

Abstract
The refined NMR structure of the mouse prion protein domain mPrP(121–231) and the recently reported NMR structure of the complete 208-residue polypeptide chain of mPrP are used to investigate the structural basis of inherited human transmissible spongiform encephalopathies. In the cellular form of mPrP no spatial clustering of mutation sites is observed that would indicate the existence of disease-specific subdomains. A hydrogen bond between residues 128 and 178 provides a structural basis for the observed highly specific influence of a polymorphism in position 129 in human PrP on the disease phenotype that segregates with the mutation Asp-178–Asn. Overall, the NMR structure implies that only part of the disease-related amino acid replacements lead to reduced stability of the cellular form of PrP, indicating that subtle structural differences in the mutant proteins may affect intermolecular signaling in a variety of different ways.
A novel class of infectious pathogens, the prions, have been proposed to be the cause of transmissible spongiform encephalopathies (TSE) (1, 2). Prions are distinct from bacteria, viruses, or viroids in that nucleic acids are apparently not essential for the propagation of the infectious agent (3). TSEs include kuru, Creutzfeldt-Jakob disease (CJD), fatal familial insomnia (FFI), and Gerstmann-Sträussler-Scheinker syndrome in humans, scrapie in sheep, and bovine spongiform encephalopathy. They have been reported as sporadic and inherited as well as infectious disorders. According to the “protein-only” hypothesis (4–6), prion diseases are linked with the presence of the prion protein (PrP) (7), which is ubiquitous in mammalian cells in the benign cellular form (PrPC) and is in rare instances transformed into the disease-related scrapie form (PrPSc).
The infectious, disease-related form of the PrP, PrPSc, has so far only been observed as an insoluble oligomer that displays partial resistance to proteinase K digestion and has characteristics of an amyloid (1, 2, 8). Based on Fourier transform reflection infrared spectroscopy it was concluded that a significant percentage of the polypeptide chain in PrPSc forms β-sheet secondary structure (9, 10). Attempts to identify posttranslational modifications of the covalent structure that would be related to the rare conversion of the ubiquitous PrPC into PrPSc have been unsuccessful. Although this finding indicates that prion diseases might indeed be disorders of protein conformation, no one has succeeded as yet to generate infectious PrPSc in vitro, either from previously denatured infectious material, natural PrPC, or from recombinant or synthetic PrP or fragments thereof (see also ref. 11).
The cellular form of mammalian PrP, PrPC, consists of a single polypeptide chain that contains two glycosylation sites and is attached to the cell surface by a glycosyl-phosphatidyl-inositol anchor at its carboxyl terminus (12). After separation from the cell membrane, mammalian PrPC is a water-soluble, protease K-sensitive protein. The NMR structures of intact recombinant mouse PrPC, mPrP(23–231) (13), and its C-terminal domain, mPrP(121–231) (14), have been determined, and this paper presents a refinement of the mPrP(121–231) structure.
Correlations between the molecular structure of prion proteins and their role in the pathology of TSEs previously have been discussed on the basis of structure predictions (15–17). The NMR structure of mPrPC (Figs. 1 and 2, refs. 13 and 14) now provides a basis for such work. The mouse and human prion proteins have identical global folds for the domain of residues 121–231 (R. Zahn, R.R., G.W., and K.W., unpublished work), as expected from the 93% sequence identity (18). All eight sequence positions in this domain for which amino acid substitutions have been related to human genetic TSEs (1) contain identical amino acids in the wild-type mouse and human proteins (18), and all amino acids that are in direct contact with these residues in the refined three-dimensional structure of mPrPC are also identical in the two species. On this basis we use the NMR structure of mPrPC to investigate likely structural and functional consequences of the disease-related amino acid substitutions in human PrP (hPrP) and to critically evaluate a previously advanced general concept that inherited TSEs might be related to destabilization of the three-dimensional structure of PrPC (15–17).
Figure 1.

