, Quantitative analysis of TUNEL-labeled sympathetic neurons treated with 200 nm K-252a or 1 μM nifedipine (NIF) in the presence of 10 ng/ml NGF (10N) or 50 mM KCl (50K) for two days. Data are expressed as the percentage of TUNEL-negative cells/total Hoechst-positive nuclei. Each point represents combined data from three separate experiments, each performed in triplicate, and is the mean ± SD. *Indicates those values that are significantly different from 10 ng/ml NGF (*P < 0.01) or 50 mM KCl (**P < 0.01).
We then determined whether TrkA activation was necessary for KCl-mediated survival using a recombinant adenovirus that expresses a kinase-inactive form of TrkA that dimerizes with, and inhibits the activity of, endogenous TrkA (dnTrkA; Ferrari et al. 1995). NGF-selected neurons were infected with this dnTrkA adenovirus at various MOIs, and were then switched to 10 ng/ml NGF or 50 mM KCl. MTT assays revealed that dnTrkA caused a concentration-dependent decrease in NGF-mediated survival, but had no effect on KCl-mediated survival (Fig. 4 b). In contrast, infection with a control adenovirus had no effect on survival in either condition (Fig. 4 b).
We confirmed that Trk activity is not necessary for KCl-induced survival using the pharmacological Trk antagonist, K-252a (Tapley et al. 1992); neurons were exposed to varying concentrations of NGF or KCl plus or minus 200 nm K-252a, and were then analyzed biochemically for TrkA activation, using MTT assays and TUNEL to monitor survival. Western blot analysis of anti-panTrk immunoprecipitates with antiphosphotyrosine (Fig. 4 c) revealed that, as predicted, K-252a completely eliminated NGF-mediated TrkA autophosphorylation. MTT assays revealed that 200 nM K-252a decreased NGF-dependent sympathetic neuron survival by 60%, but had no effect on survival mediated by KCl (Fig. 4 d). In addition, 200 mM K252a did not support survival in the absence of NGF or KCl (Fig. 4 d). TUNEL confirmed these results; in 10 ng/ml NGF, K-252a treatment led to the same percentage of TUNEL-negative neurons as did NGF withdrawal (10%), while having no effect on the number of TUNEL-negative nuclei in 50 mM KCl (82%; Fig. 4 e).
Finally, we confirmed (Franklin et al. 1995) that KCl-mediated, but not NGF-mediated, sympathetic neuron survival requires activation of L-type calcium channels, using the L-type calcium channel blocker, nifedipine (Fox et al. 1987; Triggle et al., 1990). TUNEL-labeling of sympathetic neurons maintained in NGF or KCl plus or minus 1 μM nifedipine revealed that this drug completely inhibited KCl-, but not NGF-mediated survival (Fig. 4 e). Moreover, biochemical analysis confirmed that nifedipine had no effect on Trk receptor activation in the presence of NGF or KCl (Fig. 4 c). Thus, KCl-dependent survival requires activation of L-type calcium channels, and NGF-dependent survival requires TrkA.
Depolarization Activates ERK and Akt Kinases in Sympathetic Neurons
To determine potential intracellular convergence points for KCl and NGF-mediated survival, we examined two downstream substrates; the ERKs (MAP kinases), which are activated in sympathetic neurons by depolarization (Franklin et al. 1995) and by NGF (Creedon et al. 1996), and the serine–threonine kinase Akt, which is activated by TrkA (Andjelkovic et al. 1998), and is required for NGF-mediated sympathetic neuron survival (Crowder and Freeman 1998). To perform these experiments, NGF-selected sympathetic neurons were acutely stimulated with varying concentrations of NGF or KCl for 15 min, and then analyzed biochemically. Western blot analysis of neuronal lysates with phospho-specific ERK antibodies revealed that both NGF and KCl caused a dose-dependent increase in ERK phosphorylation, as monitored by phospho-specific antibodies for tyrosine and threonine (tyr/thr) or for tyrosine alone (Fig. 5 a). We also performed in vitro kinase assays using myelin basic protein as a phospho-acceptor substrate to monitor ERK activity. This analysis revealed that while both NGF and KCl induced increased phosphotransferase activity of ERK in a concentration-dependent fashion, higher levels of activation were observed with NGF than with KCl (Fig. 5 b).
Figure 5.

Activation of ERK and Akt kinases by NGF and KCl. a and b, ERK activation in response to increasing concentrations of NGF or KCl. a, Western blot analysis of equal amounts of protein derived from sympathetic neurons treated for 15 min with increasing concentrations of NGF (ng/ml) or KCl (mM), as detected with antibodies to tyrosine/threonine-phosphorylated ERKs (P-tyr/thr ERKs), tyrosine-phosphorylated ERKs (P-tyr ERKs), or to total ERK protein (total ERKs). The upper band in the doublet is ERK1 and the lower is ERK2. Note that all three panels are reprobes of the same blot. b, In vitro ERK activation assay using myelin basic protein (MBP) as a substrate in lysates of sympathetic neurons treated for 15 min with increasing concentrations of NGF (ng/ml) or KCl (mM). All samples were normalized for equal amounts of protein. c–e, Akt activation in response to increasing concentrations of NGF or KCl. c, Western blot analysis of equal amounts of protein derived from sympathetic neurons treated for 15 min with increasing concentrations of NGF (ng/ml) or KCl (mM), as detected with antibodies to serine-phosphorylated Akt (P-ser Akt) or to total Akt protein (Total Akt). Note that both panels are reprobes of the same blot. d, In vitro Akt activation using histone H2B as a substrate in lysates of sympathetic neurons treated for 15 min with increasing concentrations of NGF (ng/ml) or KCl (mM). All samples were normalized for equal amounts of protein. e, Quantitative in vitro Akt activity assay using an Akt-specific substrate in lysates of sympathetic neurons treated for 15 min with increasing concentrations of NGF (ng/ml) or KCl (mM). In sister cultures, neurons were coincidently treated with 100 μM of the PI3-kinase inhibitor LY294002 (LY). Bars represent the mean ± SD. Note that inhibition of PI3-kinase with LY294002 abolishes Akt activity induced by NGF or KCl.
Similar results were obtained when Akt activity was assessed. Western blot analysis with a phospho-specific (serine 473) Akt antibody revealed that NGF and KCl both caused a concentration-dependent increase in Akt phosphorylation, with NGF being more effective than KCl (Fig. 5 c). To more accurately measure Akt activity, we also performed in vitro kinase assays using two substrates, histone H2B (Franke et al. 1995) and the Akt specific substrate PRPAATF. With histone H2B, results were similar to those obtained using the phospho-Akt antibody (Fig. 5 d). Results differed slightly, however, with the Akt-specific substrate; in this case, both NGF and KCl increased Akt phosphotransferase activity in a dose-dependent fashion to approximately similar levels (Fig. 5 e). Thus, somewhat surprisingly, depolarization was able to increase Akt activity to levels similar to NGF-induced TrkA activation at doses where both NGF and KCl mediate maximal survival.
