

Abstract
Nitroalkene derivatives of linoleic acid (LNO2) and oleic acid (OA-NO2) are present; however, their biological functions remain to be fully defined. Herein, we report that LNO2 and OA-NO2 inhibit lipopolysaccharide-induced secretion of proinflammatory cytokines in macrophages independent of nitric oxide formation, peroxisome proliferator-activated receptor-γ activation, or induction of heme oxygenase-1 expression. The electrophilic nature of fatty acid nitroalkene derivatives resulted in alkylation of recombinant NF-κB p65 protein in vitro and a similar reaction with p65 in intact macrophages. The nitroalkylation of p65 by fatty acid nitroalkene derivatives inhibited DNA binding activity and repressed NF-κB-dependent target gene expression. Moreover, nitroalkenes inhibited endothelial tumor necrosis factor-α-induced vascular cell adhesion molecule 1 expression and monocyte rolling and adhesion. These observations indicate that nitroalkenes such as LNO2 and OA-NO2, derived from reactions of unsaturated fatty acids and oxides of nitrogen, are a class of endogenous anti-inflammatory mediators.
Reactive oxygen species and NO-derived oxidizing, nitrosating and nitrating products mediate diverse cell signaling and pathologic processes in cardiovascular, pulmonary, and neurodegenerative diseases (1, 2). These reactive inflammatory mediators chemically modify carbohydrates, DNA bases, amino acids, and unsaturated fatty acids to form oxidized, nitrosated and nitrated derivatives. For example, accumulation of inflammatory-induced protein tyrosine nitration products represents a shift from the physiological signal-transducing actions of NO to an oxidative, nitrative, and potentially pathogenic pathway (1).
Recently, it has been reported that nitration products of unsaturated fatty acids (nitroalkenes) are formed via NO-dependent oxidative reactions (3–5). These derivatives were initially viewed to be, like nitrotyrosine, a “footprint” of NO-dependent redox reactions (3, 6). More recently, we have observed that nitrolinoleate (LNO2)7 mediates pluripotent cell signaling actions, since it induces relaxation of phenylephrine-preconstricted rat aortic rings, inhibits thrombin-induced Ca2+ elevations and aggregation of human platelets, and attenuates human neutrophil superoxide generation, degranulation, and integrin expression. These cell responses are mediated by both cGMP- and cAMP-dependent and -independent mechanisms (7–9).
LNO2 positional isomers, including 9-, 10-, 12-, and 13-nitro-9,12-cis-octadecadienoic acids, have been identified as free acids in human plasma and red blood cells and as esterified components of plasma lipoproteins and red blood cell membranes (10). In addition, plasma and red cell free and esterified nitrooleate (OA-NO2, isomers 9- and 10-nitro-9-cis-octadecenoic acid) was also identified in healthy human blood (11).
Current knowledge indicates that enzymatically oxidized unsaturated fatty acid-derived products, such as prostaglandins, thromboxanes, leukotrienes, epoxyeicosatrienoic acids, hydroxyeicosatetraenoic acids, lipoxins, and resolvins serve as lipid mediators or autacoids. These signaling mediators act within a local microenvironment to orchestrate both physiological and pathological events, including platelet aggregation, constriction of vascular smooth muscle, neonatal development, wound healing, and resolution of inflammation (12, 13). In this context, endogenous nitrated fatty acid derivatives, such as OA-NO2 and LNO2, represent an emerging class of NO and fatty acid-derived signaling molecules (14). At present, the cell signaling mechanisms and detailed structure-function relationships of this family of fatty acid derivatives are an object of interest. Recently, nitrated unsaturated fatty acids were shown to be potent electrophiles that mediate reversible nitroalkylation reactions with both glutathione and the Cys and His residues of proteins. This occurs both in vitro and in vivo and is viewed to transduce redox- and NO-dependent cell signaling by inducing a covalent, thiol-reversible post-translational modification that regulates protein structure, function, and subcellular distribution (15).
Herein, we report the effects of LNO2 and OA-NO2 on inflammatory responses in vascular cells, including monocytes/macrophages and endothelial cells. These data indicate that the nitroalkene derivatives of linoleic acid and oleic acid (LNO2 and OA-NO2) are a novel class of endogenous, electrophilic mediators in the vasculature that can exert adaptive anti-inflammatory signaling reactions via the post-translational modification of transcriptional regulatory proteins.
Experimental Procedures
Materials
LNO2 and OA-NO2 were synthesized, purified, and quantitated as previously described (10, 11). Briefly, OA-NO2 and LNO2 were synthesized using a nitrosenylation reaction. OA-NO2 and LNO2 were purified by preparative TLC developed twice using silica HF plates and a solvent system consisting of hexane/ether/acetic acid (70:30:1, v/v/v). The regions corresponding to OA-NO2 and LNO2 were scraped and extracted. Stock concentrations of OA-NO2 and LNO2 were quantitated by chemiluminescent nitrogen analysis, using caffeine as a standard, and confirmed spectrophotometrically using an extinction coefficient (E268) of 8.22 mm−1 cm−1 for OA-NO2 in 100 mm phosphate buffer at pH 7.4. Lipopolysaccharide (LPS), phorbol 12-myristate 13-acetate (PMA), zinc protoporphryin-IX, 3,3-bis(aminoethyl)-1-hydroxy-2-oxo-1-triazene (DETA-NONOate), 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide (cPTIO), sodium nitrite (NaNO2), diethylenetriaminepentaacetate (DTPA), and Na2HPO4 were from Sigma. Rosiglitazone, 15-deoxy-Δ12,14-PGJ2, Wy14643, and GW501516 were from Cayman Chemical (Ann Arbor, MI). Streptavidin-coated plates were from Pierce. Recombinant NF-κB p65 protein and TransAMTM NF-κB p65 Chemi kit were from Active Motif (Carlsbad, CA).
Fatty Acid Biotinylation
The synthesis of biotinylated OA, LA, OA-NO2, and LNO2 was performed as described for the synthesis of biotinylated (15S)-hydroxyeicosatetraenoic acids (16). Briefly, 1-methylpiperidine (5.3 μl, 48 μmol) was added to a solution of either OA, LA, OA-NO2, or LNO2 (15 mg in 1.5 ml of methylene chloride). After cooling the mixture to −78 °C, isobutylchloroformate (6.2 μl, 48 μmol) was added. The reaction mixture was maintained at −78 °C for 45 min and then warmed to −20 °C. After 35 min, a heated (80 °C) solution of biotin hydrazide (48.6 mg, 188 μmol) in dimethylformamide (2 ml) was added. The mixture was immediately cooled and maintained at −20 °C for 40 min and then warmed to room temperature. After extraction with methylene chloride, the products were chromatographed and purified by preparative TLC developed twice using silica HF plates, using a solvent system consisting of methylene chloride/methanol (80/20, v/v). The regions corresponding to the biotinylated OA, LA, OA-NO2, and LNO2 were scraped and extracted. Stock concentrations of biotinylated OA, LA, OA-NO2, and LNO2 were quantitated by chemiluminescent nitrogen analysis using caffeine as a standard. The characterization of the biotinylated OA, LA, OA-NO2, and LNO2 was done by direct infusion into a ESI MS (ion trap) at both positive and negative mode or by ESI-liquid chromatography-MS/MS in the negative ion mode, using multiple reaction monitoring and an enhanced product information scan mode on a 4000 hybrid triple quadrupole/linear ion trap mass spectrometer (Q-Trap) (Applied Biosystems, Foster City, CA). The ESI HPLC analysis was performed by C-18 reverse phase chromatography using a MercuryMS chromatography system equipped with a Luna C18 column (Phenomenex, Belmont, CA) equilibrated in 70% A (aqueous formic acid (0.01%)) and 30% B (0.01% formic acid in acetonitrile). One minute after injection, a 2-min gradient was initiated to reach a composition of 60% A and then to 7% A over an additional 4 min, held for 1 min at 7% A, and then back to 70% A and re-equilibrated. The retention times (min) and specific multiple reaction monitorings used for each species were as follows: biotinylated LA 7.50, 519/476; biotinylated OA 7.94, 521/478; biotinylated LNO2 6.90, 564/517; biotinylated OA-NO2 7.30, 566/519.
Cell Culture
Bone marrow cells were isolated from 8–12-week-old heme oxygenase-1 (HO-1) knockout mice or wild-type mice as previously (17). Briefly, bone marrow macrophages were prepared by culturing isolated bone marrow cells in α-minimal essential medium (Invitrogen) containing 10% heat-inactivated fetal bovine serum (FBS) in the presence of 0.1 volume of culture supernatant from macrophage colony-stimulating factor-producing cells for 2 days. Human umbilical vein endothelial cells (HUVEC) were purchased from BioWhittaker (San Diego, CA). The cells were cultured in endothelial cell growth medium-2 (BioWhittaker), containing 5% fetal calf serum, human basic fibroblast growth factor, insulin-like growth factor, human epithelial growth factor, vascular endothelial growth factor, 50 μg/ml gentamicin, 50 ng/ml amphotericin-B, hydrocortisone, and ascorbic acid. Early passages (passages 3–5) of HUVEC were used for all experiments. THP-1 cells (a human monocyte cell line from ATCC, Manassas, VA; catalog number TIB-202™) were cultured in RPMI 1640 (ATCC, catalog number 30-2001) supplemented with 10% FBS. THP-1 monocytes were differentiated into macrophages with PMA (0.1 μm) for 5 days. RAW264.7 cells (a murine macrophage cell line from ATCC, catalog no. TIB-71™) were cultured in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% FBS. Human peripheral blood mononuclear cells were purchased from Cambrex Bio Science (East Rutherford, NJ; catalog number CC-2702) and cultured with RPMI 1640 supplemented with 10% FBS. The nonadherent cells were removed with PBS before treatment. CV-1 cells (African green monkey kidney fibroblast cell line from ATCC; catalog number CCL-70™) were cultured in Dulbecco's modified Eagle's medium/F-12 supplemented with 10% FBS.