Global features of the refined NMR structure of mPrP(121–231). (a) Backbone of residues 126–226 of the mean structure of mPrP(121–231) as a spline function through the Cα positions. The variable radius of the yellow cylinder is proportional to the mean global backbone displacement per residue evaluated after superposition of the 20 conformers used to represent the solution structure for best fit of the atoms N, Cα, and C′ of residues 126–166 and 172–226. The chain ends and some sequence locations are indicated. (b) Backbone 126–226 represented with helices as red ribbons, the β-sheet as yellow ribbons, and the loop regions as orange tubes. The disulfide bond Cys-179–Cys-214 is indicated in green. The hydrophobic core containing residues 134, 137, 139, 141, 158, 161, 175, 176, 179, 180, 184, 198, 203, 205, 206, 209, 210, and 213–215 (see text) is shown in a translucent envelope. Outside of the core all hydrogen bonds involving side chains (blue stick models) are represented by dashed cyan cylinders and labeled by a code of lowercase letters that refers to Table 2. (c) Superposition for best fit of the backbone atoms of residues 126–219 of the refined structure in b with the structure from ref. 14, which is drawn in gray. The three helices are identified as α1, α2, and α3. (d) Location of mutations that in the homologous hPrP have been associated with inherited prion diseases (see text). Helices 2 (on the right) and 3 are drawn as gray cylinders, the connecting loop as a gray tube, and the two-stranded β-sheet as a yellow plane. The disulfide bond is identified by green coloring. The mutation sites that segregate with CJD are colored blue, the Gerstmann-Sträussler-Scheinker syndrome mutation sites are cyan, and the site of the polymorphism M/V129 is red. The amino acids in the mouse sequence, which are shown here, are in all cases identical to those in the human protein. This figure and Fig. 2 were prepared with the program molmol (45).
Figure 2.
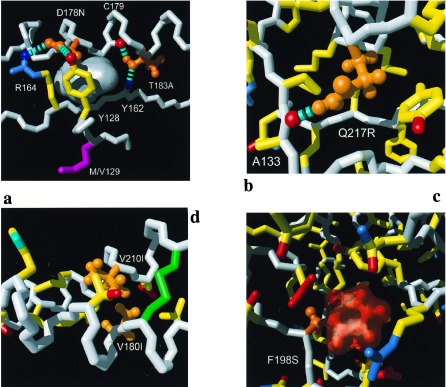
Close-ups of individual mutation sites: (a) D178N and T183A, (b) Q217R, (c) F198S, and (d) V180I and V210I. Color code: white, polypeptide backbone; orange, side chains for which a mutation has been associated with inherited human prion diseases; yellow, side chain carbon—carbon bonds; blue, bonds to side-chain nitrogen atoms and selected backbone amide groups; red, bonds to side-chain oxygen atoms and selected backbone carbonyl oxygens. The hydrogen bonds are represented by broken cyan cylinders. (a) An empty cavity observed in wild-type mPrP(121–231) is shown as a gray surface. (b) All bonds within a slice of approximately 18 Å thickness and (c) those within an 10 Å slice. (c) The orange transparent surface represents the empty space that would be left after the amino acid replacement F198S in the absence of any subsequent structural rearrangement. (d) All side chains of helices 2 and 3 are shown for which the inter-helix distance between at least one pair of side-chain heavy atoms is shorter than 5 Å.
MATERIALS AND METHODS
Isotope-labeled mPrP(121–231) was prepared as described previously (14, 19). NMR measurements were performed on Bruker AMX500 and AMX600 and Varian Unity+750 spectrometers. For data processing and spectral analysis we used the programs prosa (20) and xeasy (21), respectively. Stereospecific assignments for the methyl groups of Val and Leu were obtained with 10% biosynthetically directed 13C-labeling of the protein (22).
