

Abstract
DNA vaccination has emerged as a promising strategy for cancer immunotherapy. However, since DNA vaccines have low immunogenicity, various strategies have been developed to enhance the potency of DNA vaccines. In the current study, we aim to determine whether the potency of the DNA vaccine encoding human papillomavirus type 16 (HPV-16) E7 antigen can be enhanced by IL-2. We have generated a DNA vaccine encoding IL-2 linked to HPV-16 E7 antigen. Our results indicate that the DNA vaccine encoding a fusion of IL-2 and E7 proteins generated the highest frequency of E7-specific CD8+ T cells. We also found that the DNA vaccine encoding a fusion of IL-2 and E7 proteins generated the strongest protective as well as therapeutic anti-tumor effect against E7-expressing tumors. In addition, it was observed that CD8+ T cells were mainly responsible for the antitumor effect generated by the DNA vaccine encoding a fusion of IL-2 and E7 proteins. Thus, we conclude that the linkage of IL-2 to HPV-16 E7 antigen significantly enhances the DNA vaccine potency against E7-expressing tumors. Our strategy may potentially be used in other antigenic systems to control infectious diseases and/or cancer.
Keywords: HPV, DNA vaccine, E7, IL-2
Introduction
DNA vaccination poses as an attractive approach for cancer immunotherapy (for review, see [1]). Compared to live viral or bacterial vectors, naked DNA plasmid vaccines are relatively safe and can be easily administered. Furthermore, DNA vaccines are easy to prepare on a large scale with high purity and high stability and can be engineered to express antigenic peptides or proteins (for reviews, see [2,3]). While DNA has considerable advantages, one major drawback is its limited potency. DNA vaccine potency may be enhanced by targeting DNA to the antigen presenting cells. Among different routes of administration of DNA vaccines, intradermal administration by gene gun has emerged as one of the most efficient routes of delivering DNA into professional APCs.
Intradermal administration via gene gun consists of the acceleration of gold particles coated with DNA encoding a protein of interest, i.e., a tumor antigen, into the skin of the recipient, using high-pressure inert gas as the propellant. Within the epidermal tissue, the key professional antigen-presenting cells (APCs), the Langerhans cells, receive the DNA and express the encoded antigen [4]. These antigen-expressing cells mature, and are then able to migrate to draining lymph nodes, where they interact with specific naïve T cells, resulting in their activation to effector T cells specific for the antigen encoded by the DNA vaccine [5]. Thus, the administration of DNA vaccines via gene gun serves as an efficient method to directly deliver DNA into the professional APCs.
Other strategies to improve DNA vaccine potency include the employment of cytokines related to T cell proliferation, such as IL-2. IL-2 is an important cytokine that is produced by activated T-cells and is responsible for clonal T-cell proliferation (for review, see [6]). Previous studies have shown that DNA vaccines employing IL-2 co-expressed with the Hepatitis B surface antigen has led to significant enhancement of the HBsAg-specific immune responses [7]. Furthermore, another study has shown that DNA vaccines encoding a fusion of HER2/Neu to IL-2 significantly enhanced the therapeutic efficacy of the DNA vaccine against HER2/Neu-expressing tumors [8]. In addition, co-expression of IL-2 has also been shown to enhance the immune response to the HSV1 glycoprotein D antigen in DNA vaccines [9]. Taken together, these studies suggest that DNA vaccine encoding IL-2 in conjunction with antigen may potentially enhance the antigen-specific immune responses resulting in improved vaccine potency.
In the current study, we have developed a DNA vaccine encoding IL-2 linked to human papillomavirus type 16 (HPV-16) E7 antigen. HPV is one of the most common sexually transmitted diseases in the world and HPV infection is a necessary factor for cervical cancer [10]. Thus, effective vaccination against HPV is essential for the control of cervical cancer. Although the current available preventive HPV vaccine is highly effective in preventive HPV infections, it only protects up to 75% of all cervical cancers (for review, see [11]). Furthermore, there are a significant number of patients with existing HPV infections and HPV-associated lesions. The current vaccine does not have therapeutic effect against established HPV infections. Thus, it is important to develop a therapeutic HPV vaccine. The HPV E7 protein is essential for transformation and is co-expressed in a majority of HPV-associated lesions. Thus, the E7 protein represents an ideal target for the development of HPV therapeutic vaccines. (for review, see [12]).
In the current study, we aim to determine whether the potency of the DNA vaccine encoding HPV-16 E7 antigen can be enhanced by IL-2. We have observed that DNA vaccines encoding a fusion of IL-2 and E7 proteins generated the highest frequency of E7-specific CD8+ T cells. Furthermore, DNA vaccines encoding a fusion of IL-2 and E7 proteins also generated the strongest protective as well as therapeutic anti-tumor effect against E7-expressing tumors. In addition, we also showed that the CD8+ T cells were mainly responsible for the antitumor effect generated by the DNA vaccine encoding a fusion of IL-2 and E7 proteins. Thus, we conclude that the linkage of IL-2 to HPV-16 E7 antigen significantly enhances the potency of the DNA vaccine against E7-expressing tumors. Our DNA vaccine has potential for future clinical translation.
Materials and Methods
Mice
C57BL/6 mice (6 to 8 weeks old) were purchased from the National Cancer Institute (Frederick, MD, USA). All animals were maintained under specific pathogen-free conditions at the Johns Hopkins Hospital (Baltimore, MD, USA). All procedures were performed according to approved protocols and in accordance with recommendations for the proper care of laboratory animals.
