

Abstract
Cell migration and wound contraction requires assembly of actin into a functional myosin motor unit capable of generating force. However, cell migration also involves formation of actin-containing membrane ruffles. Evidence is provided that actin-myosin assembly and membrane ruffling are regulated by distinct signaling pathways in the migratory cell. Interaction of cells with extracellular matrix proteins or cytokines promote cell migration through activation of the MAP kinases ERK1 and ERK2 as well as the molecular coupling of the adaptor proteins p130CAS and c-CrkII. ERK signaling is independent of CAS/Crk coupling and regulates myosin light chain phosphorylation leading to actin-myosin assembly during cell migration and cell-mediated contraction of a collagen matrix. In contrast, membrane ruffling, but not cell contraction, requires Rac GTPase activity and the formation of a CAS/Crk complex that functions in the context of the Rac activating protein DOCK180. Thus, during cell migration ERK and CAS/Crk coupling operate as components of distinct signaling pathways that control actin assembly into myosin motors and membrane ruffles, respectively.
Keywords: adaptor proteins, cell migration, mitogen-activated protein kinase, myosin, signal transduction
During development, wound repair and inflammation cells migrate in response to growth factors/cytokines and adhesive proteins present in the extracellular matrix (ECM)1 (Lauffenburger and Horwitz 1996; Keely et al. 1998). In many cases, these events are mediated by cytokine and integrin receptors that transmit a cascade of signals important for regulation of the migration machinery (Lauffenburger and Horwitz 1996; Aplin et al. 1998; Keely et al. 1998). Initiation of migration is characterized by the rapid reorganization of actin to the cell edge. This results in the protrusion of a leading lamellipodium with new adhesive contacts necessary for traction. Membrane ruffles are often found on the cell surface and at the advancing front of a lamellipodium. They are thought to serve as sites of actin polymerization, endocytosis, receptor tyrosine kinase signaling, and protease activity (Lauffenburger and Horwitz 1996; Mitchison and Cramer 1996). However, for a cell to move, it must also organize actin into a functional actin-myosin motor unit capable of generating contractile force. This propels the cell forward and contributes to the release of adhesive contacts at the rear of the cell (Lauffenburger and Horwitz 1996; Mitchison and Cramer 1996). Myosin II is the best-characterized myosin isoform and is well known to promote contraction in non-muscle cells (Kolega et al., 1993; Jay et al. 1995). The ability of myosin II to associate with actin and mediate contraction are modulated by the phosphorylation of the regulatory light chain by the Ca2+/calmodulin-dependent enzyme myosin light chain kinase (MLCK) as well as dephosphorylation by myosin phosphatase (Adelstein 1983; Yoshioka et al. 1998).
While significant progress has been made in identifying biochemical signals involved in cell migration, the relationship of these events and how they impact the migration machinery of cells are not well defined. Given the complexity of cell motility, it is not surprising that multiple signals regulate this process. Indeed, PLCγ (Chen et al., 1994), FAK (Ilic et al., 1995; Parsons, 1996; Cary and Guan, 1998), c-Src (Rodier et al., 1995; Boyer et al. 1997; Fincham and Frame 1998), PI3 kinase (Keely et al. 1997), MAP kinase (Anand-Apte et al. 1997; Klemke et al. 1997; Wei et al. 1998), as well as Tiam1 (Michiels et al. 1995; Sander et al. 1998) and the Rho family of small GTPases have all been shown to modulate cell migration (Keely et al. 1997, Keely et al. 1998; Tapon and Hall 1997).
Recently, the adaptor proteins p130CAS (CAS) and c-CrkII (Crk) have been shown to be involved in cell migration (Cary et al. 1998; Klemke et al. 1998). Adaptor proteins (Nck, Crk, Shc, Grb2) do not contain catalytic activity but consist primarily of src homology 2 (SH2) and 3 (SH3) domains that serve to assemble signal generating complexes and target them to discrete sites within the cell (Bar-Sagi et al., 1993). For example, activation of cytokine and integrin receptors promotes tyrosine phosphorylation of the substrate domain of CAS and its association with the SH2 domain of Crk (Vuori et al. 1996; Casamassima and Rozengurt, 1997, 1998; Ojaniemi and Vuori 1997). Crk, in turn, binds though its SH3 domain to several downstream effector molecules including the small GTPase activating proteins DOCK180, C3G, and SOS (Matsuda et al. 1994; Feller et al., 1995; Hasegawa et al., 1996). While the coupling of CAS to Crk can serve as a “molecular switch” leading to cell migration and metastasis (Klemke et al. 1998), it is not yet known how these proteins impact the migration machinery of cells. That CAS and Crk localize to focal contacts as well as membrane ruffles suggest this protein complex may regulate these processes in migratory cells. However, CAS and Crk may also facilitate cell migration through its ability to couple to NCK and/or SOS which is known to facilitate activation of the Ras/ERK pathway (Schlaepfer et al. 1997). Activated ERK can modulate integrin affinity and phosphorylate myosin light chain kinase leading to increased MLC phosphorylation (Hughes et al. 1997; Klemke et al. 1997). Both of these signaling events may be important for cell migration as MLC phosphorylation facilitates contraction and remodeling of adhesive structures (Adelstein 1983; Lauffenburger and Horwitz 1996; Chrzanowska-Wodnicka and Burridge 1996; Mitchison and Cramer 1996; Rosenfeldt et al. 1998). Furthermore, cell contraction and integrin aggregation can lead to activation of FAK (Chrzanowska-Wodnicka and Burridge 1996). Since FAK and/or c-src that associates with FAK can tyrosine phosphorylate CAS, this could also facilitate CAS/Crk complexes and Rac activation (Vuori et al. 1996; Tachibana et al. 1997). Thus, it is feasible that CAS/Crk and ERK may cooperate to promote both actin-myosin function and membrane ruffling in migratory cells. In fact, v-Crk has recently been shown to promote activation of Rho which can facilitate MLC phosphorylation and migration (Altun-Gultekin et al., 1998; Yoshioka et al. 1998). Alternatively, these events may not be coupled, but represent separate biochemical pathways capable of regulating these cellular processes.
In this report, we investigate whether CAS/Crk coupling and ERK activation are the same or parallel signaling pathways involved in the regulation of actin assembly into membrane ruffles as well as myosin motors. Evidence is provided that CAS/Crk coupling regulates the migration machinery by promoting membrane ruffling independent of ERK signaling. In contrast, ERK, but not CAS/Crk, controls MLC phosphorylation leading to actin/myosin-mediated cell contraction. These findings suggest that during cell migration CAS/Crk and ERK signaling operate as distinct biochemical pathways necessary for membrane ruffling and cell contraction, respectively.
Materials and Methods
Expression Vectors and Reagents
The expression plasmid pUCCAGGS containing either full-length DOCK180, myc-tagged c-CrkII, c-CrkII cDNA with a mutated amino-terminal SH3 domain (tryptophan 109 to cysteine), or Crk with a mutated SH2 domain (arginine 38 to valine) have been previously described (Matsuda et al. 1993, Matsuda et al. 1994; Tanaka et al. 1993; Kiyokawa et al. 1998b). The pEBG expression plasmid containing glutathione-S-transferase–tagged or untagged wild-type p130CAS or CAS with an in frame deletion of its substrate domain (CAS-SD, amino acids 213–514) have been previously described (Mayer et al. 1995). Myc-tagged dominant negative Rac 1 (N17) and mutationally activated MEK in pcDNA3 has been previously described (Minden et al. 1995; Klemke et al. 1997). PD98059 (2-[2′-amino-3′methoxyphenyl]-oxanaphthalen-4-one; Calbiochem-Novobiochem). Anti-myosin antibody that recognizes the myosin IIB isoform present in COS7 cells was a gift from Dr. Robert Adelstein (Molecular Cardiology, National Lung Heart and Lung Institute, National Institutes of Health, Bethesda, MD). The phosphoERK antibody was from Promega. The anti-Rac, DOCK180, and myc antibodies were from Santa Cruz Biotechnology. FG-C are human pancreatic carcinoma cells stably overexpressing c-CrkII as previously described (Klemke et al. 1998).