The input of nuclear Overhauser effect (NOE) distance constraints for the structure calculation was obtained from the following 750-MHz data sets: three-dimensional 13C-resolved [1H,1H]-NOE spectra (23) with τm = 50 ms, using 13C/15N-labeled protein; three-dimensional 15N-resolved [1H,1H]-NOE spectra (24) with τm = 50 ms, using 15N-labeled protein; and two-dimensional [1H,1H]-NOE spectra (25) in H2O and 2H2O solution with τm = 50 ms, using unlabeled protein. The NOE intensities were calibrated with the tools of the program dyana (26). Other tools of dyana were used to eliminate NOEs that represent no true conformational constraints and, where required, pseudo-atoms were introduced with appropriate corrections (27). Supplementary constraints were: From Cα chemical shifts deviating from the random coil values, −120° < φ < −20° and −100° < ψ < 0° for Δδ(Cα) > 1.5 ppm; −200° < φ < −80° and 40° < ψ < 220° for Δδ(Cα) < −1.5 ppm (28). With these constraints and intraresidual and sequential NOEs, habas (29) yielded 229 constraints on dihedral angles φ, ψ, and χ1. The usual limits were used for the disulfide bond Cys-179—Cys-214 (30).
When using asno (31) a final input for the structure calculation of 1,592 NOE upper-limit distance constraints, 229 dihedral angle constraints and the six disulfide bond constraints (30) were obtained. The calculation was started with 50 randomized conformers, and the 20 best dyana conformers were energy-minimized in a water bath using the amber force field (32) in the program opal (33). The mean structure of mPrP(121–231) used for the illustrations was obtained by averaging the coordinates of the 20 refined conformers after superposition of the backbone atoms of residues 124–166 and 172–226 and subsequent energy minimization.
RESULTS AND DISCUSSION
The NMR structure of the soluble form of the mouse PrP mPrP(23–231) consists of a globular C-terminal domain of residues 126–226 (Fig. 1a), a flexible unstructured 103-residue N-terminal tail, and a flexibly disordered C-terminal segment 227–231 (13, 34). Suitably isotope-labeled mPrP(121–231) preparations were used to collect NMR data for a structure refinement of the globular domain. Although the initial structure determination (14) described the global fold and the spatial distribution of the amino acid side chains, the refined structure further affords a detailed description of the side-chain packing (Figs. 1b and 2).
The Refined NMR Structure of mPrP(121–231).
Table 1 gives a survey of the solution conditions used, the input of conformational constraints, and the quality of the structure determination. Key data are that the structurally defined polypeptide segments include residues 124–166 and 172–226, as compared with 125–166 and 177–219 in the previous work (14), and that the rms deviation values for the backbone and all heavy atoms of these residues are 0.8 Å and 1.2 Å, respectively, as compared with 1.4 Å and 2.0 Å in ref. 14. The thickness of the yellow cylindrical rod in Fig. 1a represents the variable precision of the backbone structure determination along the sequence from residues 124–226.
Table 1.
Characterization of the energy-minimized NMR structure of mPrP(121–231)
| Quantity | 20 conformers | Mean structure* |
|---|---|---|
| Number ≥0.1 Å | 2.8 ± 1.6 | 0 |
| Maximum (Å) | 0.10 ± 0.0 | 0.08 |
| Residual dihedral angle constraint violations | ||
| Number ≥1.5 deg. | 1.7 ± 1.0 | 0 |
| Maximum, deg. | 1.9 ± 0.3 | 1.3 |
| AMBER energies, kcal/mol | ||
| Total | −5,061 ± 67 | −4,777 |
| Van der Waals | −330 ± 19 | −348 |
| Electrostatic | −5,679 ± 73 | −5,303 |
| rms deviation from ideal geometry | ||
| Bond lengths, Å | 0.0078 ± 0.002 | 0.0074 |
| Bond angles, deg. | 2.33 ± 0.05 | 1.62 |
| Peptide bonds, deg. | 9.7 ± 2.3 | 10.7 |
| rms deviation to the averaged coordinates, Å | ||
| N, Cα, C′ (124–166, 172–226) | 0.8 ± 0.1 | |
| Same plus best defined side chains† | 0.8 ± 0.1 |
mPrP(121–231) was studied in aqueous solution containing 0.8 mM protein, pH = 4.5, T = 20°C. The structure determination was based on sequence-specific assignments for 98% of all 1H, 13C, and 15N nuclei. The missing 1H assignments include all resonances of Asp-167, the backbone resonances of Gln-168, 1HN of Tyr-169, Ser-170, and Asn-171, the nonaromatic resonances of Phe-175, and the side-chain resonances of Glu-220. The input for the final structure calculation consisted of 1,592 NOE upper distance limits (388 intraresidual, 428 sequential, 413 medium range, 363 long range) and 229 dihedral angle constraints. The average residual target function value for the 20 best DYANA conformers was 1.02 ± 0.48 Å2.
