

Abstract
The rate-limiting step in transcriptional initiation typically is opening the promoter DNA to expose the template strand. Opening is tightly regulated, but how it occurs is not known. These experiments identify an activity, recognition of specific DNA fork junctions, and suggest that it is critical to bacterial promoter opening. This activity is both sequence and structure specific; it recognizes the bases that constitute the upstream double-stranded/single-stranded boundary of the open complex. Promoter mutations known to reduce opening rates lead to comparable reductions in fork junction binding affinity. The activity acts to establish the upstream boundary of melted DNA and works in conjunction with two single-stranded DNA binding activities that recognize separately the two melted strands. The junction binding activity is contained within the sigma factor component of the holoenzyme. The activity occurs in both a typical prokaryotic transcription system and in a eukaryotic-like bacterial system that responds to enhancers and needs ATP. Thus DNA opening catalyzed by fork junction binding may occur in a variety of systems in which DNA must be opened to be copied.
DNA must be locally melted to be transcribed. Most transcription regulators act at the steps leading up to melting (1–3), but the range of activities required for melting (1, 4, 5) has not been established. In the simplest cases in bacteria, appropriate double-stranded promoter consensus sequences are recognized by the sigma 70 subunit of the RNA polymerase (RNAP) holoenzyme, and then a short segment is melted to make the template strand accessible to the catalytic core (α2ββ′) (6). In this process, holoenzyme first binds to the promoter to form a closed complex and then opens a segment roughly from position −11 to +3. Chemical modifications reveal structural distortions in closed complexes near position −11, suggesting that nucleation of promoter opening occurs near the upstream boundary of the open region (7–10). The activity responsible for this critical step is not known. Opening then may extend unidirectionally to expose the transcriptional start site (7, 8). The sequences on the nontemplate strand of the −10 consensus element, which extends from −12 to −7, are known to have an important influence (11).
Both the sigma and core components of the holoenzyme may contribute to this targeted melting reaction. Mutations in both core and sigma can affect promoter melting (12–17). Sigma 70 has an established role in binding to the consensus promoter elements and appears to include a single-stranded DNA recognition component (5). This recognition occurs with the nontemplate strand and requires DNA elements at the upstream edge of the −10 consensus (5). However, this activity alone appears not to be sufficient to account for melting as it is highly localized and does not confer resistance to inactivation by the polyanion heparin; this heparin resistance is the hallmark of stable open complex formation. Thus we initiated a search for new activities that might trigger specific melting to form heparin-resistant complexes.
All open complexes contain two double-stranded segments and two single-stranded segments. Fork junctions are created where double and single strands join. These junctions are distinctive structures, and it is possible that components of the holoenzyme might recognize them and contribute to promoter opening. The upstream junction may be particularly relevant because, as just discussed, its formation appears to nucleate promoter melting. Below, we show experiments indicating that sigma factors contain an activity that recognizes this upstream fork junction and acts with other activities to open the promoter.
MATERIALS AND METHODS
Proteins and DNAs.
Escherichia coli RNAP core enzyme and holoenzyme containing sigma 70 are commercial products of Epicentre Technologies (Madison, WI). Sigma 54 was purified as reported (18). Oligonucleotides were made by using a Beckman DNA synthesizer. DNA probes were prepared as follows. The strand whose length was kept unchanged was labeled with γ-[32P]ATP. The 40-μl mixture, containing 4.0 pmol kinased DNA and 6.0 pmol complementary strand in 20 mM Tris⋅HCl, pH 7.5/80 mM NaCl, was boiled briefly and slowly cooled to room temperature. The resulting annealed probes then were diluted in Tris-EDTA buffer containing 80 mM NaCl to the desired concentration. Proper annealing was monitored by electrophoresis.
Electrophoresis Mobility-Shift Assay (EMSA).
