

Abstract
PURPOSE
The onset of pantothenate kinase-associated neurodegeneration (PKAN) occurs in the first and second decade of life and a pigmentary retinal degenerationis a feature of the disorder. Since the neuro-ophthalmologic and electroretinographic (ERG) features have never been well delineated, we describe them in 16 patientswith PKAN.
DESIGN
Observational case series.
METHODS
Sixteen patients with genetic and neuroimaging-confirmed PKAN were examined. Ten underwent neuro-ophthalmologic examination and all had ERGs.
RESULTS
Of the 10 who underwent neuro-ophthalmologic examination, all showed saccadic pursuits and eight showed hypometric or slowed vertical saccades. Seven of eight had inability to suppress the vestibulo-ocular reflex; two patients could not cooperate. Two had square wave jerks and four had poor convergence. Vertical optokinetic responses were abnormal in five, and two patients had blepharospasm. Eight patients had sectoral iris paralysis and partial loss of the pupillary ruff consistent with Adie’s pupils in both eyes. Only four of 10 examined patients showed a pigmentary retinopathy, but 11 of 16 had abnormal ERGs ranging from mild cone abnormalities to severe rod-cone dysfunction. No patient had optic atrophy. The PANK2 mutations of all of the patients were heterogeneous.
CONCLUSIONS
Adie’s-like pupils, abnormal vertical saccades, and saccadic pursuits were very common. These findings suggest that mid-brain degeneration occurs in PKAN more frequently than previously thought. ERG abnormalities were present in approximately 70% and no patient had optic atrophy. Although genotype-ocular phenotype correlations could not be established, allelic differences probably contributed to the variable clinical expression of retinopathy and other clinical characteristics in these patients.
Pantothenate kinase-associated neurodegeneration (PKAN), formerly known as Hallervorden-Spatz syndrome, is a progressive autosomal recessive disorder that classically occurs early in the first decade of life or atypically beginning in the second and later decades. It is characterized by dystonia, parkinsonism, iron accumulation in the brain, and occasionally retinopathy or optic neuropathy.1,2 Although a peripheral retinal degeneration is commonly associated with this disorder, electrophysiologic details are routinely missing.3 All patients have mutations in the gene encoding pantothenate kinase 2 PANK2).4 Pupillary and ocular motor findings have never been described and attention to iron accumulation in the posterior fossa has only been rarely reported.5-7 Since neuro-ophthalmologic features have not been described previously, we delineate these in 10 patients with PKAN. We also report the electroretinogram (ERG) findings in these plus six additional cases.
METHODS
The research, in this observational case series study, was reviewed and approved by the Institutional Review Board of the Oregon Health & Science University. Ten patients aged 7 to 69 years with neuroimaging-confirmed PKAN underwent neuro-ophthalmologic and ERG examination; six additional patients were examined electrophysiologically. All patients had defined genetic mutations in PANK2. Neuro-ophthalmologic examination consisted of Snellen acuity at 20 feet and Hardy-Rand-Rittler color plate testing. Ocular motility was examined by vertical and horizontal saccades and pursuit, response to a vertical and horizontal optokinetic strip, horizontal suppression of the vestibular ocular reflex (VOR), and presence of intact fixation or misalignment. Saccades were assessed by observing a fast eye movement from an eccentric position back to the position of the examiner’s face. Pursuit was tested by having the patient follow a slow moving target in the horizontal or vertical plane. The VOR was tested by observing extraneous eye movements as the patient fixated on a distant target while the examination chair was rotated from side to side. Suppression of the VOR was assessed by observing extraneous eye movements as the patient fixated on their outstretched thumb while the chair was rotated from side to side. Convergence, but not accommodation, was also assessed by degree of bilateral adduction associated with pupillary constriction. Pupils were assessed with a pupil light and neutral density filters and then examined under a biomicroscope. Visual fields were performed using Goldmann perimetry with the device calibrated to 1000 apostilb. Each patient was tested using the I4e, I2e, and I1e isopters; the V4e isopter was used only if the I4e isopter was abnormal. Patients were dilated with tropicamide 1% and phenylephrine 2.5% and the posterior poles and peripheral fundi were viewed. All fundi were photographed. Some patients were re-checked 24 hours later by instilling dilute pilocarpine 0.125% in each eye and noting whether constriction occurred. Pupil size before and after pilocarpine instillation was documented photographically.
