

Abstract
The widely-held view that neurogenic placodes are vertebrate novelties has been challenged by morphological and molecular data from tunicates suggesting placodes predate the vertebrate divergence. Here, we examine requirements for the development of the tunicate atrial siphon primordium, thought to share homology with the vertebrate otic placode. In vertebrates, FGF signaling is required for otic placode induction and for later events following placode invagination, including elaboration and patterning of the inner ear. We show that results from perturbation of the FGF pathway in the ascidian Ciona support a similar role for this pathway: inhibition with MEK or Fgfr inhibitor at tailbud stages in Ciona results in a larva which fails to form atrial placodes; inhibition during metamorphosis disrupts development of the atrial siphon and gill slits, structures which form where invaginated atrial siphon ectoderm apposes pharyngeal endoderm. We show that laser ablation of atrial primordium ectoderm also results in a failure to form gill slits in the underlying endoderm. Our data suggest interactions required for formation of the atrial siphon and highlight the role of atrial ectoderm during gill slit morphogenesis.
Keywords: ascidian, chordate, placode, atrial siphon primordium, gill slits, FGF signaling
Introduction
Vertebrate neurogenic placodes give rise to specialized features of the sensory system including the organs of the inner ear, the lateral line system, and cranial sensory ganglia. Neurogenic placodes, along with neural crest, have been regarded as important innovations unique to the vertebrate clade and, in fact, as defining characters which allowed for a wide range of adaptive behaviors including an increasingly predatory lifestyle (Northcutt and Gans, 1983). Phylogenies which had placed the cephalochordates as the sister group to vertebrates, which together with the tunicates comprise the chordate phylum, were consistent with this claim as placodes have not been observed in the filter feeding amphioxus. An increasing body of morphological and molecular evidence shows, however, that neurogenic placodes are present in the tunicata (Bassham and Postlethwait, 2005; Manni et al., 2001; Manni et al., 2004; Mazet et al., 2005), implying that these structures predate vertebrates. Proposed phylogenies in which cephalochordata and vertebrata are sister taxa – e.g., those based on morphology and 18S rDNA, 28S rDNA, and mitochondrial gene sequence (Berril, 1955; Cameron et al., 2000; Glenner et al., 2004; Turbeville et al., 1994; Winchell et al., 2002) – are consistent with the appearance of placodes in the chordate ancestor, followed by an independent loss in the cephalochordate lineage. Recent phylogenetic models based on concatenated protein sequences conflict with this view and suggest that tunicates, not cephalochordates, are the true sister group to vertebrates (Bourlat et al., 2006; Delsuc et al., 2006; Philippe et al., 2005; Vienne and Pontarotti, 2006). Under this scenario, neurogenic placodes were present before the vertebrate-tunicate divergence, while their apparent absence in cephalochordates may represent the ancestral state.
Tunicate structures which have been homologized to vertebrate placodes show the same basic morphology, thickenings within the embryonic ectoderm, and include the paired atrial siphon primordia, the oral or stomodeal siphon primordium, the neurohypophyseal organ, and the adhesive organ precursor or rostral placode (Manni et al., 2004). In vertebrates, different inductive cues for different placodes lead to the upregulation of a similar suite of genes (Baker and Bronner-Fraser, 2001; McCabe et al., 2004). Vertebrate placodes are defined by combinatorial gene expression patterns including genes from the six, pax, eya, pitx, fox and dll families. Similar gene expression patterns have been observed in putative placode homologs of ascidians, and in the larvacean, Oikopleura (Bassham and Postlethwait, 2005; Mazet et al., 2005; Mazet and Shimeld, 2005).
A one-to-one correspondence between tunicate and vertebrate placodes may not always be easy to ascertain – vertebrates have more placodes than tunicates, so homology relationships are not likely to be direct in all cases. Vertebrate placode number may be the result of morphological duplications of any one of the ancestral placodes, in which case a single ascidian placode may share homology to more than one vertebrate placode, as has been proposed for the neurohypophysial duct which may be homologous to vertebrate olfactory, adenohypophysial, and hypothalamic placodes (Manni et al., 2001). Alternatively, ascidians may have fewer placodes than the common ancestor, a loss perhaps explained by a comparatively simplified larval body plan and a sessile adult phase. Finally, certain authors posit the existence of a pan-placodal field from which all neurogenic placodes develop – some vertebrate placodes may have evolved through specification of novel territories within this field (Baker and Bronner-Fraser, 2001; Schlosser, 2002).
Combined morphological, positional, and expression data can provide at least a first approximation of homology between vertebrate placodes and tunicate structures (Bassham and Postlethwait, 2005; Manni et al., 2001; Mazet and Shimeld, 2005; Schlosser, 2005). Several authors have proposed homology between the atrial siphon primordia of ascidians, which form the adult atrial siphon, or exhalant siphon, out of which water, waste, and gametes are directed, and the otic placodes of vertebrates, which contribute to structures of the ear (Bassham and Postlethwait, 2005; Jefferies, 1986; Katz, 1983; Manni et al., 2004; Mazet and Shimeld, 2005; Schlosser, 2005; Wada et al., 1998). In Ciona savignyi and Ciona intestinalis, the atrial siphon primordia are evident in larvae as paired ring-shaped structures of approximately six cells in the lateral ectoderm of the trunk, just anterior to the notochord and posterior to the sensory vesicle (Katz, 1983). During metamorphosis each primordium undergoes invagination to form an atrial siphon rudiment. Although details of the morphological movements of the primordia leading to siphon formation are not well described for Ciona, the end result for each primordium is an atrial opening, leading to an atrial cavity which connects to the pharynx through ciliated gill slits. Later, right and left atrial siphons fuse in Ciona post-metamorphic juveniles, leading to the single opening with a shared atrium that is observed in the sessile adult.
Some authors postulate a direct relationship between otic and atrial structures, while others favor a compound homology of atrial primordia to both otic placode and lateral line (Bassham and Postlethwait, 2005; Jefferies, 1986; Katz, 1983; Manni et al., 2004; Mazet and Shimeld, 2005; Schlosser, 2005; Wada et al., 1998). Both otic and atrial siphon precursors appear during development as paired rings of columnar cells, at about the level of the hindbrain in vertebrates and its equivalent in ascidians. A similar morphogenesis also characterises these structures. The otic placode invaginates and pinches off forming the otic vesicle. The process which initiates the formation of the atrial siphon also begins with invagination of the atrial primordium, and, as described in the colonial species Botryllus, the invagination pinches off as a vesicle separate from the overlying ectoderm (Manni et al., 2002). The ascidian atrial primordium is also neurogenic, a feature of most vertebrate cranial placodes: Bone and Ryan (Bone, 1978) described the cupular organ of the atrial siphon of Ciona, a structure similar to the vertebrate mechanosensory hair cell, except that the ascidian cupular organ is not secondarily innervated as are vertebrate hair cells; Mackie and Singla (Mackie and Singla, 2003) describe the hydrodynamic capsular organ of Chelyosoma as consistent with an atrial-otic homology, but like the cupular organ, the capsular organ is not secondarily innervated. In the larvacean tunicate Oikopleura, however, the proposed otic placode homolog, the Langerhans organ, receives mechanosensory stimulation through ciliated cells that are secondarily innervated (Bassham and Postlethwait, 2005).
In vertebrates, functional studies have shown that the fibroblast growth factor (FGF) pathway plays a role in the specification and formation of the otic placode and, as well, that this pathway is important after placode formation during and following the invagination phase, including the morphogenetic movements which give rise to the inner ear. Before otic placode formation, members of the Fgf family are expressed in the periotic mesenchyme beneath the future site of the otic placodes. Studies in fish, mouse and chick have shown that, in particular, members of the Fgf 3/7/10 and Fgf 8/17/18 families play a prominent role during placode specification and induction (Ladher et al., 2005; Leger and Brand, 2002; Liu et al., 2003; Maroon et al., 2002; Phillips et al., 2001; Solomon et al., 2004; Wright and Mansour, 2003). Later in vertebrate embryogenesis, the placode invaginates and forms the otic cyst, and FGF from the hindbrain helps to direct patterning and morphogenesis of ear formation (Liu et al., 2003; Phillips et al., 2001; Wright and Mansour, 2003).