The Ras–PI3-kinase–Akt Pathway Is Involved in Depolarization-induced Neuronal Survival
Together, these results indicate that depolarization leads to robust activation of both the ERKs and Akt in sympathetic neurons. Since both of these substrates are known downstream targets of Ras (Rosen et al. 1994; Datta et al. 1996), and because Ras previously has been shown to be activated by calcium influx (Farnsworth et al. 1995), we hypothesized that depolarization-induced neuronal survival required Ras-dependent stimulation of either the PI3-kinase–Akt pathway or the MEK–ERK pathway. To test this hypothesis, we selectively blocked Ras and/or each of these two signaling pathways.
We first determined whether KCl-mediated survival required Ras activation, using an adenovirus expressing a dominant-inhibitory form of Ras (dnRas, N17Ras; Howe and Marshall 1993). NGF-selected neurons were infected with 200 MOI of adenovirus, expressing either dnRas or green fluorescent protein (GFP), and then were switched to KCl for two days. MTT assays revealed that dnRas expression decreased survival 60% in the presence of 50 mM KCl, a decrease similar to that observed in 10 ng/ml NGF (Fig. 6 a). The GFP adenovirus had no effect on neuronal survival.
Figure 6.

Ras, PI3-kinase, and Akt activity are important in KCl-mediated sympathetic neuron survival. a, Ras is involved in KCl-mediated survival. NGF-selected sympathetic neurons were infected with 200 MOI of adenovirus expressing either dnRas or GFP, and one day later were switched to 10 ng/ml NGF (10N) or 50 mM KCl (50K). 48 h later, survival was monitored by MTT assay. Results derive from one representative assay performed in triplicate; values represent the mean ± SD. Similar results were obtained in three separate experiments. *Indicate values that are significantly different (P < 0.01) from 10 ng/ml NGF (*) or 50 mM KCl (**). b and c, PI3-kinase, but not MEK activity, is involved in KCl-mediated survival. NGF-selected neurons were maintained for two days in 10 ng/ml NGF (10N) or 50 mM KCl (50K), plus or minus 100 μM of the PI3-kinase inhibitor LY294002 (LY) and/or 75 μM of the MEK inhibitor PD98059 (PD). Survival was then monitored by MTT assay (b) or TUNEL (c). In b, results represent data from one representative experiment performed in triplicate, and each point is the mean ± SD. In c, results represent the mean percentage of TUNEL-negative cells/total Hoechst-positive nuclei from three separate experiments, each performed in triplicate. *Asterisks indicate values that are significantly different (P < 0.01) from 10 ng/ml NGF (*) or 50 mM KCl (**). d, Akt is essential for a portion of KCl-mediated survival. NGF-selected sympathetic neurons were infected with 10–100 MOI of an adenovirus expressing dnAkt and one day later were switched into 10 ng/ml NGF (10N) or 50 mM KCl (50K). All neurons were also coinfected with 50 MOI of an adenovirus expressing the transactivator TTA. As controls, neurons were infected with 200 MOI of the TTA adenovirus alone. Neuronal survival was then measured two days later by MTT assay. Results represent data from one representative experiment performed in triplicate, and each point is the mean ± SD. Similar results were obtained in three separate experiments. *Indicate values that are significantly different (P < 0.01) from 10 ng/ml NGF (*) or 50 mM KCl (**). e, Western blot analysis for ERKs and Akt kinase in sympathetic neurons treated for 15 min with NGF (ng/ml) or KCl (mM), as analyzed using antibodies specific to tyrosine/threonine-phosphorylated ERKs (P-tyr/thr ERKs), tyrosine phosphorylated ERKs (P-tyr ERKs), serine-phosphorylated Akt (P-ser Akt), or total ERK or Akt protein. Some cultures were coincidently treated with 100 μM LY294002 (LY) or 75 μM PD98059 (PD). All samples were normalized for equal amounts of protein. The three ERK panels are reprobes of the same blot, as are the two Akt panels. f, Ras activity is necessary for KCl-mediated Akt phosphorylation. Western blot analysis for ERKs and Akt kinase in sympathetic neurons infected with 200 MOI dnRas, and then induced for 15 min with KCl (mM), using the same antibodies as in e. All ERK panels derive from reprobes of one blot, and the Akt panels derive from reprobes of another.
We next determined if either of the two downstream Ras pathways, PI3-kinase–Akt or MEK–ERK, were also essential for KCl-induced survival using two pharmacological agents; LY294002, which selectively inhibits PI3-kinase (Vlahos et al. 1994), and PD98059, which selectively inhibits MEK (Alessi et al. 1995). NGF-selected neurons were maintained for 48 h in 50 mM KCl or 10 ng/ml NGF plus or minus 100 μM LY294002 or 75 μM PD98059, and were then analyzed by MTT assays and TUNEL. MTT assays revealed that inhibition of PI3-kinase with LY294002 reduced KCl-induced sympathetic neuron survival by ∼40%, while treatment with PD98059 had no effect (Fig. 6 b). The effect of cotreatment with both drugs was similar to LY294002 alone (Fig. 6 b). In contrast, LY294002 completely inhibited survival mediated by NGF (Fig. 6 b), as previously reported (Bartlett et al. 1997; Crowder and Freeman 1998). TUNEL confirmed these results; PD98059 had no effect on numbers of TUNEL-negative neurons in either KCl or NGF, whereas LY294002 decreased the proportion of TUNEL-negative neurons to 42 and 10% respectively, in the presence of 50 mM KCl and 10 ng/ml NGF (Fig. 6 c).
To confirm that this concentration of PD98059 inhibited MEK, in spite of having no effect on neuronal survival, we examined the downstream MEK substrates, the ERKs. Western blot analysis of sympathetic neurons treated with 10 ng/ml NGF or 50 mM KCl plus or minus 75 μM PD revealed that, as predicted, this drug inhibited both tyrosine/threonine and tyrosine phosphorylation of the ERKs, but had no effect on Akt phosphorylation (Fig. 6 e). Thus, the MEK–ERK pathway is apparently not required for depolarization-induced sympathetic neuron survival.