Cytokine Assay
Macrophages in fresh 1% FBS or 1% delipidated FBS (Cocalico Biologicals; catalog number CBX4234)-containing culture medium were treated as indicated. Medium was collected from triplicate wells 18–20 h after treatment. The concentrations of human and mouse TNFα, IL-6, and MCP-1 released from cells into medium was measured by enzyme-linked immunosorbent assay (ELISA) kits using protocols supplied by the manufacturer (R&D Systems, Minneapolis, MN). The half-maximal concentration for LNO2 and OA-NO2 inhibition of LPS-induced cytokine synthesis (IC50) was calculated using software obtained from GraphPad Software, Inc.
Quantitative Real Time Reverse Transcription-PCR
Total RNA from cell pellets was extracted using RNeasy kits (Qiagen Inc., Valencia, CA), and reverse transcription reactions (Advantage RT for PCR kit; Clontech) were performed with 0.5–1 μg of DNase I (Qiagen)-treated RNA. Quantitative real time reverse transcription-PCRs were carried out using the LightCycler thermocycler and the SYBR green I kits (Roche Applied Science) according to the manufacturer's recommendations. Cycle numbers obtained at the log-linear phase of the reaction were plotted against a standard curve prepared with serially diluted control samples. Expression levels of target genes were normalized by concurrent measurement of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA levels.
Transfection and Reporter Gene Assay
CV-1 cells at ∼85% confluence in 24-well plates were transiently co-transfected using Lipofectamine 2000 (Invitrogen) with a plasmid containing the luciferase gene under the control of three tandem PPAR-response elements (3× PPRE TK-luciferase) in pGL3 basic vector (Promega, Madison, WI) and human PPARγ1 in pcDNA3.1 vector (Invitrogen), respectively. Green fluorescent protein expression plasmid was co-transfected as an internal control for transfection efficiency. Twenty-four hours after transfection, cells were cultured for 4 h in Opti-MEM I (Invitrogen) and then treated with various stimuli as indicated for an additional 16–20 h. RAW264.7 cells were transiently co-transfected with NF-κB-luciferase reporter (NF-κB-luc) (Stratagene, La Jolla, CA) and green fluorescent protein expression plasmids. Twenty-four hours after transfection, cells were pretreated with nitrated fatty acids or control fatty acids in 1% FBS Eagle's minimal essential medium for 16–20 h and then stimulated with LPS (1 μg/ml) for 6 h. Each transfection was performed in triplicate on at least 3 occasions. Reporter luciferase assay kits (Promega) were used to measure the luciferase activity of cells with a luminometer, according to the manufacturer's instructions (Victor II; PerkinElmer Life Sciences). Luciferase activity was normalized by green fluorescent protein units.
NF-κB Activity Assay in Vitro
Partially purified human recombinant p65 protein (470 units/ml) was incubated in phosphate buffer 100 mm containing 100 μm DTPA (pH 7.4) with different concentrations of LNO2, OA-NO2, LA, or OA for 1 h at room temperature. The remaining p65 activity was measured using the TransAM™ NF-κB p65 Chemi kit from Active Motif following the manufacturer's instructions. A modification was introduced into the protocol by avoiding any addition of dithiothreitol to sample and buffer preparations. The amount of p65 added to each well was 10 ng. The p65 activity was not affected by native fatty acids (LA and OA) or vehicle. Thus, the activity of p65 incubated with 1 μm of corresponding native fatty acids was considered maximal (100%).
Analysis of p65 Alkylation by Nitrated Fatty Acids
Partially purified human recombinant p65 protein (470 units/ml) was incubated in 100 mm phosphate buffer containing 100 μm DTPA (pH 7.4) with different concentrations of biotinylated LNO2, OA-NO2, LA, or OA for 1 h at room temperature. The reaction mixture (25 ng of p65) was added to streptavidin-coated plates that were previously blocked for 1 h with Tween Tris-buffered saline (TTBS: 0.5% Tween 20, 30 mm Tris, 150 mm NaCl) containing 3% albumin, incubated for 1 h at room temperature, and washed three times using TTBS containing 1% albumin. The plates were then incubated with anti-p65 in 1% albumin containing TTBS for 1 h at room temperature, washed three times, incubated with goat anti-rabbit IgG for 1 h at room temperature, and then washed four times. Color development was followed at 650 nm using tetramethyl benzidine as substrate.
Immunoprecipitation and Immunoblotting Analysis
Cells treated under different experimental conditions were washed quickly with ice-cold PBS containing 1 mm Na3VO4, frozen in liquid nitrogen, scraped off, and lysed in Nonidet P-40 lysis buffer (1% Nonidet P-40, 25 mm HEPES (pH 7.5), 50 mm NaCl, 50 mm NaF, 5 mm EDTA, 10 nm okadaic acid, 1 mm sodium orthoavanadate, 1 mm phenylmethylsulfonyl fluoride, and 10 μm aprotinin) for 10–15 min on ice. Insoluble material was removed by centrifugation at 14,000 × g for 20 min at 4 °C. Protein concentration was measured in the cleared supernatant via Coomassie dye binding (Bio-Rad). The cell lysates (200 μg) were precipitated overnight with 50 μl of UltraLink Immobilized NeutrAvidin Plus (Pierce), or 50 μl of a 50% slurry of protein G-Sepharose 4 Fast Flow (Sigma) was added to 1 ml of cell lysate with equal amounts of protein and incubated at 4 °C for 1 h with gentle shaking. The precleared lysates (200 μg) were incubated with anti-p65 with constant agitation at 4 °C overnight and then further incubated with protein G-Sepharose 4 Fast Flow for 1 h. These precipitates were washed four times with Nonidet P-40 lysis buffer. The whole cell lysates (20 μg) or precipitates were subjected to SDS-PAGE and electro-transferred onto Hybond-ECL nitrocellulose membrane. Immunoblotting was done using anti-p65 (sc-372), anti-vascular cell adhesion molecule-1 (anti-VCAM-1; sc-13160), anti-actin (sc-1616) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), and anti-HO-1 (SPA-895) (Stressgen Biotechnologies, Inc., San Diego, CA).
Analysis of THP-1 Monocyte Adhesion and Rolling on HUVEC
HUVEC were grown until 95% confluent in 48-well fibronectin-coated plates (for static adhesion analyses) or 25-mm dishes (for laminar flow analyses) and then incubated for 2 h with or without LNO2. Subsequently, HUVEC were activated with TNFα (2 ng/ml) for 16 h.
Static Adhesion of THP-1 Cells on HUVEC
Fluorescently labeled monocytes treated with Cell Tracker Green (Molecular Probes, Eugene, OR) were added at a leukocyte/endothelial ratio of 10:1. After a 30-min incubation at 37 °C, unbound monocytes were removed by washing (nonadherent fraction). Both fractions (adherent and nonadherent cells) were lysed using 1 m NaOH (20 min), and fluorescence was measured at excitation = 480 nm and emission = 515 nm in a PerkinElmer Life Sciences fluorescent plate reader.
Adhesion and Rolling of THP-1 Monocyte Cell on HUVEC in Laminar Flow
THP-1 cell adhesion to and rolling on HUVEC during flow were determined using a Glycotech flow chamber system (Rockville, MD) at flow rates corresponding to a wall shear rate of 0.5–1.5 dynes/cm2. Fluorescently labeled THP-1 cells were viewed on a Leica inverted fluorescence microscope equipped with differential interference contrast optics and a Hamamatsu Orca ER digital CCD camera (Compix Inc., Cranberry Township, PA). Real time images of each field were captured at 30 frames/s for 5 min using the automated image capture feature of Simple PCI software (Compix). Resulting time lapse images were analyzed by the motion tracking and analysis feature of Simple PCI software to calculate rolling velocities and the number of firmly bound cells. Cells not moving for 5 s or more were considered firmly bound cells and were counted. The rolling velocities for THP-1 cells were calculated using Simple PCI software.
Statistics
Values are expressed as mean ± S.D. throughout. The data were analyzed using analysis of variance with the Newman-Keuls test unless specified. Values of p < 0.05 were considered to be statistically significant.
