

Abstract
Nitric oxide (˙NO)-derived reactive species nitrate unsaturated fatty acids, yielding nitroalkene derivatives, including the clinically abundant nitrated oleic and linoleic acids. The olefinic nitro group renders these derivatives electrophilic at the carbon β to the nitro group, thus competent for Michael addition reactions with cysteine and histidine. By using chromatographic and mass spectrometric approaches, we characterized this reactivity by using in vitro reaction systems, and we demonstrated that nitroalkene-protein and GSH adducts are present in vivo under basal conditions in healthy human red cells. Nitro-linoleic acid (9-, 10-, 12-, and 13-nitro-9,12-octadecadienoic acids) (m/z 324.2) and nitro-oleic acid (9- and 10-nitro-9-octadecaenoic acids) (m/z 326.2) reacted with GSH (m/z 306.1), yielding adducts with m/z of 631.3 and 633.3, respectively. At physiological concentrations, nitroalkenes inhibited glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which contains a critical catalytic Cys (Cys-149). GAPDH inhibition displayed an IC50 of ∼3 μm for both nitroalkenes, an IC50 equivalent to the potent thiol oxidant peroxynitrite (ONOO−) and an IC50 30-fold less than H2O2, indicating that nitroalkenes are potent thiol-reactive species. Liquid chromatography-mass spectrometry analysis revealed covalent adducts between fatty acid nitroalkene derivatives and GAPDH, including at the catalytic Cys-149. Liquid chromatography-mass spectrometry-based proteomic analysis of human red cells confirmed that nitroalkenes readily undergo covalent, thiol-reversible post-translational modification of nucleophilic amino acids in GSH and GAPDH in vivo. The adduction of GAPDH and GSH by nitroalkenes significantly increased the hydrophobicity of these molecules, both inducing translocation to membranes and suggesting why these abundant derivatives had not been detected previously via traditional high pressure liquid chromatography analysis. The occurrence of these electrophilic nitroalkylation reactions in vivo indicates that this reversible post-translational protein modification represents a new pathway for redox regulation of enzyme function, cell signaling, and protein trafficking.
Nitric oxide (˙NO)5 exerts a broad influence on cell and inflammatory signaling via both cGMP-dependent and -independent oxidative, nitrosative, and nitrative reactions (1, 2). The nitration of polyunsaturated fatty acids present in both membranes and lipoproteins is now emerging as a novel mechanism for transducing ˙NO-dependent redox signaling (3, 4). Recent evidence indicates that all major unsaturated fatty acids present in human blood contain some proportion of alkenyl nitro derivatives (R1HC=C(NO2)R2), also termed nitroalkenes. Because of the prevalence of fatty acid nitroalkenes in healthy humans, these species are now appreciated as an abundant pool of bioactive oxides of nitrogen in the vasculature (5). The two most clinically abundant nitroalkene fatty acid derivatives, nitro-oleic acid (9- and 10-nitro-9-cis-octadecaenoic acids; OA-NO2) and nitro-linoleic acid (10-nitro-9,12-octadecadienoic and 12-nitro-9,12-octadecadienoic acids; LNO2) are present in net concentrations of >1 μm in membrane and lipoprotein lipid extracts prepared from healthy human blood. These nitroalkene concentrations far exceed the <10 nm concentrations reported for other ˙NO derivatives, including S-nitrosothiols, nitrosyl heme and 3-nitrotyrosine, (6).
Nitroalkenes display pluripotent cell signaling capabilities, acting via both cGMP-dependent and -independent mechanisms (7–9). First, nitroalkenes such as LNO2 decay via the hydrophobically regulated Nef reaction to release ˙NO, thus acting as endogenous ˙NO donors that mediate cGMP-dependent cell signaling responses such as relaxation of vascular smooth muscle cells (2). Second, fatty acid nitroalkene derivatives are potent endogenous peroxisome proliferator-activated receptor (PPAR) ligands that act within physiological concentration ranges to modulate key PPAR-regulated signaling events (9). Third, nitroalkenes inhibit human peripheral blood neutrophil activation ( production and formyl-Met-Leu-Phe-stimulated Ca2+ influx, degranulation, and CD11b expression) via non-cAMP, non-cGMP-dependent mechanisms (7). Fourth, thrombin-induced platelet aggregation is inhibited by nitroalkene-induced attenuation of cAMP-dependent Ca2+ mobilization and activation of the phosphorylation of vasodilator-stimulated phosphoprotein at Ser-157 (8). Current evidence supports a dual regulation of platelet adenylyl cyclase and phosphodiesterase E activities by nitroalkenes. Finally, fatty acid nitroalkene derivatives, as opposed to native unsaturated fatty acids, potently regulate the expression of key inflammatory, cell proliferation, and cell differentiation-related proteins (5, 9).
The broad impact of nitroalkenes on differentiated characteristics of cells and tissues motivated analysis of additional chemical reactivities that would account for their pluripotent signaling capabilities. NMR, MS/MS, and spectroscopic analysis of nitrated fatty acids indicate the presence of predominantly nitroalkene rather than nitroalkane derivatives in clinical specimens, with this structural configuration conferring unique biochemical and pharmacological qualities. The alkenyl nitro configuration of endogenous nitrated fatty acids indicates potential electrophilic reactivity of the β-carbon adjacent to the nitro-bonded carbon. This would promote nitroalkene reactivity with nucleophiles (i.e. Cys and His residues) via Michael addition reactions, yielding new carbon-carbon or carbon-heteroatom bond frameworks (2, 10). This electrophilic property of nitroalkenes was first suggested by the biological detection of α,β-nitro-hydroxy fatty acid derivatives (5). Also, synthetic nitroalkenes generated hydroxy derivatives under aqueous conditions, presumably induced by reaction with the low levels of hydroxide ion present at physiological pH (2).
Other electrophilic lipids, in particular 15-deoxy-Δ12,14-PGJ2 and 4-hydroxy-2-nonenal (HNE), are reactive and form adducts with proteins that contain nucleophilic centers (11). The electrophilic HNE reacts with sulfhydryl groups (12), the imidazole of histidine (13) and the ε-amino of lysine (14). Covalent modification by electrophilic lipids has been shown to alter the structure and activities of cathepsin B (15), Keap1 (16), and insulin (13).
Herein, the reactions of nitroalkenes with glutathione (GSH) are characterized, and GAPDH is evaluated as an exemplary target protein for nitroalkene reaction. GAPDH is of additional relevance as follows: (a) catalytic thiols are essential for GAPDH activity (17); (b) GAPDH can be inhibited by electrophilic lipids (18, 19); and (c) the ˙NO-dependent post-translational modification and translocation of GAPDH to specific cellular microenvironments impact the roles of GAPDH in intermediary metabolism and apoptotic signaling (20, 21).
We reveal here, via model reaction systems and the proteomic analysis of human red cells, that nitroalkenes readily undergo covalent, thiol-reversible post-translational modification of nucleophilic amino acids in GSH and proteins. The occurrence of these electrophilic nitroalkylation reactions in vivo indicates that fatty acid nitroalkene derivatives serve to regulate protein function and trafficking.
Experimental Procedures
Materials and Chemicals
Rabbit muscle GAPDH, dl-glyceraldehyde 3-phosphate (GAP), glutathione (reduced and oxidized forms), ascorbic acid, DTPA, dithiothreitol (DTT), hydrogen peroxide (H2O2), and sodium arsenite were from Sigma. Nicotinamide adenine dinucleotide (oxidized) was from Roche Applied Science. Sodium pyrophosphate was from Mallinckrodt. Nitrolinoleic acid (LNO2), nitro-oleic acid (OA-NO2), and their corresponding internal standards [13C18]LNO2 and [13C18]OA-NO2 were prepared as described previously (5, 22). Hitrap desalting columns were from Amersham Biosciences. Pipette tips for sample preparation (ZipTip C18, P10) were from Millipore Corp. (Bedford, MA). Sequencing grade modified trypsin was from Promega (Madison, WI). Peroxynitrite (ONOO−) was handled as described previously (23, 24). Peroxynitrite concentration was determined at 302 nm in 1 m NaOH (ε = 1670 m−1·cm−1). GAPDH concentration was determined at 280 nm (ε = 1.46 × 105 m−1·cm−1) (25).
GSH-Nitroalkene Reactions
GSH (1 mm) solvated in 50 mm sodium phosphate buffer, pH 7.4, was treated with equimolar LNO2 or OA-NO2 at 20 °C for 30 min. The reaction mixture was analyzed by ESI-MS with and without liquid chromatography. When the adduct was analyzed by reverse phase HPLC-MS, the elution protocol used for tryptic peptide resolution was employed (see below). Synthetic nitroalkene adducts of GSH were used as standards for LC separations and quantitative MS analysis, including GS-OA-NO2, GS-[13C18]OA-NO2, GS-LNO2, and GS-[13C18]LNO2. These derivatives were prepared by reacting the corresponding nitroalkene (2 mm) with GSH (20 mm) in sodium borate buffer (0.1 m), pH 10.3, for 30 min at 20 °C. GS-nitroalkene adducts were purified from nonreacted GSH by reverse phase chromatography. Samples were loaded onto PrepSep™ C18 columns (Fisher) equilibrated with 0.1% formic acid. After washing, adducts were eluted with 0.1% formic acid in methanol and fractions concentrated in vacuo. The concentration of purified standards was determined by elemental nitrogen analysis following pyrolysis, using an Antek chemiluminescent nitrogen detector (Houston, TX).
GAPDH Activity Analysis
Enzyme assay mixtures contained 100 mm sodium pyrophosphate, 0.1 mm DTPA, 0.25 mm NAD+, 10 mm sodium arsenite, and 0.5 mm GAP, pH 7.4. GAPDH activity (0.05 μm final GAPDH concentration) was assessed at 20 °C by following NAD+ reduction at 340 nm for the first 20 s after addition of GAP (Shimadzu, UV 2401-PC) (25). As a control, GAPDH was fully activated in 50 mm pyrophosphate, 0.1 mm DTPA, 20 mm DTT, pH 8.5, for 30 min at 4 °C (25). Excess thiol was removed by size exclusion HPLC using a HiTrap column equilibrated with 50 mm pyrophosphate, 0.1 mm DTPA, pH 7.4.