Mobility assays were done as follows. (a) One unit of RNAP holoenzyme (1.2 μg) was added to a 10-μl reaction mixture containing 1× buffer T (50 mM Hepes-HCl, pH 7.9/100 mM KCl/10 mM MgCl2/0.1 mM EDTA/1 mM DTT/0.05 μg/ml BSA/2.8% polyethylene glycol 8,000/6.0 ng/μl poly dI:dC; Pharmacia) with 1 nM annealed DNA probe. After a 30-min incubation on ice, samples were directly loaded onto prechilled 5% PAGE with 1× TBE buffer (45 mM Tris-borate/1 mM EDTA). After electrophoresis, the radioactive bands were visualized by PhosphorImaging. (b) For heparin challenge experiments, heparin was added into samples after 30-min incubation to final concentration of 100 μg/ml. Samples then were incubated for another 10 min on ice before electrophoresis. (c) When heparin is added before probes, 1 unit of RNAP was incubated with 1× buffer T and 100 μg/ml of heparin for 10 min on ice before probes were added to final concentration of 1 nM. After another 30-min incubation on ice, samples were electrophoresed. (d) For assays with sigma 54 alone, experiments were done as in c except that 0.36 μg of sigma 54 was added into the reaction instead of holoenzyme.
RESULTS
E. coli RNAP Holoenzyme Interacts Preferentially with Specific Fork Junction Structures.
To test the role of an upstream junction structure in the formation of stable open complexes, fork junction probes were designed based on the λ late gene promoter PR′ from −41 to +1 (11) (Fig. 1 Top). The parent double-stranded probe includes the −10 recognition sequence from −12 to −7, which becomes largely melted when open complexes form (19). Pairs of oligonucleotides of differing lengths were annealed to create fork junction probes. Each of these contains an upstream double-stranded region, extending from position −41 to various downstream positions within the promoter. The double-stranded region in each probe is joined to one downstream single-stranded arm, whose length varies according to where the junction is created. The use of one arm at a time avoids the potentially confusing effects of choosing mutant sequences for the second arm in heteroduplex probes. An EMSA binding assay is used under closed complex conditions to minimize melting of the double-stranded part of the probes.
Figure 1.

EMSA of E. coli RNAP holoenzyme with bottom single-stranded fork structures. (Top) The sequence of the parent probe that is derived from the λ late gene promoter PR′, The consensus −10 element is in bold. (Middle) Schematic of the various fork probes with • representing the terminal bases on the top strand used in various probes. The identical bottom strand was annealed to each indicated top strand probe. The 32P- labeled bottom strand is marked with ∗. (Bottom) Results of EMSA using fork probes. In all cases the position of last 3′ base on the top strand (T) is indicated above the appropriate lane. In this and subsequent experiments, the unbound DNA probes were run off the gel. The appropriate shifted band, representing the complex of holoenzyme and DNA fork probe, is marked with an arrow. ssBot refers to a bottom single-stranded probe. Core refers to enzyme-lacking sigma 70. (A) Without heparin. (B) Heparin challenge after holoenzyme incubation with probes. (C) Heparin added before probes. (D) Control as in B.
In the first experiment the bottom (template) strand is left intact and the top (nontemplate) strand is “cut back” from +1 to create various potential transcription forks (see Fig. 1 Middle, with dots representing the terminal bps of various probes). The results show that complexes exist that survive a heparin challenge assay and show strong junction specificity (Fig. 1B, arrow). The preferred structure has bp number −11 as the terminal pair (T−11 in Fig. 1B) at a fork junction. Either adding (T−10) or removing (T−12) one more bp is inhibitory, and binding decreases further as more bps are either added or removed. Note that the binding is much stronger than that to either double-stranded DNA (T+1) or to single-stranded DNA containing the same sequences (ssBot in Fig. 1B and see below) and that it strongly depends on the presence of the sigma subunit (see Fig. 1D). We infer that E. coli RNAP can strongly bind to a structure in which −11 is the terminal bp and which contains a bottom single-stranded tail. In the absence of heparin challenge, all of the probes assayed were bound efficiently (see examples in Fig. 1A). For the optimal structure, approximately 45% of the complexes survive the 10-min heparin challenge.