In this report, we test the hypothesis that the otic placode and atrial primordium share homology by perturbing FGF signaling. We find that inhibitors of MAP kinase kinase (MEK) or the Fgf receptor (Fgfr) block formation of the atrial siphon primordium when applied at early tailbud stages, and that similar treatments inhibit siphon and gill slit formation when applied at the onset of metamorphosis. We also use a dye laser to ablate single cells comprising the atrial primordium, resulting in disruption of atrial siphon and gill slit formation. The data presented here allow us to conclude that major aspects of patterning in the mature atrial siphon complex, including gill slit formation, require an intact atrial primordium, and that the development of these structures depends on signaling via FGF.
Materials and methods
Study organisms
C. savignyi were collected from the Santa Barbara yacht harbor or provided by the Ascidian Stock Center (http://www.ascidiancenter.ucsb.edu/). Gametes were collected from gravid adults and combined in sea water to produce embryos (Hendrickson, 2004). Embryos were staged according to a published developmental staging series for C. intestinalis (Hotta et al., 2007).
In situ hybridization
In situ hybridization experiments were carried out as previously described (Corbo et al., 1997), except that the labeled RNA probe was not hydrolyzed prior to hybridization.
Pharmacological inhibitors
U0126 and its inactive analog, U0124, were diluted in DMSO to 10 μg/μL and added directly to embryos at concentrations of 1–50 μM. DMSO-only served as an additional negative control. SU5402 treatments were as for U0126, except that embryos also were treated at late neurula stage. Dechorionated animals showed that same response to treatment as animals with intact chorions, but those with intact chorions had better survival post-metamorphosis, as was the case with untreated control animals.
FGF treatment
Basic human recombinant FGF (Calbiochem) was reconstituted in a solution of PBS + 1 mM DTT + 1% BSA to 25 μg/mL. Embryos were dechorionated and then placed in FGF at concentrations ranging from 625 ng/μL to 5 μg/μL. A control treatment containing PBS solution without FGF was performed alongside each treatment. Animals were allowed to develop until shortly after hatching stage. Larvae were stained for acetylcholinesterase activity (see below) and scored for atrial placode morphology.
Histochemical staining
For acetylcholinesterase (AchE) staining, animals were fixed in 5% formaldehyde in sea water for 5 minutes, then placed in AchE buffer (10 mM Na citrate pH 5.0, 3 mM CuSO4, 0.5 mM KFe cyanide, 65 mM NaOAc) for 10 minutes. Animals were placed in AchE buffer with acetylthiocholine iodide (15 μg/mL) for staining, then placed in PBS and imaged. Staining for alkaline phosphatase activity was performed as described previously (Whittaker and Meedel, 1989) with 5-bromo-4-chloro-3-indolyl phosphate (BCIP) as the substrate.
An antibody against diphosphorylated extracellular signal-related kinase (anti-dpERK1/2, Sigma M8159) has been used previously for Ciona embryos (Hudson et al., 2003; Yasuo and Hudson, 2007). We used Tyramide Amplification Kit #2 (Invitrogen, T20912) following manufacture’s instructions. The mouse anti-dpERK was used at 1:1000 dilution; the kit-supplied HRP-anti-mouse IgG secondary was used at 1:100. Staining was carried out on DMSO-treated and U0126-treated animals; additionally, a negative control omitting the anti-dpERK1/2 primary was run to ensure that the secondary alone did not yield signal. The tyramide substrate reaction was allowed to proceed for 15 minutes.
Tubulin staining
For staining with the anti-acetylated tubulin antibody (Sigma T6793), juveniles were allowed to sit in 10% saturated menthol in seawater for 10 minutes. These animals were placed in 100% MeOH for 5 minutes at room temperature, transferred to 100% EtOH at room temperature for 5 minutes, then washed three times in PBST (PBS + 0.1% Triton-X100). Juveniles were transferred to a 1.5 mL eppendorf tube containing anti-acetylated tubulin antibody in PBS for 1 hour at room temperature or overnight at 4°C. For imaging, anesthetized animals were placed on a depression slide containing seawater mixed with 3% methyl cellulose, and observed with a fluorescein filter set.
Laser ablations
Larvae were collected in the first few hours after hatching and anethetized with 0.05% MS-222 in seawater. Anesthetized larvae were transferred to a glass slide and placed under a cover slip, with a self-adhesive reinforcement acting as a spacer between cover slip and slide. These larvae were observed under DIC optics using a Nikon Microphot-SA microscope. Right or left side atrial siphon primordia were ablated using a dye laser (Coumarin 440 nm; Photonic Instruments, Inc.) attached to the epifluorescence port. Briefly, atrial placode cell nuclei were individually illuminated with the laser until the cell was seen to be pycnotic or rupture.
Results
MEK and Fgfr inhibitors block atrial primordia and siphon development
Vertebrate otic placodes require the activity of FGF family members during induction and invagination/elaboration phases. Pharmacological inhibitors of MEK (U0126) and Fgf receptor (SU5402) were used to test for a similar requirement of FGF signaling in Ciona atrial siphon development. The inhibitors have a significant advantage compared to such treatments as translational inhibition by morpholino oligonucleotides: pharmacological agents may be applied during a discrete developmental window and, therefore, their effects are likely to be proximal to the formation of the atrial primordia. Morpholino-knockdown of fgf’s by injection at blastomere stages, by contrast, results in early, more global defects rendering the later anatomy uninterpretable and a causal link to the absence of primordia tenuous. Two treatment regimes were followed for the pharmacological inhibitors (Fig. 1). Early treatment assessed the effect of the inhibitors on the development of the atrial siphon primordia, evident in hatching-stage tadpoles [approximately 18 hours post fertilization (hpf) at 18°C] (Fig. 2A). In these treatments, embryos were soaked in the inhibitors from the tailbud to hatched tadpole stages. The late treatment (Fig. 1) assessed the effects of the inhibitors on the development of the atrial siphon proper and pharyngeal gill slits during metamorphosis. For these treatments animals were placed in the inhibitor solutions from the beginning of metamorphosis, which corresponds to the time of attachment of the larvae, and cultured until untreated siblings had formed gill slits (approximately 48 hpf).
Fig. 1.

Three separate treatments were used to assess gill slit and atrial primordium formation. 1) Early treatment – inhibitor (U0126 or SU5402) was added at early tailbud stage and remained until a few hours after hatching at which time larvae are scored for presence or absence of atrial siphon primordia (asp). Cartoon indicates only the single right side primordium, which normally forms posterior to the pigmented ocellus and otolith (depicted as crescent and circle). 2) Late treatment began just after larvae attached and began metamorphosis. Animals were scored at approximately 48 hpf, at which time small paired atrial siphons (as) and 1° gill slits had formed. The juvenile depicted is tilted so that both left and right side siphons and gill slits are seen. Also indicated is the flow of water, through the oral or inhalant siphon (os), into the pharynx (ph), out through the atrial or exhalant siphons. 3) Laser ablation of cells comprising a single primordium was carried out after hatching; the contralateral, unablated side served as a control.
Fig. 2.