Exposure of GAPDH to Oxidants and Reductants
GAPDH solvated in 100 mm sodium pyrophosphate, 0.1 mm DTPA, pH 7.4, was treated at 20 °C with nitroalkenes (0–20 μm), ONOO− (0–100 μm) or H2O2 (0–500 μm), and GAPDH activity was immediately measured. Oxidant-treated GAPDH was incubated with GSH (0.01–20 mm), DTT (20 mm), or ascorbic acid (20 mm) for 30 min at room temperature, and enzymatic activity was re-determined. The pH profile of GAPDH inactivation by nitroalkenes was analyzed using GAPDH maintained for 5 min in 50 mm sodium pyrophosphate buffer adjusted from pH 5 to 10, followed by nitroalkene addition. Aliquots were then removed for GAPDH activity assay. For quantitation of GAPDH thiol content, GAPDH preparations were added to 50 mm Tris-HCl, 5 mm EDTA, 1% SDS, and 1 mm 5,5′-dithiobis(2-nitrobenzoic) acid, pH 8.0, using ε = 1.36 × 104 m−1·cm−1 at 412 nm for the thionitrobenzoate anion (24, 25).
Matrix-assisted Laser Desorption and Ionization Time of Flight Mass Spectrometry (MALDI-TOF MS) Analysis
MALDI-TOF MS was performed using a Voyager DE PRO system (Applied Biosystems, Foster City, CA), equipped with a N2 laser source (337 nm). Mass spectra were acquired for positive ions in linear and reflector mode. For whole molecular mass determinations, native and nitroalkene-treated GAPDH (0.5 μm) was desalted using C18-ZipTip (P10) (Millipore Corp., Bedford, MA), following the manufacturer's protocol. Proteins were eluted by adding the matrix solution (sinapinic acid, 10 mg/ml in 50% acetonitrile (ACN), 0.2% trifluoroacetic acid) and directly applied to the stainless steel sample plate. GAPDH-derived peptide masses were measured in the reflector mode with an accuracy of ∼50 ppm, attained by internal mass calibration using characteristic GAPDH tryptic peptides as mass standards and α-cyano-4-hydroxycinnamic acid as the matrix. To obtain complete post-source decay (PSD) spectra, a series of reflectron mass spectral segments were acquired, each optimized to focus fragment ions within different mass/charge (m/z) ranges. Each segment was stitched together using Bio-spectrometry work station software to generate a composite PSD spectrum (26).
Peptide Mapping and Electrospray Ionization Liquid Chromatography-Mass Spectrometry Analysis (ESI-LC-MS)
Native and nitroalkene-modified GAPDH (10 μm) were digested with sequencing grade trypsin in 50 mm pyrophosphate buffer, pH 7.4, at 37 °C for 20 h using an enzyme:substrate ratio of 1:50 (w/w). Peptide samples were initially analyzed by ESI-LC-MS using a LCQ ion trap mass spectrometer (LCQ Deca; Thermo Electron Corp., San Jose, Ca). A reversed-phase column (5 μm, 2.1 × 150 mm, 300 Å, from Grace Vydac, (Hesperia, CA)) was eluted with solvent A (0.1% formic acid) and solvent B (0.08% formic acid in acetonitrile). Peptides were eluted at 40 °C with a linear gradient of solvent B (2–60% in 105 min) at a flow rate of 0.25 ml·min−1. Electrospray voltage was 5 kV, and capillary temperature was 260 °C. Peptides were detected in the positive ion mode using a mass range of 100–2000, following mass calibration with the GS-OA-NO2 adduct. MS/MS peptide analyses was performed by nanospray ion trap mass spectrometry (LTQ; Thermo Electron Corp.) using a BioBasic C18 picofrit column, 5 μm, 75 μm × 10 cm (Thermo Electron Corp.), in line with a trap column (Zorbax 300SB-C18, 5 μm, 0.3 × 50 mm, Agilent Technologies, Chicago). A flow rate of 700 nl·min−1 and similar gradient conditions were used.
GAPDH-Liposome Interactions
A lipid film was prepared by dissolving soybean phosphatidylcholine (200 mg) and cholesterol (50 mg) in chloroform and removal of solvent under a stream of nitrogen. Multilamellar vesicles were prepared by hydrating the lipid film with 10 ml of 10 mm pyrophosphate buffer, pH 7.4, at 25 °C for 1 h. After vortexing, the suspension was placed in a 25 °C sonicating water bath for 30 min. Subsequently, control or nitroalkene-treated GAPDH preparations (100 μl of 10 mg/ml protein) were incubated with liposome suspensions (1 ml) for 30 min at 25 °C. Liposome-associated GAPDH was sedimented by ultracentrifugation at 100,000 × g for 30 min. Soluble GAPDH in the supernatant and sedimented liposome-associated GAPDH were analyzed by SDS-PAGE (4–12% bis-Tris, Criterion™ XT Precast Gel; Bio-Rad), stained with Coomassie Blue, and quantitated by densitometry using AlphaInotech software.
Identification of Endogenous Nitroalkylated GSH in Human Red Blood Cells
All human studies were reviewed and approved by the University of Alabama at Birmingham Institutional Review Board (protocol X040311001). Human red blood cells (RBCs) were obtained by centrifugation (800 × g for 10 min) of freshly drawn heparinized blood from a healthy donor. RBCs were washed twice with 0.15 m NaCl, and an aliquot of packed RBCs (1.5 ml) was lysed by diluting with 5 volumes of 0.1% formic acid in water, acidified to inhibit further nitroalkylation reactions during sample processing. Samples were centrifuged at 100,000 × g for 20 min at 4 °C, and supernatants were collected and GS-[13C18]LNO2 (10 μl, 155 nm) and GS-[13C18]OA-NO2 (10 μl, 116 nm) added as internal standards. Samples were then loaded onto PrepSep™ C18 columns (Fisher) equilibrated with 0.1% formic acid. After washing, samples were eluted with 0.1% formic acid in methanol. The eluted fractions were concentrated in vacuo, and GSH-nitroalkene adducts were measured by ESI-LC-MS/MS in the positive ion mode using a multiple reaction monitoring (MRM) scan mode on a 4000 Q-Trap (Applied Biosystems, Foster City, CA). The MRM transitions of m/z 635.3/506.3 (GS-OA-NO2), 653.3/524.3 (GS-[13C18]OA-NO2), 633.3/504.3 (GS-LNO2), and 651.3/522.3 (GS-[13C18]LNO2) were used, consistent with the generation of the y ion of adducted glutathione (27). The identity of the GSH-nitroalkene adduct was further confirmed by performing enhanced product ion (EPI) analysis and comparing fragmentation patterns with those obtained from EPI analysis of the synthetic standard GS-OA-NO2.
Identification of Endogenous Nitroalkylated GAPDH in Human Red Blood Cells
300 μl of freshly obtained, packed RBCs were lysed as before, and the cytosolic and membrane fractions were separated by ultracentrifugation (100,000 × g for 20 min at 4 °C). Membranes were resuspended in 100 μl of phosphate-buffered saline, and aliquots of both cytosolic and membrane fractions (50 μl) were resolved by electrophoresis under nonreducing and denaturing conditions (4–15% gradient gel, Bio-Rad) and then stained with Coomassie Blue. The 36-kDa band corresponding to purified GAPDH was excised and in-gel digestion with trypsin performed. Peptides were eluted and analyzed by nano-LC-ESI-MS/MS ((700 nl/min; Finnigan LTQ; Thermo Electron Corp.) in line with a trap column (Zorbax 300SB-C18, 5 μm, 0.3 × 50-mm column, Agilent Technologies, Chicago).
Crystal Structure of the Rabbit Muscle GAPDH
The image of GAPDH structure obtained by x-ray diffraction methods and produced using MolScript and Raster3D was downloaded from Pubmed, Protein Data Bank (1J0X) (28).
Results
The in vitro reaction of LNO2 with GSH was monitored by ESI-ion trap MS, which revealed a GS-LNO2 adduct (m/z 631.3), indicative of the nitroalkylation of GSH (Fig. 1A). Either source fragmentation of the m/z 631.3 adduct or residual LNO2 (m/z 324.2) and GSH (m/z 306.1) accounted for the other ions present in the spectrum. Collision-induced dissociation (CID) of the GS-LNO2 adduct precursor ion (m/z 631.3) yielded a main product ion with m/z of 306.1, corresponding to GSH (Fig. 1B). This was further confirmed by MS/MS/MS of the pair m/z 631.3/306.1 (Fig. 1C), which yielded the specific product ions characteristic of GSH (Fig. 1C), as reported previously (29). Similar results were obtained for OA-NO2 reaction with GSH (not shown).
FIGURE 1. Mass spectrometric analysis of the alkylation of glutathione by nitroalkenes and its impact on GSH lipophilicity.
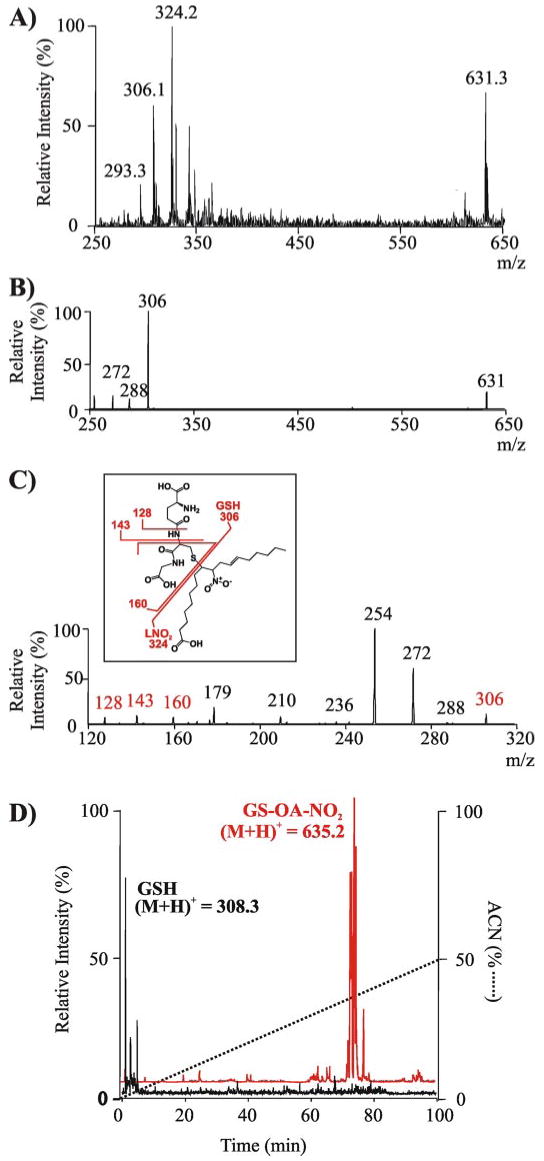
A, ESI-ion trap MS spectrum in negative ion mode (LCQ Deca; Thermo Electron Corp.) of the reaction product generated by LNO2 (300 μm) reaction with GSH (300 μm) in 50 mm sodium phosphate buffer, pH 7.4, at 20 °C for 30 min. Previous to the MS analysis, the reaction mixture was diluted in methanol, 0.1% NH4OH. B, MS/MS spectrum in negative ion mode of the GS-LNO2 adduct (m/z = 631.3). C, MS/MS/MS spectrum of fragment ion 306 m/z from GS-LNO2 adduct (m/z = 631.3). Inset, structural scheme of the adduct showing main fragmentation sites. The mass of main fragment ions is shown in red as detected in the negative ionization mode. D, total ion chromatogram in positive ion mode shows a significant increase in the lipophilicity of GSH following alkylation by OA-NO2, as indicated by the change in retention times of GSH ((M + H)+ = 308.3) (D, black) versus the GS-OA-NO2 adduct ((M + H)+ = 635.3) (D, red).