We altered the experiment by using probes in which parts of the top strand were exposed as single stranded (Fig. 2). Previous studies showed that RNAP preferentially recognizes the top strand (5, 11). When the downstream part of the top strand is exposed to create a fork junction, heparin-resistant binding becomes apparent (Fig. 2B; compare double-stranded B+1 to fork B−7). The binding strengthens somewhat as the −10 consensus element becomes fully exposed (B−8 to B−12). The binding is stronger than to the bottom strand fork as 60% of the complexes survive the 10-min heparin challenge (see Fig. 2A).
Figure 2.

EMSA of E. coli RNAP holoenzyme with top single-stranded fork structures. The experiment is as in Fig. 1 except that the top strand was kept intact and was annealed to various bottom strand (B) probes with last 5′ base as indicated. The arrow indicates the appropriate shifted band. (A) Without heparin. (B) Heparin challenge after holoenzyme incubation with probes. (C) Heparin added before DNA probes. (D) Control as in B.
The data suggest some preference for junctions within the upstream edge of the consensus element. To reveal stronger preferences within the set we added heparin before the probes to require exceptionally strong binding. The data show that such preferences exist (Fig. 2C). Complexes appear when the double strand is “cut back” enough to expose the top strand −7T (see B−8). The binding is strongest in structures that contain a fork junction at either base pair −12 or −11, leaving most of the consensus element exposed on the top single strand. These structures bind much better than either single-stranded (ssTop) or double-stranded DNA (B+1) containing the same sequences. They also bind much better than probes with fork junctions that begin either upstream or downstream of position −11/−12. In this heparin pre-addition protocol the strongest signal strength is approximately 10% of the corresponding signal in an assay without heparin challenge (Fig. 2C). When the heparin pre-addition protocol is repeated on the weaker bottom strand forks assayed above, the same preference for a −11 fork is apparent but the signal is only at the 2% level (Fig. 1C). We infer that the highest affinity binding involves a structure containing a fork junction near the upstream edge of the −10 consensus element and a top single-stranded tail.
Exposure of the Pivotal −11 Nucleotide and the Top Strand Consensus Sequences.
Based on the above results, we created three sets of probes (Fig. 3A) with junctions at the strongly preferred positions and progressively cut away the single-stranded part to test the importance of single-stranded sequence for highly stable heparin-resistant binding. The first set of probes had fork junction at bp −12 (Fig. 3A Top) and “cut back” the top strand. The structure with no top strand tail showed no binding (T−12) in this heparin challenge assay. This finding confirms in another context that heparin-resistant binding requires the single-stranded part of the fork. Inclusion of a single unpaired base at the −11 top strand position restored heparin-resistance (T−11). Binding is strongest when the remaining top consensus sequence is exposed (T−7).
Figure 3.

Identification of three DNA elements for promoter opening by using EMSA. (A) Effect of top single-stranded sequences. The terminal bp of the fork junction was placed at either −12, −11, or −13 (■). Top strands of differing lengths (•) were annealed to labeled bottom strands. The EMSA heparin challenge results on each series are shown. (B) Effect of mutations at top strand −7 and −11 on probes with the optimal fork junction structure (−12 bp terminus). The top strand base present is indicated above each lane. (C) Effect of bottom single-stranded sequences.
We altered the experiment by using sets of probes terminated at nearby positions. When the last bp is at −11 bp (Fig. 3A Middle) the double-stranded probe can bind somewhat (Fig. 3A Middle, lane T−11). The binding is significantly strengthened by inclusion of the single-stranded consensus sequence on the top strand of the fork (Fig. 3A, T−7 vs. T−11). The experiment was repeated with a set of probes with bp −13 as the fork terminus (Fig. 3A Bottom). The double-stranded probe does not bind (T−13). Addition of a single base on the top strand does not restore binding in this case (T−12). Binding is restored only when base −11 is present on the top strand of the fork (T−11) as was the case with the fork terminated at bp −12 (above). As in all of the above cases, binding is strongest when the entire consensus −10 sequence is present in single-stranded form (T−7).