Results
LNO2 and OA-NO2 Inhibit LPS-induced Secretion of Proinflammatory Cytokines in Macrophages
To explore the physiological actions of LNO2 and OA-NO2, we investigated the impact of synthetic LNO2 and OA-NO2 (Scheme 1), identical to that detected in the healthy human circulation (10, 11), on inflammatory responses of monocytes/macrophages. The concentration-dependent effects of LNO2 and OA-NO2 on LPS-induced proinflammatory cytokine secretion by macrophages were first defined. LNO2 and OA-NO2 dose-dependently inhibited the LPS-induced secretion of proinflammatory cytokines, including IL-6, TNFα, and MCP-1, in THP1 and RAW264.7 macrophages in either lipid-rich serum (L-serum) or delipidated serum (D-serum). In THP-1 macrophages, the half-maximal concentration for LNO2 inhibition of LPS-induced proinflammatory cytokine synthesis (LNO2 IC50) was 1.2–1.9 μm (L-serum) and 1.2–1.8 μm (D-serum) for IL-6, 0.4–0.8 μm (L-serum) and 0.5–0.8 μm (D-serum) for TNFα, and 0.5– 0.6 μm (L-serum) and 0.5–0.7 μm (D-serum) for MCP-1 (Fig. 1A). The half-maximal concentration for OA-NO2 inhibition of LPS-induced proinflammatory cytokine synthesis (OA-NO2 IC50) was 1.1–1.7 μm (L-serum) and 1.0–1.9 μm (D-serum) for IL-6, 0.3–0.9 μm (L-serum) and 0.5–0.8 μm (D-serum) for TNFα, and 0.4–0.6 μm (L-serum) and 0.5–0.8 μm (D-serum) for MCP-1 (Fig. 1A). In RAW264.7 cells, the LNO2 IC50 was 1.1–1.7 μm (L-serum) and 1.3–1.8 μm (D-serum) for IL-6, 1.4–1.8 μm (L-serum) and 1.2–1.8 μm (D-serum) for TNFα, and 1.1–1.7 μm (L-serum) and 1.1–1.8 μm (D-serum) for MCP-1 (Fig. 1B). The OA-NO2 IC50 was 1.8 –2.5 μm (L-serum) and 1.9–2.4 μm (D-serum) for IL-6, 1.7–2.2 μm (L-serum) and 1.4–1.9 μm (D-serum) for TNFα, and 1.2–1.9 μm (L-serum) and 1.3–2.2 μm (D-serum) for MCP-1 (Fig. 1B). Similar responses were observed for the IC50 values of LNO2 and OA-NO2 in the inhibition of LPS-induced proinflammatory cytokine synthesis in primary cultures of human macrophages: LNO2 IC50, 0.5–0.7 μm (d-serum) for IL-6, 0.4–0.6 μm (D-serum) for TNFα, and 0.6–0.8 μm (D-serum) for MCP-1; OA-NO2 IC50, 0.7–0.8 μm (D-serum) for IL-6, 0.5–0.7 μm (D-serum) for TNFα, and 0.6–0.8 μm (D-serum) for MCP-1 (Fig. 1C). In addition, there were no statistically significant differences in the anti-inflammatory effects of these nitroalkenes in the presence or absence of the lipidic components of serum. The native fatty acid precursors of LNO2 and OA-NO2, LA or OA, also displayed no inhibitory effects toward LPS-induced proinflammatory cytokine secretion (Fig. 1). These results affirm that the anti-inflammatory actions of LNO2 and OA-NO2 are direct and not mediated by hydrophobic interactions or via reaction products generated by the interaction of nitroalkenes with the lipophilic components of serum or macrophage membranes. In some control experiments, it was observed that the native fatty acids LA and OA slightly inhibited LPS-induced proinflammatory cytokine synthesis (10–20% inhibition) at concentrations greater than 5 μm (not shown). The subsequent nitration of LA and OA added to cultured cells remains a subject of further investigation. Higher concentrations of LNO2 (∼5 μm) and OA-NO2 (∼5 μm) did not induce cytotoxic effects in our experimental conditions, as evidenced by an absence of alterations in cellular morphology and no detectable release of cellular lactate dehydrogenase (not shown). Collectively, our results indicate that LNO2 and OA-NO2 exert strong anti-inflammatory effects in macrophages. The close correspondence between IC50 values for suppressing LPS-induced cytokine expression and nitroalkene concentration suggests a common mechanism of action.
SCHEME 1. Structures of LNO2 and OA-NO2.
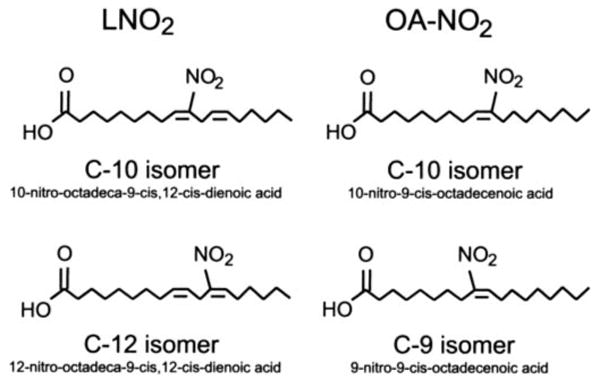
Structures of LNO2 and OA-NO2 used in this investigation are identical to those detected in the healthy human circulation (10, 11)
FIGURE 1. Nitrated fatty acids inhibit LPS-induced inflammatory cytokine secretion in macrophages.
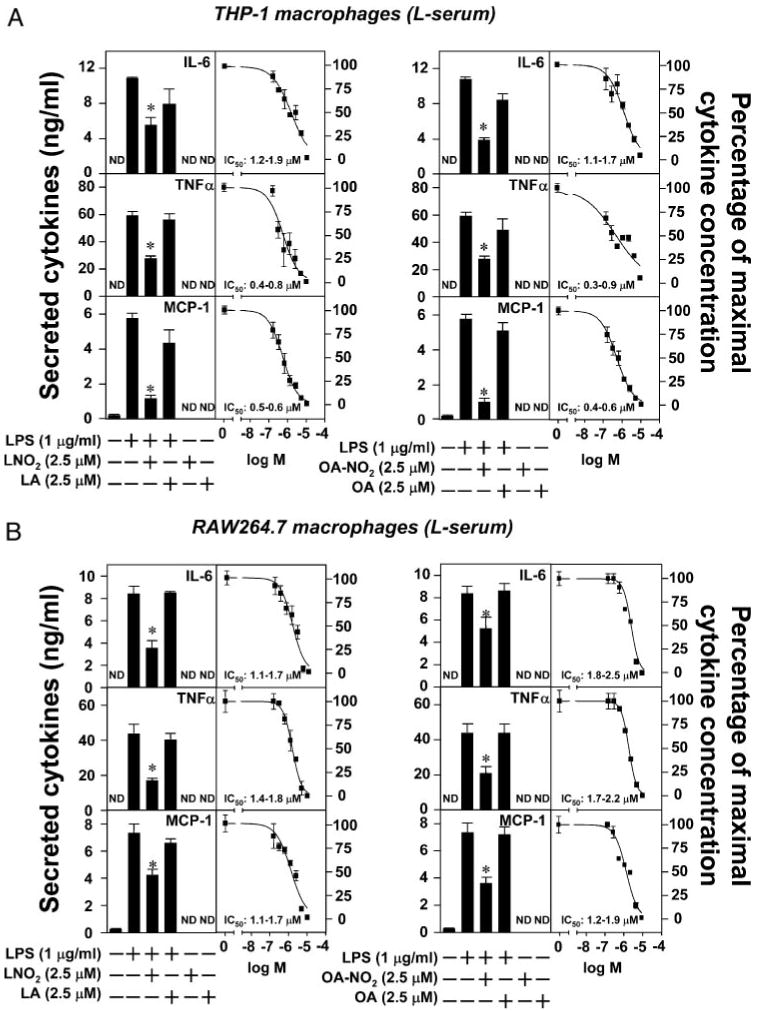
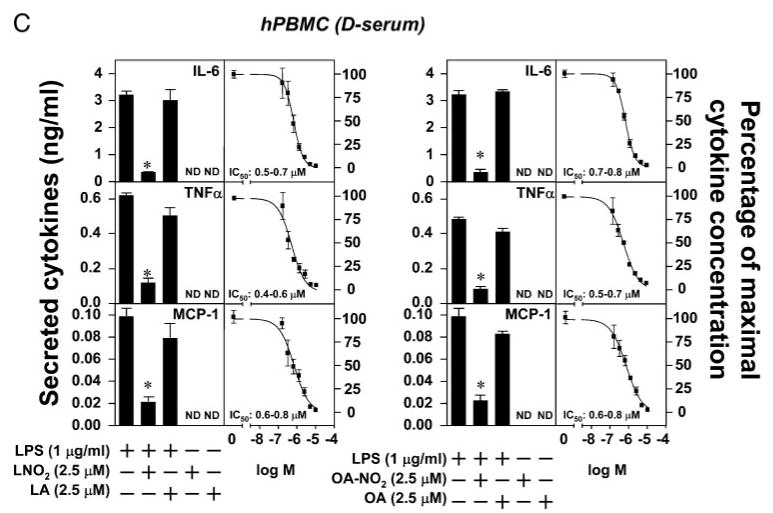
A–C, LNO2 and OA-NO2 inhibited LPS-induced inflammatory cytokine secretion in THP-1 and RAW264.7 macrophages and human peripheral blood mononuclear cells (hPBMC) in either lipid-rich or delipidated serum. Cells were stimulated as indicated overnight in fresh culture medium with 1% lipid-rich normal serum (L-serum) (A and B) or with 1% delipidated serum (D-serum). The secretion of proinflammatory cytokines was assessed by ELISA. Values are expressed as mean ± S.D. (n = 6). *, p < 0.05 versus LPS alone. ND, nondetectable.
The Inhibition of Proinflammatory Cytokine Secretion by LNO2 and OA-NO2 in Macrophages Is NO-independent
Nitrated fatty acids can undergo Nef-like decay reactions in aqueous milieu that yield NO (18). The partition coefficient of LNO2 is ∼1,500:1 (hydrophobic versus aqueous compartments). Thus, only 1 in 1,500 molecules of LNO2 are expected to decay to yield NO in serum lipoprotein-containing media and cell models, yielding at the most femtomolar concentrations of NO. OA-NO2 is relatively more stable in aqueous milieu and only minimally decays to release NO (not shown). Furthermore, it has been reported that very high concentrations of the NO donor DETA-NONOate (0.1–1.0 mm) are required to inhibit LPS-induced expression of TNFα and IL-1 in human alveolar macrophages (19). Similarly, a 0.2–2.0 mm concentration of the NO donor SNAP is needed to attenuate cytokine-induced expression of inducible NO synthase in the NR8383 rat alveolar macrophage cell line (19). In addition, a 200 –1000 μm concentration of the NO donor SNAP is needed to attenuate cytokine-induced expression of inducible NO synthase in the NR8383 rat alveolar macrophage cell line (20). Thus, it is unlikely that the anti-inflammatory effects of LNO2 and OA-NO2 are due to NO release from nitroalkenes being employed at a maximal concentration of ∼5 μm. Nevertheless, we investigated the potential involvement of NO or NO-derived species in LNO2- and OA-NO2-mediated anti-inflammatory signaling actions by comparing with NO donors, nitrite , and NO scavengers. As shown in Fig. 2, LPS-induced inflammatory cytokine production was not affected by the NO donor DETA-NONOate (100 μm) or nitrite (50 μm), the primary end product of NO oxidation, at concentrations far exceeding those observed in vivo (21). In addition, the NO scavenger cPTIO (100 μm) also had negligible effects on the inhibitory actions of LNO2 or OA-NO2 toward LPS-induced cytokine expression (Fig. 2). These results indicate that the cytokine-suppressive actions of LNO2 and OA-NO2 are NO-independent.
FIGURE 2. The inhibitory effect of LNO2 and OA-NO2 on LPS-induced proinflammatory cytokine secretion is NO-independent.