Transfection of COS-7 and Analysis of Cell Migration
Transient transfection of COS cells and Transwell migration assays were performed as previously described (Klemke et al. 1998). In brief, COS-7 cells were cotransfected with lipofectamine and expression vectors containing cDNAs encoding wild-type and/or mutant forms of MEK, Crk or CAS, Rac, DOCK180, along with a reporter construct encoding β-galactosidase (pCMV5-β-gal) or green fluorescent protein (pEGFP-C1; Clontech). Control cells were mock-transfected with the appropriate amount of the empty expression vectors along with the β-gal reporter. Cells were allowed to incorporate the cDNA constructs for 6–8 h, washed, and then allowed to incubate for 40 h which provides optimal transient expression in these cells. COS cells were then prepared for haptotaxis cell migration using X-gal as a substrate and analyzed for expression of specific proteins by immunoprecipitation and immunoblotting as previously described (Klemke et al. 1998). Transfection efficiency and cell adhesion of these cells to purified extracellular matrix proteins were monitored as described below. The migration of FG cells, metabolic labeling of transfected cells with [32P]orthophosphate, immunoprecipitation of myosin light chains, SDS-PAGE, and autoradiography were performed as previously described (Klemke et al. 1997).
Cell Adhesion and Transfection Efficiency
Controls for transfection efficiency and cell adhesion to ECM proteins were performed as previously described (Klemke et al. 1998). In brief, an aliquot of cells from the migration experiments above was allowed to attach to culture dishes coated with purified ECM proteins. The dishes were washed and adherent cells transfected with the β-gal reporter gene were detected using X-gal as a substrate according to the manufacturer's recommendation (Promega). In typical transfection experiments with these cells, we obtain 70–75% efficiency, as determined by counting the number of β-gal positive cells relative to the total number of cells attached per field (200×). It is important to note that in an individual experiment, transfection efficiently varies <10%. The efficiency and adhesion control assures that changes observed in cell migration is not simply the result of differences in transfection efficiency or expression of the β-gal reporter gene or differences in the ability of transfected cells to attach to the ECM.
Laser Confocal Imaging of Membrane Ruffles, Actin, and Myosin IIB
COS-7 cells were cultured on glass coverslips and then transfected as described above along with a reporter construct encoding either GFP or β-gal. Cells were serum-starved for 24 h then treated with or without insulin (10 μg/ml) or IGF-1 (20 ng/ml) for 15 min. In some cases, MEK or mock-transfected cells were exposed to 50 μM PD98059 for 2 h before being treated with insulin or IGF-1 as described above. To visualize F-actin containing membrane ruffles, cells were rinsed in PBS then fixed in 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and stained with rhodamine-conjugated phalloidin. Cells with prominent actin-rich membrane ruffles were quantified blindly by two independent investigators and represents the average number of transfected cells (i.e., GFP cells) with ruffles of eight high powered fields (400×). To observe F-actin filaments and myosin IIB colocalization in cells, we used a procedure that preserves the association of these proteins in whole cells. In brief, cells were extracted with 0.1% Triton X-100 detergent, fixed in 4% paraformaldehyde, then stained with rhodamine-conjugate phalloidin and rabbit anti-myosin IIB, followed by FITC-conjugated secondary antibodies as previously described (Cramer and Mitchison 1995). In some cases, cells transfected with the β-gal reporter construct were detected by staining with mouse anti-β-gal and donkey anti-Cy5-conjugated secondary antibodies along with three color cell fluorescence and a laser confocal microscope (Bio-Rad 1024 and a Zeiss axiovert microscope). Myosin content per cell was obtained by determining the fluorescence intensity (sum of total green pixels × intensity) in the green/FITC channel per cell area (μm2) using Adobe Photoshop and IPLab Spectrum P computer software.
Cell Contraction of Collagen Gels
Cell contraction of a three-dimensional collagen matrix was performed as previously described (Rosenfeldt et al. 1998). In brief, 1 × 106 cells/ml collagen were cultured for 2 d in DME containing 10% FBS. Cells were serum-starved for 4 h then exposed for 60 min to serum-free culture media containing PD98059 (50 μM), M7 (1 μM; [(5-iodonaphthalene-1-sulfonyl)homopiperazine, HCL; Calbiochem-Novabiochem], or BDM (10 mM; butanedione monoxime; Sigma Chemical Co.). To initiate contraction, collagen gels were mechanically released from the culture dish in the continued presence of the inhibitors. The change in diameter in millimeters was measured with a ruler at various times after release. In some cases, cells were washed to remove the inhibitors then cultured an additional 24 h in drug-free media before initiation of contraction as described above.
Results
CAS/Crk Coupling and ERK Activation Represent Distinct Signals Necessary for Cell Migration
Recently, we reported that CAS/Crk coupling as well as ERK activation facilitate cell migration (Klemke et al. 1997, Klemke et al. 1998). Each of these events were shown to be required for cell migration, yet it remains unclear whether these signals represent the same or parallel pathways involved in regulation of this process. To address this issue, cells were transfected with CAS lacking its substrate domain (CAS-SD) or Crk with a mutated SH2 domain (CRK-SH2), either of which are capable of preventing CAS/Crk coupling and downstream signaling events (Feller et al. 1994; Matsuda et al. 1994; Klemke et al. 1998). Cells containing these cDNAs were treated with the cytokine insulin and examined for migration as well as ERK activity. Expression of either CAS-SD or Crk-SH2 blocked insulin-induced cell migration, yet had no effect on ERK activity in these cells (Fig. 1A). Similar findings were observed in cells expressing Crk with a mutated amino-terminal SH3 domain that retains its ability to couple to CAS, but is unable to link to downstream effector molecules such as DOCK180 or C3G (data not shown; Matsuda et al. 1994; Kiyokawa et al. 1998a,Kiyokawa et al. 1998b). These findings reveal that disruption of CAS/Crk coupling or its binding to downstream effectors can suppress cell migration without influencing ERK activity.
Figure 1.