Inhibitor treatment disrupts atrial primordium formation. Early treatment with U0126 or SU5402 disrupts atrial primordium formation. (A) Lateral view, anterior is right. An untreated animal a few hours after hatching. Arrow indicates single, right-side atrial siphon primordium (asp). Cartoon at bottom right shows area under view. (B) Larva which has undergone early treatment with 10 μM U0126. Atrial siphon primordia are absent. Acetylcholinesterase-stained untreated (C) and treated (D) larvae, about 5 hrs after hatching, dorsal view. Untreated control shows left and right side primordia (arrows), whereas in the treated larva the atrial primordia are absent, although the oral siphon primordium is present (arrow). Dll-A in situ hybridization of pre-hatching stage Ciona savignyi tailbud embryos (dorsal view, anterior to left) shows message is detected in control embryo (E) in patches of ectoderm which precede formation of visible atrial primordia, whereas in drug-treated tailbud embryos this expression is absent (F). Likewise, an antibody which recognized diphosphorylated ERK, the activated form of MEK, shows staining in bilaterally paired patches of ectoderm before the primordia are detectable (G), but in U0126-treated animals is not detected (H). (I) Post-hatching-stage larvae show ERK continues to stain atrial primordia (short arrows) as well as the oral siphon primordium (long arrow). (J) After invagination, faint signal is still detectable in the atrial ectoderm (short arrows, dotted lines) and is also observed around the oral siphon primordia (down arrow) and within the sensory vesicle (up arrow). Signal is detectable at the extreme rostral extent of the animal and in the forming palps, but is not highlighted here.
Early treatment of embryos with U0126 at 20 μM and 10 μM resulted in tadpoles that failed to form atrial siphon primordia (100%, n=86 at 20 μM; n=43 at 10 μM), as determined by both light microscopy and acetylcholine esterase staining (Fig. 2B, 2D). However, the oral siphon primordium, which also stains for acetylcholine esterase, was not disrupted by UO126 treatment (Fig. 2D, arrow; see also Fig. 3D). Untreated embryos always formed both siphons (n=175). As a negative control, an inactive analog of U0126, U0124, was used; no defect in atrial siphon development or any other tissue was observed at concentrations up to 100 μM (n=82). Likewise, DMSO alone had little or no effect on development (n=60). At 5 μM U0126, 19.5% of larvae were seen to have either one or both siphon primordia (n=41), and at 1 μM, no effect was observed (n=27). The homeobox gene distalless A (dll-A) marks atrial primordia and the atrial ectoderm preceding visible placode formation; in DMSO-treated embryos, dll-A is present in late tailbud-stage embryos whereas its expression is absent in U0126-treated embryos (Fig. 2E, F). An antibody against activated extracellular signal related kinase (dpERK), a component of the Fgf-signaling pathway, showed staining in the ectoderm prefiguring the atrial primordia in control animals, but no staining in inhibitor-treated animals (for control animals n=24, with 19/24 animals showing signal; for drug-treated animals n=37, with signal absent in all) (Fig. 2G, H). This signal was detectable in post hatching stages in the atrial primordia proper (Fig. 2I) and was also detectable, although attenuated, after the atrial primordia had begun invagination (Fig. 2J); as with the earlier drug-treated individuals, signal was not at detectable levels in later stage drug-treated animals (not shown).
Fig. 3.

Inhibitor treatment disrupts siphon/gill slit formation in juveniles. (A, B) Control juveniles right-side view, oral siphon (os) is to the right. Post-metamorphosis animals show characteristic 1° gill slits. Staining for acetylated tubulin shows gills slits and peripharyngeal band (pp) and endostyle (es). (C, D) Early-treated animals which are allowed to recover and undergo metamorphosis do not develop gill slits. Tubulin staining is absent where gill slits normally form, but is still present in peripharyngeal band and endostyle. (E,F) Late treatment with U0126; Gill slits and atrial siphon are absent in the treated animal. Tubulin staining shows that while structures such as the peripharyngeal band and endostyle are present, well-formed gill slits are not. (G,H) Fgf8/17/18 in situ hybridization of juvenile Ciona savignyi. (G) Specific staining was observed around the atrial siphon opening (as), within the atrium (at), and in the neural gland (ng). (H) Control sense-strand probe does not show staining in neural gland or atrial siphon. (os, oral siphon.)
Early treatment with the Fgfr inhibitor, SU5402, gave similar results as for U0126. At 40 μM, SU5402 resulted in tadpoles with no atrial primordia (100%, n=40). In addition many tadpoles also lacked the characteristic thickening at the anterior end of the animal where the palps form. A lower concentration, 20 μM, was effective in eliminating the primordia, with no effect on the palps, if animals were treated at late neurula stages (100%, n=25). We conclude that the SU5402 treatment shows a similar effect to the MEK inhibitor. Aside from the atrial primordia phenotype, and the palp phenotype at high SU5402 concentration, animals treated with U0126 or SU5402 appeared normal in other respects. The sensory vesicle, the oral siphon primordium (similar in structure to an atrial primordium, but positioned at the dorsal midline anterior to the sensory vesicle), the notochord, and the tail all appeared normal.
Animals treated early with U0126 were transferred to U0126-free, DMSO-free seawater to recover, and allowed to undergo metamorphosis. The resulting juveniles had largely normal morphology (Fig. 3C). However, closer inspection of the juveniles revealed the absence atrial siphons and gill slits; a tubulin antibody, which recognizes cilia, confirmed a lack of gill slits (Fig. 3D). Consistent with our observation that early U0126 treatment did not disrupt the oral siphon primordium (Fig. 2D), the oral siphon of these juveniles appeared to form properly (Fig. 3C). Perhaps due to the absence of the outflow (i.e., atrial) siphon, the pharyngeal cavity showed an enlarged, “gaping” phenotype.
To determine a more precise temporal window during which exposure to the MEK inhibitor was effective, we performed a time course. U0126 treatment at 10 μM for each of the first four hours beginning at early tailbud [early tailbud I, stage 19 (Hotta et al., 2007)] resulted in hatched larvae with no atrial primordia (Fig. 4A). We were unable to find an earliest time when drug treatment had no effect on atrial primordia because earlier treatments resulted in abnormal embryos that included many other defects including lack of palps (and an inability to settle, as was seen with highest concentrations of SU5402), lack of trunk elongation, and improper migration of the pigmented precursors of the otolith and ocellus, and inability to hatch. Treatment after the first four hours of tailbud development had no apparent effect on the atrial primordia even at longer duration of U0126 exposure, including treatment from approximately 5 hours after early tailbud until hatching. We conclude that MEK dependence in atrial primordia development persists until a few hours after the beginning of the early tailbud stage.
Fig. 4.

Time course with U0126 during tailbud and post-settling stages reveals temporal differences in MEK dependence. (A) Early tailbud (stage 19) treatment with U0126 at 10 μM shows that after hr 4, atrial primordia development is unaffected by the inhibitor. Control DMSO-only treatment has no effect. (B) Post-settling treatment with 40 μM U0126 reveals that, except for first 12-hr period following attachment, treatment during any of the next three 12-hr periods results in a loss of gill slits. Likewise, treatment for either of the 24-hr periods following attachment result in a loss of gill slits. DMSO-treatment during any of these periods does not result in gill slit loss. Numbers above bars indicate number of affected animals over the total observed.
Inhibitor treatment at metamorphic/juvenile stages disrupts siphon and gill slit development
Vertebrates require FGF signaling after the formation of the otic placode, at the invagination stage, and for the formation of the inner ear. A strict one-to-one homology between the ascidian atrial placode and the vertebrate inner ear or other structures is not easily made. Nevertheless, we reasoned that some of the same signaling requirements may still be in place if the otic placode and atrial siphon primordia share a basic level of homology. Ciona has a biphasic lifestyle which includes a swimming larval phase followed by a sessile, adult filter-feeding phase. The larva finds a suitable substrate and attaches anterior end first, via its adhesive palps, then resorbs its tail, undergoes rearrangement of viscera, and forms many of the adult structures associated with its filter-feeding mode. Among these structures are an oral siphon leading into a pharynx which is perforated by numerous gill slits through which, in turn, an out current flows through the atrium and out the atrial siphon (Fig. 1).