Quantitation of the entire data set (not shown) indicates that the strongest binding occurs when the last bp is at −12 and the −11 to −7 consensus sequence is exposed on the top strand of the fork. When the fork junction is moved upstream from −12, a specific position, −11, needs to be present in single-stranded form to detect binding. Thus −11 exposure appears to be critical as it is at the junction of the optimal fork and must be present in single-stranded form for binding when the fork junction is in an upstream position. In view of these observations the low, but detectable, level binding to the double-stranded structure terminated at bp −11 is probably a consequence of low-level exposure of base −11 caused by fraying at the helix terminus. In these experiments and those of Figs. 1 and 2 strong binding requires that the top strand base −11 be present either in single-stranded form or at a terminal helix position where it would have very substantial single-stranded character.
This bp −11 is known to be critical for open complex formation. It is highly conserved in promoters (6, 20), and substitutions there have by far the strongest effect in diminishing rates of open complex formation (11). Thus we substituted at −11 to see whether the identity of the base was also important for forming tight heparin-resistant complexes with fork junction probes. Fig. 3B shows that substitution of the top strand −11 A with either G, C, or T eliminates strong binding in a heparin challenge assay, with substitution of T retaining some binding (Fig. 3B, +Heparin). In analysis of promoter opening using both double-stranded and heteroduplex mutants, all mutations at this base greatly reduce the rate; the defect caused by G substitution is strongest and the T substitution defect is weaker (11). The comparison shows that the effect of mutation on functional opening is mimicked in the binding affinity to fork junctions. It is known that substitution at position −7 has a much smaller affect on open complex formation (11), and Fig. 3B shows that the same is true of fork junction binding. Thus fork junction binding and open complex formation have similar nucleotide sequence requirements.
Single-Stranded Binding Elements Act in Conjunction with Binding to the Upstream Fork Junction.
As discussed above, the data show that removal of the top strand bases in the segment including −7 to −10 reduces binding (compare lanes T−11 and T−7 in Fig. 3A). There is no reduction in any of the three cases when sequences from +1 to −6 are removed (compare lanes T+1 and T−7 in Fig. 3A). Thus only the upstream half of the top melted single strand in the open complex contributes additional important binding determinants. The contribution of this segment depends on use of the correct −12/−11 fork junction determinant; when the consensus −11A is replaced with G, the presence of the single-stranded segment does not lead to heparin resistance [Fig. 3B, Bottom, lane T−7(−11G)].
As the sequences downstream from −7 also are melted in open complexes, there should be an additional activity that accounts for this. Because the top strand sequences in the −6 to +1 region do not seem to be important (see above), we tested the bottom template strand sequences. A probe was created in which the bottom single strand protruded and was progressively “cut back.” Fig. 3C shows that removal of the sequences upstream from the +1 start site strongly reduces binding affinity (compare lanes B+1 and B−7). Progressive removal of individual bases showed that the sequences downstream from −4 contribute most to enhancement of heparin resistance (data not shown). However, as in the case of the top single-stranded determinant this bottom strand element is subsidiary to the use of the fork junction. That may be seen in Fig. 1B. When this bottom strand segment is exposed in the absence of an optimal fork there is little heparin-resistant binding (see lane T−7, for example). This lack of strong heparin resistance occurs in the context of a probe that contains the full −35 and −10 sequence elements in double-stranded form. Thus it is apparent that exposure of the bottom strand determinant is not sufficient to achieve heparin-resistant complex formation.
Taken together the data demonstrate that there are three contributing elements to forming high affinity heparin-resistant complexes. The upstream fork junction between bps −12 and −11 is dominant. Two single-stranded sequences contribute, one on the nontemplate strand through the consensus element and the other on the template strand up to the start site. Together these three elements cover the melted region end to end.
