

Abstract
The key transcription factor that regulates the cellular responses to hypoxia is hypoxia inducible factor-1 (HIF-1). The signaling mechanisms that regulate the hypoxic activation of HIF-1 are not fully understood. Our objective here was to test whether AMP-activated kinase (AMPK) was an upstream regulator of HIF-1. Our results show that AMPK is not required for the hypoxic activation of HIF-1. Interestingly, the AMPK inhibitor, Compound C, inhibits the hypoxic activation of HIF-1 independent of AMPK. Furthermore, we demonstrate that Compound C functions as a repressor of HIF-1 by inhibiting respiration and suppressing mitochondrial generated ROS.
Keywords: Hypoxia inducible factor 1, AMP-activated kinase, Compound C, hypoxia, mitochondria, reactive oxygen species
1. Introduction
Hypoxia is the reduction in normal oxygen tension and occurs during acute and chronic vascular disease, pulmonary disease and cancer. HIF-1 is the master transcription factor that regulates the cellular responses to hypoxia [1]. HIF-1 is composed of two subunits, an oxygen-sensitive HIF-1α subunit, and a constitutively expressed HIF-1β subunit. HIF-1 activity is dependent on the availability of the HIF-1α subunit. Under normal oxygen conditions (21% O2), HIF-1α is targeted for ubiquitin-mediated degradation by an E3 ubiquitin ligase complex [2]. This process is dependent on the hydroxylation of two proline residues by a family of prolyl hydroxylase (PHD) enzymes [3–5]. Under hypoxic conditions, hydroxylation is inhibited and HIF-1α is stabilized [6]. To date the upstream regulators of HIF-1 are poorly understood. The reactive oxygen species (ROS) generated within complex III of mitochondria during hypoxia has been shown by multiple groups to be required and sufficient to activate HIF-1 [6–10]. However, the direct targets of the mitochondrial ROS or the upstream regulators of HIF-1 remain unresolved. Alternatively, it has been proposed that the mitochondria transduce hypoxic signals by regulating the availability of oxygen for the PHDs to hydroxylate HIF-1α [11,12].
AMPK is a heterotrimeric serine/threonine kinase consisting of a catalytic α subunit and two regulatory β and γ subunits [13–15]. AMPK is ubiquitously expressed and functions as an intracellular fuel sensor by maintaining energy balance. Hypoxia, ischemia, and ROS are potent activators of AMPK[16–18]. Previous work has genetically shown that AMPK is not necessary for HIF-1α protein stability [18]. Yet, other reports have suggested that Compound C prevents HIF-1 activation [19,20]. To resolve this discrepancy and to investigate whether AMPK is an upstream regulator of HIF-1 we used both Compound C and AMPKα null cells. Here we show that Compound C prevents the hypoxic activation of HIF-1 by suppressing mitochondrial generated ROS independent of AMPK.
2. Materials and methods
2.1 Cell culture and reagents
Ampkα WT, Ampkα1−/−, Ampkα2−/−, and Ampkα1−/−2−/− mouse embryonic fibroblasts (MEFs), obtained from B. Viollet, were grown in Dulbecco’s modified Eagle’s medium (DMEM) with 4.5 g/L glucose, L-glutamine, and sodium pyruvate, supplemented with 10% heat inactivated fetal bovine serum (Gibco), 100U/ml penicillin, 100μg/ml streptomycin, 0.25μg/ml amphotericin B, and 20mM HEPES. The following reagents were used: Dimethyloxaloylglycine (DMOG) (Frontier Scientific, Inc.), Compound C (Calbiochem), and AICA-Riboside (AICAR) (Calbiochem).
2.2 Oxygen conditions
Hypoxic conditions (1.5% O2 and 5% CO2 balanced with N2) were achieved in a humidified variable aerobic workstation (INVIVO O2, BioTrace), which contains an oxygen sensor that continuously monitors the chamber oxygen tension.
2.3 Immunoblot analysis
HIF-1α protein was analyzed in nuclear extracts as previously described [21]. The anti- HIF-1α antibody (Cayman Chemical) was used and an anti-POL II antibody (Santa Cruz Biotechnology, Inc.) as the loading control. For total cell lysates, the following antibodies were used for immunoblotting: AMPKα, Acetyl CoA Carboxylase (ACC), phospho- Acetyl CoA Carboxylase (Ser79) (all Cell Signaling Technology, Inc.), and an anti-α-tubulin antibody (Sigma-Aldrich, Inc.) to control for loading. A representative blot is shown of three independent experiments.