The energy-minimized mean structure differs from the average of the atom coordinates of the 20 conformers by a rms deviation of 0.5 Å calculated for the backbone atoms of residues 124–166 and 172–226.
Best-defined side chains are those with a displacement of the heavy atoms smaller than 1.0 Å and include residues 130, 132–134, 137, 139, 141, 146, 149, 153, 158–161, 178–184, 188–192, 199, 201–203, 205–207, 209, 210, and 213–216.
The global polypeptide fold of mPrP(121–231) contains three α-helices with residues 144–154, 175–193, and 200–219, two β-strands with residues 128–131 and 161–164, and a short segment of helix-like structure with residues 222–226 (Fig. 1a). Comparison with the initial NMR structure (14) (Fig. 1c) shows that helices 2 and 3 have been extended by four residues at the N terminal end and by two residues at the C-terminal end, respectively (Fig. 1c) (35), which is a consequence of the improved definition of these segments (36), and the residues 220–226 are somewhat better defined (see below). During the refinement the axis of helix 3 has been slightly shifted relative to the other regular secondary structures.
Although the local precision of the structure determination generally was improved during the refinement, some short peptide segments remain poorly defined. In addition to the flexibly disordered chain-terminal segments 121–125 and 227–231 (14), these segments include residues 167–171 in the loop connecting the second β-strand with helix 2 and the last two turns of helix 2 with the sequence –Thr188–Val–Thr–Thr–Thr–Thr193–. With regard to the pathology-related conformational polymorphism in position 129 of hPrP (37), the β-sheet in PrPC attracts particular interest. Although residues 129, 131, 161, 163, and 164 show typical β-sheet deviations of the 13Cα chemical shifts from the random coil values (38), the Hα chemical shifts and the 3JHNα coupling constants in the first β-strand are close to the random coil values, which may indicate local conformational averaging. On the other hand, there is a hydrogen bond from the amide proton of Met-134 to the carbonyl oxygen of Asn-159 that would be compatible with an elongation of the β-sheet toward the first helix, with a β-bulge at residue 132, which is again indicative of dynamic plasticity of the β-sheet.
The mPrP(121–231) preparations used in the initial study (14) showed proteolytic cleavage at Tyr-226–Asp-227, which gave rise to multiple NMR lines for the C-terminal region so that the structure could not be characterized beyond residue 219. The presently used more stable protein preparations enabled a structural characterization to residue 226. Helix 3 is well characterized up to Thr-219, but residues 220 and 221 gave broad peaks and only a small number of NOEs could be observed, indicating structural disorder and possibly increased mobility. For residues 222–225 typical α-helix NOE distance constraints (36) were observed in mPrP(23–231) (13) as well as in mPrP(121–231). The structure calculation using the ensemble of all conformational constraints yielded the structure shown in Fig. 1, with helix 3 linked to a distorted helical turn by a nonregular dipeptide. However, because all long-range constraints of the segment 222–226 are with residues in or adjacent to the disordered loop of residues 167–171, the three-dimensional arrangement of segment 220–226 remains uncertain.