The above data reveal another feature of potentially high relevance to open complex formation using these three determinants. When an unpaired top strand −10 nucleotide is added to any fork junction probe, binding is strongly inhibited (Fig. 3A, compare T−10 with T−11). However, when the additional downstream nucleotides are added, the inhibition is progressively overcome and binding eventually reaches even higher levels. Thus unpaired nucleotide −10 can act as an inhibitor of formation of heparin-resistant complexes. We note that −10 is the boundary nucleotide between the fork junction and the nontemplate strand recognition elements discussed above. Thus it can be seen as a kind of gate controlling the ability to connect two adjacent independent interaction sites.
The DNA Fork Junction Binding Activity Is Contained in a Sigma Factor.
Of these three determinants it is only the fork junction activity that involves bases that are known targets of interaction with the sigma factor, strongly suggesting that it is sigma that recognizes the fork junction (6, 21). This hypothesis cannot be tested directly as the binding potential of isolated sigma 70 is masked (22). However, binding by the enhancer receptor protein sigma 54 is not as strongly masked (23). Preliminary experiments (unpublished work) showed that the sigma 54 holoenzyme forms a strong heparin-resistant complex with the specific fork junction probe shown in Fig. 4. Thus we used isolated sigma 54 to test for specific fork junction recognition. Fig. 4 shows that the isolated protein binds the fork junction probe far more tightly than it binds to either double-stranded or single-stranded DNA containing the same sequences. Note that it is only the structural feature of a double-stranded/single-stranded boundary that is preserved here from the sigma 70 system; the recognition sequences at the junctions are entirely different for sigma 70 and sigma 54 (6). Sigma 54 is sequence-unrelated to sigma 70 and is used for eukaryotic-like enhancer-dependent bacterial transcription (24). Thus very diverse opening reactions, catalyzed by sequence-unrelated proteins recognizing unrelated sequences, both involve recognition of specific fork junctions.
Figure 4.
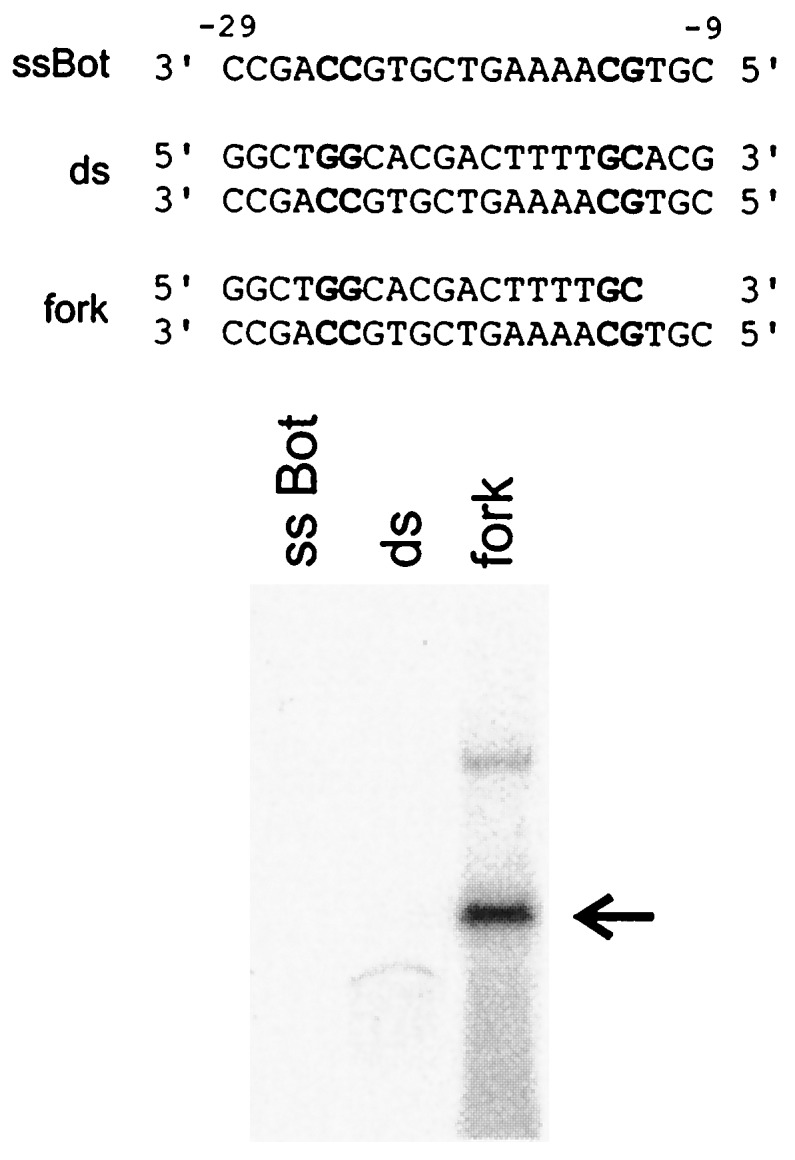
Isolated sigma 54 binds very tightly to a specific fork junction. EMSA (as in Fig. 1C) used the three probes shown. Critical consensus nucleotides of the sigma 54-dependent promoter nifH are in bold. The arrow indicates the sigma 54-fork DNA complex. Control lanes lacking sigma showed no band (not shown). Weak binding to the double-stranded probe can be seen under these EMSA conditions. Unbound DNA probes were run off. Similar results were obtained in the absence of heparin.
DISCUSSION