The ERG was recorded on all subjects using a custom full-field stimulator of previously described design8 according to published methods.9,10 The ERG protocol included, but was not confined to, the ISCEV standard stimulus conditions.11 Pupils were fully dilated and recordings begun after 45 minutes of dark adaptation. The ERGs were recorded with 0.1 to 1000 Hz bandpass using a bipolar Burian-Allen contact lens electrode and six intensities of white light distributed over a 3.7 log range (+0.6 to −3.1 log cd-s/m2). Scotopically matched blue and red stimuli tested dark-adapted rods and cones. Oscillatory potentials were isolated from the full frequency bright flash scotopic response using a digital filter (100 Hz to 300 Hz bandpass). Flicker 30 Hz responses were recorded initially without background. The subject was light-adapted for 10 minutes at 34 cd/m2 after which white light flashes and 30 Hz flicker were recorded with the rod-suppressing background. Naka-Rushton parameters were derived from the b-wave amplitudes for the scotopic responses, generating Rmax, an indicator of maximum response, and log K, an indicator of retinal sensitivity.
Neuroimaging of the brain was performed by magnetic resonance imaging (MRI) using standard sequences. The “eye of the tiger” sign refers to dark signal in the globus pallidus, darker than typical, that surrounds a small area of bright signal (Figure 1). Since the “eye of the tiger” sign has been associated in a 1:1 correlation with PANK2 mutations, MRI was used to confirm the diagnosis of PKAN in all patients.12
FIGURE 1.
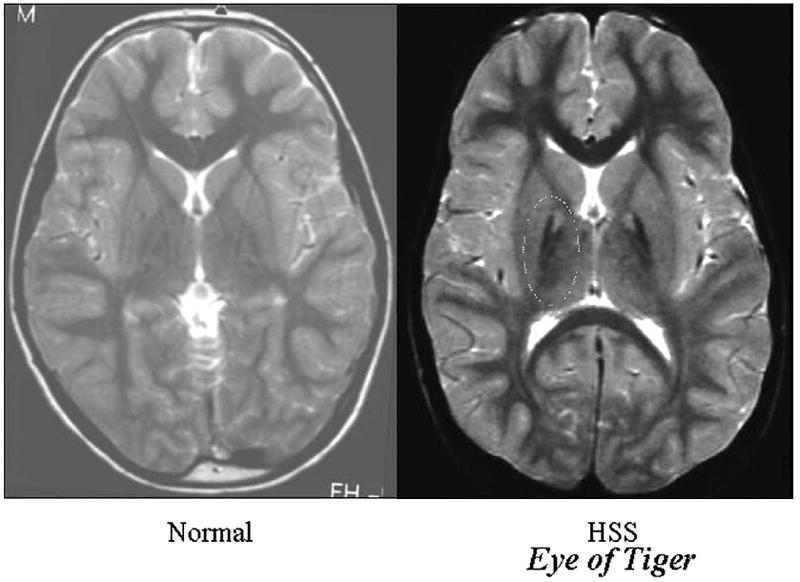
T2-weighted axial magnetic resonance imaging (MRI) scan of the brain through the globus pallidus in a normal individual and in an individual with pantothenate kinase-associated neurodegeneration (PKAN). Note that the globus pallidus of the patient with PKAN is much darker than typical with a bright spot in the center bilaterally. This is a typical “eye of the tiger sign” of a patient with PKAN12 (used with permission from Hayflick SJ, Westaway SK, Levinson B et al. Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N Engl J Med 2003;348:33-40).
DNA was isolated from lymphoblast cell lines or from buccal brushings. For mutation detection, PCR products representing exons and flanking intron sequences for exons 1C-7 were amplified from patient DNA and visualized on agarose gels to check size and specificity, purified to remove primers, quantitated, and submitted for sequencing in both directions using the same primers as for PCR amplification. DNA sequence determination was performed either by the Oregon Health & Science University MMI Research Core Facility (http://www.ohsu.edu/core) on a model 377 Applied Biosystems Inc automated fluorescence sequencer or by the University of Chicago Genetic Services laboratory using the ABI PRISM BigDye cycle sequencing kit (Applied Biosystems, Inc, Foster City, California, USA) on an ABI 3100 fluorescence sequencer (Applied Biosystems, Inc) and analyzed using the Sequencher software (Gene Codes, Inc, Ann Arbor, Michigan, USA). Complete exons including intronic splice signals were sequenced.