The nitroalkylation adduct of OA-NO2 and GSH was also analyzed by reverse phase LC-MS and compared with the elution characteristics of GSH (Fig. 1D). GSH ((M + H)+ 308.3) was not retained on column and eluted with the void volume (Fig. 1D, black tracing). In contrast, the GS-OA-NO2 adduct ((M + H)+ 635.2) eluted at ∼74 min when the solvent gradient reached 38% acetonitrile (Fig. 1D, red tracing). This increased organic phase requirement for elution by reverse phase HPLC reveals that the nitroalkylation of peptides and proteins confers strong hydrophobic character. The elution profile of the GS-OA-NO2 adduct, revealing multiple resolving peaks (Fig. 1D, red tracing), suggests that different isomers are being formed by the reaction of OA-NO2 with GSH.
GAPDH, which contains a critical redox and electrophile-sensitive catalytic cysteine (Cys-149 (17)), was incubated with nitro fatty acids. There was a dose-dependent inactivation of GAPDH (0.5 μm) upon incubation of the enzyme with either LNO2 or OA-NO2 (0–10 μm) for 15 min (Fig. 2A). The loss of GAPDH activity as a function of nitroalkene concentration was sigmoidal. The nitroalkene concentration required to inactivate 50% of initial activity (IC50) was ∼3 and ∼4 μm for OA-NO2 and LNO2, respectively. Compared with other recognized biological thiol oxidants, this concentration of nitroalkenes is 30 times less than that of H2O2 and similar to that of ONOO− for inducing similar extents of GAPDH inactivation (Fig. 2B) (25, 30). The time course of OA-NO2-mediated GAPDH inactivation was fast, with 50% inactivation occurring within 2 min under the conditions studied (Fig. 2C). The inactivation of GAPDH by OA-NO2 was strongly pH-dependent, with maximal inactivation obtained at alkaline pH (Fig. 2D).
FIGURE 2. The inhibition of GAPDH following alkylation by nitrolinoleate, nitro-oleate, peroxynitrite, and hydrogen peroxide.
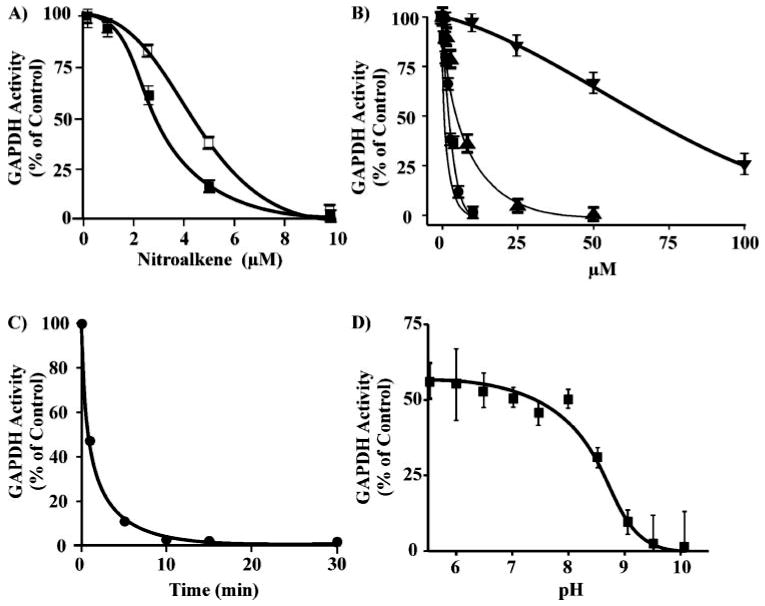
A, GAPDH (0.5 μm) was incubated with increasing concentrations of LNO2 (0–10 μm) (☐) or OA-NO2 (■) in 100 mm sodium pyrophosphate, 100 μm DTPA, pH 7.4, at 20 °C for 15 min. Aliquots were removed, and GAPDH activity was determined. B, the relative inactivation of GAPDH by LNO2 (●), OA-NO2 (■), ONOO− (0–50 μm; ▲), and H2O2 (0–100 μm, ▼). C, time course of OA-NO2-mediated GAPDH inhibition. GAPDH (0.5 μm) was incubated with OA-NO2 (10 μm) in 0.1 m pyrophosphate 0.1 mm DTPA, pH 7.4, at 25 °C. At the indicated time points aliquots were removed, and enzyme activity was determined. D, pH profile of the inhibition of GAPDH by OA-NO2. After preincubation of GAPDH (0.5 μm) for 5 min in 50 mm sodium pyrophosphate buffer adjusted to pH 5.5–10 at 20 °C, OA-NO2 (7.5 μm) was added, and after 15 min GAPDH activity was assessed as before. The percentage of control GAPDH activity at each pH was determined.
The biochemical nature of nitroalkene-GAPDH interactions was investigated in more detail. GAPDH thiol content was determined after OA-NO2-mediated enzyme inactivation (Fig. 3A). Nitroalkene-induced GAPDH thiol depletion paralleled the loss of enzyme catalytic activity, with 50% thiol depletion occurring at nitroalkene concentrations inducing a 50% loss of initial enzyme activity (Fig. 3A).
FIGURE 3. OA-NO2-induced thiol oxidation in GAPDH and reversibility of enzyme inactivation by thiol reagents.
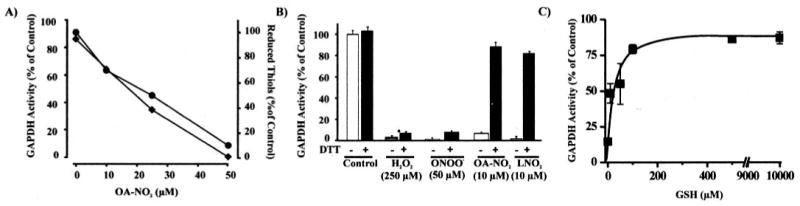
A, GAPDH (2 μm) was incubated for 15 min in 0.1 m pyrophosphate, 0.1 mm DTPA, pH 7.4, at 25 °C with OA-NO2 (0–50 μm). Aliquots were removed, and GAPDH activity was determined (♦). Reduced thiol content was determined by 5,5′-dithiobis(2-nitrobenzoic) acid reaction of GAPDH denatured in 1% SDS (●). B, GAPDH (0.5 μm) was incubated with H2O2 (250 μm), ONOO− (50 μm), OA-NO2 (10 μm), or LNO2 (10 μm) in 0.1 m pyrophosphate, 0.1 mm DTPA, pH 7.4, at 25 °C for 15 min, and GAPDH activity was determined before (open bars) and after (solid black bars) treatment of the samples with DTT (10 mm) for 30 min at 25 °C. As a control, GAPDH was pretreated with DTT and activity was measured. C, OA-NO2-inactivated GAPDH was treated with increasing concentrations of GSH (0–10 mm) and enzyme activity determined.
In order to gain further insight into the mechanisms by which nitroalkenes inactivate GAPDH and the oxidation state of the critical thiol, the reversibility of nitroalkene-mediated GAPDH inhibition by other thiol-containing reducing agents was studied. The inhibition of GAPDH by nitroalkenes was reversed by incubation with low concentrations of DTT or GSH (Fig. 3, B and C). Of significance, inactivation of GAPDH by ONOO− and H2O2 was not reversible by DTT or GSH, as described previously (25), whereas both reducing agents restored ∼85% of initial GAPDH activity following OA-NO2 reaction (Fig. 3B). Extremely low concentrations of thiol (10 μm GSH) reversed GAPDH nitroalkylation and restored ∼50% of initial GAPDH activity, emphasizing the reversibility of protein adduction by nitroalkenes under biological conditions (Fig. 3C). In contrast, incubation of nitroalkene-inactivated GAPDH with high concentrations of ascorbic acid (20 mm) did not restore enzymatic activity (not shown).
MALDI-TOF MS analysis of nitroalkene-treated GAPDH showed a shift in the mass of the enzyme (Fig. 4A), revealing covalent modification by nitroalkenes. GAPDH displayed a broader mass distribution following reaction with OA-NO2 (10 μm, OA-NO2:GAPDH molar ratio 20:1), with an increase in mass of up to ∼2.0 kDa (∼36 kDa to a maximum of ∼38 kDa; Fig. 4B). This profile of mass shifts indicates that GAPDH was adducted by up to ∼7 molecules of OA-NO2. Reduction of GAPDH-OA-NO2 adducts with GSH (10 mm) eliminated the OA-NO2-induced higher mass species of GAPDH and restored the precursor protein mass (Fig. 4C).
FIGURE 4. Mass spectrometric analysis of GAPDH alkylation by OA-NO2 and its reversal by GSH.
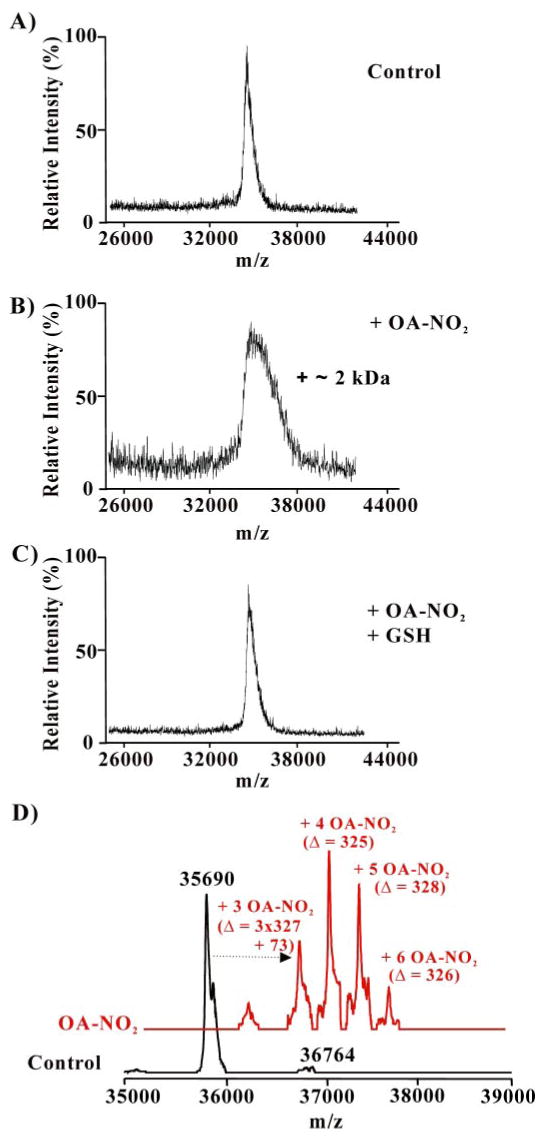
GAPDH (0.5 μm) was incubated with OA-NO2 (10 μm) in 0.1 m pyrophosphate, 0.1 mm DTPA, pH 7.4, at 25 °C for 15 min. GSH (10 mm) was then added for 15 min. After desalting, aliquots were analyzed by MALDI-TOF MS (A–C; Voyager DE PRO, Applied Biosystems, Foster City, CA) or by LC-ESI two-dimensional linear ion trap MS (D; LTQ; Thermo Electron Corp.). A, spectrum of native GAPDH; B, OA-NO2-treated GAPDH before, and C, after addition of GSH; D, native GAPDH (black trace) and OA-NO2-treated GAPDH (red trace).