Cells
Briefly, TC-1 cells were obtained by co-transformation of primary C57BL/6 mouse lung epithelial cells with HPV-16 E6 and E7 and an activated ras oncogene as described previously [13]. The expression of E7 in TC-1 cells has also been characterized previously by He et al [14].
DNA Constructs
The generation of pcDNA3-E7 has been described previously [15]. For the generation of pcDNA3-IL2, DNA fragments encoding the full length of mouse IL-2 were generated by RT-PCR using E7 specific T cells [16] and a pair of primers, 5′-tttgcggccgcatgtacagcatgcagctcgca-3′ and 5′-aaagaattcttgagggcttgttgagatgat-3′. The amplified DNA was further cloned into the NotI and EcoRI sites of pcDNA3. To generate pcDNA3-IL2-E7, the E7 fragment was isolated from pcDNA3-E7 and further cloned into the EcoRI and BamHI sites of pcDNA3-IL2.
Transfection
A human embryonic kidney 293 cell line expressing the Db and Kb (293 Db,Kb), two C57BL/6 mouse MHC class I molecules, was kindly provided by Dr. James C. Yang (National Cancer Institute, NIH, Bethesda, MD) [15,17]. pcDNA3 (1 ug), pcDNA3-E7 (0.5ug)+pcDNA3 (0.5ug), pcDNA3-IL2 (0.5ug)+pcDNA3 (0.5ug), pcDNA3-E7 (0.5ug)+pcDNA3-IL2(0.5ug), or pcDNA3-IL2E7(0.5ug)+pcDNA3(0.5ug) were transfected into 293 Db, Kb cells using Lipofectamine 2000 (Life Technologies, Inc., Rockville, MD). Cells were collected 24 h after transfection. Transfected 293 Db, Kb cells were washed with complete RPMI-1640 containing 10% fetal bovine serum, and coincubated with HPV-16 E7 aa49–57 peptide-specific T cells (with a ratio 1:5) at the presence of 1 ml/ml of GolgiPlug (BD Pharmingen) at 37C overnight. Intracellular staining of IFN-g, flow cytometry and data analysis were performed.
RT-PCR
RNA was extracted from the transfected 293 Db, Kb cells by TRIZOL (Invitrogen, Carlsbad, Calif). RT-PCR was performed using the Superscript One-Step RT-PCR Kit (Invitrogen). One microgram of total RNA was used. Sequences of primers for E7 and GAPDH were as follows: E7-F (5′-atgcatggagatacacctaca -3′), E7-R (5′-ttatggtttctgagaacagat-3′), GAPDH-F (5′-CCGGATCCTGGGAAGCTTGTCATCAACGG -3′), and GAPDH-R (5′-GGCTCGAGGCAGTGATGGCATGGACTG -3′). The reaction condition for E7 was 1 cycle (94°C, 30 sec), 30 cycle (94°C, 30 sec; 55°C, 30 sec; 72°C, 30 sec), and 1 cycle (72°C, 10 min). The reaction condition for GAPDH was similar except that amplification was repeated for 20 cycles. The products were analysed by electrophoresis on a 1.5% agarose gel containing ethidium bromide.
CFSE labeling of T cells and proliferation experiment
E7-specific CD8+ T cells were labeled at 1 × 107 cells/ml with 5 μM CFSE (Molecular Probes, Carlsbad, CA) in PBS for 5 min at room temperature followed by incubation with 5% FBS-PBS (5 mM EDTA) for 10 min at 37°C. After three washes with 5%FBS-PBS, 5 × 105/ml of the labeled cells in 1000 μl of media were mixed with 100ul medium from various DNA transfected 293 Db, Kb cells in a 24-well plate. After 4 days culture, flow cytometry acquisition was done.
DNA vaccination by gene gun
DNA-coated gold particles were prepared, and gene gun particle-mediated DNA vaccination was performed, according to a protocol described previously [18]. Gold particles coated with DNA vaccines were delivered to the shaved abdominal regions of mice by using a helium-driven gene gun (Bio-Rad Laboratories Inc., Hercules, CA, USA) with a discharge pressure of 400 lb/in2. Mice were immunized with 2μg of the DNA vaccine and received one boost with the same dose at 1-week interval. Splenocytes were harvested 1 week after the last vaccination.
Intracellular cytokine staining and flow cytometry analysis
Pooled splenocytes from the vaccinated mice were harvested 1 week after the last vaccination and incubated overnight with 1 μg/ml E7 peptide (aa49–57) in the presence of GolgiPlug (BD Pharmingen, San Diego, CA, USA) (1 μl/ml). The stimulated splenocytes were then washed once with FACScan buffer and stained with phycoerythrin-conjugated monoclonal rat anti-mouse CD8a (clone 53.6.7). Cells were subjected to intracellular cytokine staining using the Cytofix/Cytoperm kit according to the manufacturer’s instruction (BD Pharmingen, San Diego, CA, USA). Intracellular IFN-γ was stained with FITC-conjugated rat anti-mouse IFN-γ. All antibodies were purchased from BD Pharmingen. Flow cytometry analysis was performed using FACSCalibur with CELLQuest software (BD Biosciences, Mountain View, CA, USA).