RESULTS
All patients harbored at least one deleterious mutation in PANK2; for three patients, a second mutation has yet to be found. One patient was homozygous for a 460 CT transition predicted to change an arginine to a tryptophan at codon 154. Clinical, retinal, and genetic features are listed in Table 1. The mutations in these patients were heterogeneous. The neuro-ophthalmologic features are listed in Table 2. Eight patients had sectoral iris paralysis and partial loss of the pupillary ruff consistent with Adie’s pupils in both eyes. Dilute pilocarpine was given to six of 10 patients. Positivity was confirmed in four of these and negativity was confirmed in two. Ocular motility showed hypometric and slowed vertical saccades in eight and saccadic pursuits in all studied. Horizontal saccades were normal. Seven of eight had inability to suppress the horizontal VOR in a rotating chair; the other two patients could not cooperate with this maneuver. Two had square wave jerks and four had poor convergence. Vertical optokinetic responses were abnormal in five, and two patients had blepharospasm. No patient had optic atrophy. All patients had dysarthria and gait abnormalities.
TABLE 1.
Clinical Characteristics of Patients With Pantothenate Kinase-Associated Neurodegeneration
| Patient | Age (y)/Gender | Onset Age (y) | VA | Color | GVF | Dysar | Abnl Gait | Bone Spicules | ERG Abnl? | ERG loss pattern | Eye of Tiger | Gene Defect |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 7F | 2 | CNP | CNP | CNP | Yes | Yes | Yes | Yes | Severe | Yes | 885C>G; 1231G>A |
| 2 | 11 M | 9 | NL | Abnl | NL | Yes | Yes | No | Yes | Mild cone | Yes | 460C->T × 2 |
| 3 | 15F | 1 | NL | NL | CNP | Yes | Yes | Yes | Yes | Severe | Yes | 71A>G; IVS4-1G>T |
| 4 | 16F | 13 | — | — | — | — | — | — | Yes | Mild cone | Yes | 987delT; 1253C>T |
| 5 | 16M | 14 | — | — | — | — | — | — | NL | — | Yes | 1231G>A; 1255A>G |
| 6 | 18F | 2 | CNP | CNP | CNP | Yes | Yes | Yes | Yes | Severe | Yes | IVS4-1G>T; 1231G>A |
| 7 | 19F | 17 | — | — | — | — | — | — | NL | — | Yes | 1231G>A; 1255A>G |
| 8 | 20 F | 14 | NL | NL | NL | Yes | Yes | No | Yes | Mild cone | Yes | 614_622del; 1397C>T |
| 9 | 20 M | 5 | NL | NL | CNP | Yes | Yes | No | Yes | Mild cone | Yes | 1253C>T; UNK |
| 10 | 21 F | 10 | — | — | — | — | — | — | Yes | Mod. rod-cone | Yes | 1231G>A; 1061C>G |
| 11 | 23 F | 8 | — | — | — | — | — | — | Yes | Mild rod-cone | Yes | 1231G>A; 1061C>G |
| 12 | 29 M | 14 | NL | NL | NL | Yes | Yes | No | NL | — | Yes | 370A>G; IVS4-1G>T |
| 13 | 33 M | 6 | CNP | CNP | CNP | Yes | Yes | Yes | Yes | Severe | Yes | 767delC; UNK |
| 14 | 34 M | 14 | NL | NL | nasal | Yes | Yes | No | Yes | Mod. rod-cone | Yes | 285delG; 370A>G |
| 15 | 43 F | 17 | NL | NL | NL | Yes | Yes | No | NL | — | Yes | 1231G>A; UNK |
| 16 | 69 F | 23 | — | — | — | — | — | — | NL | — | Yes | 1231G>A; 568A>G |
Abnl = abnormal; Bone Spicules = patient 2 likely had congenital color blindness; CNP = the patient could not perform; Color = color vision; Dysar = dysarthria; GVF = Goldmann visual fields; mod. = moderate; nasal = nasal depression; NL = normal; severe = ERG responses were severely sub-normal for both rod and cone stimuli or were indistinguishable from noise; UNK = unknown; VA = visual acuity.
Patients 4, 5, 7, 10, 11, and 16 did not have neuro-ophthalmologic examinations. Patient 2 had two copies of his mutation likely from consanguinity. Patients 5 and 7 are siblings. Patient 16 is the grandmother of patients 10 and 11, who are siblings. Patient 2 was homozygous for his mutation. Mutation numbering is based on counting starting from the ATG start codon of the PANK2 gene.
TABLE 2.