Animals treated at metamorphosis (Fig. 1, late treatement) with U0126 at 10 μM were found to lack both atrial siphons and gill slits, when observed at 48 hpf (Fig. 3E, F). The same effects were observed at 20 μM. The absence of the atrial siphon in treated animals was always associated with the absence of gill slits (i.e., treated animals were never observed to have gill slits but no siphon openings, or siphon openings but no gill slits). In contrast to the animals treated at tailbud stages which showed a “gaping” pharynx and oral siphon, the late-treated animals were characterized by an oral siphon aperture and a pharyngeal cavity that were more constricted. Like the animals treated at earlier stages, however, overall development of the animals was not grossly abnormal, and the formation of tissues such as the endostyle, esophagus and stomach, was not disrupted. Treatment with SU5402 at 40 μM yielded results that were similar to those seen with addition of U0126 at the same stage.
A time course with UO126 for 12 hour or 24 hour periods during the first 48 hours after attachment showed that at 40 μM U0126, treatment during the first 12-hr period after attachment did not result in a loss of gill slits, but treatments over the any of next three 12-hr periods did (Fig. 4B; animals were scored at 72 hours post-metamorphosis). At lower concentrations, 10 μM or 20 μM U0126, no single 12-hr period resulted in the loss of gill slits, but treatment for the first or second 24-hr periods did (data not shown). These treatments indicate that MEK-dependent events in gill-slit morphogenesis occur during both the first and second days after settling. Moreover, results from the highest concentration treatment suggest that these events occur after the first 12-hr period following attachment and tail resorption. These data are consistent with the temporal appearance of nascent gill slits as described for C. intestinalis (Chiba et al., 2004).
Expression of Fgf orthologs in the trunk lateral mesenchyme and early atrial siphon
Having demonstrated that MEK and Fgfr inhibitors block atrial primordium formation early and atrial siphon and gill slit formation at metamorphosis, we next determined where candidate Fgf genes might be expressed during Ciona development. Fgf genes have undergone extensive duplication in the vertebrate lineage. Ascidian Fgf family genes are named based on their similarities to vertebrate Fgf’s. For example, Fgf8/17/18 is thought to be the single ascidian Fgf with orthology to vertebrate Fgf8, Fgf17, and Fgf18, respectively.
Fgf8/17/18 expression has been previously described for early and larval stages for Ciona intestinalis (Imai et al., 2004; Imai et al., 2002). Fgf8/17/18 transcript is localized to the bilaterally paired A7.6 cells, which fate to trunk lateral mesenchyme cells, by the 64-cell stage. Expression persists through early tailbud stages, at which time staining is seen in a broad area in the mesenchyme underlying the future site of the atrial siphon primordia. Fgf 8/17/18 is also found in some epidermal lineages (but not in the atrial primordia), the tip of the tail of the tailbud embryo, and in cells of the nervous system thought by some authors to lie in a region homologous to the vertebrate midbrain-hindbrain boundary (Jiang and Smith, 2002; Wada et al., 1998; Wada and Satoh, 2001), but whose functional and anatomical equivalence have yet to be demonstrated rigorously (Canestro et al., 2005).
We examined the expression of Fgf8/17/18 in animals during early metamorphosis. Fgf8/17/18 transcript was localized to the outer ring forming the entrance to the atrial siphon and faintly within the atrium itself (Fig. 3G). We also saw localization of the Fgf 8/17/18 message to the cells at the entrance of the oral siphon and in the neural gland.
Because members of the Fgf3/7/10 family have also been implicated in vertebrate otic placode formation, we cloned a C. savignyi Fgf 3/7/10 and performed in situ hybridization at tailbud stages preceding atrial primordium formation and during metamorphosis. Fgf3/7/10 staining has also been described previously (Imai et al., 2004). Fgf3/7/10 transcript appears to be restricted to the developing tail at tailbud stages. Staining is never observed in the trunk epidermis, but low levels of expression (relative to Fgf 8/17/18) are observed the mesenchyme underlying the future site of atrial primordium formation, as has been observed for Fgf8/17/18 (results not shown). We did not observe staining for Fgf3/7/10 in post-metamorphic juveniles. We conclude that the mesenchymal expression in the trunk for Fgf3/7/10 is consistent with a role in initial specification of the atrial placodes, but that our inability to detect it in juveniles suggest that it is unlikely to have a later role in the formation of the atrium or gill slits.
We reasoned that treatment with exogenous FGF protein might have an effect on atrial primordia development, resulting in larger or ectopic atrial primordia. Animals treated with the highest concentration of FGF (5 μg/μL) were uninterpretable; they failed to undergo trunk elongation, had shortened tails, were missing palps and pigment cells, and lacked any AchE staining in the trunk, where it normally marks not only siphon primordia, but also trunk epidermal neurons, palps and heart primordia, indicating massive cell fate transformations. Lower concentrations had no observable effect in atrial primordium morphology, neither in cell number nor in size. Additionally, we never observed ectopic patches of AChE staining that may have indicated ectopic primordia. We believe that this result indicates that the presumptive atrial ectoderm likely responds to a localized signal of FGF and that relative levels of FGF may also be important. Likewise, this may indicate that the competence of the Ciona embryonic ectoderm to respond to inducing signal is also spatially restricted. Implantation of an FGF-coated bead, a difficult manipulation in Ciona but perhaps feasible in an ascidian with a larger embryo, could help to resolve this.
Laser ablation of atrial siphon primordia results in lack of atrial siphon complex
Laser ablation was used to eliminate the atrial primordium ectoderm to determine the capacity of the animal to regulate for loss of atrial primordia. The ablation procedure allowed us to examine the correlation between the perforation of pharyngeal endoderm during gill slit formation and the invagination of siphon primordium ectoderm suggested by the close apposition of pharyngeal endoderm and siphon primordium ectoderm and by the absence of gill slits in drug-treated animals in which atrial primordium formation failed to occur (Fig. 1). We ablated the atrial siphon primordium soon after its first appearance, just after hatching, at about 20 hpf. We ablated individual cells of either the left or right primordium, allowing the untreated side to serve as a control. These animals were allowed to reach attachment/settling stage and to undergo metamorphosis. The treated, ablated side of an animal failed to form atrial siphons and gill slits (Fig. 5A, B). Absence of atrial siphon openings was confirmed by staining for acetylcholinesterase which shows a characteristic ring pattern around the entry to the atrium in untreated animals; ablated animals lacked this staining pattern (data not shown). Absence of gill slits was observed under transmitted light and by fluorescence using a tubulin antibody which recognizes gill slit cilia (Fig. 5D, F). In a small number of cases, a small patch positive for the anti-tubulin antibody on the ablated side could be found, though well-formed gill slits were not evident. If unilaterally ablated animals which showed loss of siphon and gill slits were allowed to develop further, no gill slits were ever seen on the ablated side (up to 7 days).
Fig. 5.