The capacity of OA-NO2 to form covalent adducts with GAPDH was confirmed by LC-MS using a two-dimensional linear ion trap. The greater mass resolution of the two-dimensional linear ion trap resolved the expected mass of native GAPDH (Fig. 4D, black trace) and revealed multiple GAPDH-OA-NO2 derivatives that all differed by the neutral mass of OA-NO2 (m/z 327 Da). These nitroalkylated GAPDH derivatives had 3–6 OA-NO2 adducts (Fig. 4D, red trace). A difference of 73 Da between the theoretical mass and the measured mass of GAPDH adducted with 3 OA-NO2 was observed (Fig. 4D, red trace), suggesting that other modifications (i.e. Cys and Met oxidations) were formed. Indeed, Cys and Met were both detected in the native and oxidized form in the tryptic peptide mapping.
To determine the precise sites of GAPDH nitroalkylation, native and OA-NO2-treated GAPDH were digested with trypsin and analyzed by different mass spectrometric analytical approaches, including LC-MS, nanospray LC-MS/MS, and MALDI-TOF. Tryptic peptide mass mapping by LC-MS analysis of native GAPDH identified peptides covering 75% of the primary sequence (supplemental Fig. 1 and supplemental Table 1). LC-MS analysis of the peptide map of OA-NO2-treated GAPDH showed marked differences (Table 1 and Fig. 5). There were five peptides displaying modified MS and chromatographic behavior in OA-NO2-treated GAPDH, although all remaining peptides had similar relative ion intensities and HPLC retention times in both native and treated GAPDH. As an example, peptides 7 and 17, which do not contain Cys or His, are shown to retain similar ion intensity and retention time in OA-NO2-treated GAPDH (Table 1 and Fig. 5A). Of the five uniquely behaving peptides, three contained His and displayed significantly less relative ion intensity in OA-NO2-treated GAPDH (peptides 2, 18, and 23; see Table 1 and Fig. 5B). The other two modified peptides contained Cys as the nucleophilic residue and were undetectable in their nonmodified form in OA-NO2-treated GAPDH. Peptide 19 contains 2 Cys residues, the catalytic active Cys-149 and the Cys-153. The other peptide (peptide 21) contains the Cys-244 (Table 1 and Fig. 5C). Of note, the catalytic thiol-containing peptide (peptide 19) contained an intramolecular disulfide bond between both Cys residues in control GAPDH, showing a loss of 2 Da with respect to the expected mass of the peptide (m/z = 1705.86 to 1703.86) (Table 1 and Fig. 5C). This disulfide was probably formed during trypsin digestion and/or LC-MS analysis, as described previously (18).
TABLE 1. OA-NO2-modified GAPDH tryptic peptides identified by ESI-LC-MS.
RT indicates retention time, and bold letters indicate modified residues.
| Peak/peptide | RT | Sequence | Monoisotopic mass | z | Theoretical
m/z |
Measured
m/z |
Amount detected | |
|---|---|---|---|---|---|---|---|---|
| Control | OA-NO2-treated | |||||||
| min | Da | |||||||
| 2 | 2.06 | AGAH108LK | 595.35 | 1 | 596.35 | 596.3 | ✓ | ↓ |
| 18 | 27.86 | VVDLMVH327MASK | 1228.64 | 1 | 1229.64 | 1229.5 | ✓ | ↓ |
| 119 | 28.02 | IVSNASC149TTNC153LAPLAK | 1704.86 | 1 | 1705.86 | 1703.8 | ✓ | ↓ ↓ ↓ |
| 21 | 36.36 | VPTPNVSVVDLTC244R | 1498.79 | 1 | 1499.79 | 1499.7 | ✓ | ↓ ↓ ↓ |
| 23 | 39.39 | VIISAPSADAPMFVMGVNH134EK | 2212.11 | 2 | 1107.05 | 1107.1 | ✓ | ↓ |
FIGURE 5. ESI-HPLC-MS analysis of tryptic peptides from native and OA-NO2-alkylated GAPDH.
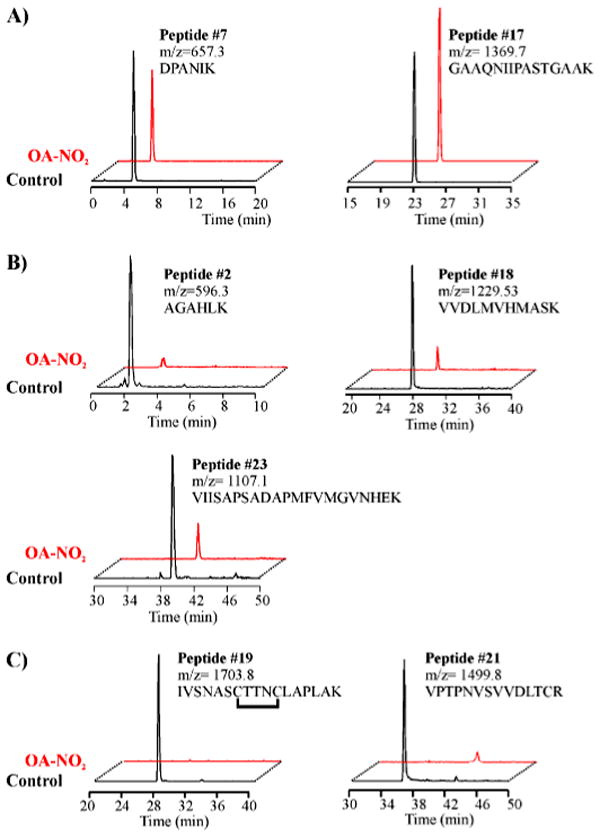
Selective ion chromatograms from native or OA-NO2-treated GAPDH digested with sequence-grade trypsin and analyzed by ESI-LC-MS (LCQ-Deca; Thermo Electron Corp.). Nonalkylated peptides (A) and peptides alkylated by OA-NO2 (B and C) are numbered as in the supplemental Fig. 1 and Table 1. A, similar relative ion intensities of non-nucleophilic peptides 7 and 17 (m/z 657.3 and 1369.7) were generated by both control and OA-NO2-nitroalkylated GAPDH. B, peptides 2, 18, and 23 containing the nucleophilic OA-NO2-reactive amino acid His were present at lower ion intensities in the tryptic digest of OA-NO2-treated GAPDH. C, peptides 19 and 21, containing OA-NO2-reactive nucleophilic amino acid Cys, were absent in tryptic digests of OA-NO2-treated GAPDH.
Furthermore, relative to the theoretical calculated masses of the unmodified peptides, four of these five peptides (peptides 2, 18, 21 and 23) were detected by nanospray LC-MS/MS with an increased mass of 327.3 Da, corresponding to the addition of a single molecule of OA-NO2. In particular, a new peptide eluting at 76 min ((M + H)+ = 1556.7 Da) was identified in the tryptic digest of OA-NO2-treated GAPDH (Fig. 6A). The mass of this peptide, not present in native GAPDH, corresponded to the mass of peptide 18 (sequence 321–331) plus 327.3 Da, the neutral mass of OA-NO2. Nanospray LC-MS/MS analysis revealed that His-327 was adducted to OA-NO2 in this particular peptide (Fig. 6A and its table). The MS/MS spectrum and the [M + 2H]2+ ion at m/z 844.34 from this OA-NO2-modified peptide are shown (Fig. 6B and Table 2). The full series of singly charged y and b fragments were detected, with y and b fragments corresponding to the ions that retain the charge at the C- and N-terminal groups of the peptide, respectively. The mass difference between y5 and y6 and also between b6 and b7 fragments corresponded to the mass of the His residue (137.06 Da) increased by the neutral mass of OA-NO2 (Fig. 6B and the corresponding inset table). These results demonstrate that His-327 is the modified residue in this peptide. Modified peptide 18 was also found in the analysis of trypsin-digested GAPDH by MALDI-TOF MS (Fig. 6C). PSD sequencing analysis of this peptide showed a principal, unique product ion with a mass of 437.1 Da. This fragment corresponds to the immonium ion of His (H = 110.1 Da) adducted to OA-NO2 (327.3 Da; see Fig. 6, D and E). This adducted immonium ion was also detected by MS/MS analysis performed via nanospray LC ion trap MS (Fig. 6B). The adducted immonium ion does not appear as a major fragment in this spectrum, likely due to differences in the way ions are generated and fragmented in the two different techniques.
FIGURE 6. Mass spectrometric analysis of nitroalkylation patterns following in vitro treatment of purified GAPDH with OA-NO2.