Neuro-ophthalmologic Characteristics of Patients With Pantothenate Kinase-Associated Neurodegeneration
| Patient | Adie’s Pupils | Abnl Saccades | Vertical Saccadic Pursuit | Abnl VOR Suppression | Sq. Wave Jerks | Abnl Conv | Abnl OKN | Skew Dev | Bleph |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Yes* | Yes | Yes | Yes | No | No | Yes | No | No |
| 2 | No* | No | Yes | Yes | No | No | Yes | No | No |
| 3 | Yes* | Yes | Yes | Yes | Yes | Yes | No | No | No |
| 4 | — | — | — | — | — | — | — | — | — |
| 5 | — | — | — | — | — | — | — | — | — |
| 6 | Yes | Yes | Yes | ND | No | Yes | Yes | No | No |
| 7 | — | — | — | — | — | — | — | — | — |
| 8 | Yes | Yes | Yes | No | No | No | No | No | No |
| 9 | Yes* | No | Yes | Yes | No | Yes | No | No | Yes |
| 10 | — | — | — | — | — | — | — | — | — |
| 11 | — | — | — | — | — | — | — | — | — |
| 12 | No* | Yes | Yes | Yes | Yes | No | No | No | No |
| 13 | Yes | Yes | Yes | ND | No | No | Yes | Yes | Yes |
| 14 | Yes* | Yes | Yes | Yes | No | No | Yes | No | No |
| 15 | Yes | Yes | Yes | Yes | No | Yes | No | No | No |
| 16 | — | — | — | — | — | — | — | — | — |
Abnl = abnormal; Bleph = blepharospasm; Conv = convergence; NT = not tested.
Patients 4, 5, 7, 10, 11, and 16 did not have neuro-ophthalmologic examinations.
Pilocarpine 0.125% given to confirm supersensitivity.
One patient, the most severely affected, exhibited periodic alternating skew deviation. This patient (patient 13) was a 33-year-old man who maintained most commonly a left hypertropia which would alternate with a right hypertropia every 10 to 30 seconds. In low light conditions, this cycling occurred faster. He also had blepharospasm. He had posterior sub-capsular cataracts as well as a maculopathy in each eye (Figure 2). His ERG showed severe loss of rod and cone responses to the level of noise for the recordings. His mutation was a cytosine deletion at position 767 and was unique among more than 100 patients with PKAN. The mutation on his other allele has not been identified.