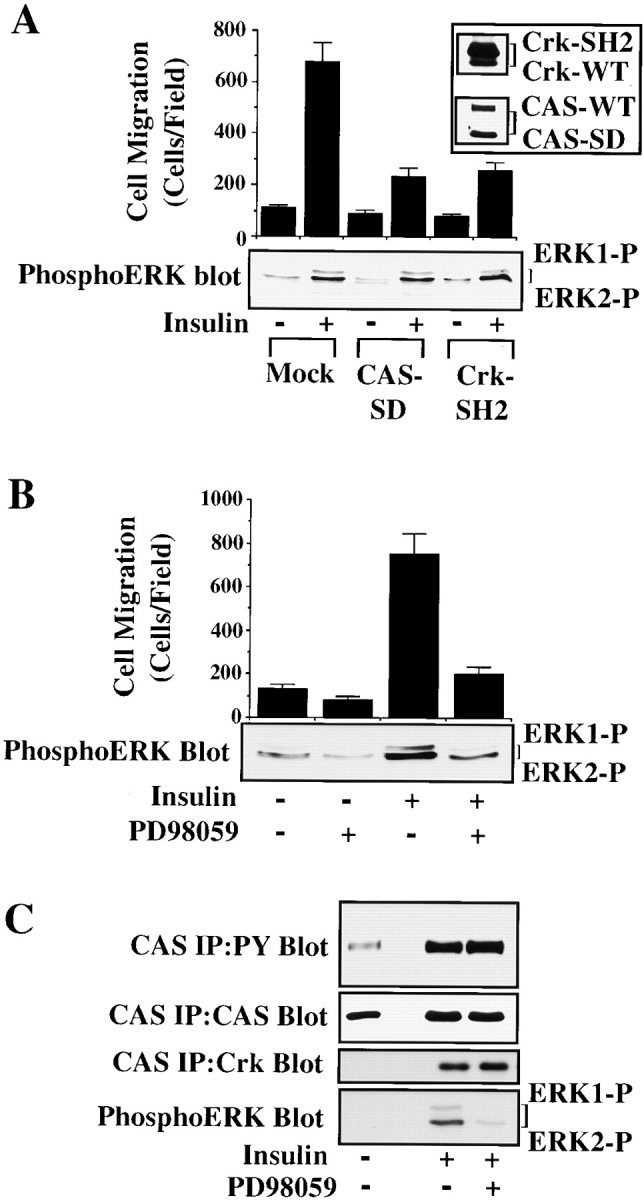
ERK activation and CAS/Crk coupling are separate signaling events necessary for cytokine-induced cell migration. (A) Serum-starved COS-7 cells were allowed to migrate for 3 h on vitronectin-coated membranes in the presence or absence of insulin (10 μg/ml) after transient transfection with a β-gal reporter construct, along with either the empty expression vector (Mock) or with expression vectors encoding gst-tagged CAS without its substrate domain (CAS-SD), or myc-tagged Crk with a mutated SH2 domain (Crk-SH2). CAS-SD and Crk-SH2 have been shown to prevent CAS/Crk coupling and downstream signals (Matsuda et al. 1993; Feller et al. 1994; Klemke et al. 1998). The number of transfected cells migrating were enumerated by counting cells on the underside of the membrane that coexpress the β-gal vector as described in Materials and Methods. An aliquot of cells treated as described for the migration experiment above was lysed in detergent and immunoblotted with antibodies to either the phosphorylated/activated form of ERK1/ERK2 (lower panel), Crk, or CAS (top right). Note that Crk-SH2 shows reduced mobility compared with wild-type endogenous Crk (Crk-WT) as the result of the molecular tag. (B) Serum-starved COS-7 cells were allowed to migrate in the presence or absence of the MEK inhibitor PD98059 (25 μM) with or without insulin (10 μg/ml). Cell migration and ERK1/ERK2 activity in these cells were determined as described above. Similar findings were obtained with fibronectin and collagen type I–coated membranes (data not shown). Each bar represents the mean ± SEM of at least three independent experiments. (C) Serum-starved COS-7 cells pretreated with or without the MEK inhibitor PD98059 (50 μM) for 2 h were exposed to insulin (10 μg/ml) for 5 min before being lysed in detergent. CAS was immunoprecipitated and then immunoblotted with antibodies to either phosphotyrosine, Crk, or CAS. The detergent lysates from these cells were also examined for changes in ERK1/ERK2 activity as described above. The result shown is representative from at least three independent experiments.
To determine whether ERK signaling represents an independent pathway necessary for cell migration, cells were induced to migrate with insulin and then exposed to the compound PD98059 that blocks ERK kinase (MEK), and thereby prevents ERK activation (Dudley et al. 1995). These cells were then examined for cell migration, ERK activity, and formation of CAS/Crk complexes. While the MEK inhibitor prevented insulin-induced cell migration and ERK1/ERK2 activation, it did not impact CAS tyrosine phosphorylation or the formation of CAS/Crk complexes in these cells (Fig. 1B and Fig. C). Similar findings were observed when cells were stimulated to migrate with either EGF or IGF-1 (data not shown). Thus, CAS/Crk coupling and ERK activation appear to represent components of distinct signaling pathways necessary for cytokine-induced cell migration.
To investigate directly whether formation of a CAS/Crk complex could activate ERK, serum-starved COS-7 cells were transiently transfected with vectors encoding CAS and Crk or mutationally activated MEK. These cells were then examined for ERK activity and migration. Expression of MEK in these cells promoted a four- to fivefold increase in cell migration and significantly increased ERK activity compared with mock-transfected control cells (Fig. 2 A). However, while CAS/Crk transfected cells showed a fourfold increase in cell migration, there was no change in ERK activity (Fig. 2 A). Similar findings were obtained in FG carcinoma cells stably transfected with c-Crk (FG-C). These cells also showed significantly enhanced migration, yet ERK activity was the same as control cells (Fig. 2b and Fig. C). Together, these findings indicate that CAS/Crk-induced cell migration does not result from increased ERK activity. However, since ERK signaling appeared to be a separate event necessary for cell migration, we investigated whether ERK activity was also necessary for CAS/Crk-induced cell movement. To investigate this possibility, FG-C and COS-7 cells transfected with CAS and Crk were exposed to PD98059 and analyzed for their ability to migrate on ECM proteins. In this case, PD98059 blocked cell migration induced by CAS/Crk (Fig. 2a and Fig. b) without affecting the formation of CAS/Crk complexes in these cells (data not shown). Conversely, MEK-induced cell migration was blocked by expression of CAS-SD or dominant negative RacN17 (Fig. 3A and Fig. B) without impacting ERK activity (data not shown). Thus, CAS/Crk/Rac signaling and ERK activation appear to be separate biochemical pathways necessary for cell migration.
Figure 2.
CAS/Crk-induced cell migration does not result from increased ERK activity. (A) Upper panel, serum-starved COS-7 cells were allowed to migrate for 3 h in the presence or absence of the MEK inhibitor PD98059 (25 μM) on vitronectin-coated membranes after transient transfection with a β-gal reporter construct, along with either the empty expression vector or with expression vectors encoding gst-tagged wild-type CAS and myc-tagged c-Crk or mutationally activated MEK. The number of transfected cells migrating was enumerated by counting cells on the underside of the membrane that coexpress the β-gal vector as described in Materials and Methods. Each bar represents the mean ± SEM of at least three independent experiments. Lower panels, total cell lysates prepared from cells treated as described for the migration experiments above were immunoblotted with antibodies that recognize the activated form of ERK1/ERK2 proteins, Crk, or CAS. Note that the gstCAS and myc-Crk proteins show reduced mobility compared with endogenous wild-type forms of these proteins as the result of the molecular tags. Immunoblots were exposed to film for 15 and 90 s after treatment with enhanced chemiluminescence reagents as described in Materials and Methods. (B) Serum-starved FG cells stably transfected with Crk or mock-transfected with the empty vector were allowed to migrate on vitronectin or collagen-coated membranes for 3 h in the presence or absence of PD98059 (25 μM) and quantified as described in Materials and Methods. Each bar represents the mean ± SEM of at least three independent experiments. (C) Cells treated as described for the migration experiment above were either held in suspension (SUS) or allowed to attach (ATT) to collagen-coated culture dishes for 30 min, then lysed in detergent and immunoblotted with either antibodies that specifically recognize the activated form of ERK1/ERK2 proteins (upper panel), ERK2 (middle panel), or Crk (lower panel). The upper band recognized by the ERK2 antibody represents the phosphorylated/activated form of this protein (ERK2-P) that has reduced mobility as a result of being phosphorylated. Similar results were obtained with ERK1 protein (data not shown).


Figure 3.