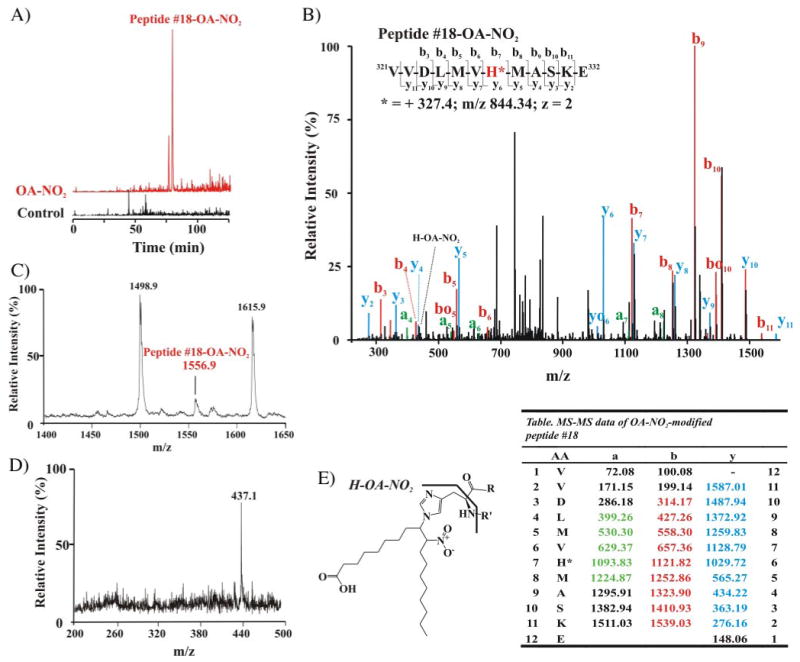
A, selective ion chromatograms from LC-ESI-MS analysis of OA-NO2-modified peptide 18 (LCQ Deca; Thermo Electron Corp.). The nitroalkylation of peptide 18 (native peptide (M + H)+ = 1229.6, room temperature 27.9 min) by OA-NO2 increased the retention time to 76 min, and the mass of the OA-NO2-modified peptide 18 was increased by 327 Da, equivalent to the neutral ion mass of OA-NO2, becoming m/z 1556.7. B, LC nanospray MS/MS spectrum of the nitroalkylated peptide 18 (LTQ; Thermo Electron Corp.). MS/MS spectrum of the doubly charged ion at m/z 844.34. Colors are annotated in the corresponding table. The yo and bo nomenclature indicates the corresponding y-H2O and b-H2O fragments, respectively. Inset, amino acid sequence of peptide 18 indicating major C- and N-terminal fragment ions detected by full-scan MS/MS. C, MALDI-TOF mass spectrum of the tryptic digest of OA-NO2-treated GAPDH (Voyager DE Pro, Applied Biosystem, Foster City, CA), focusing on nitroalkylated-peptide 18 ((M + H)+ = 1556.9). D, PSD MALDI-TOF-MS analysis of modified peptide 18 gives a main product ion at m/z 437.1, corresponding to the immonium ion of the histidine (H)-OA-NO2 adduct. E, structure and fragmentation pattern of the His-OA-NO2 adduct, showing the immonium adduct fragment (H-OA-NO2). Table list of MS/MS fragment ions m/z from peptide 18. Ions that are detected are highlighted in color (B). AA, amino acids.
TABLE 2. Retention times (RT) of the native versus OA-NO2-modified forms of GSH and GAPDH peptides.
| Peptide | Sequence | Native-modified | OA-NO2-modified | ||
|---|---|---|---|---|---|
| RT | ACN | RT | ACN | ||
| min | % | min | % | ||
| GSH | γ–ECG | 1.6 | 2.0 | 74.4 | 38.2 |
| 2 | AGAHLK | 2.1 | 2.0 | 65.6 | 33.3 |
| 18 | VVDLMVHMASK | 27.9 | 12.6 | 76.0 | 39.1 |
| 19 | IVSNASCTTNCLAPLAK | 28.0 | 12.7 | 83.6 | 43.2 |
| 21 | VPTPNVSVVDLTCR | 36.4 | 17.3 | 83.1 | 43.0 |
| 23 | VIISAPSADAPMFVMGVNHEK | 39.4 | 18.9 | 76.6 | 39.4 |
The nitroalkylation of peptide 23 (sequence 116–136) by OA-NO2 was also determined by an increased mass of 327 Da (supplemental Fig. 2). This modification accounted for a 75% decrease in ion intensity of the native peptide upon LC-MS analysis (Fig. 5B). When this modified peptide was analyzed by PSD, the immonium ion of His attached to OA-NO2 was detected, indicating that His-134 was derivatized by OA-NO2 in peptide 23 (supplemental Fig. 2). The OA-NO2-adducted immonium ion of His detected by PSD sequencing analysis was a revealing and readily detected footprint of nitroalkene-modified His-containing peptides by MALDI-TOF MS.
The MS/MS spectrum and data for the OA-NO2-modified peptide 2 ([M + 2H]2+ ion at m/z 462.06; 105Ala-Gly-Ala-His-Leu-Lys110 plus the mass addition of 327.3) are shown in supplemental Fig. 3 and its table. This modified peptide was only detectable in OA-NO2-treated GAPDH. The mass difference between the y3 and y2 fragments corresponded to the mass of His (137.06 Da) increased by the neutral mass of OA-NO2 (327.3 Da, supplemental Fig. 3 and its accompanying table), affirming that His-108 is the modified residue in this peptide.
The final peptide detected to have a single OA-NO2 adduct was peptide 21. The MS/MS spectrum and data for this peptide are shown in supplemental Fig. 4 and its accompanying table. The masses of the b9 fragment and y2* (i.e. y2-NH3), together with the mass difference between the b9 and the b8, demonstrate that Cys-244 is adducted to OA-NO2 in this peptide.
During nanospray LC-MS/MS, peptide displayed an increase in mass of 654.6 Da, corresponding to the adduction of two OA-NO2. The mass of the fragments b*4 (i.e. b4-NH3) and y7 identifies Cys-149 and Cys-153 as the two modification sites in this peptide (supplemental Fig. 5 and its accompanying table). Thus, by nanospray LC-MS/MS analysis we demonstrated that in vitro Cys-149, Cys-153, Cys-244, His-108, His-134, and His-327 are the GAPDH residues adducted to OA-NO2.
The increased hydrophobicity induced by OA-NO2 adduction of GSH was also manifested by all five OA-NO2-modified GAPDH peptides (Table 2). The nitroalkylation of peptides profoundly increased the percentage of hydrophobic solvent required to elute adducted peptides from a reverse phase C18 column. Independently of the retention time of the nonmodified native form of the peptides, all nitroalkylated peptides eluted when the concentration of ACN reached ∼ 40% (Table 2). These results reveal one explanation why this abundant post-translational protein modification has not been appreciated sooner, because most LC-MS analyses of peptides rarely utilize ACN concentrations exceeding 30% in gradient elution schemes.
The increased hydrophobicity of nitroalkylated peptides suggests that nitroalkenes may facilitate membrane trafficking following post-translational modification of target proteins. The interaction of nitroalkylated GAPDH with membrane lipids was evaluated by treating GAPDH with increasing concentrations of OA-NO2 and then incubating the treated protein with liposomes. Residual soluble GAPDH was separated from liposome-associated GAPDH by ultracentrifugation and identified by electrophoretic analysis of the supernatant (Fig. 7A). This analysis also revealed a dose-dependent increase in OA-NO2-adducted GAPDH association with sedimented liposomes (Fig. 7B). Three of the six GAPDH residues identified to be nitroalkylated in vitro (Cys-149, His-134, and His-327) are located on the surface of the protein and exposed to the solvent (28). The other three potentially modified residues (His-108, Cys-153, and Cys-244) are buried deeper within the protein (28), suggesting that nitroalkylation at these sites occurs provided that 1) the initial nitroalkylation of exposed residues induces conformational changes that result in the exposure of previously hidden domains, and/or 2) nitroalkenes are able to diffuse to and react with nonsolvent-exposed protein residues. These data support that post-translational nitroalkylation of proteins by nitrated fatty acids will impact protein hydrophobicity, membrane interactions, and consequently, subcellular distribution.
FIGURE 7. Increased membrane association of GAPDH following nitroalkylation by OA-NO2.
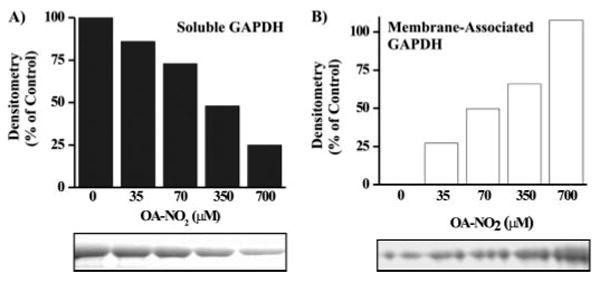
Control and OA-NO2-treated GAPDH were incubated with liposomes for 30 min at 25 °C. Liposomes were sedimented by ultracentrifugation, and the translocation of soluble GAPDH in the supernatant (A) to a liposome membrane-associated state (B) was determined as a function of OA-NO2 treatment concentration by SDS-PAGE.
To probe whether nitroalkylation occurs in vivo, we analyzed proteins, focusing on GAPDH, from red blood cells obtained from healthy humans. After separating cytosolic and membrane-associated proteins by electrophoresis under nonreducing conditions, the 36-kDa protein band (which includes GAPDH) was excised and digested in-gel with trypsin. Peptides were eluted and analyzed by nanospray LC-MS/MS (Fig. 8, A and B). The MS/MS spectrum of the human homolog of rabbit GAPDH peptide 19, encompassing the catalytically active Cys-149, showed adduction by OA-NO2 (Fig. 8A). The mass of the b fragments, particularly fragment b2, showed that Cys-149 was modified by OA-NO2 in vivo. Another His-containing GAPDH peptide was also OA-NO2-adducted (Fig. 8B). The increased mass of the peptide by 327 Da and the mass of fragment b3 identified His-303 as the modified, adducted residue. This peptide was found both in cytosolic (not shown) and membrane fractions (Fig. 8B) of red cells, affirming that GAPDH is modified by OA-NO2 in vivo (Fig. 8).
FIGURE 8. Mass spectrometric detection of endogenous nitroalkylated GAPDH in red blood cells obtained from healthy humans.