2.4 Transfections and reporter assays
Transfections were done using the TransIT Transfection reagent (Mirus) according to manufacturer’s protocol. HIF-1 mediated transcriptional activity was measured using the HRE-Luciferase reporter gene construct, a pGL2 vector containing three hypoxia response elements from the Pgk-1 gene upstream of firefly luciferase. Cells were subjected to conditions for 16 hours before lysates were collected. Luciferase values were determined using a dual-luciferase reporter assay kit (Promega). Values for firefly luciferase were normalized to Renilla luciferase under the controlof the thymidine kinase promoter in the pRLTK vector. The data presented are the result of triplicate analyses and the error bars indicate standard error of the mean (SEM).
2.5 Measurement of cell deat
Cell death was measured using a cytotoxicity detection kit (Roche Applied Science) according to manufacturer’s protocol. This kit is based on the measurement of lactate dehydrogenase (LDH) that is released into the medium by damaged cells. Cell death is presented as amount of LDH measured in the medium divided by the total LDH released after treatment with 1% Triton X-100. All cell death results are from four independent experiments and are represented as the mean value ± standard error of the mean (SEM).
2.6 Measurement of ROS
Intracellular ROS generation was assessed using 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) (Molecular Probes). ROS in the cells cause oxidation of DCFH, yielding the fluorescent product 2′, 7′-dichlorofluorescein (DCF). Cells were plated on Petri dishes and incubated with DCFH-DA (10 μM) under various conditions. The media was then removed, cells were lysed, centrifuged to remove debris, and the fluorescence in the supernatant was measured using a spectrofluorometer (excitation, 500 nm; emission, 530 nm). Data were normalized to values obtained from normoxic, untreated controls.
2.7 Oxygen consumption
Cellular O2 consumption rates were measured in aliquots of 1–2 × 106 subconfluent cells removed from flasks and studied in a magnetically stirred, water-jacketed (37°C) anaerobic respirometer fitted with a polarographic O2 electrode (Oxytherm system; Hansatech Instruments). Oxygraph Plus software was used to determine oxygen consumption rate. The data presented is the result of triplicate analyses and the error bars indicate standard error of the mean (SEM).
3. Results
3.1 Compound C inhibits hypoxic activation of AMPK and HIF-1
Compound C is known to function as an ATP-competitive inhibitor of AMPK. To first demonstrate that Compound C inhibits the hypoxic activation of AMPK, cells were exposed to 21%O2 (N), 1.5%O2 (H), DMOG (D), and AICAR (A) ) ± Compound C. To assess AMPK activation, lysates were immunoblotted using an antibody that recognizes the phosphorylation site (Ser79) on acetyl-CoA carboxylase (ACC), a direct target of AMPK [14,22] (Figure 1A). As expected, Compound C inhibits both the hypoxic and AICAR induced activation of AMPK, whereas, the hypoxic mimetic DMOG does not activate AMPK (Figure 1A). DMOG is a PHD inhibitor which serves as a hypoxic mimetic under normal oxygen conditions, whereas, AICA-Riboside (AICAR) mimics AMP, thereby acting as an activator of AMPK. To test the requirement of AMPK for the hypoxic activation of HIF-1, cells were exposed to hypoxia (1.5% O2) or normoxia (21% DMOG ± Compound C. Treatment with Compound C O2) in the presence or absence of completely suppressed the stabilization of the HIF-1α protein under hypoxia, whereas HIF-1α protein level was unaffected with DMOG treatment (Figure 1B). These results were further confirmed by examining HIF-1 activity using the HRE-Luciferase assay (Figure 1C). Hypoxia and DMOG increased HRE-dependent luciferase induction in the untreated cells (Figure 1C). In contrast, cells treated with Compound C displayed a marked attenuation of luciferase induction under hypoxia but not in the presence of DMOG during normoxia (Figure 1C).
Figure 1. Compound C inhibits hypoxic activation of HIF-1.