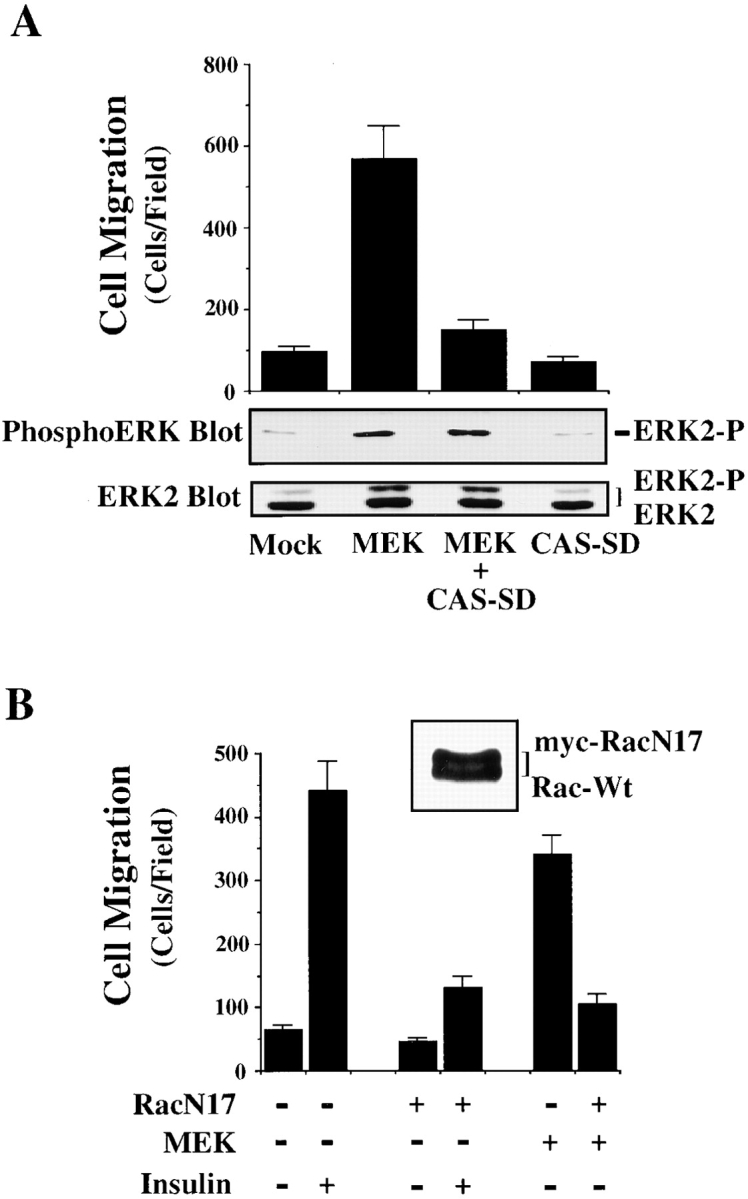
ERK-induced cell migration requires CAS/Crk and Rac activity. (A) Upper panel, serum-starved COS-7 cells were allowed to migrate for 3 h on vitronectin-coated membranes after transient transfection with a β-gal reporter construct, along with either the empty expression vector or with expression vectors encoding mutationally activated MEK, or MEK cotransfected with dominant negative CAS (CAS-SD). The number of transfected cells migrating were enumerated by counting cells on the underside of the membrane that coexpress the β-gal vector as described in Materials and Methods. Each bar represents the mean ± SEM of at least three independent experiments. Lower panels, cells treated as described for the migration experiment above were lysed in detergent and immunoblotted with antibodies to the activated form of ERK1/ERK2 (middle panel) or ERK2 (lower panel). The upper band recognized by the ERK2 antibody represents the phosphorylated/activated form of this protein (ERK2-P) that has reduced mobility as a result of being phosphorylated. Similar results were obtained with ERK1 protein (data not shown). (B) Serum-starved COS-7 cells were allowed to migrate for 3 h on vitronectin-coated membranes after transient transfection with a β-gal reporter construct, along with either the empty expression vector or with expression vectors encoding mutationally activated MEK, or MEK together with dominant negative myc-tagged Rac (RacN17) in the presence or absence of insulin (10 μg/ml) in the lower chamber. An aliquot of cells transfected with RacN17 and lysed and immunoblotted with an antibody to Rac is shown (top right). Note that RacN17 migrates slower as the result of the myc tag compared with endogenous wild-type Rac (Rac-Wt). Each bar represents the mean ± SEM of at least three independent experiments.
Membrane Ruffling Requires CAS/Crk and Rac, but Not ERK Activity
Activation of cell migration is characterized by the assembly of actin into membrane ruffles as well as cell contraction (Lauffenburger and Horwitz 1996; Mitchison and Cramer 1996). To explain how the coupling of CAS/Crk and activation of ERK might influence the migration machinery, cells expressing CAS-SD were stimulated with insulin and then examined for actin-containing membrane ruffles. Exposure of mock-transfected control cells to insulin induced prominent membrane ruffles rich in F-actin (Fig. 4 A). Approximately 18% of the control cell population showed membrane ruffling before stimulation with insulin and this was increased to 80% after cells were exposed to this cytokine (Fig. 4 B). Importantly, expression of CAS-SD in these cells completely blocked the insulin-induced membrane ruffling response (Fig. 4A and Fig. B). As expected, cells within the field of view not transfected with CAS-SD showed prominent membrane ruffles (Fig. 4 A). Expression of Crk-SH2 in cells also blocked insulin-induced membrane ruffles (data not shown). Recently, it was reported that CAS/Crk coupling facilitates Rac activity which can promote membrane ruffling (Ridely et al. 1992; Kiyokawa et al. 1998a). Therefore, we determined whether CAS/Crk-induced membrane ruffles required Rac activity. Cells were transfected with CAS and Crk along with a dominant negative form of Rac (RacN17) and then examined for F-actin containing membrane ruffles. Expression of RacN17 in these cells blocked CAS/Crk-induced ruffles (Fig. 4 C). RacN17 also blocked cell migration without impacting ERK activity (data not shown). Thus, CAS/Crk coupling promotes membrane ruffles that depend on Rac, but not on ERK activity.
Figure 4.

CAS/Crk association, but not ERK activation, is required for Rac-dependent membrane ruffling. (A) Serum-starved COS-7 cells in the presence or absence of insulin (10 μg/ml for 15 min) were stained with rhodamine-conjugated phalloidin, then analyzed by confocal imaging for F-actin (red) containing membrane ruffles after being transfected with either the empty vector (control) or the vector encoding dominant negative CAS (CAS-SD) along with a reporter vector encoding green fluorescent protein (GFP) to identify transfected cells. In some cases, control cells were pretreated for 2 h with 50 μM of PD98059 to inhibit ERK activity before being exposed to insulin as described above. Photomicrographs were taken with a Bio-Rad Labs 1024 laser and a Zeiss Axiovert microscope (400×). Arrowheads indicate cells with prominent F-actin membrane ruffles. (B) COS-7 cells treated as described above were scored for membrane ruffles as described in Materials and Methods. Results are expressed as the percentage of total transfected cells (i.e., green cells) that displayed prominent F-actin membrane ruffles and are the mean ± SEM of three separate experiments. (C) COS cells were transfected with expression vectors encoding CAS, Crk, and dominant negative RacN17, along with a reporter vector encoding GFP, then examined for actin membrane ruffles as describe above. Photomicrographs of CAS/Crk cells (400×) and RacN17 expressing cells (600×) were taken with a Bio-Rad Labs 1024 laser and a Zeiss Axiovert microscope. Arrowheads indicate cells with prominent F-actin membrane ruffles. (D) COS-7 cells transfected with either wild-type DOCK180, gst-tagged CAS and myc-tagged Crk, or CAS and Crk, together with DOCK180 and/or myc-tagged RacN17 were examined for cell migration as described above. An aliquot of cells transfected with CAS/Crk and DOCK180 (lane 2) or cells mock-transfected with the empty vectors (lane 1) as described for the migration experiment above were lysed in detergent and immunoblotted with antibodies to the phosphorylated/activated form of ERK1/ERK2 as described above (top left). Note that in these experiments cells were transfected with CAS/Crk vectors at DNA levels that give half-maximal migration. Each bar represents the mean ± SEM of at least three independent experiments. (E) An aliquot of cells treated as for the migration experiment above were lysed in detergent then immunoblotted with antibodies to CAS, Crk, DOCK180, or myc to detect myc-tagged RacN17. Lane 1, control cells transfected with the empty vectors. Lane 2, cells transfected with the vector containing the cDNA as indicated. Note that the gstCAS and mycCrk proteins show reduced mobility compared with endogenous wild-type forms of these proteins as the result of the molecular tag. Bars, 10 μm.