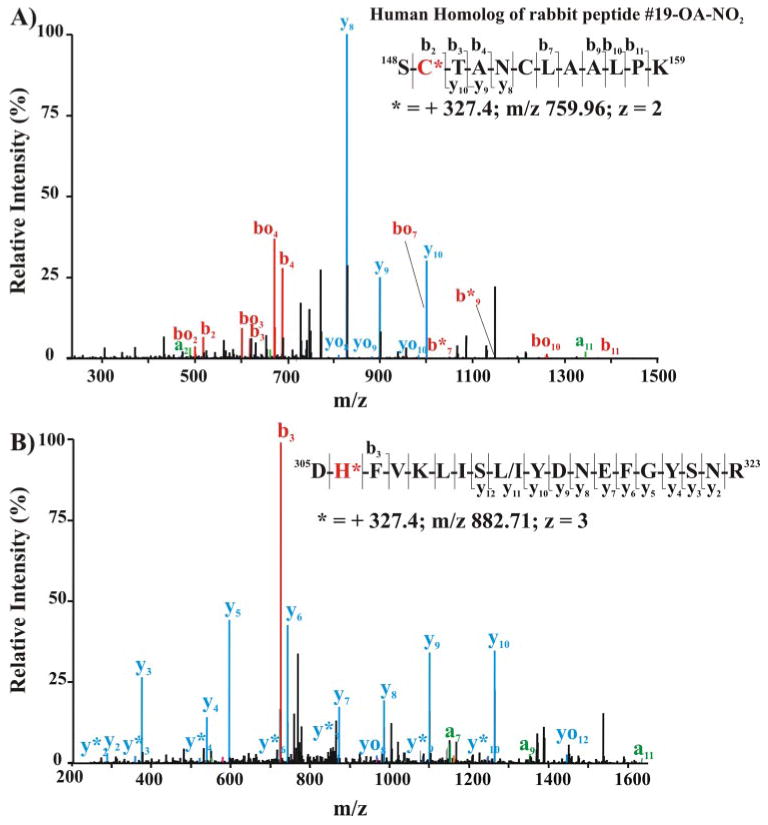
The cytosolic (A) and membrane-associated (B) protein fractions from lysed red cells were separated by SDS-PAGE using nonreducing, denaturing conditions (4–15% gradient gel). The 36-kDa Coomassie dye-binding band corresponding to the Rf of GAPDH was excised and digested in-gel with sequencing grade trypsin. Peptides were extracted, separated, and analyzed by LC nanospray linear ion trap MS/MS (LTQ; Thermo Electron Corp.). A, MS/MS of the doubly charged ion at m/z 759.96 corresponding to the human homolog of rabbit nitroalkylated peptide 19. Inset, amino acid sequence indicating major C- and N-terminal fragment ions detected by full-scan MS/MS. B, MS/MS spectrum of triply charged human GAPDH ion at m/z 882.71 corresponding to the human peptide sequence 305–323. Inset, amino acid sequence indicating major C- and N-terminal fragment ions detected by full-scan MS/MS.
Finally, because of (a) the reversibility of in vitro protein nitroalkylation reactions by GSH, (b) the identification of endogenous OA-NO2-adducted GAPDH in human red cells, and (c) the fact that GSH is present in high concentrations in red cells (∼5 mm), the potential presence of nitroalkene-adducted GSH in red cells was examined (Fig. 9). Multiple reaction monitoring (MRM) analysis of partially purified cytosolic fractions of red cells revealed endogenous species that co-chromatographed with synthetic GS-[13C18]OA-NO2 (Fig. 9A) and GS-[13C18]LNO2 (Fig. 9B) and displayed identical mass transitions (Fig. 9, A and B, red versus black traces). The identity of endogenous OA-NO2- and LNO2-adducted GSH was further confirmed by EPI analysis and comparison of fragmentation patterns with those obtained from the EPI analysis of synthetic GS-OA-NO2 (Fig. 10, A and B). Fragmentation of both the synthetic standard (Fig. 10A) and the endogenous molecules (Fig. 10B) gave the corresponding b2 (m/z 560.3), y2 (m/z 506.3), y2-HNO2 (m/z 506.3) and Cys-immonium ion (C1), all adducted with OA-NO2. To further characterize the GS-OA-NO2 adduct, the y2 fragment of the synthetic adduct was analyzed by MS/MS/MS (Fig. 10C). The CID of this product ion resulted in product ions, including those indicative of the loss of the NO2 group, supporting that the lipid backbone of the adduct was bonded to the cysteinyl-glycine portion of the molecule and not to the γ-Glu. Moreover, CID of the y2 ion also induced the formation of the cysteinyl immonium ion (C1) adducted to the OA-NO2 (m/z 403.3). The loss of the NO2 group from this ion (−HNO2; m/z 356.3) further confirmed that the nitroalkene was attached to the cysteinyl residue of GSH. Based on this fragmentation pattern, it is not possible to determine the location of the NO2 group on the fatty acid backbone and thus to which carbon the GS is adducted.
FIGURE 9. Mass spectrometric detection of endogenous nitroalkylated GSH in healthy human red blood cells.
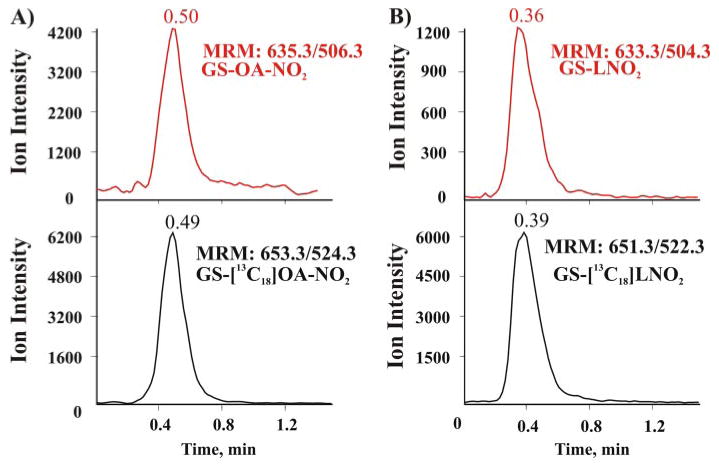
Red cells were lysed, and membranes sedimented by centrifugation and the soluble fraction were supplemented with the internal standards GS-[13C18] OA-NO2 and GS-[13C18 (LNO2) before purification by reverse phase chromatography using a preparative C18 column. The eluted fraction was concentrated and analyzed by LC-ESI-MS/MS (Q-Trap 4000; Applied Biosystem, Foster City, CA). A, mass spectra of the eluent produced by monitoring the MRM transition 635.3/506.3 (corresponding to the generation of the y2-adducted fragment) for endogenous GS-OA-NO2 (red trace) and 653.3/524.3 for the added internal standard GS-[13C18] OA-NO2 (MRM 653.3/524.3 (black trace). B, similar to A, but monitoring the transitions for GS-LNO2. The injection peak in Fig. 9 occurred at 0.13 min.
FIGURE 10. Mass spectrometric characterization of nitroalkylated GSH.
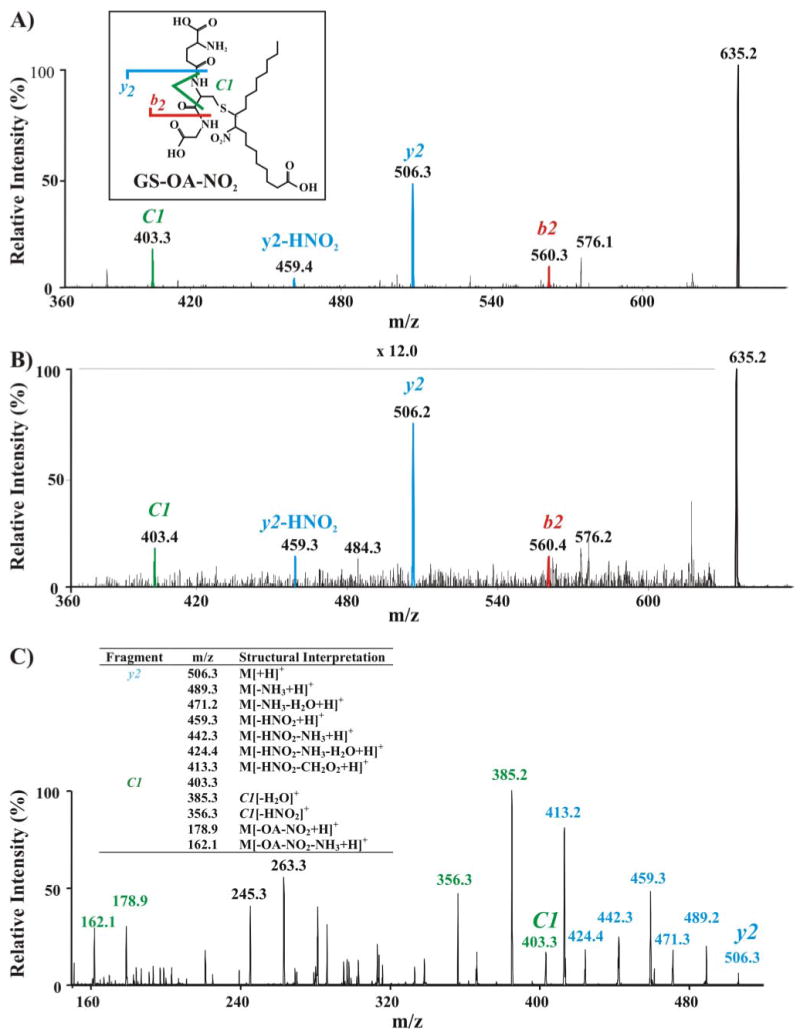
A, EPI analysis (e.g. MS/MS fragmentation pattern) of the synthetic standard GS-OA-NO2 showing major C- and N-terminal fragment ions detected by full-scan MS/MS (y and b fragments, respectively). Inset, scheme showing the structure and principal EPI fragments of GS-OA-NO2. B, EPI analysis of the endogenous RBC cytosolic GS-OA-NO2 adduct, displaying a fragmentation pattern identical to that of synthetic GS-OA-NO2. C, MS/MS/MS of the fragment ion y2 (m/z 506.3) from GS-OA-NO2 adduct (m/z = 635.2). Table, list and structural interpretation of fragment ions generated.
The fragmentation profile of the GS-OA-NO2 adduct strongly depends on the MS mode used to analyze the molecule. In the negative ion mode, CID of the GS-LNO2 adduct precursor ion yielded GSH and LNO2 as principal product ions (Fig. 1). No ions formed by the combination of GSH with the lipid were observed. In contrast, in the positive ion mode all of the principal product ions (y2, b2, and C1) maintained the lipid backbone of the molecule. In aggregate, these results reveal the existence of nitroalkylated GSH in vivo. GS-OA-NO2 and GS-LNO2 were present at ∼3.3 and ∼1.3 nm, respectively, in healthy human red cells.
Discussion
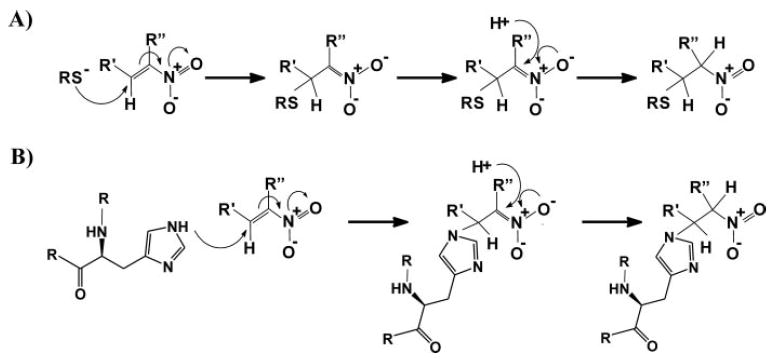
Chromatographic and mass spectrometric analyses reveal that nitrated unsaturated fatty acids are potent electrophiles that mediate reversible nitroalkylation reactions with both GSH and the Cys and His residues of proteins. This occurs both in vitro and in vivo (Scheme 1) and is viewed to transduce redox- and NO-dependent cell signaling by a covalent, thiol-reversible post-translational modification that regulates protein structure, function, and subcellular distribution.