NMR structures also have been presented of the polypeptide fragments of residues 90–231 and 29–231 of the PrP from Syrian hamster, shPrP(90–231) (ref. 39; PDB ID code 2PRP) and shPrP(29–231) (40). The polypeptide backbone fold in shPrP coincides with that of Fig. 1a, except that the loop of residues 167–171 is reported to be well defined based on complete resonance assignments, that helix 3 is described as a straight helix extending from residues 200–227, and that the polypeptide segment 132–144 between the first β-strand and helix 1 shows locally somewhat different arrangements in the two proteins.
The major result of the present structure refinement for mPrP(121–231) is the precise structural definition of a large fraction of the amino acid side chains. Twenty residues form a tightly packed hydrophobic core. In Fig. 1b the outer confines of the core are indicated by a translucent sheet. Thirteen residues in the hydrophobic core come from the antiparallel helices 2 and 3, which have mutual interactions through the residues Phe-175, Val-176, Val-180, Ile-184, Val-203, Met-206, Val-210, and Met-215, and the disulfide bridge Cys-179–Cys-214, whereas Met-205, Val-209, and Met-213 interact with the four core residues Met-134, Pro-137, Ile-139, and Phe-141 in the loop between the first β-strand and the first helix (Fig. 1b). Pro-158 and Val-161 are adjacent and within the β-sheet, respectively, and Phe-198 is located in the loop between helices 2 and 3.
The hydrophobic core is surrounded by an outer shell of protein structure that contains 22 hydrogen bonds with amino acid side chains (Fig. 1b and Table 2), which include medium-range interactions in all three helices and a variety of longer-range interactions. The N terminus of the first helix contains the hydrogen bond Asn-143Hδ– OɛGlu-146 (b in Fig. 1b), which is directly supported by NOEs between δNH2 of Asn-143 and γCH2 of Glu-146. A hydrogen bond between the backbone oxygen of Asn-171 and the side chain of Asn-174 (l in Fig. 1b) initiates the second helix. The side chain of Thr-183, which also acts as an acceptor for a long-range hydrogen bond with the amide proton of Tyr-162, forms a hydrogen bond to the carbonyl oxygen of Cys-179, as is supported by the fact that of the hydroxyl proton of Thr-183 exchanges sufficiently slowly to be observed by 1H NMR. The N terminus of the third helix is stabilized by a capping box (41) of Thr-199HN–OδAsp-202 and Asp-202HN–OγThr-199, and the side chain–side chain hydrogen bond Thr-199OHγ–OδAsp-202 (r, s, and t in Fig. 1b). Among the longer-range hydrogen bonds connecting different groups within the outer shell there are the two hydrogen bonds Tyr-128Hη–OδAsp-178 and Tyr-162HN–OγThr-183 (a and j in Fig. 2b), and the salt bridge Arg-164–Asp-178 (k in Fig. 1b), which hold the β-sheet against helix 2. The salt bridge Arg-156–Glu-196 (g in Fig. 1b) orients the C-terminal end of helix 1 toward the loop connecting the other two helices.
Table 2.