FIGURE 2.
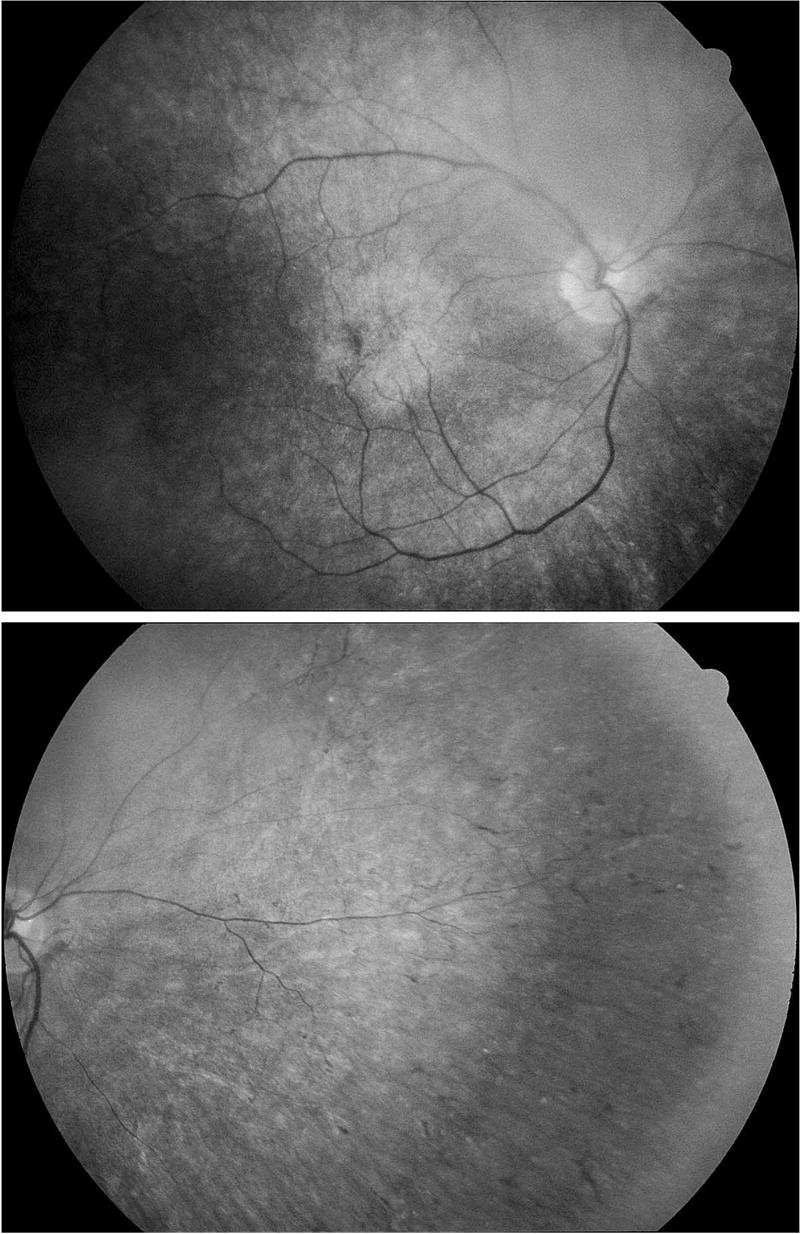
Posterior pole (top) and nasal retina (bottom) of patient 13 with PKAN at age 33 years. The retinal vessels are markedly attenuated and the nasal retina shows scattered bone spicule formations. The macula shows a soft-bordered atrophic lesion with a small area of focal hyperpigmentation. The left fundus was similar in appearance.
Only four patients showed a pigmentary peripheral retinopathy, but 11 had abnormal ERGs. These four patients had severely sub-normal rod and cone single flash responses with residual averaged cone responses that were either reduced to within a few microvolt ranges or indistinguishable from noise. The age of onset of the oldest of these four was only age 6 years. Figure 3 shows typical pigmentary retinopathy in affected cases. For several of the subjects, their movement disorder and limited ability to comply with testing led to a higher level of noise; none of the patients was sedated. Five patients had totally normal ERGs and the youngest age of onset of these was 14. Four patients had only mild cone abnormalities consisting of decreased dark-adapted or light-adapted cone amplitudes and prolonged cone implicit times. One patient had mild and two patients had moderate rod-cone amplitude losses with prolonged cone implicit times.
FIGURE 3.
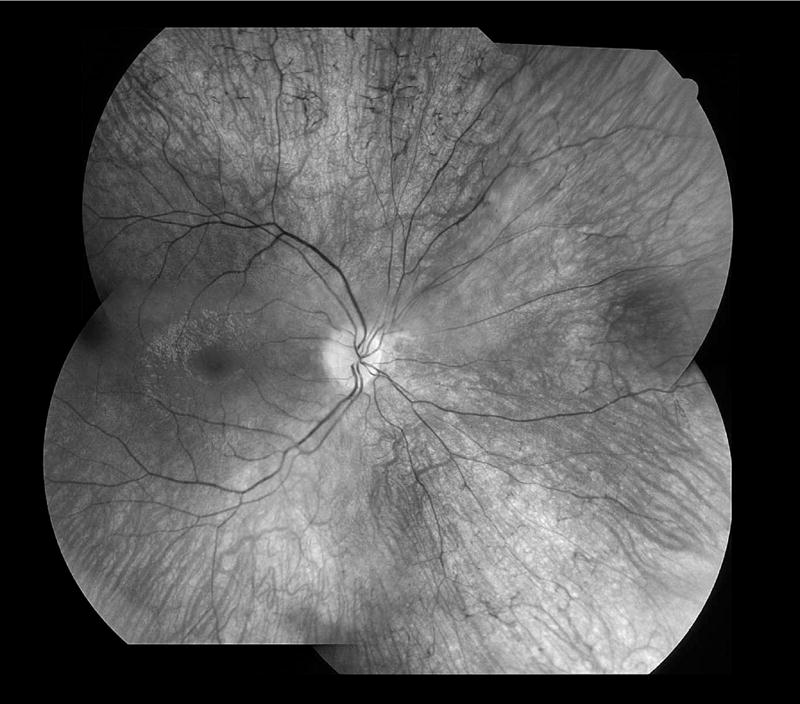
Fundus montage of right eye of patient 3 with PKAN at age 15 years. The optic disk is waxy with mild pallor, the retinal vessels are mildly attenuated with perivenous pigmentary cuffing, and throughout all quadrants there are scattered bone-spicule formations. The reflexes off the inner surface of the retina in the macula suggest retinal swelling or edema. The appearance of the left eye was similar.