Since PI3-kinase is necessary for 40–50% of KCl-mediated neuronal survival, we next determined whether KCl-induced survival also required the PI3-kinase substrate Akt, using an adenovirus expressing dnAkt from a TTA-inducible promoter (Boudreau, M., C. Tudan, and D.R. Kaplan, unpublished data). NGF-selected sympathetic neurons were infected with 10 to 100 MOI dnAkt adenovirus and 50 MOI TTA-expressing adenovirus (which transactivates the promoter for dnAkt), and were then maintained in 50 mM KCl for two days. As a control, neurons were infected with 50 or 200 MOI of the TTA adenovirus alone. MTT analysis revealed that dnAkt reduced KCl-mediated survival ∼40%, whereas the TTA adenovirus on its own had no effect (Fig. 6 d). In contrast, similar experiments with 10 ng/ml NGF revealed that, as previously reported (Crowder and Freeman 1998), dnAkt completed inhibited NGF-induced survival (Fig. 6 d).
Together, these data indicate that blocking either Ras, PI3-kinase, or Akt is sufficient to reduce KCl-mediated survival by 40–50%. To test whether these results reflect the presence of a linear Ras–PI3-kinase–Akt survival pathway, we determined whether inhibiting Ras or PI3-kinase blocked the KCl-induced phosphorylation of Akt. Initially, we examined Ras: neurons were infected with the dnRas adenovirus and were then acutely activated with 50 mM KCl or 10 ng/ml NGF for 15 min. Western blot analysis with antiphospho-Akt revealed that dnRas blocked Akt activation in response to 50 mM KCl (Fig. 6 f), suggesting that Akt activity is dependent upon Ras activity. Similarly, inhibition of PI3-kinase with 100 μM LY294002 revealed that treatment with LY294002 completely blocked the KCl-induced phosphorylation of Akt, as assayed either by Western blot analysis with antiphospho-Akt (Fig. 6 e) or Akt kinase assays using the Akt-specific substrate (Fig. 5 e). Thus, calcium influx through L-type calcium channels causes activation of a Ras–PI3-kinase–Akt pathway that is essential for ∼50% of KCl-induced neuronal survival.
Depolarization and NGF Converge on Akt and ERKs, but only Akt Is Necessary for Synergistic Survival
On the basis of these data, we hypothesized that the synergistic survival seen with NGF and KCl might be mediated by a convergent stimulation of the Ras–PI3-kinase–Akt pathway. To test this hypothesis, NGF-selected neurons were induced for 15 min with 5 ng/ml NGF plus 10 mM KCl, a combination that mediated survival synergistically (Fig. 1 a), and were then analyzed biochemically. Western blot analysis with antiphospho-Akt, followed by quantitative densitometry, revealed that while 10 mM KCl had no detectable effect on Akt phosphorylation (Fig. 7, a and c), the addition of 10 mM KCl to either 2.5 or 5 ng/ml NGF led to levels of Akt phosphorylation that were higher than either of these concentrations of NGF alone (Fig. 7, a and c). Similar results were obtained when ERK phosphorylation was examined. 10 mM KCl did not detectably increase ERK tyrosine/threonine phosphorylation above controls, but the addition of 10 mM KCl to either 2.5 or 5 ng/ml NGF led to significantly higher levels of ERK phosphorylation than did treatment with either concentration of NGF alone (Fig. 7, a and b).
Figure 7.
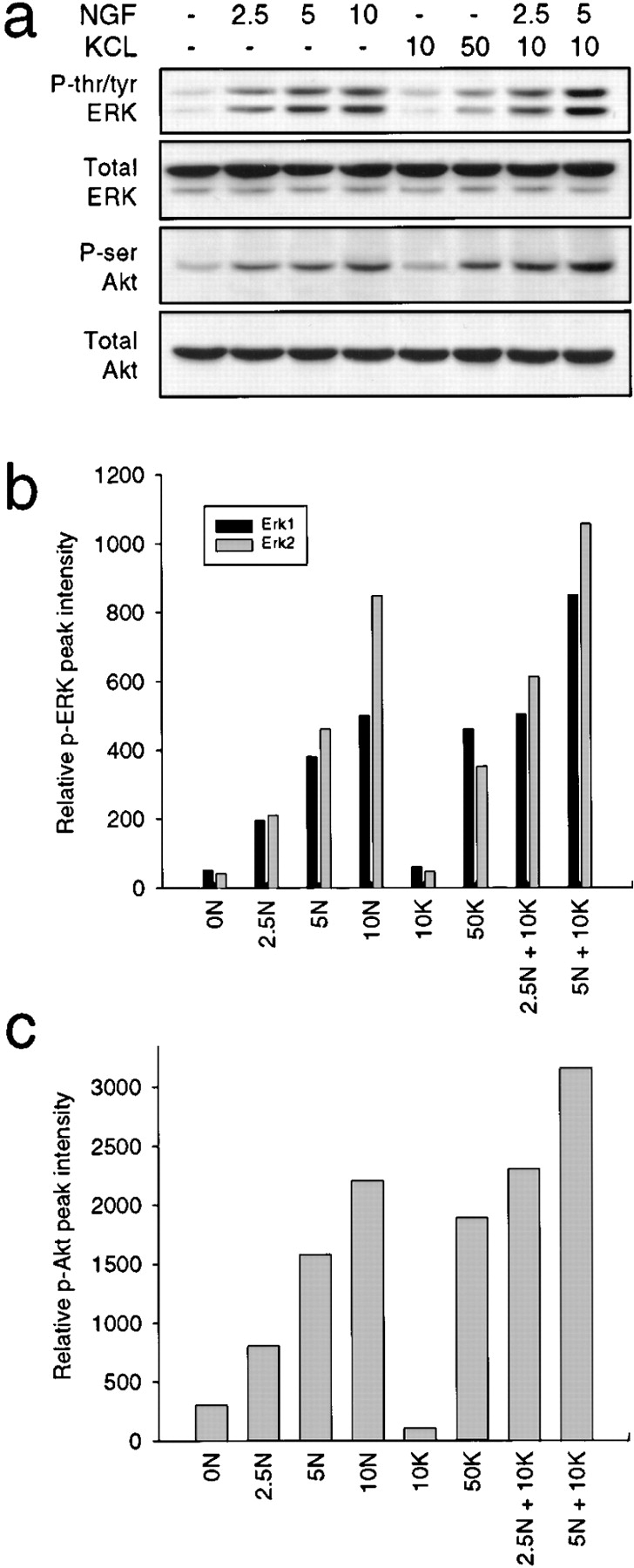
Suboptimal concentrations of NGF and KCl synergistically increase ERK and Akt kinase phosphorylation. a, Western blot analysis of sympathetic neurons induced for 15 min with various concentrations of NGF (ng/ml) and/or KCl (mM), using antibodies specific to tyrosine/threonine-phosphorylated ERKs (P-tyr/thr ERK), serine-phosphorylated Akt (P-ser Akt) or total ERK or Akt protein. All samples were normalized for equal amounts of protein. All ERK panels derive from reprobes of one blot, and the Akt panels reprobes of another. b and c, Quantitative analysis of ERK (b) or Akt (c) phosphorylation in response to various concentrations of NGF and/or KCl, obtained by scanning densitometry of Western blots similar to those shown in a. Note that 10 mM KCl (10K) does not produce signals that are detectably increased over control (0N) neurons, but treatment with 2.5 ng/ml NGF (2.5N) and 5 ng/ml NGF (5N) plus 10 mM KCl results in levels of ERK and Akt phosphorylation that are higher than either 2.5 or 5 ng/ml NGF alone, respectively.