Hydrogen bonds with amino acid side chains in the NMR structure of mPrP(121–231)
| Donor | Acceptor | n* | |
|---|---|---|---|
| a | Tyr-128Hη | OδAsp-178 | 11 |
| b | Asn-143Hδ | OɛGlu-146 | 16 |
| c | Tyr-149Hη | OδAsp-202 | 16 |
| d | Tyr-150Hη | O′Pro-137 | 17 |
| e | Arg-151Hη | OɛGlu-152 | 8 |
| f | Asn-153Hδ | O′Tyr-149 | 18 |
| g | Arg-156Hη | OɛGlu-196 | 13 |
| h | Tyr-157Hη | O′Asp-202 | 7 |
| i | Gln-160Hɛ | O′Gly-131 | 19 |
| j | Tyr-162HN | OγThr-183 | 12 |
| k | Arg-164Hη | OδAsp-178 | 7 |
| l | Asn-174Hδ | O′Asn-171 | 12 |
| m† | Thr-183OHγ | O′Cys-179 | 6 |
| n | Thr-188OHγ | O′Ile-184 | 12 |
| o | Thr-191OHγ | O′His-187 | 8 |
| p | Thr-192OHγ | O′Glu-196 | 7 |
| q | Lys-194Hζ | OɛGlu-196 | 7 |
| r | Thr-199HN | OδAsp-202 | 17 |
| s | Thr-199OHγ | OδAsp-202 | 14 |
| t | Asp-202HN | OγThr-199 | 15 |
| u | Thr-216OHγ | O′Gln-212 | 10 |
| v | Gln-217Hɛ | O′Ala-133 | 8 |
All hydrogen bonds are listed that occur in more than 35% of the 20 energy-refined conformers used to represent the solution structure. The notations a through v relate to the visualization of the hydrogen bonds in Fig. 1b.
n is the number of conformers, out of the group of 20 conformers used to represent the NMR structure, that contain the hydrogen bond.
This hydrogen bond is listed although not all criteria for H-bond identification were satisfied.
Most of the residues located in the hydrophobic core as well as those involved in outer shell hydrogen bonds of mPrP(121–231) are strictly conserved among mammalian species (18, 35). Exceptions are position 139, which contains Ile in all but three mammalian species and Met in Syrian hamster, and seems to be a key residue in the species barrier between mouse and hamster (42); Ile-184 and Val-203, which are exchanged in a correlated manner so that the local packing requirements are preserved (35); and Met-205 and Val-215. The only nonconserved hydrogen bond-forming side chains listed in Table 2 are Asn-143, Arg-164, and Asn-174; all three, however, are exchanged conservatively so that corresponding hydrogen bonds may be maintained in the prion proteins from all different species.
Local Structures Near Mutation Sites that Segregate with Inherited Human TSEs.
According to a previously advanced hypothesis the amino acid replacements in hPrP that segregate with inherited human TSEs should result in reduced stability of the three-dimensional structure of the PrPC form and thus enhance the tendency of PrPC to undergo transitions to other conformational states, including states that may lead to PrPSc formation (15–17). In hPrP the polypeptide segment 121–231 contains eight of 11 amino acid replacements that have been associated with familial TSEs (1, 43), which all are located within or sequentially adjacent to helices 2 and 3 (Fig. 1d).
The most interesting observations relate to the amino acid replacement Asp-178–Asn (Fig. 2a), which removes the salt bridge between the strictly conserved residues Asp-178–Arg-164 (18, 35) (k in Fig. 1b). We thus predict that this variant protein has reduced thermodynamic stability when compared with wild-type mPrP(121–231). The phenotype of the prion disease that segregates with the mutation in position 178 has been shown to be determined by the nature of the amino acid residue in position 129, i.e., Met-129/Asn-178 correlates with fatal familial insomnia and Val-129/Asn-178 with CJD (44). Two hydrogen bonds in wild-type mPrP(121–231) (Fig. 2a) link the side chain of Asp-178 with two side chains in the β-sheet, one of which is sequentially adjacent to position 129. Thus, the Asp-178–Asn exchange may affect the hydrogen bonding network involving Arg-164, Tyr-128, and Asp-178 somewhat differently depending on the nature of the amino acid residue in position 129. The observed specific interactions between positions 129 and 178 (Fig. 2a) then would also provide a rationale for the observation that the Val/Met-129 polymorphism does not affect the phenotypes of the inherited TSEs that segregate with the other known mutations (Fig. 1d).
The mutation Thr-183–Ala (Fig. 2a) eliminates two hydrogen bonds that establish a link between helix 2 and the β-sheet, i.e., Thr-183OHγ–O′Cys-179 and Tyr-162HN–OγThr-183 (j and m in Fig. 1b), indicating reduced stability for this protein variant.