DOCK180 Potentiates CAS/Crk-mediated Cell Migration
Crk is known to bind to a number of downstream effector molecules via its SH3 domain, including c-Abl, SOS, C3G, Eps15, and DOCK180 (Matsuda et al. 1994; Feller et al., 1995; Hasegawa et al., 1996). Among these Crk-binding proteins, DOCK180 has been associated with Rac activation (Kiyokawa et al. 1998a). Moreover, in Caenorhabditis elegans and Drosophila melanogaster, the DOCK180 homologue ced-5 and mbc, respectively, control cell migration events associated with development (Erickson et al. 1997; Wu and Horvitz 1998). Therefore, we were prompted to investigate the role of DOCK180 in cell migration. As shown in Fig. 4D and Fig. E, expression of DOCK180 was able to potentiate CAS/Crk-induced cell migration in a Rac-dependent manner, yet it had no effect on ERK activity in these cells. Together these findings suggest that CAS/Crk in conjunction with DOCK180 can form a signaling module involved in Rac-mediated membrane ruffling and cell movement that is independent of ERK activation.
To investigate the role of ERK-dependent signaling in actin assembly, cells exposed to the cytokine insulin were treated in the presence or absence of the MEK inhibitor, PD98059. This inhibitor, which blocks cell migration, but not adhesion or spreading on collagen or vitronectin substrates (Fig. 1 B), failed to disrupt membrane ruffling in response to insulin (Fig. 4 A). In fact, 70% of cells exposed to PD98059 and insulin showed membrane ruffling, compared with 80% of cells exposed to insulin alone (Fig. 4 B). Together these findings indicate that CAS/Crk coupling is necessary for membrane ruffling, whereas ERK activity is not.
ERK Activity, but Not CAS/Crk Coupling, Is Necessary for Actin-myosin Assembly and Cell Contraction of a Three-dimensional Collagen Matrix
Cell migration also involves myosin light chain phosphorylation leading to actin-myosin association and cell contraction. While ERK can phosphorylate MLCK leading to increased MLC phosphorylation, it is not yet known if this event promotes assembly of a functional actin-myosin motor capable of generating force necessary for cell contraction (Klemke et al. 1998). This, and the fact that ERK did not influence membrane ruffling, yet appeared critical for cell migration, prompted us to examine its role in actin-myosin assembly and contractile function. We also investigated the role of CAS and Crk in this process since it is not known if these proteins regulate actin-myosin activity independent of their ability to organize actin into membrane ruffles. Cells were stimulated with insulin or transfected with mutationally activated MEK and examined for phosphorylation of MLC, which facilitates actin-myosin binding and motor activity (Adelstein 1983). These cells were also examined for actin-myosin colocalization by immunofluorescent staining and laser confocal microscopy. Cells exposed to insulin or those expressing MEK+ showed increased phosphate incorporation into MLC compared with unstimulated or mock-transfected control cells (Fig. 5 A). Exposure to insulin or transfection with MEK also promoted increased actin-myosin colocalization compared with control cells (Fig. 5B and Fig. C). In fact, the amount of insoluble myosin associated with these cells after extraction in detergent was increased by two- to threefold (Fig. 5 D). Importantly, PD98059 blocked insulin and MEK-induced MLC phosphorylation as well as actin-myosin colocalization, indicating that ERK activity was required for this response (Fig. 5, A–D). In contrast, transfection of cells with CAS-SD, which blocks membrane ruffling (Fig. 4), failed to block MLC phosphorylation and actin-myosin colocalization in response to insulin or expression of MEK+ (Fig. 5, A–D). Therefore, ERK activation selectively promotes MLC phosphorylation and actin-myosin colocalization, whereas CAS/Crk coupling facilitates membrane ruffling.
Figure 5.

ERK, but not CAS/Crk signaling, promotes myosin light chain phosphorylation and actin-myosin colocalization. (A) Myosin light chains (MLC) immunoprecipitated with an anti-myosin IIB antibody from COS-7 cells metabolically labeled with [32P]orthophosphate and treated with or without insulin (10 μg/ml) for 5 min as described in Materials and Methods. In some cases, 32P-labeled cells were pretreated for 2 h with or without the MEK inhibitor PD98059 or transfected with mutationally activated MEK, and/or CAS-SD before immunoprecipitation of myosin IIB and SDS-PAGE as described above. MLC-P denotes phosphorylated light chain. MLC shows light chains stained with Coomassie before autoradiography to confirm that equal amounts of protein were precipitated in these experiments. (B) Serum-starved COS-7 cells either pretreated with the MEK inhibitor PD98059 (25 μM) or transfected with CAS-SD along with a β-gal reporter construct to identify transfected cells. Cells were stimulated with insulin (10 μg/ml) for 10 min, then exposed briefly to detergent to remove insoluble actin-myosin, fixed, and costained for actin and myosin IIB under conditions that preserve the association of these protein in cells as described in Materials and Methods (Cramer and Mitchison 1995). Immunofluorescent laser confocal imaging was performed as described in Materials and Methods. Rhodamine-phalloidin (red) was used to visualize F-actin, whereas rabbit anti-myosin IIB and secondary FITC-conjugated goat anti–rabbit specific antibodies were used to visualize myosin (green). Yellow staining is the colocalization of red and green staining in the merged image. An asterisk indicates transfected cells as detected by immunostaining with a mouse anti-β-galactosidase antibody and a secondary antibody conjugated with Cy5 (blue, not shown). Arrowhead shows actin-containing membrane ruffles. (C) COS-7 cells transfected with mutationally activated MEK and/or CAS-SD along with a β-gal reporter construct to identify transfected cells and examined for actin-myosin association as described above. In some cases, MEK+ transfected cells were exposed to 25 μM PD98059 for 2 h to block MEK activation of ERK before staining for actin and myosin. Photomicrographs were taken with a Bio-Rad Labs 1024 laser and Zeiss Axiovert microscope (600×). An asterisk indicates transfected cells and arrow shows membrane ruffles. (D) Insoluble myosin content of COS-7 cells treated as described in B and C. Cells were stained with anti-myosin IIB and FITC-conjugated secondary antibodies and the total amount of green fluorescence intensity (×106) determined per cell area (μm2) as described in Materials and Methods. Each bar represents the mean ± SEM of 30–40 cells of 6–8 different fields of three independent experiments. Bars: (B and C) 10 μm.
While cells require actin-myosin motor function for motility, these same events generate mechanical force necessary for wound contraction (Grinnell 1994). Therefore, we investigated the role of ERK and CAS/Crk coupling in cell-mediated contraction of the extracellular matrix. Cells were transfected with CAS-SD or exposed to PD98059 and then examined for their ability to contract a three-dimensional collagen matrix. Exposure of cells to the MEK inhibitor significantly reduced the rate of cell contraction (Fig. 6 A). In fact, the time necessary to achieve half-maximal contraction was increased from 16 min in control cells to 36 min in cells exposed to the MEK inhibitor. Similar findings were obtained when cells were transfected with the ERK phosphatase MKP-2 which blocks ERK activity (data not shown). The time for half-maximal contraction was also significantly increased in cells exposed to the MLCK inhibitor M7 or in cells transfected with a dominant negative form of MLCK (MLCK−) (Fig. 6a and Fig. b), which prevents ERK-induced cell migration and myosin light chain phosphorylation (Klemke et al. 1997). In this case, half-maximal contraction time was increased to 76 and 48 min in M7 and MLCK− cells, respectively, whereas control cells responded within 16 min. The difference in cell contraction between M7 and those transfected with MLCK− is likely due to the fact that ∼75% of transfected cells express the MLCK− construct, whereas 100% of the cells are exposed to M7. In support of these results, cells exposed to BDM, a general inhibitor of myosin ATPase activity (Cramer and Mitchison 1995), showed minimal contraction indicating that myosin plays a central role in this process (Fig. 6). In contrast, contraction was not altered in cells transfected with CAS-SD (Fig. 6 B) which blocks membrane ruffling, but not actin-myosin colocalization (Fig. 4 and Fig. 5). Similar findings were observed when cells were transfected with Crk-SH2 (data not shown). Importantly, the inhibition of cell contraction induced by these compounds was not due to nonspecific cell toxicity since their effect was reversed when they were removed from the culture media (Fig. 6 C). Together, these findings support the notion that ERK activation represents a distinct signaling pathway involved in the regulation of actin-myosin assembly and cell contraction, whereas CAS/Crk coupling represents an independent pathway that regulates actin membrane ruffling in migratory cells. However, while inhibition of ERK or MLCK activity decreased cell contraction, it did not completely block this event, suggesting that additional signals may exist to regulate MLC phosphorylation and actin-myosin contraction. Consistent with this possibility, Rho kinase is known to promote MLC phosphorylation independent of MLCK activity (Yoshioka et al. 1998).