We then determined the functional necessity of this convergent activation for sympathetic neuron survival. We first examined Ras: NGF-dependent neurons were infected with the dnRas adenovirus, were switched to 5 ng/ml NGF plus 10 mM KCl, and survival was determined two days later by MTT assays. This analysis (Fig. 8 a) revealed that inhibition of Ras decreased the synergistic survival ∼50%, a result similar to that seen with NGF or KCl alone (Fig. 6 a). Coincident with the decrease in neuronal survival observed with dnRas, we also observed a partial decrease in the downstream activation of Akt and the ERKs, as assessed by Western blots with phosphorylation-specific antibodies (Fig. 8 b).
Figure 8.

The Ras–PI3-kinase–Akt pathway is required for the synergistic survival observed with NGF and KCl together. a, Ras activity is necessary for ∼50% of synergistic survival. NGF-selected sympathetic neurons were infected with 200 MOI of dnRas or GFP adenovirus, and one day later were switched into 5 ng/ml NGF (5N) plus or minus 10 mM KCl (10K). Neuronal survival was monitored two days later by MTT assay. Results represent the mean ± SD of one representative experiment performed in triplicate. Similar results were obtained in three separate experiments. *Indicates those results that are significantly different (P < 0.01) from 5 ng/ml NGF plus 10 mM KCl. b, Dominant-negative Ras partially inhibits Akt and ERK phosphorylation induced by NGF plus KCl. Western blot analysis for Akt and ERK phosphorylation in sympathetic neurons infected with 200 MOI dnRas adenovirus and then maintained in 5 ng/ml NGF (5N) and/or 10 mM KCl (10K). Lysates were analyzed with antibodies specific for serine-phosphorylated Akt (P-ser Akt), tyrosine/threonine-phosphorylated ERKs (P-tyr/thr ERKs), or total Akt or ERK protein. All samples were normalized for equal amounts of protein. All ERK panels are reprobes of one blot, and Akt panels reprobes of another. c, PI3-kinase is necessary for all of the survival seen with NGF plus KCl. MTT assays of sympathetic neurons maintained in various concentrations of NGF (ng/ml) and/or KCl (mM) treated coincidently with 100 μM LY294002 (LY) and/or 75 μM PD98059 (PD). Results represent data obtained from one representative experiment performed in triplicate, and each point is the mean ± SD. Similar results were obtained in three separate experiments. *Indicates those values that are significantly different from 5 ng/ml NGF plus 10 mM KCl (*) or 10 ng/ml NGF plus 10 mM KCl (**). d, Akt activity is necessary for all of the survival seen with NGF plus KCl. NGF-selected sympathetic neurons were infected with 10–100 MOI dnAkt adenovirus and, one day later, were switched to 5 ng/ml NGF plus 10 mM KCl. All neurons infected with dnAkt adenovirus were also infected with 50 MOI of an adenovirus expressing the TTA transactivator. Control neurons were infected with 100 MOI of the TTA adenovirus alone. Results represent the mean ± SD of one representative experiment performed in triplicate. Similar results were obtained in three separate experiments. *Indicates those values that are significantly different (P < 0.01) from 5 ng/ml NGF plus 10 mM KCl.
We then examined the necessity for PI3-kinase or MEK in synergistically-mediated survival using the pharmacological inhibitors LY294002 and PD98059. MTT assays of neurons maintained in 50 ng/ml NGF with 10 mM KCl plus or minus one of these inhibitors revealed that inhibition of PI3-kinase with LY294002 completely blocked neuronal survival, whereas inhibition of MEK with PD98059 had no effect (Fig. 8 c). Cotreatment with both LY294002 and PD98059 gave results similar to those observed with LY294002 alone (Fig. 8 c). Thus, PI3-kinase activity is essential for all of the synergistic survival effects seen with NGF plus KCl.
Finally, we examined the necessity for Akt using the dnAkt adenovirus: neurons were infected with 50 MOI of the TTA-expressing adenovirus and 10 to 100 MOI of the dnAkt adenovirus, and then switched to 5 ng/ml NGF plus 10 mM KCl for two days. MTT assays revealed that inhibition of Akt completely blocked the ability of NGF plus KCl to maintain sympathetic neuron survival (Fig. 8 d). Together with the LY294002 experiments, these data indicate that the synergistic survival observed with depolarization and NGF is mediated via convergence onto the PI3-kinase–Akt pathway.
CaMKII Activity Is Required for Depolarization-induced Survival, but Not Synergistic Survival Mediated by NGF and Depolarization
Together, these data indicate that Ras–PI3-kinase–Akt is one of the survival pathways induced by activation of neuronal L-type channels, and that this pathway is essential for the synergy between NGF and depolarization. A second, calcium-activated pathway that might also be involved in neuronal survival involves calcium/calmodulin-dependent protein kinase II (CaMKII; Hanson and Schulman 1992; Ghosh and Greenberg 1995). To determine the potential importance of this pathway for sympathetic neuron survival, neurons were maintained in various concentrations of NGF and/or KCl plus or minus 10 μM KN-62, a specific pharmacological blocker of CaMKII (Tokumitsu et al. 1990). MTT assays two days later revealed that 10 μM KN-62 dramatically reduced neuronal survival in 50 mM KCl, but had no effect on survival mediated by 10 ng/ml NGF or by 5 ng/ml NGF plus 10 mM KCl (Fig. 9 a). TUNEL assays confirmed this result: the number of TUNEL-negative cells was similar in NGF plus or minus KN-62, but a large decrease in TUNEL-negative cells was observed when KN-62 was added to 50 mM KCl (Fig. 9 b). Thus, in addition to a Ras–PI3-kinase–Akt pathway, calcium influx through L-type channels also mediates neuronal survival through a CaMKII-dependent pathway.
Figure 9.