Fig. 5 shows how the three activities of holoenzyme together likely specify the nucleotides that will be melted in the open complex. Of these three activities it is binding to the −12/−11 fork junction that best mimics the properties of open complexes. Recognition of this structure confers heparin resistance, which is commonly used as an indication of open complex formation. This optimal junction is at the same position that occurs within natural open complexes at this same promoter; the −11 nucleotide is the most upstream position accessible to permanganate attack (19). Mutations that reduce fork junction binding strength also reduce the opening rate (11). We also note that the part of sigma 70 that is implicated in binding to the −10 consensus sequences contains a structure suggested to recognize aspects of bp −12 and an adjacent melted DNA tail (25), consistent with a prominent role for fork junction recognition. In this view the role of the important −35 promoter element is primarily to assist closed complex formation and position the holoenzyme so as to interact stereospecifically with the incipient fork junction in the −10 region; replacement of the −35 region sequences lowers the stability sufficiently to inhibit binding to fork junction probes (data not shown).
Figure 5.

Three activities proposed to be involved in open complex formation by sigma 70 RNAP. Fork junction binding by sigma is proposed to initiate melting. Two subsidiary activities are proposed to lead to exposure of the entire length of the melted region.
The data on the three activities can be combined with the extensive literature to suggest the pathway of promoter opening (see model in Fig. 5). Melting by both sigma 70 and sigma 54 holoenzymes begins near −11 and spreads downstream (7–10), suggesting that sigma binding to the optimal −12/−11 fork junction probably starts the opening process. The adjacent nontemplate strand (in the case of sigma 70) then should be bound, perhaps only when conditions allow the inhibitory −10 position gate to be unlocked. The melting finally would spread downstream by using the interaction with the template strand. If there are two channels in polymerase to accommodate the two melted DNA strands, then opening could be viewed as filling these channels. The establishment of the upstream fork by sigma factor can be seen as initiating this channel filling. Fork formation and channel filling would progressively stabilize the open complex.
At least two of the three activities that open the DNA appear to be in different components of the holoenzyme. As shown above, sigma likely binds the fork junction. The downstream region bound on the template strand corresponds to a segment that cannot be opened when mutations are made in the core polymerase (12). Thus core polymerase likely interacts here. The third interaction, with the template strand from −10 to −7, cannot yet be assigned definitively to a component of the holoenzyme (21). The role of sigma factor is, of course, pivotal to this process; it recruits polymerase to the promoter, initiates the subsequent opening reaction, and acts in conjunction with the two subsidiary activities to stabilize the open complex.