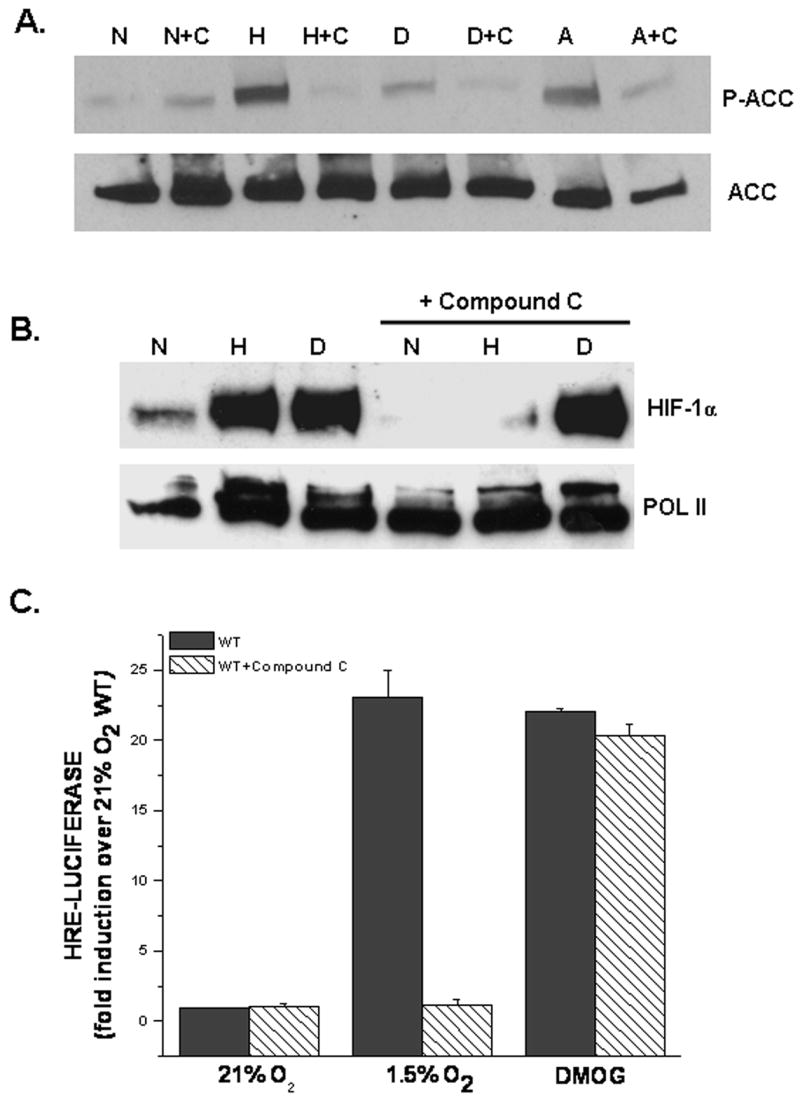
A. AMPK activation assessed by phospho-ACC protein in WT cells exposed to 21%O2 (N), 1.5%O2 (H), 1mM DMOG (D), or 0.5 mM AICAR (A) ± 20μM Compound C (C) for 30 mins. Anti-ACC antibody used as loading control.
B. HIF-1α protein levels in WT cells exposed to 21% O2 (N)± 1mM DMOG (D) or to 1.5% O2 (H) ± Compound C for 2 hours. Anti-POL II antibody used as loading control.
C. WT cells transfected with a HRE-Luciferase reporter gene construct and exposed to 21% O2 ± 1mM DMOG or to 1.5% O2 ± Compound C for 16 hours before being harvested. Firefly luciferase activity was normalized to renilla luciferase activity and data reported as fold induction over 21%O2.
3.2 AMPK is not required for hypoxic activation of HIF-1
To genetically examine the requirement of AMPK for the hypoxic activation of HIF-1, the double knockout cells for AMPKα1 and AMPKα2, as well as, the individual null cells for AMPKα1 and AMPKα2 were used. First, Ampkα WT and the Ampkα1−/−2−/− cells were exposed to hypoxia (1.5% O2) or normoxia (21% O2) in the presence or absence of DMOG. Interestingly, the loss of both AMPKα1 and AMPKα2 did not affect HIF-1α protein levels under hypoxia (Figure 2A). Moreover, Ampkα1−/− and Ampkα2−/− cells also displayed no differences in HIF-1α protein levels when subjected to hypoxia (Figure 2B). Lastly, the HRE-Luciferase assay displayed no difference between the Ampkα WT and the Ampkα1−/−2−/− cells under hypoxia (Figure 2C), providing genetic evidence that the hypoxic activation of HIF-1 is not dependent on AMPK, coinciding with data published by Laderoute et al. [18].
Figure 2. AMPK is not required for hypoxic activation of HIF-1.