In vivo tumor protection experiment
For in vivo tumor protection experiment, C57BL/6 mice (five per group) were immunized via gene gun with 2 μg/mouse of the various DNA vaccines. Mice were boosted once using the dose and vaccination regimen. One week after the last vaccination, mice were challenged with 5×104 TC-1 tumor cells/mouse subcutaneously in the right leg and monitored twice a week by inspection and palpation.
In vivo tumor treatment experiment
For in vivo tumor treatment experiment, 5×104 TC-1 tumor cells were injected into 5–8 week-old C57BL/6 mice (five per group) subcutaneously in the right leg. After 3 days, the mice were immunized with the DNA vaccines as described above. After 1 week, these mice were boosted once with the same immunization regimen. Mice were monitored once a week by inspection and palpation.
Depletion of lymphocyte subsets in vivo
The mice vaccinated with the various DNA constructs were injected intraperitoneally (i.p.) with blocking antibody using a protocol similar to one described previously [18]. Mice were injected with 100 μg of purified rat monoclonal antibody GK1.5 (anti-CD4), mAb 2.43 (anti-CD8), and mAb PK136 (anti-NK1.1). Depletion was started one week after cell-based vaccination and continued every other day for the first week and then once every week. These mice were challenged with TC-1 tumor cells two weeks after the first vaccination.
Statistical Analysis
All data expressed as means ±s.d. are representative of at least two different experiments. Data for intracellular cytokine staining with flow cytometry analysis were evaluated by ANOVA. Comparisons between individual data points were made using a Student’s t-test. For statistical analysis of the tumor protection experiment, we used Kaplan-Meier analysis.
Results
C57BL/6 mice vaccinated with the pcDNA3-IL2-E7 generate highest frequency of E7-specific CD8+ T cells
We constructed various DNA vaccines encoding the E7 protein alone (pcDNA3-E7), IL-2 alone (pcDNA3-IL2), IL-2 and the E7 protein on separate plasmids (pcDNA3-IL2+pcDNA3-E7) and the fused construct encoding IL-2 linked to the E7 protein (pcDNA3-IL2-E7). Figure 1A shows the schematic diagrams of the various DNA constructs used in the study. In order to demonstrate the expression of E7 in 293 Db, Kb cells transfected with the various DNA constructs, we performed RT-PCR analysis. As shown in Figure 1B, the quantity of E7 RNA was comparable in cells transfected with the pcDNA3-E7 (lane 2), pcDNA3-E7 mixed with pcDNA3-IL2 (lane 3) and the pcDNA3-IL2E7 (lane 4) constructs. In order to determine the E7-specific CD8+ T cell immune response in mice vaccinated with the various DNA constructs, we performed intracellular cytokine staining followed by flow cytometry analyses to determine the number of E7-specific CD8+ T cells using splenocytes from vaccinated mice. C57BL/6 mice (5 per group) were vaccinated via gene gun with the various DNA constructs. Splenocytes from vaccinated mice were collected one week after the last vaccination and stimulated with E7 peptide (aa49–57). As shown in Figure 2A, mice vaccinated with pcDNA3-IL2-E7 generated the highest number of E7-specific IFN-γ secreting CD8+ T cells compared to mice vaccinated with any of the other constructs. The observed enhancement of E7-specific CD8+ T cell immune responses in mice vaccinated with pcDNA3-IL2-E7 required the physical linkage of IL-2 to E7 in the DNA vaccine since vaccination with pcDNA3-E7 mixed with pcDNA3-IL2 did not generate appreciable enhancement of the E7-specific CD8+ T cell immune responses. Furthermore, the enhancement in the E7-specific CD8+ T cell immune response generated by pcDNA3-IL2-E7 appeared to be specific to IL2 since the linkage of E7 to irrelevant proteins, such as GFP failed to generate significant E7-specific immune responses (data not shown). A graphical representation of the number of E7-specific CD8+ T cells in each group is depicted in Figure 2B. Thus, our data indicates that mice vaccinated with pcDNA3-IL2-E7 are capable of generating a potent E7-specific CD8+ T cell immune response.
Figure 1. Characterization of the expression of E7 in cells transfected with the DNA constructs by RT-PCR.
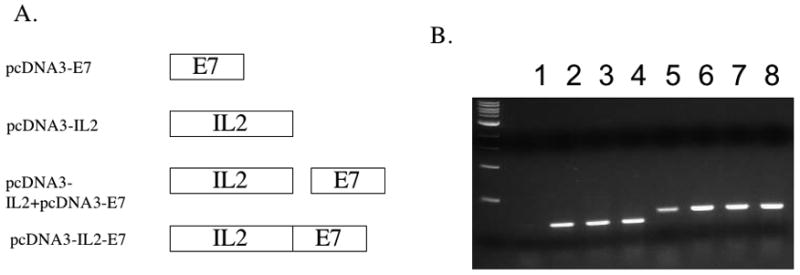
A. Diagrammatic representation of the various DNA constructs used in the current study; pcDNA3-E7, pcDNA3-IL2, pcDNA3-IL2+ pcDNA3-E7 and the fused construct pcDNA3-IL2-E7. B. RT-PCR analysis of E7 expression in cells transfected with the various DNA constructs. RNA were extracted from 293 Db,Kb cells transfected with pcDNA3 (1 ug) (lanes 1,5), pcDNA3-E7 (0.5ug)+pcDNA3 (0.5ug) (lanes 2,6), pcDNA3-E7 (0.5ug) + pcDNA3-IL2(0.5ug) (lanes 3,7), or pcDNA3-IL2-E7(0.5ug) + pcDNA3(0.5ug) (lanes 4,8) using Lipofectamine 2000. In the lanes 1–4, samples were amplified by E7 primers. In the lanes 5–8, samples were amplified by GAPDH primers.