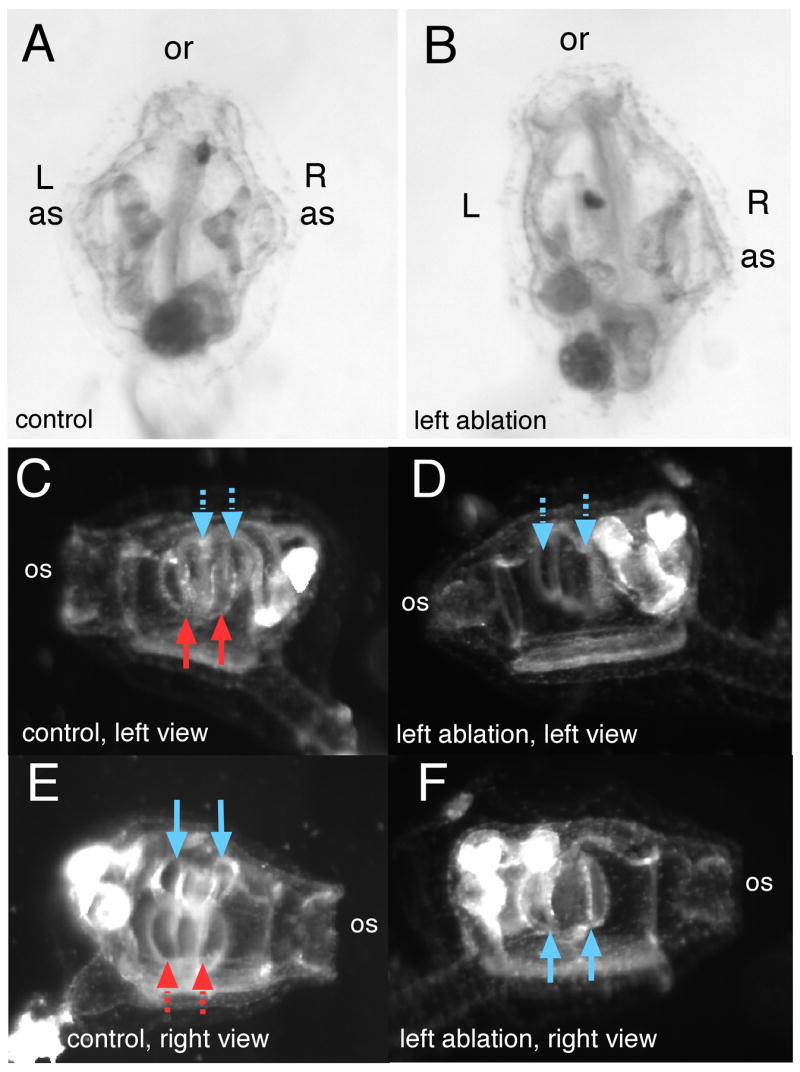
Laser ablation of an atrial siphon primordium disrupts later siphon and gill slit development. (A) Control, unablated juvenile, dorsal view, oral siphon (os) is at top. The chevron-shaped structure underlying right or left atrial siphons (as) are the primary gill slits. (B) A left-sided laser ablation of the atrial primordium at results in an animal with no atrial siphon and no primary gill slits on the ablated side. Left or right side views in dark field reveal paired primary gill slits on either side of a control animal (C,E); red arrows point to left-side primary gill slits, blue point to right-sided. Solid arrows are in the plane of focus; dashed arrows are slightly deeper, on the other side of the animal depicted in the panel. (D,F) A left-ablated juvenile reveals that primary gill slits form only on the untreated side.
Patterning of blood cavity between primary gill slits is independent of the atrial siphon complex
In early Ciona juveniles the first two primary gill slits (called “I” and “IV”) lie inside the atrium at the interface with the underlying pharyngeal cavity (Fig. 1). The primary gill slits are crescent shaped and positioned one next to the other. The small space between the two primary gill slits is occupied by a narrow coelomic-like cavity through which blood cells circulate (Fig. 6A). Tunicates are thought to lack a true, mesodermally-lined, coelom (Zeng and Swalla, 2005); we use the term here simply to refer to the cavity formed between outer epidermal and inner endodermal epithelia. The appearance of this cavity in the wildtype untreated animal coincides with the appearance of the primary gill slits themselves, so it may be a feature that shares patterning information with the gill slits or it may, in fact, be patterned by the gill slits themselves. We speculate that this structure may be a transverse bar (also known as a “gill bar”), a structure found between adjacent gill slit pairs early and later between rows of gill slits. Its appearance between gill slits I and IV may represent a slightly earlier appearance than has been previously reported (Chiba et al., 2004).
Fig. 6.

Inter-gill coelomic structure is not affected in inhibitor-treated animals. (A) An untreated juvenile view from the right-side shows characteristic inter-gill coelom (white arrowheads) in the space separating the 1° gill slits (black arrows, slightly out of plane of focus). (B) Inhibitor-treated animals retain this feature, indicating that this structure is either patterned independently of the gill slits, or can regulate after loss of the gill slits.
In animals treated with MEK or Fgfr inhibitor at early tailbud stages which are then removed from inhibitor after hatching and allowed to settle and undergo metamorphosis, the atrial siphon and gill slits do not form. However, in these treated animals a cluster of cells was observed forming a line, perpendicular to the long axis of the pharyngeal cavity on both left and right sides of the animal (Fig. 6B). These cells have a similar appearance to the cells making the inter-gill coelomic cavity of untreated animals, and moreover, seem to be in the proper orientation and proper position. We conclude that this structure is likely the inter-gill coelom and that its formation is not dependent on the FGF-signaling pathway, nor does it depend on inductive interactions from structures of the developing atrial complex, such as the primary gill slits. Supporting this notion, animals which underwent unilateral ablation also continued to show a similarly arranged structure on the ablated side (data not shown).
Discussion
The FGF-signaling cascade in atrial primordium development and siphon/gill slit formation
Inhibition of MEK and the Fgf receptor block development of atrial primordia at tadpole stages, showing that FGF signaling is required during induction or specification phases of the atrial primordia. We believe the source of signal for primordium induction is likely to be FGF from the mesenchyme underlying the future atrial primordia, where localized Fgf 8/17/18 expression is observed. Exposure of whole embryos to FGF does not result in expanded or ectopic primordia; this may indicate a requirement for localized signal in the mesenchyme or, likewise, a restricted competence in the overlying trunk ectoderm to respond to the inducing signal. Fgf expression in the ascidian mesenchyme would be homologous to Fgf in the periotic mesenchyme of vertebrates, which occupies the same position relative to the future otic placodes as Fgf8/17/18 does to the atrial primordia. In vertebrates, this domain of Fgf is required for proper otic placode induction and removal of this domain results in a loss of otic placodes, while expansion results in ectopic placodes (Ladher et al., 2005; Leger and Brand, 2002; Liu et al., 2003; Maroon et al., 2002; Phillips et al., 2001). Takuoka et al. report that in morpholino knock downs of the transcription factor Twist-like 1, mesenchyme fails to form; the resulting animals do not form gill slits, but they do form atrial siphons indicating that atrial siphon primordia formation likely occurred unimpeded, even in the absence of mesenychyme (Tokuoka et al., 2005). These experiments did not directly examine Fgf expression and did not assess the presence/absence of mesenchyme until hatched larval stages but, nonetheless, suggest the possibility that the Fgf signal may not originate solely from the mesenchyme.
MEK and Fgfr inhibitors also disrupt siphon and gill slit formation when applied shortly after attachment, during the onset of metamorphosis. We conclude that the invagination of the atrial ectoderm leading to formation of the atrial siphon also requires FGF, from within the invaginating ectoderm itself (where Fgf8/17/18 message is detected) or from another source (e.g., from the neural gland, where Fgf is also expressed).
Larvae show no ability to regulate after treatment at early or at late stages. A larva which has undergone early inhibitor treatment, leading to loss of atrial primordia, followed by recovery in untreated seawater, will undergo metamorphosis but will not recover the primordium and will not develop siphon or gill slits. An animal which has undergone late treatment and then is allowed to recover in untreated seawater will likewise never develop normal siphon or gill slit structures.
The inhibitor treated animals are apparently normal in other respects and show no gross anatomical defects. In early-treated animals the oral siphon primordia and palps form correctly, the sensory vesicle is present, rostral trunk epidermal neurons are present and the notochord and tail appear normal. Likewise, late-treated animals show normal morphology after metamorphosis with no aberrations in visceral rotation of gut, esophagus, heart, endostyle, or neural gland. These observations and previous reports suggest that much of the FGF-dependent patterning may have already taken place by the stage at which the inhibitor is added (Delaune et al., 2005; Furthauer et al., 2004; Kim and Nishida, 2001; Streit et al., 2000; Wilson et al., 2000). Nevertheless, U0126 and SU5402 may have had more subtle phenotypic effects which we were unable to detect.