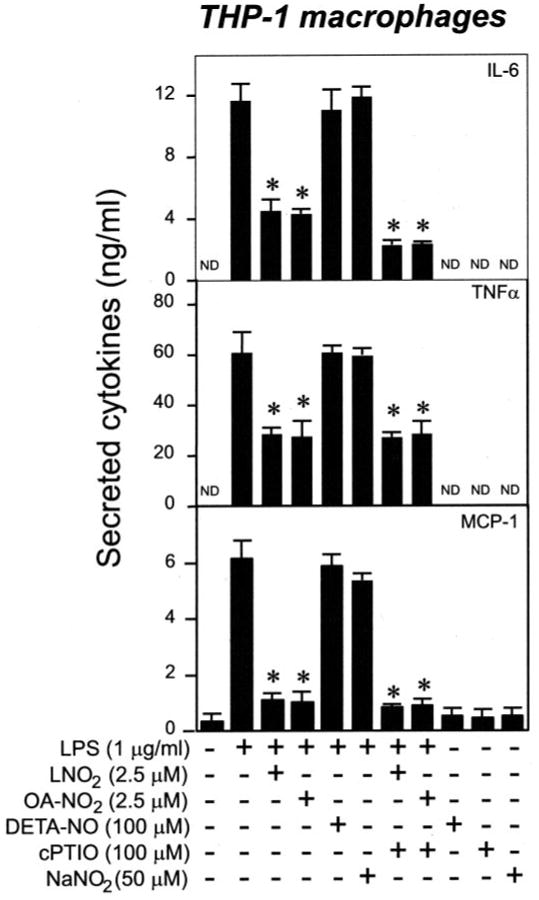
Cells were stimulated overnight as indicated in fresh culture medium with 1% lipid-rich normal serum. DETA-NO represents DETA-NONOate. The secretion of proinflammatory cytokines was assessed by ELISA. Values are expressed as mean ± S.D. (n = 6). *, p < 0.05 versus LPS alone. ND, nondetectable.
PPARγ Does Not Mediate the LNO2- and OA-NO2-mediated Inhibition of Proinflammatory Cytokine Secretion in Macrophages
We have demonstrated that LNO2 and OA-NO2 are potent PPARγ ligands (11, 22). This PPARγ ligand activity is specific for LNO2 or OA-NO2 and not mediated by LNO2 decay products, NO donors S-nitrosoglutathione (∼100 μm) or spermine-NONOate ((Z)-1-{N-[3-Aminopropyl]-N-[4-(3-aminopropylammonio)-butyl]-amino}-diazen-1-ium-1,2-diolate) (∼100 μm), LA, oxidized LA, or OA (11, 22). Because PPARγ plays a critical role in the regulation of inflammatory responses (23), the role of PPARγ in nitroalkene-induced inhibition of cytokine expression was investigated in macrophages. In RAW264.7 cells, the relative expression levels of PPARs were measured by quantitative real time reverse transcription-PCR. RAW264.7 cells expressed low levels of PPARγ1 and PPARδ and undetectable levels of PPARα and PPARγ2 (Fig. 3A). LPS-induced secretion of inflammatory cytokines was not inhibited by the high affinity PPARγ ligand rosiglitazone (2.5 μm, a concentration that maximally activated PPARγ), but was significantly inhibited by 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2; 2.5 μm, a concentration that weakly activates PPARγ). The action of 15d-PGJ2, a low abundance and low affinity PPARγ ligand, was comparable with inhibitory effects of nitroalkenes, whereas the selective PPARδ ligand (GW501516) and the PPARα ligand (Wy14643) showed no effect (Fig. 3B). Moreover, PMA-induced secretion of TNFα and MCP-1 was inhibited by LNO2 and OA-NO2, as well as 15d-PGJ2, but not by rosiglitazone (Fig. 3B), indicating that nitroalkenes inhibited the secretion of inflammatory cytokines in macrophages via PPARγ- independent pathways. These observations are also consistent with previous reports demonstrating that PPARγ-independent pathways contribute to anti-inflammatory effects of some PPARγ ligands (24).
FIGURE 3. Nitrated fatty acids inhibit secretion of proinflammatory cytokines in macrophages via PPARγ-independent pathways.
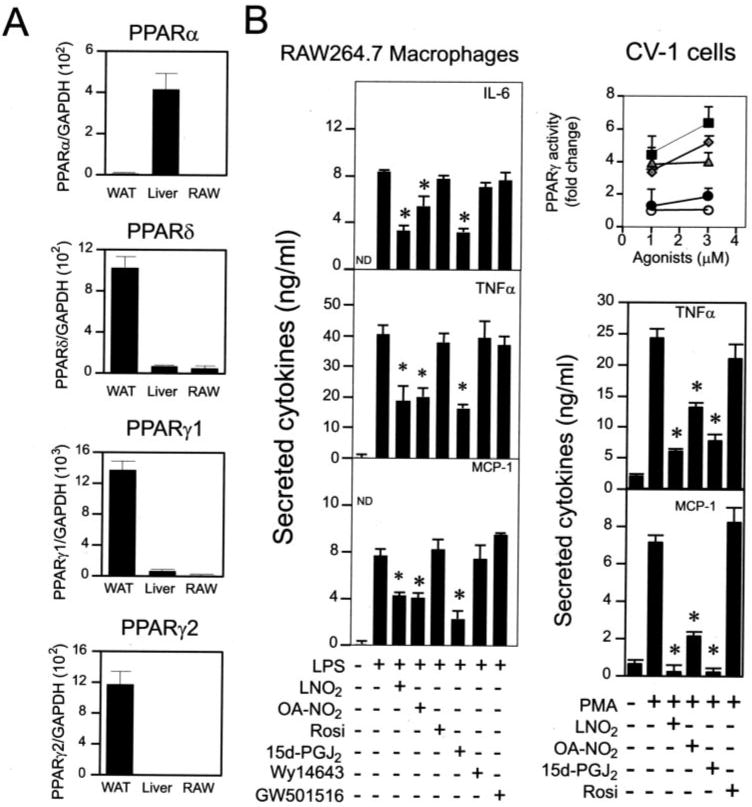
A, relative expression levels of PPARs in RAW264.7 cells. Expression of PPARs was analyzed by real time PCR. Expression of PPARs in WAT and liver was used as positive control. B, comparison of PPAR ligands and nitrated lipids on the induced secretion of proinflammatory cytokines in RAW264.7 cells. Inset, the activity of PPARγ induced by various PPARγ ligands was measured using a CV-1 reporter assay (■, OA-NO2; ◆, LNO2; ▲, rosiglitazone (Rosi); ●, 15d-PGJ2; ○, vehicle). Proinflammatory cytokine production was measured by ELISA. The concentrations used in B were: LPS, 1 μg/ml; PMA, 0.1 μm; OA-NO2, 2.5 μm; LNO2, 2.5 μm; Rosiglitazone, 3 μm; 15d-PGJ2, 3 μm; GW501516, 0.5 μm; Wy14643, 100 μm. Values are expressed as mean ± S.D. (n = 6). *, p < 0.05 versus LPS alone. WAT, white adipose tissue of C57BL/6J mice; Liver, liver of C57BL/6J mice.
HO-1 Is Not Essential for the Inhibitory Actions of LNO2 and OA-NO2 on Macrophage Cytokine Secretion
In inflammatory conditions, tissues undergo protective responses to maintain functionality and viability. A central event in the mediation of protective responses is expression of the inducible enzyme HO-1, which catalyzes the degradation of heme and yields biliverdin, carbon monoxide, and iron (25). The electrophilic derivative of arachidonic acid, 15d-PGJ2 induces HO-1 expression in various cell types, including macrophages, via mechanisms independent of PPARγ activation (26, 27). In murine J774 macrophages, the induction of HO-1 appears to be essential for the anti-inflammatory actions of 15d-PGJ2 (26). Accordingly, we examined whether HO-1 is a signaling intermediate of the cytokine-suppressive actions of nitroalkenes. LNO2 and OA-NO2 in a concentration- and time-dependent manner up-regulated HO-1 expression in RAW264.7 cells (Fig. 4A). The inhibitory action of nitroalkenes toward LPS-dependent responses was not reversible by inhibition of HO-1 catalytic activity by zinc protoporphryin-IX (Fig. 4B). To further confirm this observation, bone marrow-derived macrophages from HO-1 wild type (HO-1+/+) and knock-out (HO-1−/−) mice were utilized to further probe the mechanism of nitroalkene suppression of LPS-induced expression of IL-6, TNFα, and MCP-1. The null expression of HO-1 in HO-1−/− bone marrow macrophage was confirmed by Western blot (Fig. 4C). HO-1 deficiency did not influence the inhibitory effects of nitroalkenes toward LPS-induced secretion of cytokines in bone marrow macrophage (Fig. 4D), indicating that HO-1 does not transduce the suppression of LPS-induced cytokine expression by nitroalkenes.
FIGURE 4. HO-1 is not essential for nitrated fatty acid-mediated anti-inflammatory signaling.