Figure 2. Intracellular cytokine staining followed by flow cytometry analysis to determine the number of E7-specific CD8+ T cells in vaccinated mice.
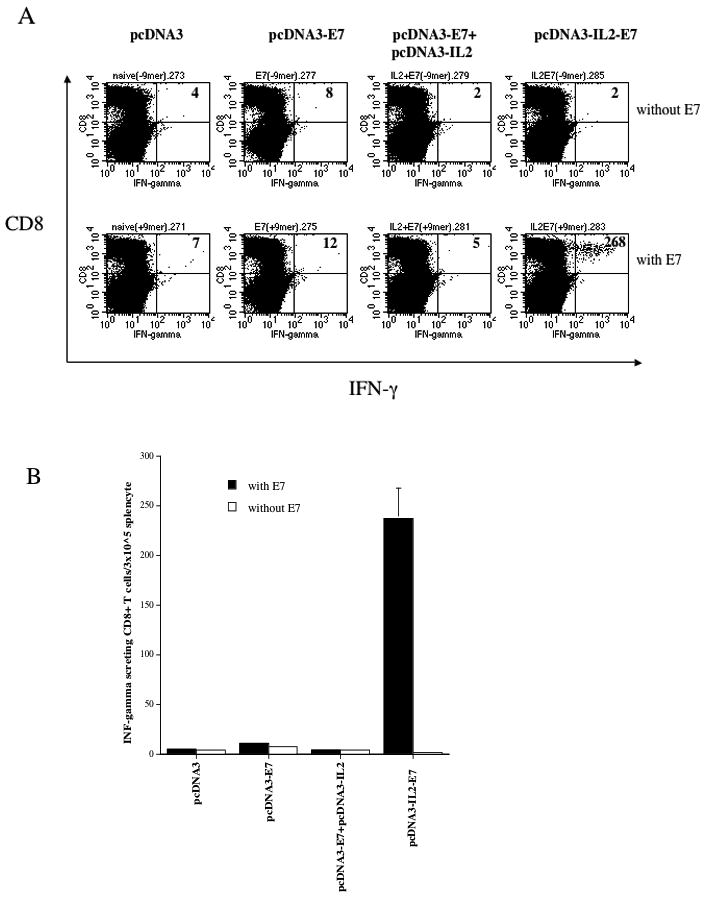
C57BL/6 mice (5 per group) were immunized twice intradermally via gene gun with 2μg/mouse of pcDNA3-E7, pcDNA3-IL2, pcDNA3-IL2+pcDNA3-E7 or pcDNA3-IL2-E7 at one-week interval. Splenocytes from vaccinated mice were harvested 1 week after the last vaccination and stimulated with E7 peptide (aa49–57). Splenocytes without peptide stimulation were used as a negative control. The splenocytes were stained for CD8 and intracellular IFN-γ. A. Representative data of intracellular cytokine stain followed by flow cytometry analysis showing the number of E7-specific IFNγ+ CD8+ T cells in the mice vaccinated with the various DNA constructs. B. Bar graph depicting the numbers of E7-specific CD8+ T-cells per 3×105 splenocytes (means±s.d.). The data presented in this figure are from one representative experiment of two performed.
C57BL/6 mice vaccinated with pcDNA3-IL2-E7 generate the best protective anti-tumor effect against E7-expressing tumors
In order to determine if the mice immunized with the various DNA constructs generate protective anti-tumor effect, we performed an in vivo tumor protection experiment. C57BL/6 mice (5 per group) were immunized twice via gene gun with 2μg/mouse of the various DNA constructs at one-week interval. One week after the last vaccination, the mice were challenged with 5 ×104 TC-1 tumor cells/mouse. The mice were monitored for evidence of tumor growth by inspection and palpation twice a week. As shown in Figure 3, the percentage of tumor-free mice was the highest in the mice immunized with pcDNA3-IL2-E7 among the mice immunized with the various DNA constructs. Thus, our data suggest that mice immunized with pcDNA3-IL2-E7 display a protective anti-tumor effect compared to the mice immunized with the other DNA constructs.
Figure 3. In vivo tumor protection experiments.