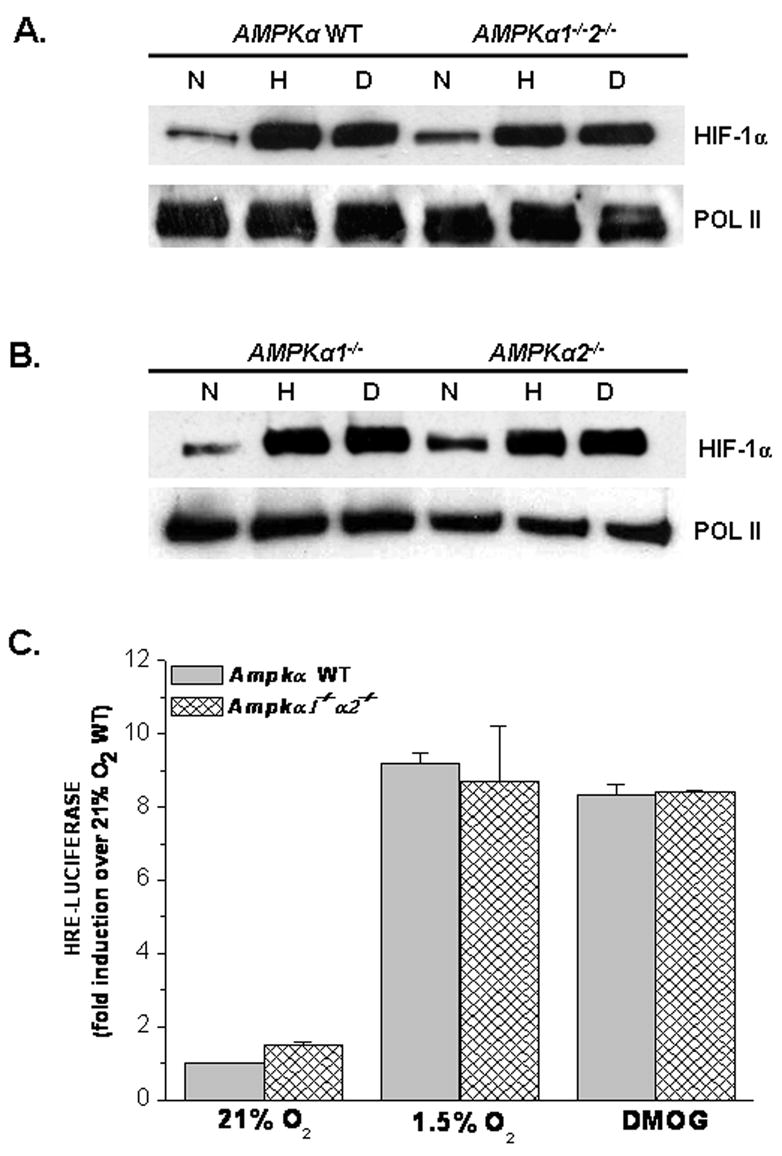
A. HIF-1α protein levels in AMPKα WT and AMPKα1−/−α2−/− cells exposed to 21% O2 (N)± 1mM DMOG (D) or to 1.5% O2 (H) for 2 hours. Anti-POL II antibody used as loading control.
B. HIF-1α protein levels in AMPKα1−/− and AMPKα2−/− cells exposed to 21% O2 (N)± 1mM DMOG (D) or to 1.5% O2 (H) for 2 hours. Anti-POL II antibody used as loading control.
C. AMPKα WT and AMPKα1−/−α2−/− cells transfected with a HRE-Luciferase reporter gene construct and exposed to 21% O2 ± 1mM DMOG or to 1.5% O2 for 16 hours before being harvested. Firefly luciferase activity was normalized to renilla luciferase activity and data reported as fold induction over 21%O2 AMPKα WT.
3.3 Compound C inhibits hypoxic activation of HIF-1 independent of AMPK
To resolve the discrepancy above, a HRE-Luciferase assay was used to examine HIF-1 activity in the Ampkα1−/−2−/− in the presence or absence of Compound C. As shown by immunoblotting, these cells are null for AMPKα (Figure 3A). Similar to Figure 2A, hypoxic and DMOG treated Ampkα1−/−2−/− cells stabilized the HIF-1α protein (Figure 3B). Whereas, treatment with Compound C in the hypoxic Ampkα1−/−2−/− cells inhibited HIF-1α protein stability (Figure 3B). Furthermore, Compound C had no affect on DMOG treated Ampkα1−/−2−/− cells (Figure 3B). Confirming the results shown in Figure 1C, WT cells treated with Compound C displayed a severe reduction in HRE- Luciferase expression when exposed to hypoxia in comparison to untreated WT cells (Figure 3C). Interestingly, hypoxic untreated Ampkα1−/−2−/− cells showed an increase in HRE-Luciferase expression, whereas the hypoxic Ampkα1−/−2−/− cells treated with Compound C displayed no induction of HRE-Luciferase under hypoxia (Figure 3C).
Figure 3. Compound C inhibits hypoxic activation of HIF-1 in AMPKα null cells.