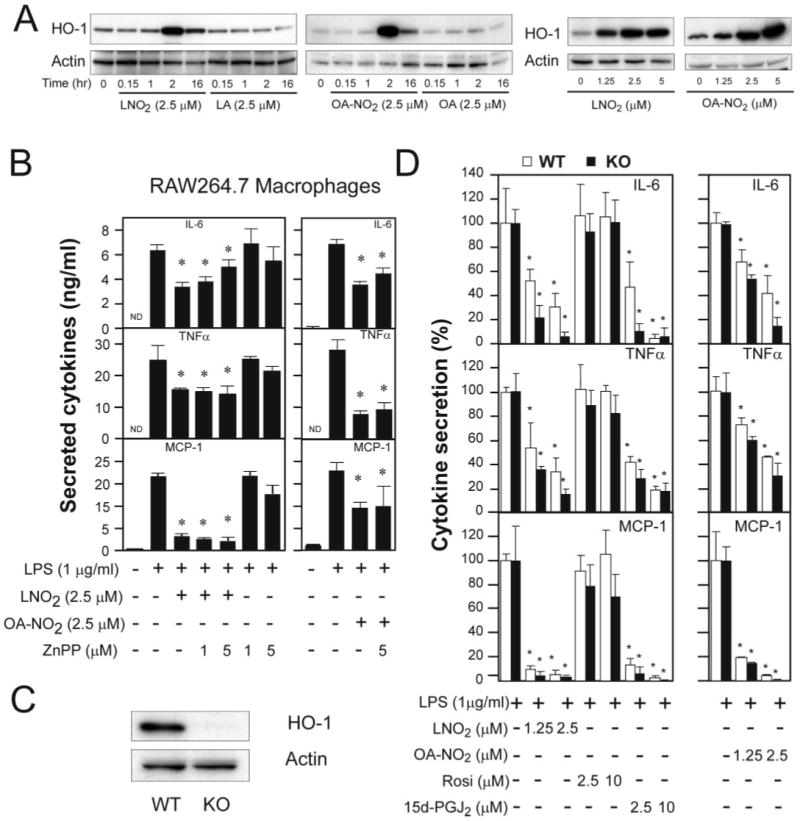
A, time course and dose responses of nitrated fatty acid-induced HO-1 expression in RAW264.7 cells. B, effect of HO-1 inhibitor on nitrated fatty acid-mediated anti-inflammatory effect. Values are expressed as mean ± S.D. (n = 4). *, p< 0.05 versus LPS alone. C, expression of HO-1 in bone marrow-derived macrophages of HO-1 wild-type (WT) and knock-out (KO) mice. D, the anti-inflammatory effects of LNO2 and OA-NO2 in macrophages differentiated from bone marrow cells of HO-1 WT and KO mice. Values are expressed as mean ± S.D. (n = 4). *, p < 0.05 versus LPS alone. HO-1 expression was examined by Western blot analysis, and proinflammatory cytokine production was measured by ELISA as described under “Experimental Procedures.”
LNO2 and OA-NO2 Inhibit NF-κB Signaling
The alkenyl nitro configuration of nitrated fatty acids indicates electrophilic reactivity of the β-carbon adjacent to the nitrobonded carbon, facilitating Michael addition reactions with biological nucleophiles such as protein Cys and His residues. This electrophilic property of nitroalkenes was first suggested by the biological detection of α,β-nitrohydroxy fatty acid derivatives (11). Also, synthetic nitroalkenes generated similar α,β-nitrohydroxy derivatives under aqueous conditions upon reaction with the low levels of hydroxide ion present at physiological pH (18). Appreciating that nitroalkenes are electrophilic, a candidate mechanism underlying LNO2 and OA-NO2 control of inflammatory cascades in macrophages can thus include the adduction (e.g. nitroalkylation) of key regulatory proteins. This nitroalkene-mediated post-translational modification of proteins, such as transcription factors, would be expected to alter protein activity, function, and anatomic distribution (15).
The transcription factor NF-κB, a dimer of members of the Rel family of proteins, plays a pivotal role in orchestrating inflammatory responses via the regulation of genes that encode proinflammatory cytokines. This includes IL-6, TNFα, and MCP-1 and adhesion molecules, including intercellular adhesion molecule-1 and VCAM-1 (28). In resting cells, NF-κB is typically sequestered in the cytoplasm by association with an inhibitory protein, IκB. In response to various stimuli, IκB kinase is activated and phosphorylates IκB on two serine residues, resulting in IκB ubiquitination, degradation by the proteasome, and facilitation of NF-κB migration into the nucleus to mediate the expression of inflammatory response genes (28). The most predominant form of NF-κB is a heterodimer of p65 and p50 subunits (28). The DNA binding activities of the p65 and p50 subunits of NF-κB are already appreciated to be inhibited by alkylation of a highly conserved Cys residue located in the DNA-binding domain (Cys62 in p50, Cys38 in p65). This alkylation reaction can be mediated by electrophiles, including 15d-PGJ2, sesquiterpene lactone, ethyl pyruvate, and N-ethyl maleimide, leading to suppression of the transactivation of NF-κB target genes (29 –32). These observations underscore a unique mechanism of NF-κB inhibition based on the post-translational modification of p65 and/or p50 through covalent alkylation of critical cysteine residues in their DNA binding domains by a Michael addition reaction.
Therefore, we tested whether LNO2 and OA-NO2 inhibit NF-κB DNA binding by alkylation of p65/Cys38 and/or p50/Cys62, thus inhibiting LPS-induced inflammatory responses. Consistent with this concept, NF-κB activation in LPS-treated RAW264.7 macrophages was inhibited by LNO2 and OA-NO2 but not by LA and OA (Fig. 5A). Additionally, LNO2 and OA-NO2 inhibited DNA binding of highly purified p65 in vitro in a dose-dependent manner (0.01–1 μm). In contrast, the native fatty acids LA and OA, even at high concentrations (1 μm), had no effect on the DNA binding activity of p65 (Fig. 5B). To explore the interaction between fatty acid nitroalkene derivatives and p65 protein further, the possible covalent alkylation of highly purified p65 protein in vitro was studied using biotinylated LA, OA, LNO2, and OA-NO2. The biotinylated fatty acids were synthesized, purified by thin layer chromatography, and structurally characterized by electrospray ionization mass spectrometry. Biotinylated fatty acids had the following m/z in the negative ion mode: 519, LA; 521, OA; 564, LNO2; 566, OA-NO2, with nitrated fatty acid derivatives presenting the characteristic neutral loss of 47 corresponding to the nitro group (Fig. 5C). These structures were confirmed by respective C–N bond fragmentations that are depicted in Fig. 5C (a). The addition of biotinylated LNO2 and OA-NO2 to purified p65 in vitro resulted in a dose-dependent association with p65. As expected, biotinylated native fatty acids (LA and OA) did not associate with p65 (Fig. 5D). To demonstrate the nature of this interaction and the relevance of this reaction in an intact cell model, RAW264.7 cells were treated with biotinylated LA, OA, LNO2 and OA-NO2. There was a specific and robust covalent nitroalkylation of p65 (Fig. 5E) and, to a lesser extent, p50 by fatty acid nitroalkene derivatives (not shown). The corresponding biotinylated native fatty acids did not react with p65. The addition of biotinylated LA and LNO2 to LPS-elicited RAW264.7 cells resulted in a significant inhibition of MCP-1 release in response to biotinylated LNO2 (p < 0.05 compared with control) (Fig. 5F). The inhibition of MCP-1 expression and release in response to LNO2 was greater than for biotinylated LNO2. There was no impact of biotinylated LA on MCP-1 release. In aggregate, the regulation of NF-κB signaling activity, via nitroalkene alkylation and consequent inhibition of p65, mediates suppression of macrophage cytokine expression and release. These results indicate that LNO2 and OA-NO2 can act endogenously as adaptive inflammatory mediators by inhibiting diverse NF-κB-mediated proinflammatory responses.
FIGURE 5. Nitrated fatty acids inhibit NF-κB activation by nitroalkylation of p65.
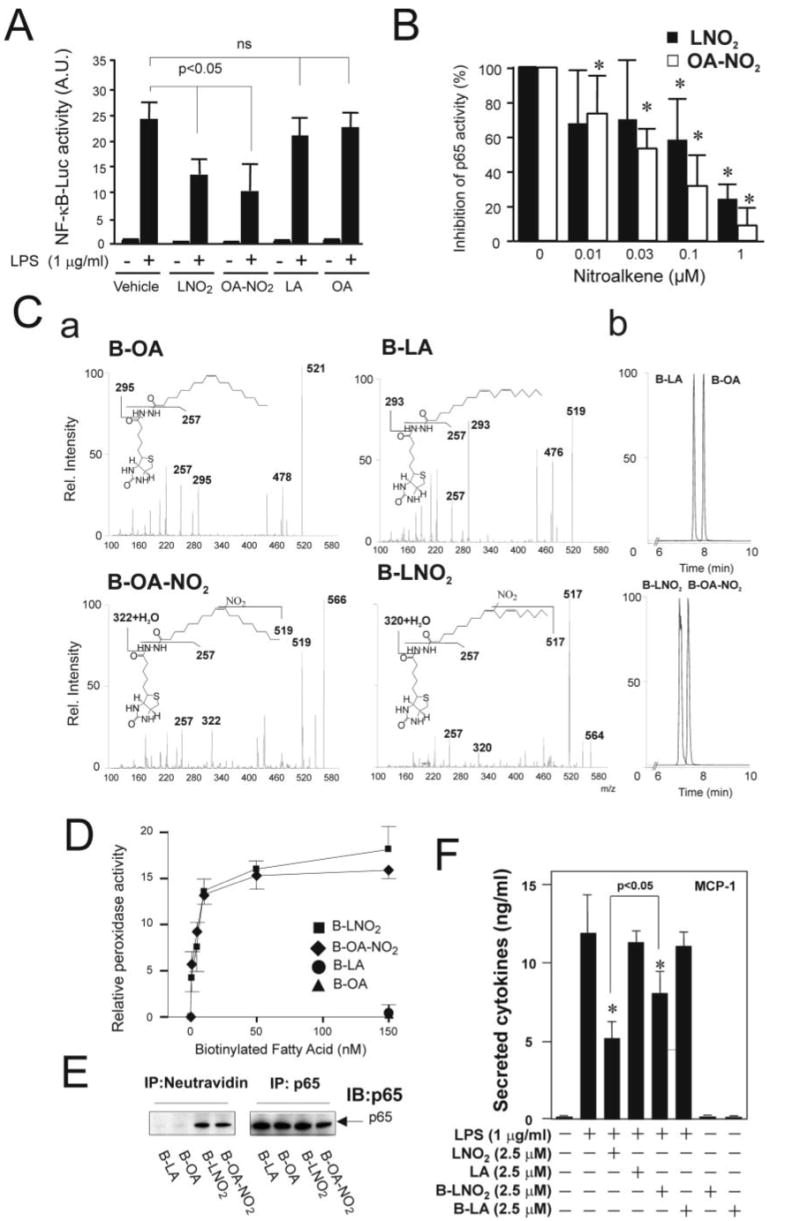
A, LNO2 and OA-NO2 inhibit LPS-induced NF-κB activation in RAW264.7 cells. Cells were pretreated with LNO2 (2.5 μm) and OA-NO2 (2.5 μm) overnight and then stimulated with LPS (1 μg/ml) for 6 h. NF-κB activity was assessed as described under “Experimental Procedures.” Values are expressed as mean ± S.D. (n = 4). The data were analyzed using analysis of variance with the Newman-Keuls test. B, LNO2 and OA-NO2 directly inhibited NF-κB activity in vitro. Purified p65 was incubated with LNO2, OA-NO2, LA, and OA at different concentrations (0.01–1 μm) and then subjected to p65 activity assay as described under “Experimental Procedures.” Native fatty acids LA and OA at 0.01–1 μm had no effect on the DNA binding activity of p65. Therefore, the activity of p65 treated with LA (1 μm) or OA(1 μm) was used as a control and set to 100%. Values are expressed as mean ± S.D. (n = 4). The data were analyzed using analysis of variance with the Newman-Keuls test. C, structural characterization and chromatographic properties of biotinylated OA, LA, OA-NO2, and LNO2. a, structural characterization of biotinylated products by direct infusion into a ESI MS (ion trap) in negative ion mode. Main fragments are identified. Biotinylated lipids were prepared and characterized as described under “Experimental Procedures.” b, the corresponding elution profiles and retention times for the different biotinylated native and nitrated fatty acids are shown. D, p65 nitroalkylation by biotinylated LNO2 and OA-NO2 (B-LNO2 and B-OA-NO2, respectively) but not by native biotinylated fatty acids. Alkylation of p65 by nitroalkenes was assessed as described under “Experimental Procedures.” E, LNO2 and OA-NO2 covalently bind to p65 in vivo. RAW264.7 cells were treated with 0.1 μm biotinylated fatty acids and their corresponding nitro-derivatives, as indicated, for ∼1.5 h. Cell lysates were then treated with anti-p65 antibody or NeutroAvidin Plus followed by immunoblotting with anti-p65 antibody. F, biotinylated LNO2 inhibits LPS-induced inflammatory cytokine secretion in RAW264.7 macrophages. Cells were stimulated as indicated overnight, and the secretion of proinflammatory cytokines was assessed by ELISA. Values are expressed as mean ± S.D. (n = 3). *, p < 0.05 versus LPS alone.