DISCUSSION
The erg has not been systematically studied in any of the disorders formerly grouped as Hallervorden-Spatz syndrome and brainstem features have been largely ignored with this disorder. We found that almost all patients studied suffered from some type of ocular motility disorder related to the brainstem, specifically the tectal mid-brain. Although several studies have documented increased iron accumulation in the substantia nigra5,13-18 none of the patients had any abnormalities suggestive of fascicular oculomotor pareses. Because the oculomotor nerve is not affected, the iron accumulation seen on MRI in the substantia nigra must be more extensive leading to dysfunction in the tectal mid-brain.
These patients also have sluggish pupils that appear similar to classic Adie’s pupils. However, the Adie’s pupil is defined as being tonic and many of these patients could not cooperate with convergence or accommodation so that tonicity could not be assessed. These irides also had sectoral paralysis and patchy loss of the pupillary ruff. Patients with these anatomic changes who were tested were all supersensitive to dilute pilocarpine. We suspect that the mid-brain deposition of iron in or near the substantia nigra may induce a proximal degeneration in the fibers destined for the iris and cause pupils identical to Adie’s pupils. It is unknown whether pathology exists at the iris or in the short ciliary nerves. The exact location of damage in Adie’s pupils is unknown but is believed to be local in the eye. It is also possible that iron deposition is occurring locally at the short ciliary nerves. The presumption that brainstem accumulation of iron causes the pupil changes supports the concept that a single region of the brain causes the most of these neuro-ophthalmologic findings.
We suspect these pupil findings may be found in other metal storage diseases. One of us (R.A.E.) has examined a 13-year-old girl with Wilson disease and Kayser-Fleischer rings who had abnormal vertical pursuit, poor convergence, and an inability to suppress the VOR (Egan RA, unpublished observation, 2004). She had sluggish pupils that exhibited sectoral paralysis, patchy loss of the pupillary ruff, and were supersensitive to dilute pilocarpine. A 31-year-old woman with non-PKAN–related neurodegeneration with brain iron accumulation was found with typical Adie’s type pupils (Egan RA, unpublished observation, 2004). The metal storage disorder in these diseases may have caused the pupillary findings through a similar mechanism to that found in our patients with PKAN.
ERG abnormalities were seen in 11 of 16 patients (69%), but five patients, including our oldest subject at age 69 years, had totally normal rod and cone amplitudes and implicit times. In those with abnormal ERGs, there appeared to be a continuum of changes with the mildest ERG findings, seen in four subjects, involving reductions of cone amplitudes and prolongations of cone implicit times. With greater involvement of the ERG, rod and cone losses became roughly equal, and for four subjects the amplitude losses were severe. We were unable to discern any genotype-phenotype correlations for the ERG responses except that severely affected patients had younger ages of onset and normal patients had older ages of onset. This age predilection was not present regarding the neuro-ophthalmologic findings.
Patient 13, the most severely affected patient, had bilateral posterior sub-capsular cataract and maculopathy. Clinically, cataract is a frequent sequela of retinitis pigmentosa, occurring in approximately 50% of cases.19,20 Alternatively, it is possible that iron deposits in the posterior capsule could contribute to the opacification. Iron storage may also precipitate ischemia, which has been postulated as the etiology of posterior subcapsular cataracts in other rare diseases.21 We are aware of only one other report of a patient with brain iron accumulation and a maculopathy.22 This other patient22 was examined before the discovery of the PANK2 gene and therefore could represent a non-PKAN form of brain iron accumulation. Patient 13 had a unique mutation which may be associated with maculopathy.
Although genotype-phenotype correlations were not apparent within this small group of patients, most of whom did not even share a common allele, the clinical and ERG findings suggest that residual gene product function, epigenetic factors, or modifying genes may alter the expression of the disease. A threshold of residual gene-product function could exist for which the retina remains protected from considerable degeneration. Also, with the study of more patients, specific clinical findings may segregate with different mutations.
None of our patients had optic atrophy, although this has been described as the presenting sign in a single genetically unconfirmed case without the “eye of the tiger.”13 Optic nerve head pallor is common in patients with retinitis pigmentosa23,24 and, in particular, cone-rod retinal degenerations.25 Although optic pallor may occur in association with advanced retinal degeneration, we feel that primary optic atrophy is not a part of the disease of PKAN but may be associated with other disorders of brain iron accumulation. In patients with primary optic atrophy and a progressive neurodegenerative disorder, we predict that genetic testing for PANK2 mutations will be negative.