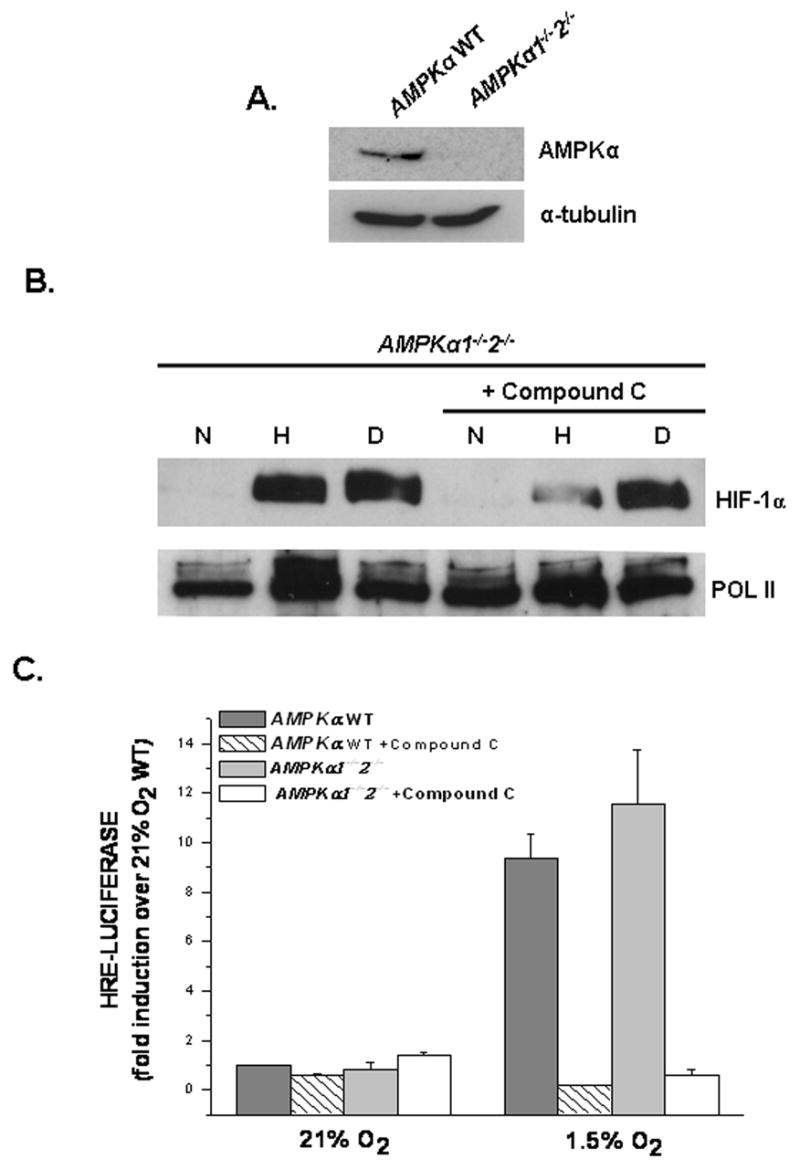
A. AMPKα protein levels in AMPKα WT and AMPKα1−/−α2−/− cells. Anti-tubulin antibody used as loading control.
B. HIF-1α protein levels in AMPKα WT and AMPKα1−/−α2−/− cells exposed to 21% O2 (N)± 1mM DMOG (D) or to 1.5% O2 (H) ± Compound C for 2 hours. Anti-POL II antibody used as loading control.
C. AMPKα WT and AMPKα1−/−α2−/− cells transfected with a HRE-Luciferase reporter gene construct and exposed to 21% O2 ± 1mM DMOG or to 1.5% O2 ± 20μM Compound C for 16 hours before being harvested. Firefly luciferase activity was normalized to renilla luciferase activity and data reported as fold induction over 21% O2 AMPKα WT.
3.4 Compound C inhibits mitochondrial function
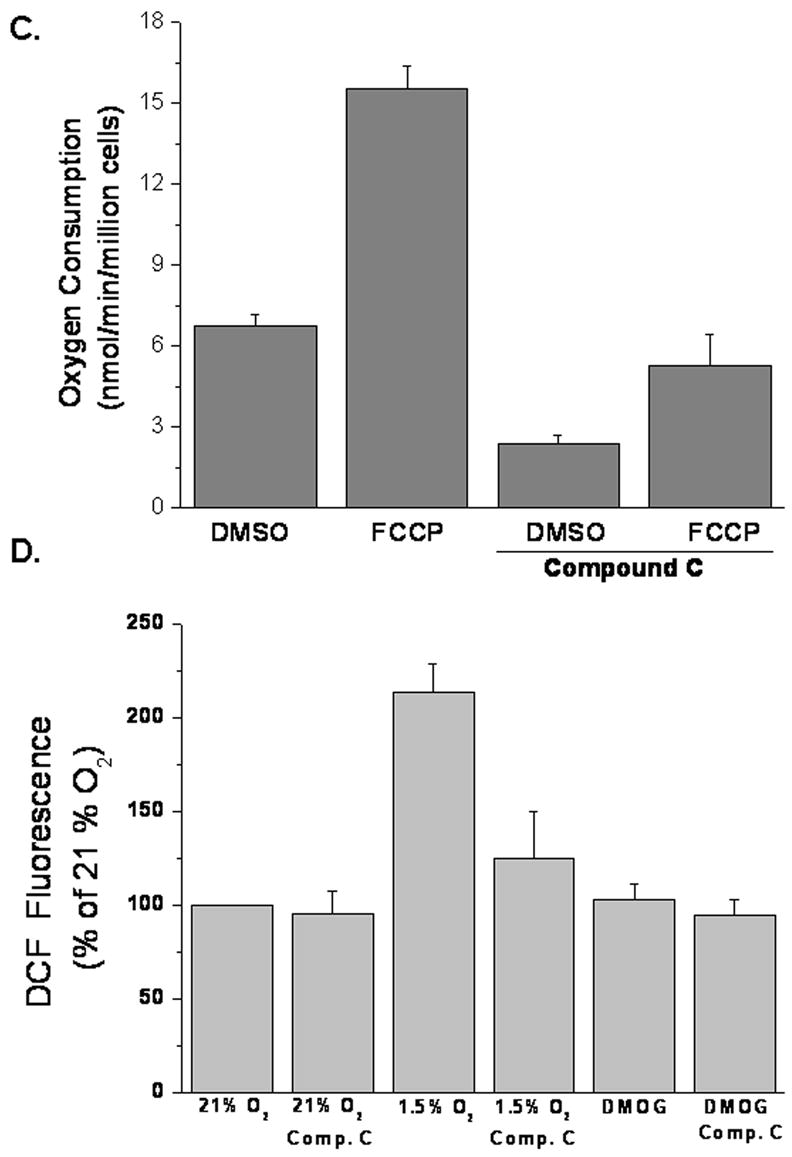
A functional electron transport chain is required for hypoxic activation of HIF-1. To examine if Compound C prevented HIF-1 activity by inhibiting oxidative phosphorylation, cells adapted to either glucose or galactose were treated with Compound C for 24, 48, and 72 hours and cell death was measured by LDH release (Figure 4A). Glucose adapted cells treated with Compound C displayed less than 20% cell death at all time points (Figure 4A). Untreated galactose adapted cells, which rely entirely on oxidative phosphorylation for survival, showed similar percentages of cell death. Galactose adapted cells display higher oxygen consumption than glucose