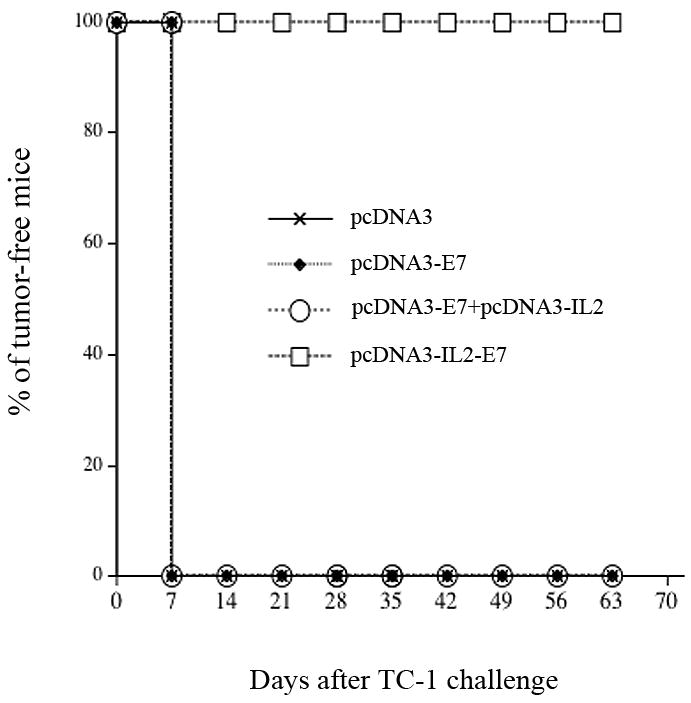
Graphical representation of the percentage of tumor-free mice over time in each group of mice immunized with the various DNA constructs. C57BL/6 mice (five per group) were immunized twice via gene gun with 2μg/mouse of pcDNA3-E7, pcDNA3-IL2, pcDNA3-IL2+pcDNA3-E7 or pcDNA3-IL2-E7 at one-week interval. One week after the last vaccination, the vaccinated mice were challenged subcutaneously with 5×104 TC-1 cells/mouse. The mice were monitored for evidence of tumor growth by inspection and palpation twice a week. The data shown here are from one representative experiment of two performed.
C57BL/6 mice vaccinated with pcDNA3-IL2-E7 generate the best therapeutic anti-tumor effect against E7-expressing tumors
In order to test the therapeutic effects in mice treated with the various DNA constructs, we challenged C57BL/6 mice (5 per group) first with 5×104/mouse of TC-1 cells and then treated tumor-challenged mice three days later with the various DNA constructs. As shown in Figure 4, we observed that mice treated with pcDNA3-IL2-E7 generated the best therapeutic effect among the mice treated with the various DNA constructs (p < 0.05). Thus, our data indicate that treatment with the pcDNA3-IL2-E7 leads to the best therapeutic anti-tumor effect against E7-expressing tumors among all the DNA constructs tested.
Figure 4. In vivo tumor treatment experiments.
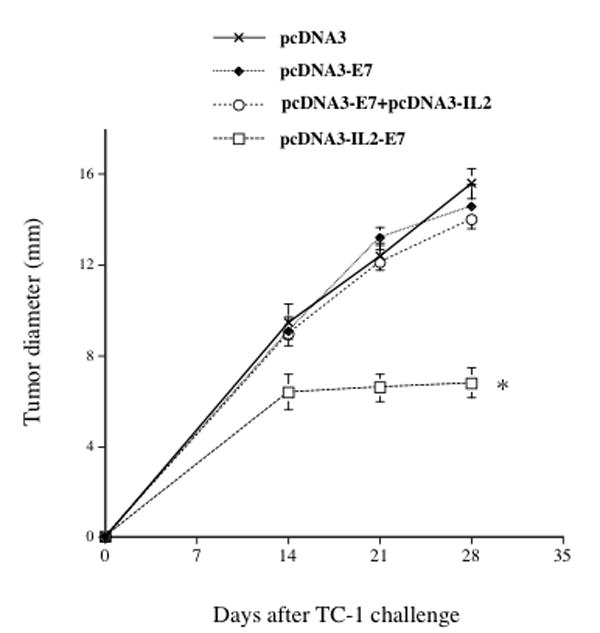
Graphical representation of the tumor size over time in mice treated with the various DNA constructs. C57BL/6 mice (5 per group) were challenged subcutaneously with 5 × 104/mouse of TC-1 cells. Three days later, the mice were treated via gene gun with 2μg /mouse of pcDNA3-E7, pcDNA3-IL2, pcDNA3-IL2+pcDNA3-E7 or pcDNA3-IL2-E7. The animals were sacrificed, the tumors were explanted and the tumor diameter was evaluated. The data was shown as mean±s.d. (* indicates p<0.05). The data shown here are from one representative experiment of two performed.
CD8+ T cells are essential for the protective anti-tumor effect generated by pcDNA3-IL2-E7 vaccine
In order to determine the subset of lymphocytes important for the protective anti-tumor effect in mice vaccinated with pcDNA3-IL2-E7 DNA construct, we performed in vivo antibody depletion experiments using monoclonal antibodies specific for CD4+ T cells (GK1.5), CD8+ T cells (2.43) or NK cells (PK136). C57BL/6 mice (5 per group) were vaccinated with the pcDNA3-IL2-E7 DNA construct at one-week interval. One week after the last vaccination, the mice were challenged with 5×104 TC-1 tumor cells/mouse. Naïve mice were also challenged with TC-1 tumor cells as a control. Depletion was initiated one week before tumor challenge. The mice were monitored for evidence of tumor growth by inspection and palpation twice a week. As shown in Figure 5, we observed that depletion of CD8+ T cells led to a completed loss of the protective antitumor effect generated by the pcDNA3-IL2-E7 DNA construct. Thus, our data suggest that the CD8+ T cells are important for protective anti-tumor immunity in mice vaccinated with pcDNA3-IL2-E7 DNA vaccine.
Figure 5. In vivo antibody depletion experiments.