We note that initiation in all transcription systems requires promoter opening and that the mammalian open complex has its upstream fork junction at approximately the same position as the bacterial complexes studied here (26, 27). Several mammalian factors required for binding DNA and polymerase and opening DNA have short stretches that have been proposed as sigma 70 homology regions (28–31). Sigma factors alone have a variety of functions typically associated with different polypeptides in mammalian transcription complexes, each apparently associated with a different domain of the single sigma polypeptide (6, 32, 33). Thus a sigma factor may be seen as a highly focused organizing center for promoter recognition and opening. The opening reaction also occurs during DNA replication, repair, and recombination, where candidate fork binding proteins have been described with their junction specificity not tested (34–37). Thus the establishment of a tightly bound fork junction followed by extension of melting could conceivably be a common mechanism in the exposure and readout of DNA. If so, fork junction binding activities could be an important target for regulation.
Acknowledgments
This work was supported by a grant from the National Institutes of Health.
ABBREVIATIONS
- RNAP
RNA polymerase
- EMSA
electrophoresis mobility-shift assay
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1. Gralla J D. Curr Opin Genet Dev. 1996;6:526–530. doi: 10.1016/s0959-437x(96)80079-7. [DOI] [PubMed] [Google Scholar]
- 2.Hochschild A, Dove S L. Cell. 1998;92:597–600. doi: 10.1016/s0092-8674(00)81126-5. [DOI] [PubMed] [Google Scholar]
- 3.Jiang Y, Triezenberg S J, Gralla J D. J Biol Chem. 1994;269:5505–5508. [PubMed] [Google Scholar]
- 4.deHaseth P L, Helmann J D. Mol Microbiol. 1995;16:817–824. doi: 10.1111/j.1365-2958.1995.tb02309.x. [DOI] [PubMed] [Google Scholar]
- 5.Marr M T, Roberts J W. Science. 1997;276:1258–1260. doi: 10.1126/science.276.5316.1258. [DOI] [PubMed] [Google Scholar]
- 6.Gross C A, Lonetto M, Losick R. In: Transcriptional Regulation. McKnight S L, Yamamoto K R, editors. Vol. 1. Plainview, NY: Cold Spring Harbor Lab. Press; 1992. pp. 129–176. [Google Scholar]
- 7.Meier T, Schickor P, Wedel A, Cellai L, Heumann H. Nucleic Acids Res. 1995;23:988–994. doi: 10.1093/nar/23.6.988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y F, Helmann J D. J Mol Biol. 1997;267:47–59. doi: 10.1006/jmbi.1996.0853. [DOI] [PubMed] [Google Scholar]
- 9.Morris L, Cannon W, Claverie-Martin F, Austin S, Buck M. J Biol Chem. 1994;269:11563–11571. [PubMed] [Google Scholar]
- 10.Spassky A, Kirkegaard K, Buc H. Biochemistry. 1985;24:2723–2731. doi: 10.1021/bi00332a019. [DOI] [PubMed] [Google Scholar]
- 11.Roberts C W, Roberts J W. Cell. 1996;86:495–501. doi: 10.1016/s0092-8674(00)80122-1. [DOI] [PubMed] [Google Scholar]
- 12.Severonov K, Darst S A. Proc Natl Acad Sci USA. 1997;94:13481–13486. doi: 10.1073/pnas.94.25.13481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Juang Y L, Helmann J D. J Mol Biol. 1994;235:1470–1488. doi: 10.1006/jmbi.1994.1102. [DOI] [PubMed] [Google Scholar]
- 14.Aiyar S E, Juang Y L, Helmann J D, deHaseth P L. Biochemistry. 1994;33:11501–11506. doi: 10.1021/bi00204a012. [DOI] [PubMed] [Google Scholar]