Figure 6.
ERK activity, but not CAS/Crk association, is necessary for cell contraction of collagen gels. Three-dimensional collagen gels containing COS-7 cells were released from the culture dish and allowed to contract for various times as described in Materials and Methods. (A) COS-7 cells in collagen gels pretreated for 60 min with the MEK inhibitor PD98059 (50 μM), the MLCK inhibitor M7 (1 μM), or an inhibitor of myosin ATPase activity (BDM, 10 mM). (B) COS-7 cells transfected with either the empty expression vector (control) or the vector containing CAS without a substrate domain (CAS-SD) or a dominant negative form of MLCK (MLCK−) (Klemke et al. 1997). Contraction is presented as change in diameter (starting-final) measured in millimeters. (C) Cells treated as described in A were washed to remove inhibitors then cultured an additional 24 h before being allowed to contract the collagen matrix for various times as described in Materials and Methods. Each point represents the mean ± SEM of three independent experiments.



Discussion
ERK activation and the molecular coupling of CAS and Crk are initiated upon cell adhesion to ECM proteins and/or exposure to various growth factors. Our findings that formation of a CAS/Crk complex and activation of the GTPase Rac are necessary for membrane ruffling, whereas ERK activity is involved in actin-myosin contraction, suggest that these signaling events regulate specific components of the migration/contraction machinery. During wound healing, the actin-myosin motor generates contractile force necessary for both cell migration as well as contraction of fibrin or collagen matrices. These cellular events appear to be independently regulated of CAS/Crk and Rac-associated events, while ERK regulates force generation and cell contraction.
Several lines of evidence suggest that CAS/Crk and ERK activation operate as components of separate signaling pathways necessary for cell migration. First, dominant negative forms of CAS and Crk that prevent CAS/Crk coupling blocked cytokine-induced cell migration without impacting ERK activation. Furthermore, inhibition of ERK activation prevented cytokine-induced cell migration without impacting CAS tyrosine phosphorylation or the formation of a CAS/Crk complex. Second, while formation of a CAS/Crk complex was sufficient to induce cell migration in serum-starved cells, it failed to promote ERK activation, indicating that this migration response was not the result of increased ERK activity. However, ERK activity was found to be a separate response necessary for CAS/Crk-induced cell migration since blocking endogenous ERK activity with PD98059 prevented CAS/Crk induced cell migration, but had no effect on the coupling of these proteins. In addition, we have observed that expression of mutationally activated MEK in cells, while sufficient to induce ERK activation and cell migration, did not impact CAS/Crk coupling (data not shown). That dominant negative forms of CAS and Crk were able to block MEK-induced cell migration, but not ERK activation, provided additional evidence that CAS/Crk and ERK are separate events required for cell migration. Finally, CAS/Crk and ERK represent distinct signals capable of regulating membrane ruffling and contraction in migratory cells. That Ras effector mutants deficient in their ability to facilitate ERK activity retain the capacity to promote membrane ruffling further support this notion (Joneson et al. 1996).
Our findings that CAS/Crk coupling was sufficient to induce membrane ruffles suggests that at least one important consequence of the molecular coupling of these proteins in cells is to facilitate Rac activation and/or its localization to the cell membrane. In fact, recent evidence indicates that CAS/Crk coupling in cells can potentiate Rac activity and cell spreading (Kiyokawa et al. 1998a). This was found to depend on the recently described Rac-activating protein DOCK180, which binds to the amino-terminal SH3 domain of Crk (Kiyokawa et al. 1998a). Our findings indicate that DOCK180 can potentiate CAS/Crk-induced cell migration, an event that depends on Rac activity. Together, these findings suggest that DOCK180 is an important downstream mediator in the CAS/Crk motility response. Interestingly, cellular expression of farnesylated, but not wild-type DOCK180, induced cell spreading and ruffling (Kiyokawa et al. 1998a), suggesting that DOCK180 requires localization to the cell surface to fully activate Rac and the associated changes in the actin cytoskeleton. During cell migration, CAS/Crk may serve to target DOCK180 to the membrane where it can interact with Rac. Indeed, CAS/Crk and Rac are known to localize to membrane ruffles of migratory cells (Tapon and Hall 1997; Klemke et al. 1998). Alternatively, the association of DOCK180 with CAS/Crk complexes may regulate signaling from integrin adhesion receptors as recently suggested (Kiyokawa et al. 1998b). In either case, this appears to be independent of ERK signaling as cells expressing CAS/Crk/DOCK180 complexes showed enhanced cell migration without significant changes in ERK activity (Fig. 4 D). The ability of CAS/Crk complexes to couple to DOCK180 may have important implications for cell migration associated with development and cell metastasis. The DOCK180 homologues, mbc and ced-5, isolated from Drosophila melanogaster and Caenorhabditis elegans, respectively, play a role in cell motility associated with the development of these organisms (Erickson et al. 1997; Wu and Horvitz 1998). In contrast, CAS/Crk coupling may contribute to the migratory/invasive behavior of tumor cells through its ability to couple to the Rac and PI3 kinase signaling pathway (Keely et al. 1998). Indeed, carcinoma cells that showed increased invasive and metastatic potential in vivo were found to have increased CAS/Crk complexes compared with non-metastatic cells (Klemke et al. 1998).
While it is not yet clear how ERK is regulated in migratory cells, our findings indicate that CAS and Crk do not play a central role in this signaling cascade. Recent evidence suggests that several signals exist to regulate ERK activity independent of CAS and Crk (reviewed by Aplin et al. 1998; Schlaepfer et al. 1999). It is known that Src can phosphorylate FAK at tyrosine 925 leading to a Grb2/SOS association and direct ERK activation (Schlaepfer et al. 1994). Furthermore, the FAK-related tyrosine kinase Pyk2 directly couples integrin signals to ERK activation independent of CAS/Crk coupling (Blaukat et al. 1999). Protein kinase C and Grb2 binding to Shc provide additional pathways capable of regulating ERK activity in response to integrin events (Wary et al. 1996; Schlaepfer et al. 1998). Alternatively, CAS-dependent mechanisms may exist to link integrin and cytokine receptors to the RAS/ERK pathway. For example, Nck couples to SOS as well as tyrosine phosphorylated CAS. This could serve as an alternate pathway to facilitate a low level of ERK activity in some cells (Schlaepfer et al. 1997). However, it is not yet known if formation of a CAS/Nck complex is necessary for ERK activation or cell migration.