The laser ablation, by contrast, is restricted in its effect to the primordium itself and so the resulting lack of structures which make up the siphon are likely a direct effect of the loss of the primordium. As with the inhibitor-treated animals, laser ablated animals show no recovery of the primordium itself, nor of the atrium or gill slits. The narrow coelomic space between the gill slits, however, is resistant to both pharmacological and laser ablation treatment and is likely to be patterned independently of the primordium, the siphon, and gill slits.
Is gill slit formation within the endoderm induced by contact with atrial ectoderm?
The leading surface of the invaginating ectoderm comes into close apposition with the underlying endodermal epithelium of the pharynx. This interaction leads to the formation of the branchial plate, and ultimately the branchial fissures that are the rudiments of the primary gill slits. A single gill slit consists of endodermally derived ciliary endodermal epithelium bordering the branchial pore (Hirano and Nishida, 2000). The ciliated pore, the gill slit proper, is the perforation between two layers of epithelia, the branchial epithelium on the inner side toward the pharyngeal cavity, and the peribranchial epithelium facing outwards towards the base of the atrium. The precise endodermal/ectodermal boundaries within the atrium/branchial complex remain uncertain, however. Much of the atrium, that portion of the ectodermal invagination which forms a channel or tube and is connected to the pharyngeal basket through the gill slits, is likely to be ectodermal, at least after the early stages of siphon formation. But opinion in the current literature is divided about where the ectodermal component ends and the endodermal component begins. In the colonial Botryllus, observations show that the inner chamber of the atrium contains tunic, which is only secreted by the epidermis (Manni et al., 2002). Scanning and transmission electron microscopy of Botryllus at stages when siphon and gill slit formation are taking place show that the peribranchial epithelium is made up of the most deeply invaginated portion of atrial ectoderm which makes contact with the endodermal branchial epithelium, thereby forming the branchial plate and fissures marking the site of gill slit formation (Manni et al., 2002). In Botryllus, the ectoderm/endoderm boundary is precisely at the gill slits themselves with the outer portion ectoderm and the inner endoderm.
In the ascidian Halocynthia, however, lineage tracing experiments following endoderm show that endodermal derivatives are found not only in the branchial side towards the inner pharyngeal cavity, but also extend to the peribranchial side towards the atrium (Hirano and Nishida, 2000). Additionally, there is a contribution to the gill slit epithelium from the A7.6-derived mesenchyme cells in Halocynthia; the same lineage contributes to the gill slit epithelium of Ciona (Tokuoka et al., 2005). Although the morphogenesis of atrial siphon and gill slit formation have not been described extensively in Halocynthia, the lineage tracing suggests that the atrial ectoderm must fuse with the endoderm within the atrium. However, the branchial fissures between peribranchial and branchial epithelia which mark the primary gill slits, are endodermal derivatives.
Our results indicate that the invaginating atrial ectoderm is required for proper pharyngeal endoderm perforation and gill slit formation, whatever the precise lineage contributions are to siphon and gill structures in Ciona. Moreover, the temporal priority of the atrial primordium – its appearance long before any overt sign of gill slits, the close association of atrial placode invagination with gill slit formation and the inability to regulate in the absence of the atrial primordia, even in those animals in which the endoderm lineages and their derivatives are normal, suggests that the atrial ectoderm could play an inductive or instructive role in gill slit formation.
Homology of atrial primordia and otic placodes
The homology assignment of the ascidian atrial primordium to the vertebrate otic placode rests primarily on similarity of position, topology, morphogenesis, combinatorial gene expression patterns. To this we can add a similar signaling requirement during induction of the placode as well as in post-induction differentiation and morphogenesis. The phylogenetic proximity of tunicates and vertebrates drives the assumption that similar characters are similar by virtue common descent, whether tunicates are sister to a vertebrate/cephalochordate clade, as has been depicted in many phylogenies (Cameron et al., 2000; Glenner et al., 2004; Winchell et al., 2002), or the sister taxon to the vertebrates, a view which has gained recent support (Blair and Hedges, 2005; Delsuc et al., 2006; Oda et al., 2002; Philippe et al., 2005; Vienne and Pontarotti, 2006). The expression of Fgf8/17/18 in the mesenchyme underlying the future site of the atrial primordium shows that this region may be homologous to the periotic mesenchyme of vertebrates. We speculate that the action of the pharmacological inhibitors on atrial primordium development is through inhibition of FGF signaling from this periplacodal mesenchyme. Minimally, this would indicate that the last common ancestor of vertebrates and tunicates had paired placodes positioned at the anterior-posterior level of the hindbrain or its equivalent and that these placodes required FGF signaling from the periplacodal mesenchyme. Subsequent morphogenetic movements, invagination, and vesicle formation also depended on the FGF pathway.
While the similarity in Fgf expression in the periplacodal region and the requirement for FGF during placode formation is likely to be homologous, it is more difficult to homologize the requirement for FGF post-placode formation. In vertebrates, FGF signaling from the hindbrain is important for inner ear formation (Kil et al., 2005; Wright and Mansour, 2003); FGF from the neural keel and cranial mesoderm of zebrafish is required during formation of the pharyngeal pouches (Crump et al., 2004). The adult ascidian body plan is specialized for sessile filter feeding and does not have obvious or direct correlates to each part of the vertebrate anatomy. A report citing expression of Phox2, however, posits that the post-metamorphic brain of Ciona corresponds to the vertebrate hindbrain (Dufour et al., 2006). Post metamorphosis, Fgf8/17/18 is expressed at the atrial opening and in the neural gland. We have not yet determined the source of signal required during atrium formation and gill slit morphogenesis/pharyngeal perforation, but speculate that although these processes retain a requirement for FGF, the radical anatomical rearrangements of ascidian metamorphosis may have led to a heterotopic change in the signal. So, while signal from the neural gland may be most closely homologous to FGF from the hindbrain or cephalic mesoderm of vertebrates, FGF at the atrial primordium itself may also have taken on a role in atrium and gill slit morphogenesis. Selective removal of the neural gland might help to test this. Nervertheless, the use of FGF during induction and post-invagination morphogenesis of atrial primordia and otic placodes argues for an early origin, before the last common ancestor of tunicates and vertebrates, of this signaling pathway in the development of this placode. The presence of gill slits in a deuterostome group outside of the chordates, the hemichordates - the sister group to the echinoderms - argues for a possible shared origin of gill slits nearer the base of the deuterostomes (Rychel et al., 2006). Although neurogenic placodes or similar structures have not been described for hemichordates such as the acorn worm Saccoglossus kowalevskii, we suggest that experiments testing the dependence of hemichordate gill slit development on FGF could be informative regarding the evolution of these structures within the deuterostome lineage.