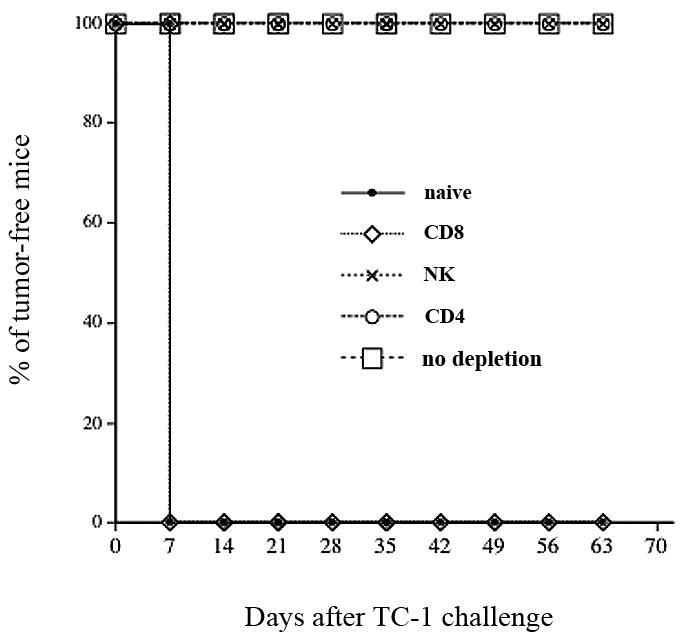
Graphical representation of the percentage of tumor-free mice over time in each group of mice. C57BL/6 mice (5 per group) were immunized twice via gene gun with 2μg/mouse of pcDNA3-IL2-E7. One week after the last vaccination, the vaccinated mice were challenged subcutaneously with 5×104 TC-1 cells/mouse. One week before the tumor challenge, the vaccinated mice were depleted of CD8, CD4 or NK cells using the 2.43, GK1.5 and PK136 monoclonal antibodies respectively every other day for 3 times for the first week and then once every week as described in the Materials and Methods section. The mice were monitored for evidence of tumor growth by inspection and palpation twice a week. The data shown here are from one representative experiment of two performed.
The linkage of IL-2 to E7 leads to enhanced MHC class I presentation of E7 antigen
In order to determine if cells transfected with the pcDNA3-IL2-E7 construct will lead to an enhanced MHC class I presentation of the E7 antigen, we transfected 293 Db, Kb cells [17] with the various DNA constructs as described in the Materials and Methods. The transfected cells were then incubated with the HPV-16 E7 peptide (aa49–57)-specific CD8+ T cell line. The activation of the E7-specific CD8+ T cells was determined using intracellular cytokine staining followed by flow cytometry analyses. As shown in Figure 6, 293 Db, Kb cells transfected with pcDNA3-IL-2-E7 were able to activate more E7-specific CD8+ T cells compared to 293 Db, Kb cells transfected with pcDNA3-E7 protein alone or pcDNA3-E7 mixed with pcDNA3-IL-2. Our results indicate that the cells transfected with the pcDNA3-IL2-E7 construct will lead to an enhanced MHC class I presentation of the E7 antigen compared to cells transfected with pcDNA3-E7 protein alone or pcDNA3-E7 mixed with pcDNA3-IL-2.
Figure 6. Intracellular cytokine staining followed by flow cytometry analysis to determine the activation of E7-specific CD8+ T cells by 293 Db, Kb cells transfected with the various DNA constructs.
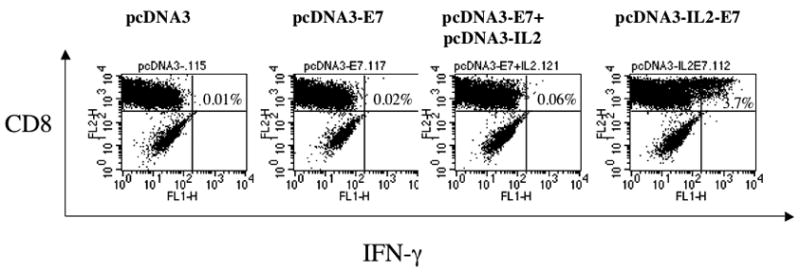
293 Db,Kb cells were transfected with the various DNa constructs and incubated with HPV-16 E7 aa49–57 peptide-specific T cells (with a ratio 1:5) at the presence of 1 ml/ml of GolgiPlug (BD Pharmingen) at 37C overnight. Intracellular staining of IFN-g, flow cytometry and data analysis were performed. A. Representative data of intracellular cytokine stain followed by flow cytometry analysis showing the number of E7-specific IFNγ+ CD8+ T cells in the cells transfected with the various DNA constructs.
In order to determine if the linkage of IL-2 to E7 will influence the function of IL-2, we incubated the CFSE-pulsed HPV-16 E7-speicifc CD8+ T cells with the supernatant derived from 293 Db, Kb cells transfected with the various DNA constructs. As shown in Figure 7, we found that the supernatant derived from 293 Db, Kb cells transfected with pcDNA3-IL2-E7 was able to induce comparable levels of proliferation and expansion of E7-specific CD8+ T cells compared to that derived from 293 Db, Kb cells transfected with pcDNA3-IL-2 alone or pcDNA3-E7 mixed with pcDNA3-IL-2. In contrast, the supernatant derived from 293 Db, Kb cells transfected with pcDNA3-E7 or pcDNA3 alone did not induce proliferation or expansion of the E7-specific CD8+ T cells. Taken together, our data suggest that the observed enhancement of E7-specific CD8+ T cell immune response generated by vaccination of pcDNA3-IL2-E7 is likely contributed by the enhanced direct priming of E7 antigen through linkage of IL-2.