LNO2 and OA-NO2 Inhibit Monocyte Adhesion to Endothelial Cells
To explore more physiological functional aspects of LNO2- and OA-NO2-mediated anti-inflammatory actions, the influence of nitroalkenes on an initial step of acute and chronic inflammation was addressed by analyzing the adhesion of monocytes to endothelial cells (33, 34)). As a consequence of endothelial cell activation by various inflammatory stimuli, monocytes adhere to activated endothelium first by rolling and then by firm adhesion to migrate ultimately into the intima, where they differentiate into macrophages and regulate the differentiation and function of vascular and nonvascular cells through the expression and secretion of cytokines, reactive species, and other chemical mediators. The transendothelial migration of monocytes is largely dependent on a class of adhesion molecules, including intercellular adhesion molecule-1, VCAM-1, E-selectin, and P-selectin. VCAM-1 is a central mediator of the selective recruitment of monocytes and lymphocytes to atherosclerotic lesions (34). Since VCAM-1 is an NF-κB target gene (28), we hypothesized that LNO2 and OA-NO2 inhibit the interaction of monocytes with endothelium by down-regulation of VCAM-1. In this regard, LNO2 and OA-NO2 dose-dependently inhibited VCAM-1 expression induced by LPS or TNFα in both THP-1 monocytes and HUVEC, whereas native fatty acids (LA and OA) had no effect (Fig. 6, A and B). The inhibition of VCAM-1 expression by nitroalkenes occurred at concentrations as low as 0.625 μm, well within the ranges of physiological LNO2 and OA-NO2 levels in the healthy human circulation (10, 11). In THP-1 monocytes, the NO donor DETA-NONOate (100 μm) had no effect on the LPS-induced VCAM-1 expression (Fig. 6A), suggesting that NO is not mediating the inhibitory effect of nitroalkenes toward VCAM-1 expression. In endothelial cells, NO inhibits NF-κB activity and VCAM-1 expression, but only at very high concentrations (34). Multiple lines of reasoning indicate that nitroalkenes do not inhibit TNFα-induced VCAM-1 expression via NO release. First, whereas LNO2 is competent to release NO in an aqueous milieu, this species is not expected to decay to yield NO in cell models due to hydrophobic stabilization, and if it did so would at best yield femtomolar levels of NO (18). Second, OA-NO2 is much more stable in an aqueous milieu and only minimally decays to release NO within the present experimental time frames. Finally, in a recent report (35), the inhibition of VCAM-1 expression by the NO donor S-nitrosoglutathione only occurred at concentrations of 100 μm. Since much lower concentrations of nitroalkenes (∼2.5 μm) inhibited VCAM-1 expression in the present report, it is viewed that any NO derived from LNO2 or OA-NO2 decay was not reaching a level that would mediate a significant impact on VCAM-1 expression.
FIGURE 6. Nitrated fatty acids inhibit the LPS- and TNF-α induced expression of VCAM-1 in THP-1 monocytes and human endothelial cells, respectively.
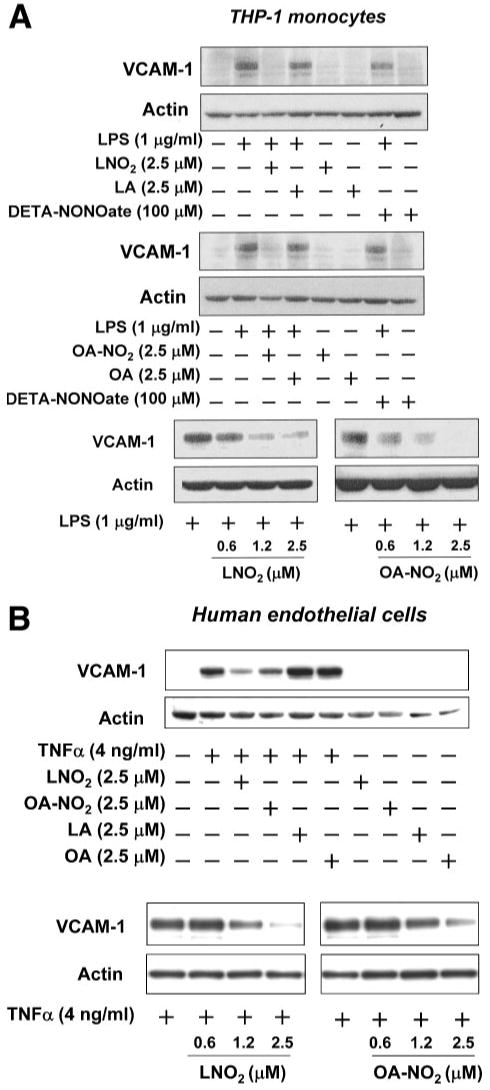
A, LNO2 and OA-NO2 inhibited LPS-induced expression of VCAM-1 in THP-1 monocytes. B, LNO2 and OA-NO2 inhibited TNFα-induced expression of VCAM-1 in human endothelial cells. VCAM-1 expression was examined by Western blot analysis as described under “Experimental Procedures.”
To evaluate the functional significance of nitroalkene-mediated down-regulation of VCAM-1 expression, the effect of LNO2 on the adhesion of THP-1 monocytes to TNFα-activated HUVEC was examined under both static and laminar flow conditions. LNO2 treatment of TNFα-activated endothelial cells fully inhibited the static adherence of 2′,7′-bis(carboxyethyl)-5(6)-carboxyfluorescein-labeled monocytes (Fig. 7). Similar responses were obtained under more physiological flow-mediated conditions (i.e. cells were cultured with a flow force ranging from 0.5 to 1.5 dynes/cm2). The numbers of THP-1 monocytes rolling on endothelium were also decreased significantly by LNO2 treatment (Fig. 7). These results indicate that endogenous and inflammatory induced fatty acid nitration products serve as adaptive mediators by inhibiting monocyte-macrophage and endothelial cell functions.
FIGURE 7. LNO2 inhibits THP-1 monocyte adhesion and rolling on human endothelial cells.
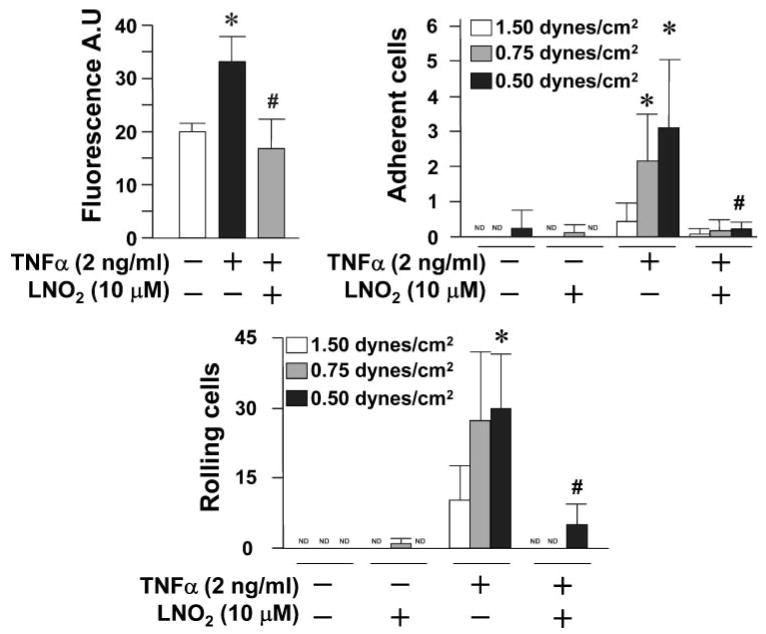
THP-1 monocyte adhesion and rolling on HUVEC was analyzed as described under “Experimental Procedures.” All values are expressed as mean ± S.D. (n = 4) and were representative of five independently performed experiments. The data were analyzed using Student's paired t test. *, p < 0.05 versus TNFα (−); #, p < 0.05 versus TNFα (+). ND, nondetectable.