adapted cells (14.4 nmol/min/million cells vs 6.34 nmol/min/million cells). Interestingly, galactose adapted cells that were treated with Compound C exhibited near 40% cell death at 48 hours and greater than 90% cell death at 72 hours (Figure 4A). Therefore, Compound C induces cell death in the presence of galactose. To further examine Compound C’s effect on respiration, oxygen consumption was measured in WT cells in the presence or absence of Compound C (Figures 4B and 4C). In fact, Compound C inhibits respiration. The likely mechanism is electron transport inhibition since compound C inhibits respiration in the presence of the uncoupler FCCP (10 uM). Previous studies have demonstrated that hypoxia increases the generation of oxidants during the ubiquinione cycle within complex III of the mitochondria and these oxidants are required and sufficient for HIF-1 activation [6–10]. Indeed, ROS is generated by hypoxia, as detected by the oxidation of the 2′,7′-dichlorodihydrofluorescein (DCFH) dye and Compound C suppresses mitochondrial generated oxidants in response to hypoxia (Figure 4D). Treatment with DMOG had no effect on ROS generation (Figure 4D).
Figure 4. Compound C inhibits oxygen consumption and ROS generation.
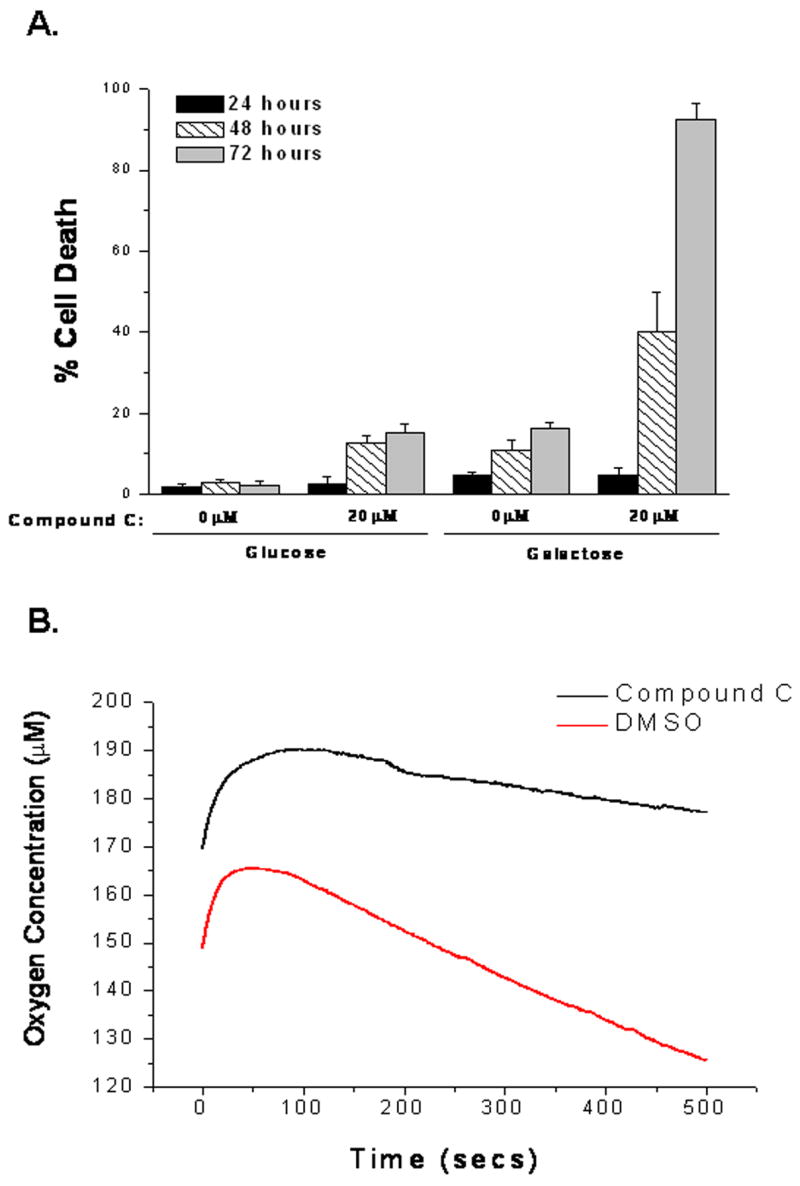
A. WT cells adapted to glucose or galactose ± 20μM Compound C for 24, 48, and 72 hours and cell death assessed by LDH release.