Figure 7. Flow cytometry analysis to characterize the proliferation and expansion of E7-specific CD8+ T cells.
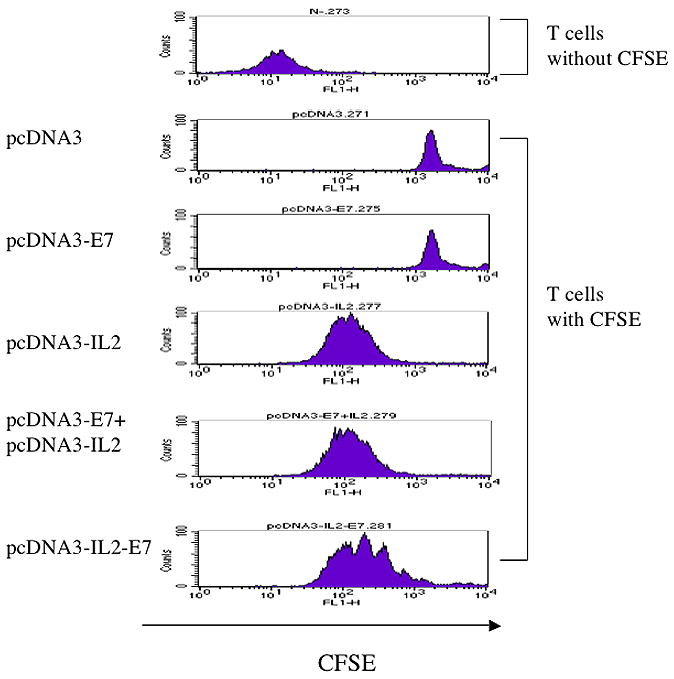
Representative data of flow cytometry analysis depicting the proliferation and expansion of CFSE-labeled E7-specific CD8+ T cells. E7-specific CD8+ T cells were labeled at 1 × 107 cells/ml with 5 μM CFSE (Molecular Probes, Carlsbad, CA) in PBS for 5 min at room temperature followed by incubation with 5% FBS-PBS (5 mM EDTA) for 10 min at 37°C. After three washes with 5%FBS-PBS, 5 × 105/ml of the labeled cells in 1000 μl of media were mixed with 100ul medium from various DNA transfected 293 Db, kb cells in a 24-well plate. After 4 days culture, flow cytometry acquisition was performed.
Discussion
In the current study, we have observed that DNA vaccines encoding a fusion of IL-2 and E7 proteins generated the highest frequency of E7-specific CD8+ T cells. In addition, DNA vaccines encoding a fusion of IL-2 and E7 proteins also generated the strongest protective and therapeutic anti-tumor effect against E7-expressing tumors. We also showed that the CD8+ T cells were responsible for the antitumor effect generated by the DNA vaccine encoding a fusion of IL-2 and E7 proteins. Thus, our data suggest that the linkage of IL-2 to the DNA vaccines encoding HPV-16 E7 antigen significantly enhances the potency of the DNA vaccine against E7-expressing tumors.
We have observed in our study that the physical linkage of IL-2 to the E7 antigen is essential in order to enhance the antigen-specific CD8+ T cell immune responses as well as the antitumor effects of the DNA vaccine. We did not observe any change in the function of IL-2 when IL-2 was linked to E7 (Figure 7). Furthermore, we observed that the linkage of IL-2 to E7 led to enhanced MHC class I presentation of E7 (Figure 6). Taken together, the observed enhancement of the E7-specific CD8+ T cell immune response in mice vaccinated with pcDNA3-IL2-E7 is most likely due to the enhanced MHC class I presentation of E7 antigen through the physical linkage to IL-2.
In comparison, previous studies have used DNA vaccines coexpressing Hepatitis B surface antigen as well as IL-2 as two separate molecules and still generated potent HBsAg-specific immune responses [7]. This study has employed intramuscular injection of the DNA vaccines. Thus, the observed discrepancy may be due to different antigens or different route of administration.
For clinical translation of the therapeutic HPV DNA vaccines, it is important to consider issues relating to safety. Concerns are raised regarding the potential for oncogenicity associated with administration of E7 as DNA vaccines into the body. Thus, it is important to use attenuated (detox) versions of E7 that has been mutated. It has been demonstrated that a mutation at E7 position 24 and/or 26 will disrupt the Rb binding site of E7, abolishing the capacity of E7 to transform cells [19]. In addition, clinical translation may preclude the use of the pcDNA3 vector, because it contains an ampicillin resistance gene. It will important to use an appropriate DNA vector such as the pNGVL4a vector, which can be obtained, from the NIH National Gene Vector Laboratory for DNA vaccine development. This vector is a second-generation plasmid derived from pNGVL-3, which has been previously used for human clinical trials [20]. The pNGVL4a vector lacks the ampicillin resistance gene and thus would be suitable for clinical translation of the current DNA vaccines.
In summary, DNA vaccines encoding IL-2 linked to the HPV-16 E7 antigen demonstrate significantly enhanced potency against E7-expressing tumors. The success of the current study warrants further exploration of other key cytokines that are important for T cell function to identify the ideal cytokine to generate the best enhancement of DNA vaccine potency. Such DNA vaccines will be highly important for future clinical translation.