Acknowledgments
This study has been supported by an unrestricted grant from Research to Prevent Blindness, New York, New York, the OHSU General Clinical Research Center (grant M01 RR000334), the National Organization for Rare Disorders (NORD), and the NBIA Disorders Association. This study was also supported in part by The Foundation Fighting Blindness, Owings Mills, Maryland (R.G.W.).
Biography

Robert A. Egan, MD, received a BS from UC Davis in genetics and then graduated from the Medical College of Wisconsin. He completed a neurology residency and a cerebrovascular disease fellowship at the Oregon Health and Science University. He trained in neuro-ophthalmology at the Massachusetts Eye and Ear Infirmary at Harvard before returning to Portland, Oregon. He is currently Associate Professor of Ophthalmology, Neurology, and Neurosurgery at OHSU.
Footnotes
This research was reviewed and approved by the OHSU Institutional Review Board.
Neuro-ophthalmologic and electroretinographic findings in pantothenate kinase-associated neurodegeneration (formerly Hallervorden-Spatz Syndrome). Robert A. Egan, MD, Richard G. Weleber, MD, Penelope Hogarth, MD, Allison Gregory, MS, Jason Coryell, MS, Shawn K. Westaway, PhD, Jane Gitschier, PhD, Soma Das, PhD, and Susan J. Hayflick, MD Sixteen patients with pantothenate kinase-associated neurodegeneration were examined. Ten had neuro-ophthalmologic examination and all had full-field electroretinograms (ERGs). Varying degrees of a dorsal mid-brain syndrome and Adie’s pupils were identified in most patients. Seventy percent of patients also displayed mild cone abnormalities to severe rod-cone dysfunction on ERG testing, although only those with severe ERG loss had pigmentary retinal changes. No patients suffered from optic atrophy.
References
- 1.Swaiman KF. Hallervorden-Spatz syndrome and brain iron metabolism. Arch Neurol. 1991;48:1285–1293. doi: 10.1001/archneur.1991.00530240091029. [DOI] [PubMed] [Google Scholar]
- 2.Dooling EC, Schoene WC, Richardson EP., Jr Hallervorden-Spatz syndrome. Arch Neurol. 1974;30:70–83. doi: 10.1001/archneur.1974.00490310072012. [DOI] [PubMed] [Google Scholar]
- 3.Thomas M, Hayflick SJ, Jankovic J. Clinical heterogeneity of neurodegeneration with brain iron accumulation (Hallervorden-Spatz syndrome) and pantothenate kinase-associated neurodegeneration. Mov Disord. 2004;19:36–42. doi: 10.1002/mds.10650. [DOI] [PubMed] [Google Scholar]
- 4.Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, Hayflick SJ. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet. 2001;28:345–349. doi: 10.1038/ng572. [DOI] [PubMed] [Google Scholar]
- 5.Neumann M, Adler S, Schluter O, Kremmer E, Benecke R, Kretzschmar HA. Alpha-synuclein accumulation in a case of neurodegeneration with brain iron accumulation type 1(NBIA-1, formerly Hallervorden-Spatz syndrome) with widespread cortical and brainstem-type Lewy bodies. ActaNeuropathol (Berl) 2000;100:568–574. doi: 10.1007/s004010000224. [DOI] [PubMed] [Google Scholar]
- 6.Sener RN. Pantothenate kinase-associated neurodegeneration: MR imaging, proton MR spectroscopy, and diffusion MR imaging findings. AJNR Am J Neuroradiol. 2003;24:1690–1693. [PMC free article] [PubMed] [Google Scholar]
- 7.Sodeyama N, Arai M, Sanjoh N, Orimo S, Tamaki M. A case of Hallervorden-Spatz syndrome with marked atrophy of the brainstem and cerebellum. Rinsho Shinkeigaku. 1993;33:525–529. [PubMed] [Google Scholar]
- 8.Weleber RG. The effect of age on human cone and rod ganzfeld electroretinograms. Invest Ophthalmol Vis Sci. 1981;20:392–399. [PubMed] [Google Scholar]