B and C. Oxygen consumption of WT cells with 20μM Compound C or DMSO control in the presence of FCCP. N=3 mean ± S.D.
D. ROS determined by incubating WT cells with DCFH-DA (10μM) exposed to 21% O2, 1.5% O2 ± 20μM Compound C, or to 1mM DMOG ± 20μM Compound C for 30 minutes. N=4 mean ± S.D.
4. Discussion
Compound C is well established and a potent inhibitor of AMPK. Hypoxia is known to activate AMPK. Consistent with previous reports Compound C prevents hypoxia activation of AMPK. Compound C also inhibits the hypoxic activation of HIF-1, correlating with other reports that have suggested that AMPK is critical for HIF-1 activity by using Compound C under hypoxic conditions or exposure to the carcinogenic metal vanadate [19,20]. In contrast to the Compound C data, cells deficient in AMPKα1 and α2 are able to still activate HIF-1 during hypoxia. The novel finding in the present study is that Compound C still prevents the hypoxic activation of HIF-1 in cells deficient in both AMPKα1 and α2. This indicates that Compound C can effectively suppress the hypoxic activation of HIF-1 independently of AMPK. Therefore, Compound C can be added to the list of known AMPK modulators, including AICAR and Metformin, which have off target effects. In addition, DN-AMPK adenoviruses have been shown to inhibit the activation of HIF-1, although these approaches also have off target effects [19,20]. Here we used AMPKα null cells, providing genetics in order to study the requirement of AMPK for the hypoxic activation of HIF-1.
Previous studies have demonstrated that a functional electron transport chain is necessary to produce an increase in ROS that subsequently stabilize HIF-1α in response to hypoxia [6,7,9,10]. Indeed we found that Compound C is also a mitochondrial inhibitor. The actual mitochondrial origin of ROS suppressed by Compound C remains unclear. Bell and colleagues have recently published that the ROS generated by the Q0 site of complex III is required for hypoxic signaling [6]. Therefore, Compound C may be acting directly or indirectly at this site in order to suppress the hypoxic activation of HIF-1. Additionally, Compound C is part of a pyrazolo-pyrimidine class of protein kinase inhibitors, therefore it is possible that this compound is also inhibiting other kinase signaling pathways that regulate HIF-1[23]. For example, p38α MAPK is required for the hypoxic activation of HIF-1 [21]. Compound C may influence the p38 MAPK pathway, as well as, inhibit mitochondrial respiration in order to suppress hypoxic signaling. Cells treated with Compound C failed to survive in media containing galactose instead of glucose. Cells adapted to galactose rely on oxidative phosphorylation for survival. Compound C inhibited respiration as measured by oxygen consumption. Furthermore, Compound C suppresses the hypoxia generated increase in ROS, thus providing a mechanistic explanation for why Compound C is effective in suppressing the hypoxic activation of HIF-1. In conclusion, this study demonstrates that Compound C is not only an inhibitor of AMPK, but it inhibits the hypoxic activation of HIF-1 by suppressing mitochondrial generated ROS. These results further highlight the importance of mitochondria as a major regulator of the hypoxic activation of HIF-1.
Acknowledgments
This work is supported in part by National Institutes of Health Grants (GM60472-08 and CA123067-01) to NSC and the Commission of the European Communities (Contract No LSHM-CT-2004-005272 EXGENESIS) to BV. BME is supported by a fellowship from the American Heart Association Grant 0610044Z. Thanks to Eric Bell for his critical reading of the manuscript and insightful comments.