The role of CaMKII in survival mediated by NGF and/or KCl. a and b, CaMKII is necessary for KCl-mediated survival, but plays no role in the synergistic survival seen with NGF plus KCl. NGF-selected sympathetic neurons maintained in NGF and/or KCl for two days with or without 10 μM KN-62 and survival was then monitored by MTT assay (a) or TUNEL (b). In a, results represent the mean ± SD of one representative experiment performed in triplicate. Similar results were obtained in three separate experiments. In c, results represent the mean percentage of TUNEL-negative cells/total Hoechst-positive nuclei as determined in three separate experiments, each of which was performed in triplicate. In both panels, *indicates values that are significantly different (P < 0.01) from 50 mM KCl. c, CaMKII is necessary for ERK, but not Akt phosphorylation in response to KCl. Western blot analysis of equal amounts of protein derived from sympathetic neurons stimulated for 15 min with NGF (ng/ml) and/or KCl (mM) with or without 10 μM KN-62. Blots were probed with antibodies specific to tyrosine/threonine-phosphorylated ERKs (P-tyr/thr ERK), serine-phosphorylated Akt (P-ser Akt), or to total ERK or Akt protein. The ERK panels derive from reprobes of the same blot, and the Akt panels from reprobes of another.
To determine whether Akt activation was also downstream of CaMKII, we assessed KCl-mediated Akt phosphorylation in the presence of 10 μM KN-62. Western blot analysis revealed that inhibition of CaMKII with KN-62 had no effect on Akt phosphorylation as induced by NGF, KCl, or combinations of NGF plus KCl (Fig. 9 c). In contrast, KN-62 dramatically reduced KCl-induced ERK tyrosine/threonine phosphorylation, but had no effect on NGF-induced ERK phosphorylation (Fig. 9 c). Interestingly, KN-62 partially blocked ERK phosphorylation induced by 5 ng/ml NGF plus 10 mM KCl (Fig. 9 c), indicating that the synergistic activation of ERKs seen in NGF plus KCl is partially due to CaMKII. Together, these data indicate that: CaMKII is a component of an important depolarization-induced survival pathway in sympathetic neurons; Akt is not downstream of CaMKII in this survival pathway; and this CaMKII pathway is not an important player in the synergistic survival observed with NGF and KCl. Instead, NGF and depolarization apparently converge onto a distinct Ras–PI3-kinase–Akt pathway to synergistically regulate survival.
Discussion
Data presented here demonstrate that combinations of trophic support (NGF) and neuronal activity (KCl-induced depolarization) have a positive, synergistic effect on the survival of peripheral sympathetic neurons at concentrations of NGF and KCl that are themselves suboptimal for neuronal survival. Moreover, these experiments demonstrate that the synergistic survival is due to convergence onto the PI3-kinase–Akt survival pathway, and that in the case of KCl, activation of this pathway is a consequence of calcium-mediated Ras activation (Fig. 10). In contrast, although KCl-mediated survival also requires concomitant activation of CaMKII, this protein is dispensable when both KCl and NGF are both present (Fig. 10), and it is not involved in activating Akt under any of the conditions studied here. This convergent activation of Akt, a signaling protein known to be essential for the survival of several neuronal populations (Dudek et al. 1997; Crowder and Freeman 1998), may provide a general mechanism for coordinating the survival effects of growth factors and neural activity throughout the developing nervous system.
Figure 10.
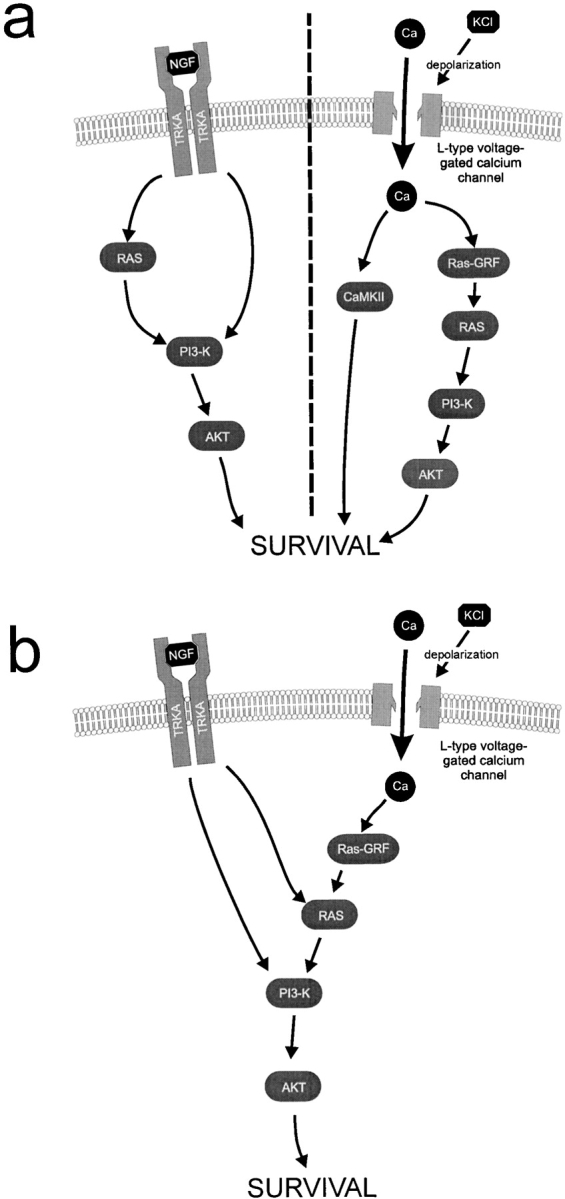
Proposed models for signal-transduction pathways mediating survival in NGF or KCl alone (a) or synergistically in the presence of both NGF and KCl (b). a, Illustrates the proposed pathways that support sympathetic neuron survival when either NGF or KCl are alone. b, Illustrates the proposed convergence onto the Ras–PI3-kinase–Akt pathway that is essential for the synergistic survival seen at concentrations of NGF and KCl, which on their own are not sufficient for maximal survival.
These findings have important implications for neuronal survival in the developing and mature injured nervous system. During development, ∼50% of central and peripheral neurons undergo apoptosis. Although neurotrophin signaling has traditionally been considered to be the major mechanism for the matching of neurons to their targets (Barde 1989; Majdan and Miller 1999), a large body of work indicates that establishment of appropriate neural activity is also important (Oppenheim 1991; Franklin and Johnson 1992). Evidence presented here demonstrates that, when NGF is limiting, levels of depolarization that themselves have no survival effect, synergize with NGF to support sympathetic neuron survival. Since NGF concentrations in vivo during naturally occuring sympathetic neuron death are limiting (Barde 1989; Levi-Montalcini, 1987), our findings imply that developing neurons that are active will have a competitive survival advantage over those that are not, even when both are exposed to similar limiting amounts of target-derived NGF. This mechanism, therefore, ensures selection of neurons that pathfind their way to an appropriate target early enough to sequester target-derived NGF and participate in a functional circuit. The validity of such a model derives from the finding that inhibition of either pre- or postganglionic activity is sufficient to enhance developmental sympathetic neuron apoptosis (Maderdrut et al. 1988).