- 9.Weleber RG. The dystrophic retina in multisystem disorders: the electroretinogram in neuronal ceroid lipofuscinoses. Eye. 1998;12(Pt 3b):580–590. doi: 10.1038/eye.1998.148. [DOI] [PubMed] [Google Scholar]
- 10.Weleber RG, Eisner A. Retinal function and physiological studies. In: Newsome DA, editor. Retinal dystrophies and degenerations. New York, NY: Raven Press; 1988. pp. 21–69. [Google Scholar]
- 11.Marmor MF, Zrenner E. Standard for clinical electroretinography (1994 update) Doc Ophthalmol. 1995;89:199–210. doi: 10.1007/BF01203373. [DOI] [PubMed] [Google Scholar]
- 12.Hayflick SJ, Westaway SK, Levinson B, et al. Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N Engl J Med. 2003;348:33–40. doi: 10.1056/NEJMoa020817. [DOI] [PubMed] [Google Scholar]
- 13.Battistella PA, Midena E, Suppiej A, Carollo C. Optic atrophy as the first symptom in Hallervorden-Spatz syndrome. Childs Nerv Syst. 1998;14:135–138. doi: 10.1007/s003810050196. [DOI] [PubMed] [Google Scholar]
- 14.Feliciani M, Curatolo P. Early clinical and imaging (high-field MRI) diagnosis of Hallervorden-Spatz disease. Neuroradiology. 1994;36:247–248. doi: 10.1007/BF00588145. [DOI] [PubMed] [Google Scholar]
- 15.Hickman SJ, Ward NS, Surtees RA, Stevens JM, Farmer SF. How broad is the phenotype of Hallervorden-Spatz disease? Acta Neurol Scand. 2001;103:201–203. doi: 10.1034/j.1600-0404.2001.103003201.x. [DOI] [PubMed] [Google Scholar]
- 16.Koeppen AH, Dickson AC. Iron in the Hallervorden-Spatz syndrome. Pediatr Neurol. 2001;25:148–155. doi: 10.1016/s0887-8994(01)00269-7. [DOI] [PubMed] [Google Scholar]
- 17.Porter-Grenn L, Silbergleit R, Mehta BA. Hallervorden-Spatz disease with bilateral involvement of globus pallidus and substantia nigra: MR demonstration. J Comput Assist Tomogr. 1993;17:961–963. doi: 10.1097/00004728-199311000-00019. [DOI] [PubMed] [Google Scholar]
- 18.Tripathi RC, Tripathi BJ, Bauserman SC, Park JK. Clinicopathologic correlation and pathogenesis of ocular and central nervous system manifestations in Hallervorden-Spatz syndrome. Acta Neuropathol (Berl) 1992;83:113–119. doi: 10.1007/BF00308470. [DOI] [PubMed] [Google Scholar]
- 19.Berson EL, Rosner B, Simonoff E. Risk factors for genetic typing and detection in retinitis pigmentosa. Am J Ophthalmol. 1980;89:763–775. doi: 10.1016/0002-9394(80)90163-4. [DOI] [PubMed] [Google Scholar]
- 20.Heckenlively J. The frequency of posterior subcapsular cataract in the hereditary retinal degenerations. Am J Ophthalmol. 1982;93:733–738. doi: 10.1016/0002-9394(82)90469-x. [DOI] [PubMed] [Google Scholar]
- 21.Egan R, Lessell S. Posterior subcapsular cataract in Degos disease. Am J Ophthalmol. 2000;129:806–807. doi: 10.1016/s0002-9394(00)00460-8. [DOI] [PubMed] [Google Scholar]
- 22.Luckenbach MW, Green WR, Miller NR, Moser HW, Clark AW, Tennekoon G. Ocular clinicopathologic correlation of Hallervorden-Spatz syndrome with acanthocytosis and pigmentary retinopathy. Am J Ophthalmol. 1983;95:369–382. doi: 10.1016/s0002-9394(14)78308-4. [DOI] [PubMed] [Google Scholar]
- 23.Heckenlively JR. Retinitis pigmentosa. Philadelphia: JP Lippincott Co; 1988. pp. 68–89. [Google Scholar]
- 24.Weleber RG, Gregory-Evans K. In: Retinitis pigmentosa and allied disorders. 3. Ryan SJ, editor. Retina St. Louis, MO: Mosby-Year Book, Inc; 2001. pp. 362–460. [Google Scholar]
- 25.Heckenlively JR, Martin DA, Rosales TO. Telangiectasia and optic atrophy in cone-rod degenerations. Arch Ophthalmol. 1981;99:1983–1991. doi: 10.1001/archopht.1981.03930020859009. [DOI] [PubMed] [Google Scholar]