Assembly of an actin-myosin motor unit is critical for cell-mediated contraction of the ECM as well as cell movement, suggesting that these processes may be related. In fact, contraction of a collagen matrix involves a rapid smooth muscle–like contraction that is associated with increased ERK activity and myosin light chain phosphorylation (Grinnell 1994; Rosenfeldt et al. 1998). Migratory cells also assemble actin-myosin motors and exert force on the ECM (Lauffenburger and Horwitz 1996). This is thought to generate the force necessary for the rapid retraction of the tail region that is known to occur in migratory cells. Previous work has shown that ERK can directly phosphorylate and, thereby, activate MLCK leading to MLC phosphorylation (Klemke et al. 1997). In this report, we have extended these findings by showing that ERK activation can promote assembly of a functional actin-myosin motor unit capable of promoting cell contraction. Based on these findings, we propose that during cell migration ERK facilitates MLCK activity and MLC phosphorylation leading to the assembly of actin-myosin motors, an event necessary for cell contraction, but not membrane ruffling. On the other hand, CAS/Crk coupling independently regulates Rac activity and membrane ruffling in migratory cells. It is likely that additional signals operate to control cytoskeletal changes involved in cell movement. In fact, Rho modulates cell migration through its ability to inactivate myosin phosphatase leading to increased myosin light chain phosphorylation and cell contractility (Yoshioka et al. 1998). Furthermore, v-Crk can regulate Rho activity, suggesting that in some cells Crk may be able to facilitate myosin contractility (Altun-Glutekin et al. 1998). p21-activated kinase (Pak1) also regulates MLC phosphorylation and cell motility in fibroblasts (Sells et al. 1999), and Ras/ERK regulates integrin affinity and modulates adhesive contacts with the ECM, which is important for cell migration (Hughes et al. 1997).
Our findings that assembly of a CAS/Crk complex and Rac activation are necessary for membrane ruffling, whereas ERK activity facilitates actin-myosin contraction, indicate that these signals regulate specific components of the migration machinery. These findings provide molecular insight as to how cellular recognition of growth factors and adhesive proteins regulate the process of cell movement during development, wound healing, and inflammation, as well as tumor cell dissemination.
Acknowledgments
We thank Dr. Robert Adelstein for providing anti-myosin IIB–specific antibodies and helpful advice on this project, and Drs. Kristiina Vuori, Primal de Lanerolle, Michiyuki Matsuda, and Patricia Gallagher for providing reagents and advice concerning this project. We also thank Dr. Kathryn S.R. Spencer and Bob Summers for assistance with confocal microscopy and Rachel Molander for technical assistance.
R.L. Klemke was supported by an award from the Joseph Drown Foundation and by National Institutes of Health (NIH) grant CA 78493-01. D.A. Cheresh was supported by NIH grants CA 50286, CA 45728, and HL 54444. J. Leng was supported by a postdoctoral fellowship from the U.S. Army Medical Research and Material Command under DAMD17-96-1-6104. This manuscript is number 12145-IMM from the Scripps Research Institute.
Footnotes
1.used in this paper: β-gal, beta galactosidase; BDM, butanedione monoxime; CAS, p130Crk-associated substrate protein; Crk, c-CrkII; Crk-SH2, Crk without a functional src-homology 2 domain; ECM, extracellular matrix; ERK, extracellular-regulated kinase; GFP, green fluorescent protein; MLC, myosin light chain; MLCK, MLC kinase; SH2, src-homology 2 domain; SH3, src-homology 3 domain
References
- Adelstein R.S. Regulation of contractile proteins by phosphorylation. J. Clin. Invest. 1983;72:1863–1866. doi: 10.1172/JCI111148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altun-Glutekin Z.F., Chandrian S., Bougert C., Ishizaki I., Narumiya S., de Graaf P., Van Bergen en Henegouwen P., Hanafusa H., Wagner J.A., Birge R.B. Activation of Rho-dependent cell spreading and focal adhesion biogenesis by the v-Crk adaptor protein. Mol. Cell Biol. 1998;18:3044–3058. doi: 10.1128/mcb.18.5.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand-Apte B., Zetter B.R., Viswanathan A., Qui R., Chen J., Ruggeri R., Symons M. Platelet-derived growth factor and fibronectin-stimulated migration are differentially regulated by the Rac and extracellular signal-regulated kinase pathways. J. Biol. Chem. 1997;272:30688–30692. doi: 10.1074/jbc.272.49.30688. [DOI] [PubMed] [Google Scholar]
- Aplin A.E., Howe A., Alahari S.K., Juliano R.L. Signal transduction and signal modulation by cell adhesion receptorsthe role of integrins, cadherins immunoglobulin-cell adhesion molecules, and selectins. Pharm. Rev. 1998;50:197–263. [PubMed] [Google Scholar]
- Blaukat A., Ivankovic-Dikic I., Gronroos E., Dolfi F., Tokiwa G., Vuori K., Dikic I. Adaptor proteins Grb2 and Crk couple Pyk2 with activation of specific mitogen-activated kinase cascades. J. Biol. Chem. 1999;274:14893–14901. doi: 10.1074/jbc.274.21.14893. [DOI] [PubMed] [Google Scholar]
- Boyer B., Roche S., Denoyelle M., Thiery J.P. Src and Ras are involved in separate pathways in epithelial cell scattering. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:5904–5913. doi: 10.1093/emboj/16.19.5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary L.A., Cho D.H., Guan J.L. Identification of p130CAS as a mediator of focal adhesion kinase-promoted cell migration. J. Cell Biol. 1998;12:211–221. doi: 10.1083/jcb.140.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrzanowska-Wodnicka M., Burridge K. Rho-stimulated contractility drives formation of stress fibers and focal adhesions. J. Cell Biol. 1996;133:1403–1415. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosamassima A., Rozengurt E. Tyrosine phosphorylation of p130CAS by bombesin, lysophosphatic acid, phorbol esters, and platelet-derived growth factor. J. Biol. Chem. 1997;272:9363–9370. doi: 10.1074/jbc.272.14.9363. [DOI] [PubMed] [Google Scholar]
- Cosamassima A., Rozengurt E. Insulin-like growth factor I stimulates tyrosine phosphorylation of p130CAS, focal adhesion kinase and paxillin. J. Biol. Chem. 1998;273:26149–26156. doi: 10.1074/jbc.273.40.26149. [DOI] [PubMed] [Google Scholar]
- Cramer L.P., Mitchison T.J. Myosin is involved in postmitotic cell spreading. J. Cell Biol. 1995;131:179–189. doi: 10.1083/jcb.131.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley D.T., Pang L., Decker S.J., Bridges A.J., Saltiel A.R. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson M.S., Galletta B.J., Abmayr S.M. Drosophila myoblast city encodes a conserved protein that is essential for myoblast fusion, dorsal closure, and cytoskeletal organization. J. Cell Biol. 1997;138:589–603. doi: 10.1083/jcb.138.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller S., Knudsen B., Hanafusa H. C-Abl kinase regulates the protein binding activity of c-Crk. EMBO (Eur. Mol. Biol. Organ.) J. 1994;13:2341–2351. doi: 10.1002/j.1460-2075.1994.tb06518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fincham V.J., Frame M.C. The catalytic activity of Src is dispensable for translocation to focal adhesions but controls the turnover of these structures during cell motility. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:81–92. doi: 10.1093/emboj/17.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinnell F. Fibroblasts, myofibroblasts, and wound contraction. J. Cell Biol. 1994;124:401–404. doi: 10.1083/jcb.124.4.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes P.E., Renshaw M.W., Pfaff M., Forsyth J., Keivens V.M., Schwartz M., Ginsberg M.H. Suppression on integrin activationa novel function of a Ras/Raf-initiated MAP kinase pathway. Cell. 1997;88:521–530. doi: 10.1016/s0092-8674(00)81892-9. [DOI] [PubMed] [Google Scholar]