- 15.Jones C H, Tatti K M, Moran C P., Jr J Bacteriol. 1992;174:6815–6821. doi: 10.1128/jb.174.21.6815-6821.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones C H, Moran C P., Jr Proc Natl Acad Sci USA. 1992;89:1958–1962. doi: 10.1073/pnas.89.5.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson C, Dombroski A J. J Mol Biol. 1997;267:60–74. doi: 10.1006/jmbi.1997.0875. [DOI] [PubMed] [Google Scholar]
- 18.Wang J T, Syed A, Gralla J D. Proc Natl Acad Sci USA. 1997;94:9538–9543. doi: 10.1073/pnas.94.18.9538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kainz M, Roberts J. Science. 1992;255:838–841. doi: 10.1126/science.1536008. [DOI] [PubMed] [Google Scholar]
- 20.Danot O, Raibaud O. J Mol Biol. 1994;238:643–648. doi: 10.1006/jmbi.1994.1324. [DOI] [PubMed] [Google Scholar]
- 21.Dombroski A J. J Biol Chem. 1997;272:3487–3494. [PubMed] [Google Scholar]
- 22.Dombroski A J, Walter W A, Record M T, Jr, Siegele D A, Gross C A. Cell. 1992;70:501–512. doi: 10.1016/0092-8674(92)90174-b. [DOI] [PubMed] [Google Scholar]
- 23.Buck M, Cannon W. Nature (London) 1992;358:422–424. doi: 10.1038/358422a0. [DOI] [PubMed] [Google Scholar]
- 24.Gralla J D. Cell. 1991;66:415–418. doi: 10.1016/0092-8674(81)90001-5. [DOI] [PubMed] [Google Scholar]
- 25.Malhotra A, Severinova E, Darst S A. Cell. 1996;87:127–136. doi: 10.1016/s0092-8674(00)81329-x. [DOI] [PubMed] [Google Scholar]
- 26.Wang W, Carey M, Gralla J D. Science. 1992;255:450–453. doi: 10.1126/science.1310361. [DOI] [PubMed] [Google Scholar]
- 27.Yan M, Gralla J D. EMBO J. 1997;16:7457–7467. doi: 10.1093/emboj/16.24.7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoffman A, Sinn E, Yamamoto T, Wang J, Roy A, Horikoshi M, Roeder R G. Nature (London) 1990;346:387–390. doi: 10.1038/346387a0. [DOI] [PubMed] [Google Scholar]
- 29.Ohkuma Y, Sumimoto H, Hoffmann A, Shimasaki S, Horikoshi M, Roeder R G. Nature (London) 1991;354:398–401. doi: 10.1038/354398a0. [DOI] [PubMed] [Google Scholar]
- 30.Sumimoto H, Ohkuma Y, Sinn E, Kato H, Shimasaki S, Horikoshi M, Roeder R G. Nature (London) 1991;354:401–404. doi: 10.1038/354401a0. [DOI] [PubMed] [Google Scholar]
- 31.Malik S, Lee D K, Roeder R G. Mol Cell Biol. 1993;13:6253–6259. doi: 10.1128/mcb.13.10.6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sasse-Dwight S, Gralla J D. Cell. 1990;62:945–950. doi: 10.1016/0092-8674(90)90269-k. [DOI] [PubMed] [Google Scholar]
- 33.Wang J T, Syed A, Hsieh M, Gralla J D. Science. 1995;270:992–994. doi: 10.1126/science.270.5238.992. [DOI] [PubMed] [Google Scholar]
- 34.Iftode C, Borowiec J A. Mol Cell Biol. 1997;17:3876–3883. doi: 10.1128/mcb.17.7.3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jezewska M J, Rajendran S, Bujalowski W. Biochemistry. 1997;36:10320–10326. doi: 10.1021/bi970712a. [DOI] [PubMed] [Google Scholar]
- 36.Rafferty J B, Sedelnikova S E, Hargreaves D, Artymiuk P J, Baker P J, Sharples G J, Mahdi A A, Lloyd R G, Rice D W. Science. 1996;274:415–421. doi: 10.1126/science.274.5286.415. [DOI] [PubMed] [Google Scholar]
- 37.McGlynn P, Al-Deib A A, Liu J, Marians K J, Lloyd R G. J Mol Biol. 1997;270:212–221. doi: 10.1006/jmbi.1997.1120. [DOI] [PubMed] [Google Scholar]