Acknowledgments
We thank the laboratories of Kathy Foltz and Patrick Lemaire for advice on pharmacological inhibitors, Joel Rothman for use of the dye laser, and Hitoyoshi Yasuo advice and protocols using the dpERK antibody. We also thank Yuki Nakatani for helpful comments on the manuscript. This work was supported by grants from the National Institutes of Health (HD038701and GM075049).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baker CV, Bronner-Fraser M. Vertebrate cranial placodes I. Embryonic induction. Dev Biol. 2001;232:1–61. doi: 10.1006/dbio.2001.0156. [DOI] [PubMed] [Google Scholar]
- Bassham S, Postlethwait JH. The evolutionary history of placodes: a molecular genetic investigation of the larvacean urochordate Oikopleura dioica. Development. 2005;132:4259–72. doi: 10.1242/dev.01973. [DOI] [PubMed] [Google Scholar]
- Berril NJ. The Origin of Vertebrates. Oxford University Press; London: 1955. [Google Scholar]
- Blair JE, Hedges SB. Molecular phylogeny and divergence times of deuterostome animals. Mol Biol Evol. 2005;22:2275–84. doi: 10.1093/molbev/msi225. [DOI] [PubMed] [Google Scholar]
- Bone AaRKP. Cupular sense organs in Ciona (Tunicata:Ascidiacea) Journal of Zoology. 1978;186:417–429. [Google Scholar]
- Bourlat SJ, Juliusdottir T, Lowe CJ, Freeman R, Aronowicz J, Kirschner M, Lander ES, Thorndyke M, Nakano H, Kohn AB, Heyland A, Moroz LL, Copley RR, Telford MJ. Deuterostome phylogeny reveals monophyletic chordates and the new phylum Xenoturbellida. Nature. 2006;444:85–8. doi: 10.1038/nature05241. [DOI] [PubMed] [Google Scholar]
- Cameron CB, Garey JR, Swalla BJ. Evolution of the chordate body plan: new insights from phylogenetic analyses of deuterostome phyla. Proc Natl Acad Sci U S A. 2000;97:4469–74. doi: 10.1073/pnas.97.9.4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canestro C, Bassham S, Postlethwait J. Development of the central nervous system in the larvacean Oikopleura dioica and the evolution of the chordate brain. Dev Biol. 2005;285:298–315. doi: 10.1016/j.ydbio.2005.06.039. [DOI] [PubMed] [Google Scholar]
- Chiba S, Sasaki A, Nakayama A, Takamura K, Satoh N. Development of Ciona intestinalis juveniles (through 2nd ascidian stage) Zoolog Sci. 2004;21:285–98. doi: 10.2108/zsj.21.285. [DOI] [PubMed] [Google Scholar]
- Corbo JC, Levine M, Zeller RW. Characterization of a notochord-specific enhancer from the Brachyury promoter region of the ascidian, Ciona intestinalis. Development. 1997;124:589–602. doi: 10.1242/dev.124.3.589. [DOI] [PubMed] [Google Scholar]
- Crump JG, Maves L, Lawson ND, Weinstein BM, Kimmel CB. An essential role for Fgfs in endodermal pouch formation influences later craniofacial skeletal patterning. Development. 2004;131:5703–16. doi: 10.1242/dev.01444. [DOI] [PubMed] [Google Scholar]
- Delaune E, Lemaire P, Kodjabachian L. Neural induction in Xenopus requires early FGF signalling in addition to BMP inhibition. Development. 2005;132:299–310. doi: 10.1242/dev.01582. [DOI] [PubMed] [Google Scholar]
- Delsuc F, Brinkmann H, Chourrout D, Philippe H. Tunicates and not cephalochordates are the closest living relatives of vertebrates. Nature. 2006;439:965–8. doi: 10.1038/nature04336. [DOI] [PubMed] [Google Scholar]
- Dufour HD, Chettouh Z, Deyts C, de Rosa R, Goridis C, Joly JS, Brunet JF. Precraniate origin of cranial motoneurons. Proc Natl Acad Sci U S A. 2006;103:8727–32. doi: 10.1073/pnas.0600805103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furthauer M, Van Celst J, Thisse C, Thisse B. Fgf signalling controls the dorsoventral patterning of the zebrafish embryo. Development. 2004;131:2853–2864. doi: 10.1242/dev.01156. [DOI] [PubMed] [Google Scholar]
- Glenner H, Hansen AJ, Sorensen MV, Ronquist F, Huelsenbeck JP, Willerslev E. Bayesian inference of the metazoan phylogeny; a combined molecular and morphological approach. Curr Biol. 2004;14:1644–9. doi: 10.1016/j.cub.2004.09.027. [DOI] [PubMed] [Google Scholar]
- Hendrickson C, Christiaen L, Deschet K, Jiang D, Joly J-S, Legendre L, Nakatani Y, Tresser J, Smith WC. Culture of adult ascidians and ascidian genetics. In: Ettensohn CA, Wessel WGMGA, editors. Development of Sea Urchins, Ascidians, and Other Invertebrate Deuterostomes: Experimental Approaches. Academic Press; San Diego: 2004. pp. 143–169. [DOI] [PubMed] [Google Scholar]
- Hirano T, Nishida H. Developmental fates of larval tissues after metamorphosis in the ascidian, Halocynthia roretzi. II. Origin of endodermal tissues of the juvenile. Dev Genes Evol. 2000;210:55–63. doi: 10.1007/s004270050011. [DOI] [PubMed] [Google Scholar]
- Hotta K, Mitsuhara K, Takahashi T, Inaba K, Oka K, Gojobori T, Ikeo K. A web-based interactive developmental table for the ascidian Ciona intestinalis, including 3D real-image embryo reconstructions: I. From fertilized egg to hatching larva. Developmental Dynamics. 2007;236:1790–1805. doi: 10.1002/dvdy.21188. [DOI] [PubMed] [Google Scholar]
- Hudson C, Darras S, Caillol D, Yasuo H, Lemaire P. A conserved role for the MEK signalling pathway in neural tissue specification and posteriorisation in the invertebrate chordate, the ascidian Ciona intestinalis. Development. 2003;130:147–59. doi: 10.1242/dev.00200. [DOI] [PubMed] [Google Scholar]
- Imai KS, Hino K, Yagi K, Satoh N, Satou Y. Gene expression profiles of transcription factors and signaling molecules in the ascidian embryo: towards a comprehensive understanding of gene networks. Development. 2004;131:4047–58. doi: 10.1242/dev.01270. [DOI] [PubMed] [Google Scholar]
- Imai KS, Satoh N, Satou Y. Region specific gene expressions in the central nervous system of the ascidian embryo. Gene Expr Patterns. 2002;2:319–21. doi: 10.1016/s0925-4773(02)00383-0. [DOI] [PubMed] [Google Scholar]
- Jefferies RPS. The ancestry of vertebrates. British Museum; London: 1986. [Google Scholar]
- Jiang D, Smith WC. An ascidian engrailed gene. Dev Genes Evol. 2002;212:399–402. doi: 10.1007/s00427-002-0255-y. [DOI] [PubMed] [Google Scholar]
- Katz MJ. Comparitive anatomy of the tunicate tadpole, Ciona intestinalis. Biological Bulletin. 1983;164:1–27. [Google Scholar]
- Kil SH, Streit A, Brown ST, Agrawal N, Collazo A, Zile MH, Groves AK. Distinct roles for hindbrain and paraxial mesoderm in the induction and patterning of the inner ear revealed by a study of vitamin-A-deficient quail. Dev Biol. 2005;285:252–71. doi: 10.1016/j.ydbio.2005.05.044. [DOI] [PubMed] [Google Scholar]
- Kim GJ, Nishida H. Role of the FGF and MEK signaling pathway in the ascidian embryo. Dev Growth Differ. 2001;43:521–33. doi: 10.1046/j.1440-169x.2001.00594.x. [DOI] [PubMed] [Google Scholar]