SCHEME 1. Michael addition reaction of fatty acid nitroalkene derivatives with (A) thiols and (B) amino groups.
GAPDH, a tetramer consisting of identical catalytically active subunits, is a key intermediary metabolism enzyme that reversibly catalyzes the oxidation and phosphorylation of GAP. Enzyme catalysis involves the formation of an hemithioacetal between GAP and the catalytic thiol (Cys-149), rendering GAPDH highly sensitive to inactivation by oxidants (25, 31, 32) and lipid peroxidation products (18, 33–35). Alkylation of Cys-149 by iodoacetate (36) or oxidation by nonphysiological peroxynitrite and hydrogen peroxide concentrations induce irreversible enzyme inhibition (18, 25, 37). Nitroalkenes are more potent inhibitors of GAPDH than H2O2, HNE, and peroxynitrite, and unlike these oxidants, nitroalkenes inactivate GAPDH within a physiological concentration range (Fig. 2B) (5, 22). Of significance, nitroalkene-induced GAPDH inhibition is reversible by low thiol concentrations, a property that is also not observed with the aforementioned oxidative by-products. GAPDH not only is inhibited by active site thiol-directed oxidation reactions but also by the modification of other nucleophilic amino acids (i.e. His, Lys, and noncatalytic Cys residues) (18). Thus, nitroalkene-induced inhibition of GAPDH could also be due to nitroalkylation of Cys-153, Cys-244, His-108, His-134, and His-327. Although GAPDH inactivation by nitroalkenes was paralleled by a loss of titratable thiols, the kinetics of enzyme inactivation followed a biphasic, sigmoidal curve (Fig. 2). This supports a differential impact of the multiple documented sites of nitroalkene adduction on enzyme activity, as shown previously for HNE-induced GAPDH inhibition (18).
Recent evidence reveals that GAPDH is a multifunctional protein that displays cell signaling activities beyond its conventionally viewed role as an intermediary metabolism enzyme. This includes an influence on DNA repair, transcriptional regulation, membrane fusion, tubulin bundling, and apoptotic signaling (38). Under physiologic conditions, a significant extent of red cell GAPDH is bound to the membrane of intact cells and is catalytically inactive (39, 40). The regulatory mechanisms whereby GAPDH fulfills its nonglycolytic functions and is targeted to different specific intracellular loci are unknown (41), but it is proposed that the functional diversity and differential subcellular distribution of GAPDH are mediated by post-translational modifications and protein-protein and protein-nucleic acid interactions (41). We reveal here that nitroalkylation of GAPDH not only directs translocation to the membrane but also inhibits catalytic activity. Thus, nitroalkylation of GAPDH provides a mechanism that can explain changes in the subcellular distribution and functional diversity of GAPDH. Future studies should reveal interesting patterns of subcellular distribution and trafficking of not only GAPDH but also other reversibly nitroalkylated proteins.
GAPDH is a target of ˙NO, which induces enzyme inhibition (42–46) and plays an undefined role in promoting the pro-apoptotic translocation of ˙NO-modified GAPDH into the nucleus and its participation in nuclear events (21). In addition to the present demonstration of nitroalkylation, Cys residues of GAPDH can be S-glutathionylated (47) and S-nitrosylated (21, 44, 45). By appreciating that (a) ˙NO-dependent mechanisms mediate fatty acid nitration (5, 48) and (b) nitrated fatty acids give spurious, false-positive reactions for S-nitrosothiols in assays qualitatively and quantitatively identifying this species (2), the contributions of protein nitroalkene derivatives to putative S-nitrosothiol adducts and downstream signaling reactions should be addressed in more detail.
In contrast to the pro-inflammatory reactions mediated by oxidized lipids in inflammatory diseases, particularly in atherosclerosis (11, 49, 50), nitrated fatty acids exert potent anti-inflammatory cell signaling actions in both vascular and non-vascular tissues (2, 5, 7–9, 51).6 These actions were initially attributed to a capacity to release ˙NO and to activate PPARs (2, 5, 9). The present observations reveal a new cell signaling reactivity of nitrated fatty acids, the capability to selectively induce reversible post-translational modification of proteins by nitroalkylation. Current data reveal that nitroalkenes induce pronounced effects on mitogen-activated protein kinase (MAPK) and c-Jun N-terminal kinase (JNK) cell signaling cascades for reasons only now becoming evident. For example, protein-tyrosine phosphatases contain an active site motif that includes an invariant Cys with a low pKa value (52), a property that promotes nucleophilic reactivity and susceptibility to nitroalkylation. Additionally, Cys residues critical for NFκB transcriptional regulation of inflammatory gene expression are potently influenced by nitroalkylation.6
There is a precedence for GSH-forming adducts with modified fatty acids that confer biological activity. The cysteinyl leukotrienes LTC4, LTD4, and LTE4 (GSH S-conjugates of leukotriene A4), recognized by the seven transmembrane-spanning G protein-coupled receptors CysLT1 and CysLT2 (53, 54), are potent pro-inflammatory lipid mediators. These oxidized fatty acid-GSH adducts are generated via the 5-lipoxygenase pathway and have been implicated in a variety of pathologic conditions (55). It is thus intriguing to speculate whether nitroalkylation of GSH induces signaling actions that parallel those already characterized for nitroalkenes (i.e. generally anti-inflammatory) and whether additional signaling events are mediated by further receptor-ligand interactions.
Nitroalkylation represents a new form of lipid-dependent protein modification, which presently includes co- or post-translational myristoylation, palmitoylation, and prenylation reactions (56). The extent and nature of protein nitroalkylation will be dependent on a number of factors summarized in Scheme 2. First, ˙NO-dependent oxidative inflammatory reactions, diet and possible enzymatic synthesis, will dictate endogenous levels of free and esterified nitroalkene derivatives (5, 48). Second, the regulated activation of lipases will mediate the hydrolytic release of fatty acid nitroalkene derivatives esterified to complex lipids, thus influencing net levels, anatomic distribution, aqueous versus hydrophobic partitioning, and reactivities of free nitroalkenes. Third, the local environment can lower the pKa value and increase the nucleophilicity of critical protein thiol and histidine residues, thus rendering greater reactivity with electrophilic nitroalkenes. Susceptibility to nitroalkylation will also be governed by steric factors and solvent accessibility. Fourth, protein nitroalkylation is thiol-reversible, indicating that the net redox status of cells and tissues will also govern the extent of biomolecule nitroalkylation. These aggregate micro-environmental and biochemical properties thus confer the following: (a) specificity to nitroalkylation of specific amino acids on particular proteins and (b) an ability of cells to regulate the extent and sites of protein adduction by nitroalkenes.
SCHEME 2. Nitroalkene-mediated post-translational modification of GAPDH and other proteins will influence protein structure, function, and subcellular distribution in a GSH-reversible manner.
The modified sites of the protein were randomly assigned.
In summary, we have described a new reactivity of endogenously present nitrated fatty acids. These recently detected derivatives mediate pluripotent signaling activities by acting as a high affinity ligand for PPARγ and PPARα, activating protein kinase signaling cascades, and serving as a hydrophobically stabilized reserve for cGMP-dependent ˙NO signaling. We now show that nitroalkenes mediate the relatively specific, reversible post-translational modification of proteins that serve to transduce redox- and ˙NO-dependent cell signaling by regulating protein function and distribution.
Supplementary Material
Acknowledgments
We thank Mutende J. Sikuyayenga, Marshall Long, and Phil Chumley (University of Alabama at Birmingham) for their helpful technical assistance.
Footnotes
This work was supported in part by National Institutes of Health Grants HL58115 and HL64937 (to B. A. F.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–5 and Table 1.
The abbreviations used are: ˙NO, nitric oxide; ˙NO2, nitrogen dioxide; LNO2, nitro-linoleic acid, 9-, 10-, 12-, and 13-nitro-9,12-octadecadienoic acids; OA-NO2 nitro-oleic acid, 9- and 10-nitro-9-cis-octadecaenoic acids; ACN, acetonitrile; ONOO−, peroxynitrite; GAPDH glyceraldehyde-3-phosphate dehydrogenase; GAP, dl-glyceraldehyde 3-phosphate; RBCs, red blood cells; GS-LNO2, GS-nitro-linoleic acid adduct; GS-OA-NO2, GS-nitro-oleic acid adduct; LC, liquid chromatography; HPLC, high pressure liquid chromatography; MS, mass spectrometry; m/z, mass/charge; ESI, electrospray ionization; MRM, multiple reaction monitoring; EPI, enhanced product information; MALDI-TOF, matrix-assisted laser desorption ionization-time of flight; CID, collision induced dissociation; PSD, post-source decay; DTT, dithiothreitol; DTPA, diethylenetriaminepentaacetic acid; PPAR, proliferator-activated receptor; HNE, 4-hydroxy-2-nonenal.
T. Cui, F. J. Schopfer, J. Zhang, K. Chen, T. Ichikawa, P. R. Baker, C. Batthyany, B. K. Chako, X. Feng, R. P. Patel, A. Agarwal, B. A. Freeman, and Y. E. Chen, submitted for publication.