- Jay P.Y., Pham P.A., Wong S.A., Elson E.L. A mechanical function of myosin II in cell motility. J. Cell Sci. 1995;108:387–393. doi: 10.1242/jcs.108.1.387. [DOI] [PubMed] [Google Scholar]
- Joneson T., White M.A., Wigler M.H., Bar-Sagi D. Stimulation of membrane ruffling and MAP kinase activation by distinct effectors of Ras. Science. 1996;271:810–812. doi: 10.1126/science.271.5250.810. [DOI] [PubMed] [Google Scholar]
- Keely P.J., Westwick J.K., Whitehead I.P., Der C.J., Parise L.V. Cdc42 and Rac1 induce integrin-mediated cell motility and invasiveness via PI 3-kinase. Nature. 1997;390:632–636. doi: 10.1038/37656. [DOI] [PubMed] [Google Scholar]
- Keely P., Parise L., Juliano R. Integrins and GTPases in tumor growth, motility and invasion. Trends Cell Biol. 1998;8:101–106. doi: 10.1016/s0962-8924(97)01219-1. [DOI] [PubMed] [Google Scholar]
- Kiyokawa E., Hashimoto Y., Kobayashi S., Sugimura H., Kurata T., Matsuda M. Activation of Rac1 by a Crk SH3-binding protein, DOCK180 Genes Dev. 12 1998. 3331 3336a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyokawa E., Hasimoto Y., Kurata T., Sugimura H., Matsuda M. Evidence that DOCK180 up-regulates signals from the CrkII-p130CAS complex J. Biol. Chem. 273 1998. 24479 24484b [DOI] [PubMed] [Google Scholar]
- Klemke R.L., Cai S., Giannini A.L., Gallagher P.J., de Lanerolle P., Cheresh D.A. Regulation of cell motility by mitogen-activated protein kinase. J. Cell Biol. 1997;137:481–482. doi: 10.1083/jcb.137.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemke R.L., Leng J., Molander R., Brooks P.C., Vuori K., Cheresh D.A. CAS/Crk coupling serves as a “molecular switch” for induction of cell migration. J. Cell Biol. 1998;140:961–972. doi: 10.1083/jcb.140.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolega J., Taylor D.L. Gradients in the concentration and assembly of myosin II in living fibroblasts during locomotion and fiber transport. Mol. Biol. Cell. 1993;4:819–836. doi: 10.1091/mbc.4.8.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffenburger D.A., Horwitz A.F. Cell migrationa physically integrated process. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- Matsuda M., Nagata S., Tanaka S., Nagashima K., Kurata T. Structural requirement of Crk SH2 region for binding to phosphotyrosine-containing proteins. J. Biol. Chem. 1993;268:4441–4446. [PubMed] [Google Scholar]
- Matsuda M., Hashimoto Y., Muroya K., Hasegawa H., Kurata T., Tanaka S., Nakamura S., Hattori S. Crk protein binds to two guanine nucleotide-releasing proteins for the Ras family and modulates nerve growth factor-induced activation of ras in PC12 cells. Mol. Cell. Biol. 1994;14:5495–5500. doi: 10.1128/mcb.14.8.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer B.J., Hirai H., Sakai R. Evidence that SH2 domains promote processive phosphorylation by protein-tyrosine kinases. Curr. Biol. 1995;5:296–305. doi: 10.1016/s0960-9822(95)00060-1. [DOI] [PubMed] [Google Scholar]
- Michiels F., Habets G.M., Stam J.C., van der Kammen R.A., Collard J.G. A role for Rac in Tiam1-induced membrane ruffling and invasion. Nature. 1995;375:338–340. doi: 10.1038/375338a0. [DOI] [PubMed] [Google Scholar]
- Minden A., Lin A., Claret F., Abo A., Karin M. Selective activation of the JNK signaling cascade and C-Jun transcriptional activity by the small GTPases Rac and Cdc4Hs. Cell. 1995;81:1147–1157. doi: 10.1016/s0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- Mitchison T.J., Cramer L.P. Actin-based cell motility and cell locomotion. Cell. 1996;84:371–379. doi: 10.1016/s0092-8674(00)81281-7. [DOI] [PubMed] [Google Scholar]
- Ojaniemi M., Vuori K. Epidermal growth factor modulates tyrosine phosphorylation of p130CAS. J. Biol. Chem. 1997;272:25993–25998. doi: 10.1074/jbc.272.41.25993. [DOI] [PubMed] [Google Scholar]
- Ridely A.J., Paterson H.F., Johnstone C.L., Diekmann D., Hall A. The small GTP-binding protein Rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- Rosenfeldt H., Lee D.J., Grinnell F. Increased c-fos mRNA expression by human fibroblasts contracting stressed collagen matrices. Mol. Cell. Biol. 1998;18:2659–2667. doi: 10.1128/mcb.18.5.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander E.E., van Delft S., ten Klooster J.P., Reid T., van der Kammen R.A., Michiels F., Collard J.G. Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell adhesion or cell migration and is regulated by phosphatidylinositol 3-kinase. J. Cell Biol. 1998;143:1385–1398. doi: 10.1083/jcb.143.5.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer D.D., Hauck C.R., Sieg D. Signaling through focal adhesion kinase. Biophys. Mol. Biol. 1999;71:435–478. doi: 10.1016/s0079-6107(98)00052-2. [DOI] [PubMed] [Google Scholar]
- Schlaepfer D.D., Jones K.C., Hunter T. Multiple Grb2-mediated integrin-stimulated signaling pathways to ERK/mitogenactivated protein kinasesummation of both c-Src and focal adhesion kinase-initiated tyrosine phosphorylation events. Mol. Cell. Biol. 1998;18:2571–2585. doi: 10.1128/mcb.18.5.2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer D.D., Broome M.A., Hunter T. Fibronectin-stimulated signaling from a focal adhesion kinase-c-src complexInvolvement of the Grb2, p130CAS and Nck adaptor proteins. Mol. Cell. Biol. 1997;17:1702–1713. doi: 10.1128/mcb.17.3.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer D.D., Hanks S.K., Hunter T., van der Geer P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature. 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- Sells M.A., Boyd J., Chernoff J. p21-activated kinase 1(Pak1) regulates cell motility in mammalian fibroblasts. J. Cell Biol. 1999;145:837–849. doi: 10.1083/jcb.145.4.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana, K., T. Urano, H. Fujita, Y. Ohashi, K. Kamiguchi, S. Iwata, H. Hirai, and C. Morimoto. 1997. Tyrosine phosphorylation of Crk-associated substrates by focal adhesion kinase. 272:29083–29090. [DOI] [PubMed]
- Tanaka S., Hattori S., Kurata T., Nagashima K., Fukui Y., Nakamura S., Matsuda M. Both the SH2 and SH3 domains of human Crk protein are required for neuronal differentiation of PC12 cells. Mol. Cell. Biol. 1993;13:4409–4415. doi: 10.1128/mcb.13.7.4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapon N., Hall A. Rho, Rac, and Cdc42 GTPases regulate the organization of the actin cytoskeleton. Curr. Opin. Cell Biol. 1997;9:86–92. doi: 10.1016/s0955-0674(97)80156-1. [DOI] [PubMed] [Google Scholar]
- Vuori K., Hirai H., Aizawa S., Ruoslahti E. Induction of p130CAS signaling complex formation upon integrin-mediated cell adhesiona role for Src family kinases. Mol. Cell. Biol. 1996;16:2606–2613. doi: 10.1128/mcb.16.6.2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wary K.K., Mainiero F., Isakoff S.J., Marcantonio E.E., Giancotti F.G. The adaptor protein Shc couples a class of integrins to the control of cell cycle progression. Cell. 1996;87:733–743. doi: 10.1016/s0092-8674(00)81392-6. [DOI] [PubMed] [Google Scholar]
- Wei J., Shaw L.M., Mercurio A.M. Regulation of mitogen-activated protein kinase activation by the cytoplasmic domain of the α6 integrin subunit. J. Biol. Chem. 1998;273:5903–5907. doi: 10.1074/jbc.273.10.5903. [DOI] [PubMed] [Google Scholar]
- Wu Y.C., Horvitz H.R. C. elegans phagocytosis and cell-migration protein ced-5 is similar to human DOCK180. Nature. 1998;292:501–504. doi: 10.1038/33163. [DOI] [PubMed] [Google Scholar]
- Yoshioka K., Matsumura F., Akedo H., Itoh K. Small GTP-binding protein Rho stimulates the actomyosin system, leading to invasion of tumor cells. J. Biol. Chem. 1998;273:5146–5154. doi: 10.1074/jbc.273.9.5146. [DOI] [PubMed] [Google Scholar]