Discussion
The observation that NO reacts with peroxyl radical intermediates of oxidizing unsaturated fatty acids, thus inhibiting the autocatalytic progression of peroxidation reactions, led to a search for novel reaction products (3, 4). In concert with this apparent antioxidant action of NO, it was also observed that unsaturated fatty acids reacting with NO and NO-derived species yielded nitrated derivatives (5). Although the detailed structural nature of all nitrated fatty acid derivatives present clinically under basal and inflammatory states remains to be defined, a significant proportion of these species are present in human blood as nitroalkenes because of nitro group bonding to olefinic carbons of fatty acids (10, 11). Due to (a) nitroalkene decay during hydrolysis of complex lipids, (b) the diversity of complex lipid classes that contain esterified nitrated fatty acids, and (c) the recent report of abundant, reversible protein alkylation by electrophilic fatty acid nitroalkene derivatives (15), net tissue concentrations of esterified and protein-adducted pools of nitroalkenes remain to be defined.
Current data reveal that nitrated fatty acids serve as mediators of diverse physiological and pathophysiological cell signaling processes, including vascular cell and inflammatory signaling. This notion was first supported by an appreciation that synthetic LNO2 activates guanylate cyclase in cultured vascular smooth muscle cells and vessel segments, inducing relaxation of vessel segments, in an NO-dependent manner (7). Subsequent analysis revealed a hydrophobically regulated Nef-like reaction of nitroalkenes that produces NO in low yields (18, 36). Also, LNO2 inhibits platelet aggregation and leukocyte function via both cGMP- and cAMP-dependent and independent mechanisms, suggesting that nitro-fatty acids display reactivities beyond serving as reserves for NO (7–9).
More recently, intact nitroalkenes, rather than their aqueous and aerobic decay products, have been observed to activate PPARγ at physiological concentrations, thus representing a significant component of endogenous ligand activity for PPARγ (11, 22). In addition to regulating many aspects of basal metabolism, PPARγ-regulated gene products play critical modulatory roles in pathophysiological processes, such as metabolic syndrome (a condition characterized by multiple related clinical disorders, including insulin resistance, obesity, hyperlipidemia, hypertension, and heart disease), diabetes, and cardiovascular disease (23, 24, 39). Thus, LNO2 and OA-NO2 are expected to exert diverse PPARγ-dependent biological effects.
From a broader perspective, nitroalkenes represent a class of pluripotent cell signaling and inflammatory mediators that transduce the signaling actions of NO via multiple reaction mechanisms. Of note, LNO2 decays in aqueous milieu with a half-life of ∼30 min, whereas OA-NO2 is relatively stable, with only minimal decay occurring over 2 h (11). In the presence of other lipids or amphiphiles at levels above the critical micellar concentration, nitroalkenes are stabilized and become highly resistant to NO release via the Nef-like reaction. This property may account for their accumulation to detectable levels in the vascular compartment. Thus, any additional signaling actions of individual nitrated fatty acid regio- and stereo-isomers will be regulated by biodistribution, pharmacokinetics, and specific secondary reactivities.
Nitrated fatty acids have been reported in a variety of tissues, with these derivatives including nitro-oleate, nitrolinoleate, nitro-arachidonate, and cholesteryl nitrolinoleate (6, 14, 36–38). With a combination of HPLC separations and ESI-MS-MS/MS, all detectable unsaturated fatty acids in healthy human serum and urine display some component of nitrated and α,β-nitrohydroxy derivatives (11). This latter derivative, formed by reaction with hydroxide at neutral pH, was the first indication that nitroalkenes might act biologically as electrophiles.
The electrophilic nature of the β-carbon to the nitroalkene bond of nitrated unsaturated fatty acids facilitates reaction with protein thiol and histidine residues (15). At physiologically relevant concentrations, nitroalkenes inhibit GAPDH (IC50 of 3 μm) in a thiol-reversible manner. Six GAPDH residues are modified by nitroalkenes in vitro, including the catalytically critical Cys149. This adduction of GAPDH and other biomolecules by nitroalkenes significantly increases hydrophobicity and facilitates translocation to membranes. Moreover, adducts between OA-NO2 and both GAPDH and glutathione have been identified in vivo in human red blood cells (15). The post-translational modification of other proteins by oxidized, electrophilic lipids has also been reported. For example, 15d-PGJ2 and 4-hydroxy-2-nonenal form adducts with proteins that contain nucleophilic centers (40). The electrophilic 4-hydroxy-2-nonenal reacts with sulfhydryl groups (41), the imidazole moiety of histidine (42), and the ε-amino of lysine (43). Covalent modification by electrophilic lipids has been shown to alter the structure and activities of a variety of proteins (i.e. GAPDH (15, 44), cathepsin B (45), Keap1 (46), and insulin (42)). Moreover, electrophiles, including 15d-PGJ2, sesquiterpene lactone, ethyl pyruvate, and N-ethyl maleimide, mediate the suppression of the transactivation of NF-κB target genes (29–32).
The inhibition of TNFα and LPS-induced monocyte cytokine expression by nitrated fatty acids displays an IC50 that falls consistently within the clinically relevant range of 500 nm. Nitrated fatty acids display apparent affinity for p65 nitroalkylation, in that potential alternative cellular targets well exceed 20 mm concentration in cells. These potential alternative targets minimally include low molecular weight thiols and proteins containing cysteines and histidines. The determinants of target molecule reactivity with nitro-fatty acids will include compartmentalization, steric restriction, the local ionic microenvironment, diffusional limitations, the rate constant for nitroalkene-nucleophile reaction, and finally, nitroalkene concentration.
The regulation of NF-κB-dependent signaling by its p65 subunit is complex and also can include phosphorylation and acetylation in the course of cytoplasmic-to-nuclear trafficking. Although these regulatory steps have been extensively described in the literature, their extents and relative contributions have yet to be elucidated. In addition, the threshold for p65 activation by various post-translational modifications (i.e. nitroalkylation, phosphorylation, and acetylation) in the context of mol % extents of target residue modification remains undefined, as with various other protein kinase-mediated signaling events.
The biotinylation of electrophilic molecules has aided in the identification of potential cellular targets, due to an ability to enrich modified proteins and enhance their sensitivity for detection via secondary labeling strategies. In this regard, biotinylated nitro-fatty acid derivatives retain the electrophilic character of the nitroalkene moiety. A limitation may be that the biotinylation of a small molecule, such as a fatty acid can influence distribution and in turn may influence reactivity due to changes in size, charge, and hydrophobic characteristics induced by derivatization. If biotinylated nitro-fatty acids induced biological responses akin to nonmodified nitrated fatty acids, it can be assumed that the same signaling mediators and pathways would be targeted. To test this hypothesis, the biotinylated derivatives of linoleic and nitro-linoleic acid were used in probing effects on MCP-1 release by LPS-elicited RAW264.7 cells. Biotinylated nitro-linoleic acid induced a significant inhibition of cytokine release by LPS-elicited RAW264.7 cells at a lower potency than the corresponding nonbiotinylated nitro-fatty acid. This indicates that a similar signaling target is being nitroalkylated (p65, Fig. 5F).
Herein, we have extended our understanding of the physiological signaling capabilities of LNO2 and OA-NO2 by reporting their potent and broad regulation of vascular inflammatory responses via post-translational modification of susceptible transcription factors, such as p65. We have demonstrated that LNO2 and OA-NO2, at clinically relevant concentrations, inhibit LPS-induced secretion of proinflammatory cytokines in macrophages and endothelial cells. Also, TNFα-induced VCAM-1 expression and monocyte rolling and adhesion are inhibited by these fatty acid nitroalkene derivatives. These particular anti-inflammatory actions of LNO2 and OA-NO2 are independent of NO formation, PPARγ activation, and induction of HO-1 expression. Rather, the electrophilic nature of fatty acid nitroalkenes facilitates the alkylation of the recombinant NF-κB p65 subunit in vitro and the corresponding alkylation of p65 in vivo, thus repressing NF-κB-dependent target gene expression. This inhibitory nitroalkylation of a key transcriptional regulatory factor in turn inhibits the down-stream expression and action of critical components in the propagation of inflammatory responses, namely proinflammatory cytokine release and integrin expression. In aggregate, these results reveal that nitroalkenes, such as LNO2 and OA-NO2, regulate the initiation and progression of inflammatory processes and represent a distinct class of endogenous cell signaling mediators.
Supplementary Material
Footnotes
This work was supported by National Institutes of Health Grants HL068878, HL075397, and S06GM08248 (to Y. E. C.), HL70146 (to R. P. P.), and HL58115 and HL64937 (to B. A. F.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1, Fig. 1, and Movie 1.
The abbreviations used are: LNO2, nitrolinoleate (positional isomers 10-nitro-9-cis,12-cis-octadecadienoic acid (C10) and 12-nitro-9-cis,12-cis-octadecadienoic acid (C12)); OA-NO2, esterified nitrooleate (9- and 10-nitro-9-cis-octadecenoic acid (C9 and C10)); LPS, lipopolysaccharide; PMA, phorbol 12-myristate 13-acetate; DETA-NONOate, 3,3-bis(aminoethyl)-1-hydroxy-2-oxo-1-triazene; cPTIO, 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide; PPARγ, peroxisome proliferator-activated receptor; HO-1, heme oxygenase 1; VCAM-1, vascular cell adhesion molecule 1; IL-6, interleukin 6; TNFα, tumor necrosis factor α; MCP-1, monocyte chemoattractant protein 1; ESI, electrospray ionization; MS, mass spectrometry; DTPA, diethylenetriaminepentaacetate; OA, oleic acid; LA, linoleic acid; HUVEC, human umbilical vein endothelial cells; FBS, fetal bovine serum; ELISA, enzyme-linked immunosorbent assay; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; 15d-PGJ2, 15-deoxy-Δ12,14-prostaglandin J2.