Does this mechanism generalize to neurons other than sympathetic neurons? Results presented here demonstrating synergistic survival effects of neurotrophins and depolarization are strikingly similar to those previously reported for central neurons, such as retinal ganglion cells (Meyer-Franke et al. 1995, Meyer-Franke et al. 1998), which require both growth factors and either depolarization or neurotransmitters for their survival in culture. Thus, central and peripheral neurons may not be as different in this regard as previously thought. It may simply be that cooperative interactions between growth factors and activity are always essential for survival of central neurons, whereas they are only essential for peripheral neurons when trophic support is suboptimal. However, since NGF concentrations are suboptimal during development (Barde 1989; Levi-Montalcini, 1987) and neural activity is essential for survival during naturally occurring sympathetic neuron death (Maderdrut et al. 1988), then these apparent differences may be a function of culture conditions, rather than a reflection of the in vivo situation. In this regard, convergent activation of Akt by growth factors and activity may be just as important to central neurons as to peripheral neurons, since Akt is a necessary survival protein for CNS neurons, such as cerebellar granule cells (Dudek et al. 1997). It is also clear, however, that in the case of central neurons, growth factors and depolarization collaborate to regulate neuronal survival by more than one mechanism (Ghosh et al. 1994; Blair and Marshall 1997; Meyer-Franke et al. 1998).
How do NGF and KCl converge to activate Akt? Neurotrophin binding to TrkA leads to activation of the PI3-kinase–Akt pathway (reviewed in Kaplan and Stephens 1994; Kaplan and Miller 1997). The activation of PI3-kinase is thought to occur both via Ras (Rodriguez-Viciana et al. 1994) and Gab-1 (Holgado-Madruga et al. 1997). Moreover, Ras, PI3-kinase, and Akt activity have all been shown to be essential for NGF-mediated survival of sympathetic neurons (Bartlett et al. 1997; Markus et al. 1997; Crowder and Freeman 1998). With regard to KCl, calcium influx has been shown to activate Ras via Ras–GRF (Farnsworth et al. 1995) and, depending on the neuronal context, may or may not activate PI3-kinase (D'Mello et al. 1997; Miller et al. 1997; Soler et al. 1998). In this paper, we demonstrate that calcium influx also activates Akt, and that this activation involves both Ras and PI3-kinase (Fig. 10). Moreover, our data demonstrate that synergistic survival is dependent on Ras, PI3-kinase, and Akt activity, indicating that NGF and KCl converge directly on these proteins to coordinately regulate neuronal survival.
The synergistic activation of signaling proteins by two growth factors previously has been observed in a number of systems, such as Erk 2 by stem cell factor and IL-3 (Pearson et al. 1998), and p38MAPK by IL-2 and IL-12 (Gollob et al. 1999). Such biochemical synergy can occur on several levels. We favor a hypothesis in which KCl causes a nonlinear activation of Ras or PI-3 kinase beyond the activation or localization observed by NGF alone, stimulating the production of PI3-kinase–derived second messenger molecules that function as activators of PDK1, PDK2, and Akt itself, and which recruit Akt to the plasma membrane, an event necessary for increasing Akt activity (Coffer et al. 1998; Downward 1998). The synergistic activation of Akt would therefore be due to the combined actions of multiple Akt activators, some of which are activated linearly and others nonlinearly.
In a recent study examining depolarization-induced survival of neuroblastoma cells, Yano et al. 1998 showed that in neuroblastoma cells, calcium influx mediates cell survival via a PI3-kinase–independent CaMK kinase–Akt pathway. In contrast, our findings indicate that depolarization-induced Akt activation in primary sympathetic neurons is totally dependent upon PI3-kinase. Moreover, although KCl-mediated sympathetic neuron survival is highly dependent on CaMKII, this protein is dispensable for the Akt-dependent synergistic survival effect seen with NGF and KCl. These findings do not imply that CaMKII is unimportant for sympathetic neurons exposed to both NGF and depolarization. In fact, data presented here indicate that CaMKII activity is essential for the synergistic ERK activation seen in response to these two extrinsic cues. Although this ERK activation is not essential for sympathetic neuron survival, it may well be essential for other neuronal responses. In particular, the MEK–ERK pathway is required for neurite extension in PC12 cells (reviewed in Kaplan and Stephens 1994), and our recent data indicate that MEK is equally important for growth of sympathetic neurons in vivo and in vitro (Zirrgeibel, U., D. Lederfein, J. Atwal, J. Toma, F. Miller, and D. Kaplan, unpublished data). Thus, the biochemical convergence of NGF and KCl on the ERKs may well be important for neuronal growth and plasticity (Impey et al. 1999). The involvement of CaMKII in this convergence fits well with its proposed role in neuronal plasticity (reviewed in Ghosh and Greenberg 1995).
In summary, these findings demonstrate that, like central neurons, NGF and depolarization synergistically regulate the survival of peripheral sympathetic neurons under conditions where trophic support is suboptimal, such as during naturally occurring neuronal death. This synergistic survival effect is mediated by intracellular convergence on a Ras–PI3-kinase–Akt pathway. Since Akt is an essential survival molecule for several neuronal populations, this intracellular convergence may represent a general mechanism for coordinating the survival effects of growth factors and neural activity throughout the nervous system.