- Ladher RK, Wright TJ, Moon AM, Mansour SL, Schoenwolf GC. FGF8 initiates inner ear induction in chick and mouse. Genes Dev. 2005;19:603–13. doi: 10.1101/gad.1273605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leger S, Brand M. Fgf8 and Fgf3 are required for zebrafish ear placode induction, maintenance and inner ear patterning. Mech Dev. 2002;119:91–108. doi: 10.1016/s0925-4773(02)00343-x. [DOI] [PubMed] [Google Scholar]
- Liu D, Chu H, Maves L, Yan YL, Morcos PA, Postlethwait JH, Westerfield M. Fgf3 and Fgf8 dependent and independent transcription factors are required for otic placode specification. Development. 2003;130:2213–24. doi: 10.1242/dev.00445. [DOI] [PubMed] [Google Scholar]
- Mackie GO, Singla CL. The capsular organ of Chelyosoma productum (Ascidiacea: Corellidae): a new tunicate hydrodynamic sense organ. Brain Behav Evol. 2003;61:45–58. doi: 10.1159/000068878. [DOI] [PubMed] [Google Scholar]
- Manni L, Lane NJ, Burighel P, Zaniolo G. Are neural crest and placodes exclusive to vertebrates? Evol Dev. 2001;3:297–8. doi: 10.1046/j.1525-142x.2001.01040.x. [DOI] [PubMed] [Google Scholar]
- Manni L, Lane NJ, Joly JS, Gasparini F, Tiozzo S, Caicci F, Zaniolo G, Burighel P. Neurogenic and non-neurogenic placodes in ascidians. J Exp Zoolog B Mol Dev Evol. 2004;302:483–504. doi: 10.1002/jez.b.21013. [DOI] [PubMed] [Google Scholar]
- Manni L, Lane NJ, Zaniolo G, Burighel P. Cell reorganisation during epithelial fusion and perforation: the case of ascidian branchial fissures. Dev Dyn. 2002;224:303–13. doi: 10.1002/dvdy.10112. [DOI] [PubMed] [Google Scholar]
- Maroon H, Walshe J, Mahmood R, Kiefer P, Dickson C, Mason I. Fgf3 and Fgf8 are required together for formation of the otic placode and vesicle. Development. 2002;129:2099–108. doi: 10.1242/dev.129.9.2099. [DOI] [PubMed] [Google Scholar]
- Mazet F, Hutt JA, Milloz J, Millard J, Graham A, Shimeld SM. Molecular evidence from Ciona intestinalis for the evolutionary origin of vertebrate sensory placodes. Dev Biol. 2005;282:494–508. doi: 10.1016/j.ydbio.2005.02.021. [DOI] [PubMed] [Google Scholar]
- Mazet F, Shimeld SM. Molecular evidence from ascidians for the evolutionary origin of vertebrate cranial sensory placodes. J Exp Zoolog B Mol Dev Evol. 2005;304:340–6. doi: 10.1002/jez.b.21054. [DOI] [PubMed] [Google Scholar]
- McCabe KL, Manzo A, Gammill LS, Bronner-Fraser M. Discovery of genes implicated in placode formation. Dev Biol. 2004;274:462–77. doi: 10.1016/j.ydbio.2004.07.012. [DOI] [PubMed] [Google Scholar]
- Northcutt RG, Gans C. The genesis of neural crest and epidermal placodes: a reinterpretation of vertebrate origins. Q Rev Biol. 1983;58:1–28. doi: 10.1086/413055. [DOI] [PubMed] [Google Scholar]
- Oda H, Wada H, Tagawa K, Akiyama-Oda Y, Satoh N, Humphreys T, Zhang S, Tsukita S. A novel amphioxus cadherin that localizes to epithelial adherens junctions has an unusual domain organization with implications for chordate phylogeny. Evol Dev. 2002;4:426–34. doi: 10.1046/j.1525-142x.2002.02031.x. [DOI] [PubMed] [Google Scholar]
- Philippe H, Lartillot N, Brinkmann H. Multigene analyses of bilaterian animals corroborate the monophyly of Ecdysozoa, Lophotrochozoa, and Protostomia. Mol Biol Evol. 2005;22:1246–53. doi: 10.1093/molbev/msi111. [DOI] [PubMed] [Google Scholar]
- Phillips BT, Bolding K, Riley BB. Zebrafish fgf3 and fgf8 encode redundant functions required for otic placode induction. Dev Biol. 2001;235:351–65. doi: 10.1006/dbio.2001.0297. [DOI] [PubMed] [Google Scholar]
- Rychel AL, Smith SE, Shimamoto HT, Swalla BJ. Evolution and development of the chordates: collagen and pharyngeal cartilage. Mol Biol Evol. 2006;23:541–9. doi: 10.1093/molbev/msj055. [DOI] [PubMed] [Google Scholar]
- Schlosser G. Development and evolution of lateral line placodes in amphibians I. Development. Zoology (Jena) 2002;105:119–46. doi: 10.1078/0944-2006-00058. [DOI] [PubMed] [Google Scholar]
- Schlosser G. Evolutionary origins of vertebrate placodes: insights from developmental studies and from comparisons with other deuterostomes. J Exp Zoolog B Mol Dev Evol. 2005;304:347–99. doi: 10.1002/jez.b.21055. [DOI] [PubMed] [Google Scholar]
- Solomon KS, Kwak SJ, Fritz A. Genetic interactions underlying otic placode induction and formation. Dev Dyn. 2004;230:419–33. doi: 10.1002/dvdy.20067. [DOI] [PubMed] [Google Scholar]
- Streit A, Berliner AJ, Papanayotou C, Sirulnik A, Stern CD. Initiation of neural induction by FGF signalling before gastrulation. Nature. 2000;406:74–78. doi: 10.1038/35017617. [DOI] [PubMed] [Google Scholar]
- Tokuoka M, Satoh N, Satou Y. A bHLH transcription factor gene, Twist-like1, is essential for the formation of mesodermal tissues of Ciona juveniles. Dev Biol. 2005;288:387–396. doi: 10.1016/j.ydbio.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Turbeville JM, Schulz JR, Raff RA. Deuterostome phylogeny and the sister group of the chordates: evidence from molecules and morphology. Mol Biol Evol. 1994;11:648–55. doi: 10.1093/oxfordjournals.molbev.a040143. [DOI] [PubMed] [Google Scholar]
- Vienne A, Pontarotti P. Metaphylogeny of 82 gene families sheds a new light on chordate evolution. Int J Biol Sci. 2006;2:32–7. doi: 10.7150/ijbs.2.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada H, Saiga H, Satoh N, Holland PW. Tripartite organization of the ancestral chordate brain and the antiquity of placodes: insights from ascidian Pax-2/5/8, Hox and Otx genes. Development. 1998;125:1113–22. doi: 10.1242/dev.125.6.1113. [DOI] [PubMed] [Google Scholar]
- Wada H, Satoh N. Patterning the protochordate neural tube. Curr Opin Neurobiol. 2001;11:16–21. doi: 10.1016/s0959-4388(00)00168-9. [DOI] [PubMed] [Google Scholar]
- Whittaker JR, Meedel TH. Two histospecific enzyme expressions in the same cleavage-arrested one-celled ascidian embryos. J Exp Zool. 1989;250:168–75. doi: 10.1002/jez.1402500208. [DOI] [PubMed] [Google Scholar]
- Wilson SI, Graziano E, Harland R, Jessel TM, Edlund T. An early requirement for FGF signalling in the acquisition of neural cell fate in the chick embryo. Curr Biol. 2000;10:421–429. doi: 10.1016/s0960-9822(00)00431-0. [DOI] [PubMed] [Google Scholar]
- Winchell CJ, Sullivan J, Cameron CB, Swalla BJ, Mallatt J. Evaluating hypotheses of deuterostome phylogeny and chordate evolution with new LSU and SSU ribosomal DNA data. Mol Biol Evol. 2002;19:762–76. doi: 10.1093/oxfordjournals.molbev.a004134. [DOI] [PubMed] [Google Scholar]
- Wright TJ, Mansour SL. Fgf3 and Fgf10 are required for mouse otic placode induction. Development. 2003;130:3379–90. doi: 10.1242/dev.00555. [DOI] [PubMed] [Google Scholar]
- Yasuo H, Hudson C. FGF8/17/18 functions together with FGF9/16/20 during formation of the notochord in Ciona embryos. Dev Biol. 2007;302:92–103. doi: 10.1016/j.ydbio.2006.08.075. [DOI] [PubMed] [Google Scholar]
- Zeng L, Swalla BJ. Molecular phylogeny of the protochordates: chordate evolution. Can J Zool. 2005;83:24–33. [Google Scholar]