References
- 1.Rubbo H, Darley-Usmar V, Freeman BA. Chem Res Toxicol. 1996;9:809–820. doi: 10.1021/tx960037q. [DOI] [PubMed] [Google Scholar]
- 2.Schopfer FJ, Baker PR, Giles G, Chumley P, Batthyany C, Crawford J, Patel RP, Hogg N, Branchaud BP, Lancaster JR, Jr, Freeman BA. J Biol Chem. 2005;280:19289–19297. doi: 10.1074/jbc.M414689200. [DOI] [PubMed] [Google Scholar]
- 3.Napolitano A, Camera E, Picardo M, d'Ischia M. J Org Chem. 2000;65:4853–4860. doi: 10.1021/jo000090q. [DOI] [PubMed] [Google Scholar]
- 4.O'Donnell VB. Antioxid Redox Signal. 2003;5:195–203. doi: 10.1089/152308603764816550. [DOI] [PubMed] [Google Scholar]
- 5.Baker PR, Lin Y, Schopfer FJ, Woodcock SR, Groeger AL, Batthyany C, Sweeney S, Long MH, Iles KE, Baker LM, Branchaud BP, Chen YE, Freeman BA. J Biol Chem. 2005;280:42464–42475. doi: 10.1074/jbc.M504212200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gladwin MT, Schechter AN, Kim-Shapiro DB, Patel RP, Hogg N, Shiva S, Cannon RO, III, Kelm M, Wink DA, Graham Espey M, Oldfield EH, Pluta RM, Freeman BA, Lancaster JR, Jr, Feelisch M, Lundberg JO. Nat Chem Biol. 2005;1:308–314. doi: 10.1038/nchembio1105-308. [DOI] [PubMed] [Google Scholar]
- 7.Coles B, Bloodsworth A, Clark SR, Lewis MJ, Cross AR, Freeman BA, O'Donnell VB. Circ Res. 2002;91:375–381. doi: 10.1161/01.res.0000032114.68919.ef. [DOI] [PubMed] [Google Scholar]
- 8.Coles B, Bloodsworth A, Eiserich JP, Coffey MJ, McLoughlin RM, Giddings JC, Lewis MJ, Haslam RJ, Freeman BA, O'Donnell VB. J Biol Chem. 2002;277:5832–5840. doi: 10.1074/jbc.M105209200. [DOI] [PubMed] [Google Scholar]
- 9.Schopfer FJ, Lin Y, Baker PR, Cui T, Garcia-Barrio M, Zhang J, Chen K, Chen YE, Freeman BA. Proc Natl Acad Sci U S A. 2005;102:2340–2345. doi: 10.1073/pnas.0408384102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ono N. In: The Nitro Group in Organic Synthesis. Feuer H, editor. John Wiley & Sons, Inc.; New York: 2002. pp. 70–119. [Google Scholar]
- 11.Ceaser EK, Moellering DR, Shiva S, Ramachandran A, Landar A, Venkartraman A, Crawford J, Patel R, Dickinson DA, Ulasova E, Ji S, Darley-Usmar VM. Biochem Soc Trans. 2004;32:151–155. doi: 10.1042/bst0320151. [DOI] [PubMed] [Google Scholar]
- 12.Esterbauer H, Zollner H, Scholz N. Z Naturforsch Sect C J Biosci. 1975;30:466–473. doi: 10.1515/znc-1975-7-808. [DOI] [PubMed] [Google Scholar]
- 13.Uchida K, Stadtman ER. Proc Natl Acad Sci U S A. 1992;89:4544–4548. doi: 10.1073/pnas.89.10.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Szweda LI, Uchida K, Tsai L, Stadtman ER. J Biol Chem. 1993;268:3342–3347. [PubMed] [Google Scholar]
- 15.Crabb JW, O'Neil J, Miyagi M, West K, Hoff HF. Protein Sci. 2002;11:831–840. doi: 10.1110/ps.4400102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levonen AL, Landar A, Ramachandran A, Ceaser EK, Dickinson DA, Zanoni G, Morrow JD, Darley-Usmar VM. Biochem J. 2004;378:373–382. doi: 10.1042/BJ20031049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harris I, Meriwether BP, Park JH. Nature. 1963;198:154–157. doi: 10.1038/198154a0. [DOI] [PubMed] [Google Scholar]
- 18.Ishii T, Tatsuda E, Kumazawa S, Nakayama T, Uchida K. Biochemistry. 2003;42:3474–3480. doi: 10.1021/bi027172o. [DOI] [PubMed] [Google Scholar]
- 19.Uchida K, Stadtman ER. J Biol Chem. 1993;268:6388–6393. [PubMed] [Google Scholar]
- 20.Chuang DM, Hough C, Senatorov VV. Annu Rev Pharmacol Toxicol. 2005;45:269–290. doi: 10.1146/annurev.pharmtox.45.120403.095902. [DOI] [PubMed] [Google Scholar]
- 21.Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD, Ferris CD, Hayward SD, Snyder SH, Sawa A. Nat Cell Biol. 2005;7:665–674. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- 22.Baker PR, Schopfer FJ, Sweeney S, Freeman BA. Proc Natl Acad Sci U S A. 2004;101:11577–11582. doi: 10.1073/pnas.0402587101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Botti H, Batthyany C, Trostchansky A, Radi R, Freeman BA, Rubbo H. Free Radic Biol Med. 2004;36:152–162. doi: 10.1016/j.freeradbiomed.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 24.Radi R, Beckman JS, Bush KM, Freeman BA. J Biol Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]
- 25.Souza JM, Radi R. Arch Biochem Biophys. 1998;360:187–194. doi: 10.1006/abbi.1998.0932. [DOI] [PubMed] [Google Scholar]
- 26.Spengler B. J Mass Spectrom. 1997;32:1019–1036. [Google Scholar]
- 27.Burford N, Eelman MD, Groom K. J Inorg Biochem. 2005;99:1992–1997. doi: 10.1016/j.jinorgbio.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 28.Cowan-Jacob SW, Kaufmann M, Anselmo AN, Stark W, Grutter MG. Acta Crystallogr Sect D Biol Crystallogr. 2003;59:2218–2227. doi: 10.1107/s0907444903020493. [DOI] [PubMed] [Google Scholar]
- 29.Piraud M, Vianey-Saban C, Petritis K, Elfakir C, Steghens JP, Morla A, Bouchu D. Rapid Commun Mass Spectrom. 2003;17:1297–1311. doi: 10.1002/rcm.1054. [DOI] [PubMed] [Google Scholar]
- 30.Little C, O'Brien PJ. Eur J Biochem. 1969;10:533–538. doi: 10.1111/j.1432-1033.1969.tb00721.x. [DOI] [PubMed] [Google Scholar]
- 31.Racker E, Krimsky I. Nature. 1952;169:1043–1045. doi: 10.1038/1691043a0. [DOI] [PubMed] [Google Scholar]
- 32.Sirover MA. Biochim Biophys Acta. 1999;1432:159–184. doi: 10.1016/s0167-4838(99)00119-3. [DOI] [PubMed] [Google Scholar]
- 33.Brodie AE, Reed DJ. Arch Biochem Biophys. 1990;276:212–218. doi: 10.1016/0003-9861(90)90028-w. [DOI] [PubMed] [Google Scholar]
- 34.Brodie AE, Reed DJ. Biochem Biophys Res Commun. 1987;148:120–125. doi: 10.1016/0006-291x(87)91084-9. [DOI] [PubMed] [Google Scholar]
- 35.Fukuda A, Osawa T, Hitomi K, Uchida K. Arch Biochem Biophys. 1996;333:419–426. doi: 10.1006/abbi.1996.0410. [DOI] [PubMed] [Google Scholar]
- 36.Harris JL, Walters M. In: The Enzymes. Boyer PD, editor. Academic Press; New York: 1976. [Google Scholar]
- 37.Hyslop PA, Hinshaw DB, Halsey WA, Jr, Schraufstatter IU, Sauerheber RD, Spragg RG, Jackson JH, Cochrane CG. J Biol Chem. 1988;263:1665–1675. [PubMed] [Google Scholar]
- 38.Kim JW, Dang CV. Trends Biochem Sci. 2005;30:142–150. doi: 10.1016/j.tibs.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 39.Kliman HJ, Steck TL. J Biol Chem. 1980;255:6314–6321. [PubMed] [Google Scholar]
- 40.Tsai IH, Murthy SN, Steck TL. J Biol Chem. 1982;257:1438–1442. [PubMed] [Google Scholar]
- 41.Mazzola JL, Sirover MA. Biochim Biophys Acta. 2003;1622:50–56. doi: 10.1016/s0304-4165(03)00117-x. [DOI] [PubMed] [Google Scholar]
- 42.McDonald LJ, Moss J. Proc Natl Acad Sci U S A. 1993;90:6238–6241. doi: 10.1073/pnas.90.13.6238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mohr S, Stamler JS, Brune B. FEBS Lett. 1994;348:223–227. doi: 10.1016/0014-5793(94)00596-6. [DOI] [PubMed] [Google Scholar]
- 44.Mohr S, Stamler JS, Brune B. J Biol Chem. 1996;271:4209–4214. doi: 10.1074/jbc.271.8.4209. [DOI] [PubMed] [Google Scholar]
- 45.Molina y Vedia L, McDonald B, Reep B, Brune B, Di Silvio M, Billiar TR, Lapetina EG. J Biol Chem. 1992;267:24929–24932. [PubMed] [Google Scholar]
- 46.Stamler JS. Cell. 1994;78:931–936. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 47.Ravichandran V, Seres T, Moriguchi T, Thomas JA, Johnston RB., Jr J Biol Chem. 1994;269:25010–25015. [PubMed] [Google Scholar]
- 48.O'Donnell VB, Eiserich JP, Chumley PH, Jablonsky MJ, Krishna NR, Kirk M, Barnes S, Darley-Usmar VM, Freeman BA. Chem Res Toxicol. 1999;12:83–92. doi: 10.1021/tx980207u. [DOI] [PubMed] [Google Scholar]
- 49.Berliner JA, Heinecke JW. Free Radic Biol Med. 1996;20:707–727. doi: 10.1016/0891-5849(95)02173-6. [DOI] [PubMed] [Google Scholar]
- 50.Steinberg D, Parthasarathy S, Carew TE, Khoo JC, Witztum JL. N Engl J Med. 1989;320:915–924. doi: 10.1056/NEJM198904063201407. [DOI] [PubMed] [Google Scholar]
- 51.Lim DG, Sweeney S, Bloodsworth A, White CR, Chumley PH, Krishna NR, Schopfer F, O'Donnell VB, Eiserich JP, Freeman BA. Proc Natl Acad Sci U S A. 2002;99:15941–15946. doi: 10.1073/pnas.232409599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barford D, Jia Z, Tonks NK. Nat Struct Biol. 1995;2:1043–1053. doi: 10.1038/nsb1295-1043. [DOI] [PubMed] [Google Scholar]
- 53.Heise CE, O'Dowd BF, Figueroa DJ, Sawyer N, Nguyen T, Im DS, Stocco R, Bellefeuille JN, Abramovitz M, Cheng R, Williams DL, Jr, Zeng Z, Liu Q, Ma L, Clements MK, Coulombe N, Liu Y, Austin CP, George SR, O'Neill GP, Metters KM, Lynch KR, Evans JF. J Biol Chem. 2000;275:30531–30536. doi: 10.1074/jbc.M003490200. [DOI] [PubMed] [Google Scholar]
- 54.Lynch KR, O'Neill GP, Liu Q, Im DS, Sawyer N, Metters KM, Coulombe N, Abramovitz M, Figueroa DJ, Zeng Z, Connolly BM, Bai C, Austin CP, Chateauneuf A, Stocco R, Greig GM, Kargman S, Hooks SB, Hosfield E, Williams DL, Jr, Ford-Hutchinson AW, Caskey CT, Evans JF. Nature. 1999;399:789–793. doi: 10.1038/21658. [DOI] [PubMed] [Google Scholar]
- 55.Lewis RA, Austen KF, Soberman RJ. N Engl J Med. 1990;323:645–655. doi: 10.1056/NEJM199009063231006. [DOI] [PubMed] [Google Scholar]
- 56.Smotrys JE, Linder ME. Annu Rev Biochem. 2004;73:559–587. doi: 10.1146/annurev.biochem.73.011303.073954. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.