Footnotes
1.used in this paper: CaMKII, calcium/calmodulin-dependent protein kinase II; dnAkt, dominant-negative Akt; ERK, extracellular signal-regulated kinase; GFP, green fluorescent protein; IGF, insulin-like growth factor; MAP, mitogen-activated protein; MEK, MAP kinase kinase; MOI, multiplicity of infection; MTT, 3[4,5-dimethylthio-zol-2-yl]2,5-diphenyltetrazolium bromide; PI3-kinase, phosphatidylinositol 3-kinase; TUNEL, TdT-mediated dUTP nick end labeling
References
- Alessi D.R., Cuenda A., Cohen P., Dudley D.T., Saltiel A. PD098059 is a specific inhibitor of the activation of mitogen activated protein kinase kinase in vitro and in vivo. J. Biol. Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Aloyz R.S., Bamji S.X., Pozniak C.D., Toma J.G., Atwal J., Kaplan D.R., Miller F.D. p53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J. Cell Biol. 1998;143:1691–1703. doi: 10.1083/jcb.143.6.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andjelkovic M., Suidan H.S., Meier R., Frech M., Alessi D.R., Hemmings B.A. Nerve growth factor promotes activation of the alpha, beta and gamma isoforms of protein kinase B in PC12 pheochromocytoma cells. Eur. J. Biochem. 1998;251:195–200. doi: 10.1046/j.1432-1327.1998.2510195.x. [DOI] [PubMed] [Google Scholar]
- Bamji S.X., Majdan M., Pozniak C.D., Belliveau D.J., Aloyz R., Kohn J., Causing C.G., Miller F.D. The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J. Cell. Biol. 1998;140:911–923. doi: 10.1083/jcb.140.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barde Y.A. Trophic factors and neuronal survival. Neuron. 1989;2:1525–1534. doi: 10.1016/0896-6273(89)90040-8. [DOI] [PubMed] [Google Scholar]
- Bartlett S.E., Reynolds A.J., Weible M., Heydon K., Hendry I.A. In sympathetic but not sensory neurons, phosphoinositide 3-kinase in important for NGF-dependent survival and the retrograde transport of 125I-βNGF. Brain Res. 1997;761:257–262. doi: 10.1016/s0006-8993(97)00329-6. [DOI] [PubMed] [Google Scholar]
- Belliveau D.J., Krivko I., Kohn J., Lachance C., Pozniak C., Rusakov D., Kaplan D., Miller F.D. NGF and NT-3 both activate TrkA on sympathetic neurons but differentially regulate survival and neuritogenesis. J. Cell Biol. 1997;136:374–388. doi: 10.1083/jcb.136.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair L.A.C., Marshall J. IGF-1 modulated N and L calcium channels in a PI 3-kinase–dependent manner. Neuron. 1997;19:421–429. doi: 10.1016/s0896-6273(00)80950-2. [DOI] [PubMed] [Google Scholar]
- Coffer P.J., Jin J., Woodgett J.P. Protein kinase B (c-Akt)a multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem. J. 1998;335:1–13. doi: 10.1042/bj3350001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creedon D.J., Johnson E.M., Lawrence J.C. Mitogen-activated protein kinase-independent pathways mediate the effects of nerve growth factor and cAMP on neuronal survival. J. Biol. Chem. 1996;271:20713–20718. doi: 10.1074/jbc.271.34.20713. [DOI] [PubMed] [Google Scholar]
- Crowder R.J., Freeman R.S. Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor dependent sympathetic neurons. J. Neurosci. 1998;18:2933–2943. doi: 10.1523/JNEUROSCI.18-08-02933.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta K., Bellacosa A., Chan T.O., Tsichlis P.N. Akt is a direct target of the phosphatidylinositol 3-kinase. Activation by growth factors, v-src and v-Ha-ras, in Sf9 and mammalian cells. J. Biol. Chem. 1996;271:30835–30839. doi: 10.1074/jbc.271.48.30835. [DOI] [PubMed] [Google Scholar]
- D'Mello S.R., Borodezt K., Soltoff S.P. Insulin-like growth factor and potassium depolarization maintain neuronal survival by distinct pathwayspossible involvement of PI3-kinase in IGF-1 signaling. J. Neurosci. 1997;17:1548–1560. doi: 10.1523/JNEUROSCI.17-05-01548.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downward J. Mechanisms and consequences of activation of protein kinase B/Akt. Curr. Opin. Cell Biol. 1998;10:262–267. doi: 10.1016/s0955-0674(98)80149-x. [DOI] [PubMed] [Google Scholar]
- Dudek H., Datta S.R., Franke T.F., Birnbaum M.J., Yao R., Cooper G.M., Segal R.A., Kaplan D.R., Greenberg M.E. Regulation of neuronal survival by the serine–threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Farnsworth C.L., Freshney N.W., Rosen L.B., Ghosh A., Greenberg M.E., Feig L.A. Calcium activation of Ras mediated by neuronal exchange factor Ras–GRF. Nature. 1995;376:524–527. doi: 10.1038/376524a0. [DOI] [PubMed] [Google Scholar]
- Ferrari F., Anderson B.L., Stephens R.M., Kaplan D.R., Greene L.A. Prevention of apoptotic neuronal death by GM1 ganglioside. J. Biol. Chem. 1995;270:3074–3080. doi: 10.1074/jbc.270.7.3074. [DOI] [PubMed] [Google Scholar]
- Fox A., Nowycky M.C., Tsien R.W. Kinetic and pharmacological properties distinguishing three types of calcium currents in chick sensory neurons. J. Physiol. 1987;394:149–172. doi: 10.1113/jphysiol.1987.sp016864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis N.J., Landis S.C. Cellular and molecular determinants of sympathetic neuron development. Annu. Rev. Neurosci. 1999;22:541–566. doi: 10.1146/annurev.neuro.22.1.541. [DOI] [PubMed] [Google Scholar]
- Franke T.F., Yang S.I., Chan T.O., Datta K., Kazlauskas A., Morrison D.K., Kaplan D.R., Tsichlis P.N. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- Franklin J.L., Johnson E.M. Suppression of programmed neuronal death by sustained elevation of cytoplasmic calcium. Trends Neurosci. 1992;15:500–508. doi: 10.1016/0166-2236(92)90103-f. [DOI] [PubMed] [Google Scholar]
- Franklin J.L., Sanz-Rodiguez C., Juhasz A., Deckworth T.L., Johnson E.M. Chronic depolarization prevents programmed death of sympathetic neurons in vitro but does not support growthrequirement for Ca2+ influx but not Trk activation. J. Neurosci. 1995;15:643–664. doi: 10.1523/JNEUROSCI.15-01-00643.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A., Greenberg M.E. Calcium signaling in neuronsmolecular mechanisms and cellular consequences. Science. 1995;268:239–246. doi: 10.1126/science.7716515. [DOI] [PubMed] [Google Scholar]
- Ghosh A., Carnahan J., Greenberg M. Requirement of BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1623. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- Gollob J.A., Schnipper C.P., Murphy E.A., Ritz J., Frank D.A. The functional synergy between IL-12 and IL-2 involves p38 mitogen-activated protein kinase and is associated with the augmentation of STAT serine phosphorylation. J. Immunol. 1999;162:4472–4481. [PubMed] [Google Scholar]