References
- 1.Baldus S, Eiserich JP, Brennan ML, Jackson RM, Alexander CB, Freeman BA. Free Radic Biol Med. 2002;33:1010. doi: 10.1016/s0891-5849(02)00993-0. [DOI] [PubMed] [Google Scholar]
- 2.Ischiropoulos H, Beckman JS. J Clin Invest. 2003;111:163–169. doi: 10.1172/JCI17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rubbo H, Radi R, Trujillo M, Telleri R, Kalyanaraman B, Barnes S, Kirk M, Freeman BA. J Biol Chem. 1994;269:26066–26075. [PubMed] [Google Scholar]
- 4.Rubbo H, Parthasarathy S, Barnes S, Kirk M, Kalyanaraman B, Freeman BA. Arch Biochem Biophys. 1995;324:15–25. doi: 10.1006/abbi.1995.9935. [DOI] [PubMed] [Google Scholar]
- 5.O'Donnell VB, Eiserich JP, Chumley PH, Jablonsky MJ, Krishna NR, Kirk M, Barnes S, Darley-Usmar VM, Freeman BA. Chem Res Toxicol. 1999;12:83–92. doi: 10.1021/tx980207u. [DOI] [PubMed] [Google Scholar]
- 6.Lima ES, Di Mascio P, Rubbo H, Abdalla DS. Biochemistry. 2002;41:10717–10722. doi: 10.1021/bi025504j. [DOI] [PubMed] [Google Scholar]
- 7.Lim DG, Sweeney S, Bloodsworth A, White CR, Chumley PH, Krishna NR, Schopfer F, O'Donnell VB, Eiserich JP, Freeman BA. Proc Natl Acad Sci U S A. 2002;99:15941–15946. doi: 10.1073/pnas.232409599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coles B, Bloodsworth A, Eiserich JP, Coffey MJ, McLoughlin RM, Giddings JC, Lewis MJ, Haslam RJ, Freeman BA, O'Donnell VB. J Biol Chem. 2002;277:5832–5840. doi: 10.1074/jbc.M105209200. [DOI] [PubMed] [Google Scholar]
- 9.Coles B, Bloodsworth A, Clark SR, Lewis MJ, Cross AR, Freeman BA, O'Donnell VB. Circ Res. 2002;91:375–381. doi: 10.1161/01.res.0000032114.68919.ef. [DOI] [PubMed] [Google Scholar]
- 10.Baker PR, Schopfer FJ, Sweeney S, Freeman BA. Proc Natl Acad Sci U S A. 2004;101:11577–11582. doi: 10.1073/pnas.0402587101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baker PR, Lin Y, Schopfer FJ, Woodcock SR, Long MH, Batthyany C, Iles KE, Baker LMS, Branchaud BP, Chen YE, Freeman BA. J Biol Chem. 2005;280:42464–42475. doi: 10.1074/jbc.M504212200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serhan CN, Oliw E. J Clin Invest. 2000;107:1481–1489. doi: 10.1172/JCI13375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith WL, Langenbach R. J Clin Invest. 2001;107:1491–1495. doi: 10.1172/JCI13271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalyanaraman B. Proc Natl Acad Sci U S A. 2004;101:11527–11528. doi: 10.1073/pnas.0404309101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Batthyany C, Schopfer FJ, Baker PRS, Durán R, Baker LMS, Huang Y, Cerveñansky C, Branchaud BP, Freeman BA. J Biol Chem. 2006;281:20450–20463. doi: 10.1074/jbc.M602814200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang LT, Vanderhoek JY. Anal Biochem. 1997;250:119–122. doi: 10.1006/abio.1997.2202. [DOI] [PubMed] [Google Scholar]
- 17.Feng X, Novack DV, Faccio R, Ory DS, Aya K, Boyer MI, McHugh KP, Ross FP, Teitelbaum SL. J Clin Invest. 2001;107:1137–1144. doi: 10.1172/JCI12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schopfer FJ, Baker PR, Giles G, Chumley P, Batthyany C, Crawford J, Patel RP, Hogg N, Branchaud BP, Jr, Lancaster JR, Freeman BA. J Biol Chem. 2005;280:19289–19297. doi: 10.1074/jbc.M414689200. [DOI] [PubMed] [Google Scholar]
- 19.Thomassen MJ, Buhrow LT, Connors MJ, Kaneko FT, Erzurum SC, Kavuru MS. Am J Respir Cell Mol Biol. 1997;17:279–283. doi: 10.1165/ajrcmb.17.3.2998m. [DOI] [PubMed] [Google Scholar]
- 20.Zhang H, Snead C, Catravas JD. Arterioscler Thromb Vasc Biol. 2001;21:529–535. doi: 10.1161/01.atv.21.4.529. [DOI] [PubMed] [Google Scholar]
- 21.van der Vliet A, Eiserich JP, Halliwell B, Cross CE. J Biol Chem. 1997;272:7617–7625. doi: 10.1074/jbc.272.12.7617. [DOI] [PubMed] [Google Scholar]
- 22.Schopfer FJ, Lin Y, Baker PR, Cui T, Garcia-Barrio M, Zhang J, Chen K, Chen YE, Freeman BA. Proc Natl Acad Sci U S A. 2005;102:2340–2345. doi: 10.1073/pnas.0408384102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daynes RA, Jones DC. Nat Rev Immunol. 2002;2:748–795. doi: 10.1038/nri912. [DOI] [PubMed] [Google Scholar]
- 24.Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM. Nat Med. 2001;7:48–52. doi: 10.1038/83336. [DOI] [PubMed] [Google Scholar]
- 25.Maines MD. Annu Rev Pharmacol Toxicol. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- 26.Lee TS, Tsai HL, Chau LY. J Biol Chem. 2003;278:19325–19330. doi: 10.1074/jbc.M300498200. [DOI] [PubMed] [Google Scholar]
- 27.Alvarez-Maqueda M, El Bekay R, Alba G, Monteseirin J, Chacon P, Vega A, Martin-Nieto J, Bedoya FJ, Pintado E, Sobrino F. J Biol Chem. 2004;279:21929–21937. doi: 10.1074/jbc.M400492200. [DOI] [PubMed] [Google Scholar]
- 28.Ghosh S, May MJ, Kopp EB. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 29.Straus DS, Pascual G, Li M, Welch JS, Ricote M, Hsiang CH, Sengchanthalangsy LL, Ghosh G, Glass CK. Proc Natl Acad Sci U S A. 2000;97:4844–4849. doi: 10.1073/pnas.97.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cernuda-Morollón E, Pineda-Molina E, Cañada FJ, Pérez-Sala D. J Biol Chem. 2001;276:35530–35535. doi: 10.1074/jbc.M104518200. [DOI] [PubMed] [Google Scholar]
- 31.Han Y, Englert JA, Yang R, Delude RL, Fink MP. J Pharmacol Exp Ther. 2005;312:1097–1105. doi: 10.1124/jpet.104.079707. [DOI] [PubMed] [Google Scholar]
- 32.Garcia-Pineres AJ, Castro V, Mora G, Schmidt TJ, Strunck E, Pahl HL, Merfort I. J Biol Chem. 2001;276:39713–39720. doi: 10.1074/jbc.M101985200. [DOI] [PubMed] [Google Scholar]
- 33.Osterud B, Bjorklid E. Physiol Rev. 2003;83:1069–1112. doi: 10.1152/physrev.00005.2003. [DOI] [PubMed] [Google Scholar]
- 34.Ley K. Cardiovasc Res. 1996;32:733–742. [PubMed] [Google Scholar]
- 35.DeCaterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Jr, Gimbrone MA, Shin WS, Liao JK. J Clin Invest. 1995;96:60–68. doi: 10.1172/JCI118074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lima ES, Bonini MG, Augusto O, Barbeiro HV, Souza HP, Abdalla DS. Free Radic Biol Med. 2005;39:532–539. doi: 10.1016/j.freeradbiomed.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 37.Lima ES, Di Mascio P, Abdalla DS. J Lipid Res. 2003;44:1660–1666. doi: 10.1194/jlr.M200467-JLR200. [DOI] [PubMed] [Google Scholar]
- 38.Balazy M, Iesaki T, Park JL, Jiang H, Kaminski PM, Wolin MS. J Pharmacol Exp Ther. 2001;299:611–619. [PubMed] [Google Scholar]
- 39.Evans RM, Barish GD, Wang YX. Nat Med. 2004;10:355–361. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- 40.Ceaser EK, Moellering DR, Shiva S, Ramachandran A, Landar A, Venkartraman A, Crawford J, Patel R, Dickinson DA, Ulasova E, Ji S, Darley-Usmar VM. Biochem Soc Trans. 2004;32:151–155. doi: 10.1042/bst0320151. [DOI] [PubMed] [Google Scholar]
- 41.Esterbauer H, Zollner H, Scholz N. Z Naturforsch. 1975;30:466–473. doi: 10.1515/znc-1975-7-808. [DOI] [PubMed] [Google Scholar]
- 42.Uchida K, Stadtman ER. Proc Natl Acad Sci U S A. 1992;89:4544–4548. doi: 10.1073/pnas.89.10.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Szweda LI, Uchida K, Tsai L, Stadtman ER. J Biol Chem. 1993;268:3342–3347. [PubMed] [Google Scholar]
- 44.Ishii T, Tatsuda E, Kumazawa S, Nakayama T, Uchida K. Biochemistry. 2003;42:3474–3480. doi: 10.1021/bi027172o. [DOI] [PubMed] [Google Scholar]
- 45.Crabb JW, O'Neil J, Miyagi M, West K, Hoff HF. Protein Sci. 2002;11:831–840. doi: 10.1110/ps.4400102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Levonen AL, Landar A, Ramachandran A, Ceaser EK, Dickinson DA, Zanoni G, Morrow JD, Darley-Usmar VM. Biochem J. 2004;378:373–382. doi: 10.1042/BJ20031049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.