Acknowledgments
We gratefully acknowledge Dr. T.-C. Wu for helpful discussions. This work was supported by the grants CMRPG34022, 34023, 350571 and the Flight Attendant Medical Research Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Boyd D, Hung CF, Wu TC. DNA vaccines for cancer. IDrugs. 2003;6:1155–1164. [PubMed] [Google Scholar]
- 2.Gurunathan S, Klinman DM, Seder RA. DNA vaccines: immunology, application, and optimization. Annual Reviews in Immunology. 2000;18:927–974. doi: 10.1146/annurev.immunol.18.1.927. [DOI] [PubMed] [Google Scholar]
- 3.Donnelly JJ, Ulmer JB, Shiver JW, Liu MA. DNA vaccines. Annu Rev Immunol. 1997;15:617–648. doi: 10.1146/annurev.immunol.15.1.617. [DOI] [PubMed] [Google Scholar]
- 4.Condon C, Watkins SC, Celluzzi CM, Thompson K, Falo LD., Jr DNA-based immunization by in vivo transfection of dendritic cells. Nature Medicine. 1996;2:1122–1128. doi: 10.1038/nm1096-1122. [DOI] [PubMed] [Google Scholar]
- 5.Porgador A, Irvine KR, Iwasaki A, Barber BH, Restifo NP, Germain RN. Predominant role for directly transfected dendritic cells in antigen presentation to CD8+ T cells after gene gun immunization. Journal of Experimental Medicine. 1998;188:1075–1082. doi: 10.1084/jem.188.6.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Waldmann TA. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nature Reviews Immunology. 2006;6:595–601. doi: 10.1038/nri1901. [DOI] [PubMed] [Google Scholar]
- 7.Chow YH, Huang WL, Chi WK, Chu YD, Tao MH. Improvement of hepatitis B virus DNA vaccines by plasmids coexpressing hepatitis B surface antigen and interleukin-2. Journal of Virology. 1997;71:169–178. doi: 10.1128/jvi.71.1.169-178.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin CC, Chou CW, Shiau AL, Tu CF, Ko TM, Chen YL, et al. Therapeutic HER2/Neu DNA vaccine inhibits mouse tumor naturally overexpressing endogenous neu. Molecular Therapy. 2004;10:290–301. doi: 10.1016/j.ymthe.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 9.Li WR, Niu B, Wang JW, Feng ZJ, Wang DX. Coexpression of interleukin-2 enhances the immunization effect of a DNA vaccine expressing herpes simplex 1 glycoprotein D. Acta Virol. 2006;50:251–256. [PubMed] [Google Scholar]
- 10.Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189:12–19. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 11.Roden R, Wu TC. How will HPV vaccines affect cervical cancer? Nat Rev Cancer. 2006;6:753–763. doi: 10.1038/nrc1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nature Rev Cancer. 2002;2:342–350. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 13.Lin KY, Guarnieri FG, Staveley-O’Carroll KF, Levitsky HI, August JT, Pardoll DM, et al. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996;56:21–26. [PubMed] [Google Scholar]
- 14.He Z, Wlazlo AP, Kowalczyk DW, Cheng J, Xiang ZQ, Giles-Davis W, et al. Viral recombinant vaccines to the E6 and E7 antigens of HPV-16. Virology. 2000;270:146–161. doi: 10.1006/viro.2000.0271. [DOI] [PubMed] [Google Scholar]
- 15.Hung CF, Hsu KF, Cheng WF, Chai CY, He L, Ling M, et al. Enhancement of DNA vaccine potency by linkage of antigen gene to a gene encoding the extracellular domain of Fms-like tyrosine kinase 3-ligand. Cancer Res. 2001;61:1080–1088. [PubMed] [Google Scholar]
- 16.Ji H, Wang T-L, Chen C-H, Hung C-F, Pai S, Lin K-Y, et al. Targeting HPV-16 E7 to the endosomal/lysosomal compartment enhances the antitumor immunity of DNA vaccines against murine HPV-16 E7-expressing tumors. Human Gene Therapy. 1999;10:2727–2740. doi: 10.1089/10430349950016474. [DOI] [PubMed] [Google Scholar]
- 17.Bloom MB, Perry-Lalley D, Robbins PF, Li Y, el-Gamil M, Rosenberg SA, et al. Identification of tyrosinase-related protein 2 as a tumor rejection antigen for the B16 melanoma. The Journal of experimental medicine. 1997;185:453–459. doi: 10.1084/jem.185.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen CH, Wang TL, Hung CF, Yang Y, Young RA, Pardoll DM, et al. Enhancement of DNA vaccine potency by linkage of antigen gene to an HSP70 gene. Cancer Res. 2000;60:1035–1042. [PubMed] [Google Scholar]
- 19.Munger K, Basile JR, Duensing S, Eichten A, Gonzalez SL, Grace M, et al. Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene. 2001;20:7888–7898. doi: 10.1038/sj.onc.1204860. [DOI] [PubMed] [Google Scholar]
- 20.Trimble C, Lin CT, Hung CF, Pai S, Juang J, He L, et al. Comparison of the CD8+ T cell responses and antitumor effects generated by DNA vaccine administered through gene gun, biojector, and syringe. Vaccine. 2003;21:4036–4042. doi: 10.1016/s0264-410x(03)00275-5. [DOI] [PubMed] [Google Scholar]