- Hanson P.I., Schulman H. Neuronal Ca2+/calmodulin-dependent protein kinases. Annu. Rev. Biochem. 1992;61:559–601. doi: 10.1146/annurev.bi.61.070192.003015. [DOI] [PubMed] [Google Scholar]
- Hempstead B.L., Rabin S.J., Kaplan L., Reid S., Parada L.F., Kaplan D.R. Overexpression of the trk tyrosine kinase receptor rapidly accelerates nerve growth factor-induced differentiation. Neuron. 1992;9:883–896. doi: 10.1016/0896-6273(92)90241-5. [DOI] [PubMed] [Google Scholar]
- Holgado-Madruga M., Moscatello D.K., Emlet D.R., Dieterich R., Wong A.J. Grb2-associated binder-1 mediates phosphatidyliositol 3-kinase activation and the promotion of cell survival by nerve growth factor. Proc. Natl. Acad. Sci. USA. 1997;94:12419–12424. doi: 10.1073/pnas.94.23.12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe L.R., Marshall C.J. Identification of amino acids in p21ras involved in exchange factor interaction. Oncogene. 1993;8:2583–2590. [PubMed] [Google Scholar]
- Impey S., Obrietan K., Storm D.R. Making new connectionsrole of ERK/MAP kinase signaling in neuronal plasticity. Neuron. 1999;23:11–14. doi: 10.1016/s0896-6273(00)80747-3. [DOI] [PubMed] [Google Scholar]
- Kaplan D.R., Stephens R.M. Neurotrophin signal transduction by the Trk receptor. J. Neurobiol. 1994;25:1404–1417. doi: 10.1002/neu.480251108. [DOI] [PubMed] [Google Scholar]
- Kaplan D.R., Miller F.D. Signal transduction by the neurotrophin receptors. Curr. Opin. Cell Biol. 1997;9:213–221. doi: 10.1016/s0955-0674(97)80065-8. [DOI] [PubMed] [Google Scholar]
- Knusel B., Rabin S.J., Hefti F., Kaplan D.R. Regulated neurotrophin receptor responsiveness during neuronal migration and early differentiation. J. Neurosci. 1994;14:1542–1554. doi: 10.1523/JNEUROSCI.14-03-01542.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1974;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Ma Y., Campenot R.B., Miller F.D. Concentration-dependent regulation of neuronal gene expression by nerve growth factor. J. Cell Biol. 1992;117:135–141. doi: 10.1083/jcb.117.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maderdrut J., Oppenheim R.W., Prevette D. Enhancement of naturally occurring cell death in the sympathetic and parasympathetic ganglia of the chicken embryo following blockade of ganglionic transmission. Brain Res. 1988;444:189–194. doi: 10.1016/0006-8993(88)90928-6. [DOI] [PubMed] [Google Scholar]
- Majdan M., Miller F.D. Neuronal life and death decisionsfunctional antagonism between the trk and p75 neurotrophin receptors. Int. J. Dev. Neurosci. 1999;In press doi: 10.1016/s0736-5748(99)00016-7. [DOI] [PubMed] [Google Scholar]
- Markus A., vonHolst A., Rohrer H., Heumann R. NGF-mediated survival depends on p21ras in chick sympathetic neurons from the superior cervical, but not from lumbosacral ganglia. Dev. Biol. 1997;191:306–310. doi: 10.1006/dbio.1997.8771. [DOI] [PubMed] [Google Scholar]
- Meyer-Franke A., Kaplan M.R., Pfrieger F.W., Barres B.A. Characterization of the signalling interactions that promote the survival and growth of developing retinal ganglion neurons in culture. Neuron. 1995;15:805–819. doi: 10.1016/0896-6273(95)90172-8. [DOI] [PubMed] [Google Scholar]
- Meyer-Franke A., Wilkinson G.A., Kruttgen A., Hu M., Munro E., Hanson M.G., Jr., Reichardt L.F., Barres B.A. Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron. 1998;21:681–693. doi: 10.1016/s0896-6273(00)80586-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller T.M., Tansey M.G., Johnson E.M., Creedon D.J. Inhibition of phosphatidylinsoitol 3-kinase activity blocks depolarization- and insulin-like growth factor I-mediated survival of cerebellar granule cells. J. Biol. Chem. 1997;272:9842–9853. doi: 10.1074/jbc.272.15.9847. [DOI] [PubMed] [Google Scholar]
- Oppenheim R.W. Cell death during development on the nervous system. Ann. Rev. Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Pearson M.A., O'Farrell A.M., Dexter T.M., Whetton A.D., Owen-Lynch P.J., Heyworth C.M. Investigation of the molecular mechanisms underlying growth factor synergythe role of ERK 2 activation in synergy. Growth Factors. 1998;15:293–306. doi: 10.3109/08977199809017484. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P., Warne P.H., Dhand R., Vanhaesebroeck B., Gout I., Fry M.J., Waterfield M.D., Downward J. Phosphatidylinositol-3–OH kinase as a direct target of Ras. Nature. 1994;370:527–532. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- Rosen L.B., Ginty D.D., Weber M.J., Greenberg M.E. Membrane depolarization and calcium influx stimulate MEK and MAP kinase via activation of Ras. Neuron. 1994;12:1207–1221. doi: 10.1016/0896-6273(94)90438-3. [DOI] [PubMed] [Google Scholar]
- Slack R.S., Belliveau D.J., Rosenberg M., Atwal J., Lochmuller H., Aloyz R., Haghighi A., Lach B., Seth P., Cooper E. Adenovirus-mediated gene transfer of the tumor suppressor, p53, induces apoptosis in postmitotic neurons. J. Cell Biol. 1996;135:1–12. doi: 10.1083/jcb.135.4.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler R.M., Egea J., Mintenig G.M., Sanz-Rodriguez C., Iglesias M., Comella J.X. Calmodulin is involved in membrane depolarization-mediated survival of motor neurons by phosphatidylinositol-3 kinase- and MAPK-independent pathways. J. Neurosci. 1998;18:1230–1239. doi: 10.1523/JNEUROSCI.18-04-01230.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapley P., Lamballe F., Barbacid M. K-252a is a selective inhibitor of the tyrosine protein kinase activity of the trk family of oncogenes and neurotrophin receptors. Oncogene. 1992;7:371–381. [PubMed] [Google Scholar]
- Tokumitsu H., Chijiwa T., Hagiwara M., Mizutani A., Terasawa M., Hidaka H. KN-62, 1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-l-tyrosyl]-4-phenylpiperazine, a specific inhibitor of Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 1990;265:4315–4320. [PubMed] [Google Scholar]
- Triggle D.J. Calcium, calcium channels and calcium channel antagonists. Can. J. Physiol. Pharmacol. 1990;68:1474–1481. doi: 10.1139/y90-224. [DOI] [PubMed] [Google Scholar]
- Vlahos C.J., Matter W.F., Hui K.Y., Brown R.F. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J. Biol. Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- Yano S., Tokumitsu H., Soderling T.R. Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature. 1998;396:584–587. doi: 10.1038/25147. [DOI] [PubMed] [Google Scholar]