Abbreviations
- HIF-1
Hypoxia inducible factor 1
- AMPK
AMP-activated kinase
- DMOG
Dimethyloxaloylglycine
- AICAR
AICA-Riboside
- ROS
Reactive Oxygen Species
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Semenza GL. Expression of hypoxia-inducible factor 1: mechanisms and consequences. Biochem Pharmacol. 2000;59:47–53. doi: 10.1016/s0006-2952(99)00292-0. [DOI] [PubMed] [Google Scholar]
- 2.Maxwell PH, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–5. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 3.Ivan M, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–8. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 4.Jaakkola P, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–72. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 5.Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ. Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. Embo J. 2001;20:5197–206. doi: 10.1093/emboj/20.18.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bell EL, Klimova TA, Eisenbart J, Moraes CT, Murphy MP, Budinger GR, Chandel NS. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol. 2007;177:1029–36. doi: 10.1083/jcb.200609074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brunelle JK, Bell EL, Quesada NM, Vercauteren K, Tiranti V, Zeviani M, Scarpulla RC, Chandel NS. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005;1:409–14. doi: 10.1016/j.cmet.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 8.Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000;275:25130–8. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 9.Guzy RD, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–8. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 10.Mansfield KD, Guzy RD, Pan Y, Young RM, Cash TP, Schumacker PT, Simon MC. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab. 2005;1:393–9. doi: 10.1016/j.cmet.2005.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doege K, Heine S, Jensen I, Jelkmann W, Metzen E. Inhibition of mitochondrial respiration elevates oxygen concentration but leaves regulation of hypoxia-inducible factor (HIF) intact. Blood. 2005;106:2311–7. doi: 10.1182/blood-2005-03-1138. [DOI] [PubMed] [Google Scholar]
- 12.Hagen T, Taylor CT, Lam F, Moncada S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science. 2003;302:1975–8. doi: 10.1126/science.1088805. [DOI] [PubMed] [Google Scholar]
- 13.Rutter GA, Da Silva Xavier G, Leclerc I. Roles of 5′-AMP-activated protein kinase (AMPK) in mammalian glucose homoeostasis. Biochem J. 2003;375:1–16. doi: 10.1042/BJ20030048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hardie DG, Scott JW, Pan DA, Hudson ER. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003;546:113–20. doi: 10.1016/s0014-5793(03)00560-x. [DOI] [PubMed] [Google Scholar]
- 15.Carling D. The AMP-activated protein kinase cascade--a unifying system for energy control. Trends Biochem Sci. 2004;29:18–24. doi: 10.1016/j.tibs.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 16.Choi SL, Kim SJ, Lee KT, Kim J, Mu J, Birnbaum MJ, Soo Kim S, Ha J. The regulation of AMP-activated protein kinase by H(2)O(2) Biochem Biophys Res Commun. 2001;287:92–7. doi: 10.1006/bbrc.2001.5544. [DOI] [PubMed] [Google Scholar]
- 17.Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 18.Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J, Foretz M, Viollet B. 5′-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol. 2006;26:5336–47. doi: 10.1128/MCB.00166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hwang JT, Lee M, Jung SN, Lee HJ, Kang I, Kim SS, Ha J. AMP-activated protein kinase activity is required for vanadate-induced hypoxia-inducible factor 1alpha expression in DU145 cells. Carcinogenesis. 2004;25:2497–507. doi: 10.1093/carcin/bgh253. [DOI] [PubMed] [Google Scholar]
- 20.Lee M, Hwang JT, Lee HJ, Jung SN, Kang I, Chi SG, Kim SS, Ha J. AMP-activated protein kinase activity is critical for hypoxia-inducible factor-1 transcriptional activity and its target gene expression under hypoxic conditions in DU145 cells. J Biol Chem. 2003;278:39653–61. doi: 10.1074/jbc.M306104200. [DOI] [PubMed] [Google Scholar]
- 21.Emerling BM, Platanias LC, Black E, Nebreda AR, Davis RJ, Chandel NS. Mitochondrial reactive oxygen species activation of p38 mitogen-activated protein kinase is required for hypoxia signaling. Mol Cell Biol. 2005;25:4853–62. doi: 10.1128/MCB.25.12.4853-4862.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ha J, Daniel S, Kong IS, Park CK, Tae HJ, Kim KH. Cloning of human acetyl-CoA carboxylase cDNA. Eur J Biochem. 1994;219:297–306. doi: 10.1111/j.1432-1033.1994.tb19941.x. [DOI] [PubMed] [Google Scholar]
- 23.Fraley ME, et al. Optimization of a pyrazolo[1,5-a]pyrimidine class of KDR kinase inhibitors: improvements in physical properties enhance cellular activity and pharmacokinetics. Bioorg Med Chem Lett. 2002;12:3537–41. doi: 10.1016/s0960-894x(02)00827-2. [DOI] [PubMed] [Google Scholar]