For the amino acid replacement Gln-217–Arg (Fig. 2b) the NMR structure indicates reduced stability of PrPC: in the wild-type protein, Gln-217 is surrounded by hydrophobic groups (Fig. 2b), and its side chain forms a long-range hydrogen bond to the carbonyl oxygen of Ala-133, thus stabilizing a distinct position of the loop between the first β-strand and helix 1 relative to the hydrophobic core. The replacement Gln-217–Arg introduces a positive charge into this otherwise uncharged region (the nearest charged group is about 10 Å away), and it appears most unlikely, for steric reasons, that a satisfactory hydrogen bond geometry with the carbonyl oxygen of Ala-133 could be achieved with the Arg side chain.
The replacement of Phe-198 in the hydrophobic core by Ser is a pronouncedly nonconservative mutation. In the absence of follow-up structural changes this mutation would lead to an empty cavity that could accommodate 2–3 water molecules (Fig. 2c). In the wild-type protein the aromatic ring is surrounded by numerous hydrogen-bonded polar side chains (g, r, s, and t in Fig. 1b) and its replacement by serine is likely to result in a modified pattern of polar interactions. These interactions could trigger a collapse of the cavity, which, in turn, would influence the surface structure and thus could also alter the ligand binding properties of PrPC. In view of the anticipated complex structural rearrangement, it is difficult to estimate the concomitant change in thermodynamic stability, but significantly reduced stability would be anticipated.
Finally, there are four amino acid replacements for which the NMR structure predicts no or at most minor variations in stability. The mutations Glu-200–Lys and Arg-208–His (Fig. 1d) are located on the protein surface, so that even the change in overall charge of the protein should not have a major impact on the global structure. For the replacement of Val by Ile in either positions 180 or 210 (Fig. 2d) there is enough space to accommodate the somewhat larger Ile side chain.
CONCLUSIONS
The refined NMR structure of mPrP(121–231) and the recent determination of the solution structure of the intact mPrP, mPrP(23–231) (13) show convincingly, in contrast to earlier suggestions (15, 16), that familial human TSEs generally cannot be rationalized by the assumption of reduced stability of the PrPC form of the mutant proteins. First, three point mutations that segregate with inherited TSEs are in the flexibly extended polypeptide segment N terminal to residue 121 (13), where it is unlikely that individual amino acid substitutions would measurably affect the global stability of PrPC. Second, for the eight protein species with disease-related amino acid exchanges in the globular C-terminal domain the refined NMR structure of wild-type mPrP(121–231) predicts largely different effects on the protein stability, from very small variations relative to the parent protein to major destabilization caused by loss of internal hydrogen bonds and salt bridges. These structure-based predictions on the relative thermodynamic stabilities of the individual variant proteins coincide qualitatively with experimental measurements using the corresponding recombinant protein variants of mPrP(121–231) (R. Glockshuber, personal communication). The refined NMR structure of mPrP(121–231) further shows that the mutation sites related to the human TSE phenotypes Gerstmann-Sträussler-Scheinker syndrome (GSS) and CJD do not form separate clusters in the three-dimensional structure of PrPC (Fig. 1d), excluding the presence of GSS- and CJD-specific subdomains, and it presents a rationale for the specific influence of the residue type in the polymorphism site 129 on the TSE phenotypes that segregate with the mutation Asp-178–Asn (44).
Acknowledgments
We acknowledge the Centro Svizzero di Calcolo Scientifico for use of the NEC SX-4 computer, the Eidgenössiche Technische Hochschule for the use of the Cray J-90 cluster, and Mrs. M. Geier and Mrs. E. Ulrich for the careful processing of the manuscript. Financial support was obtained from the Schweizerischer Nationalfonds [Projects 31.49047.96 and 438+050287 (K.W.) and 438+050285 (R.G.)]. S.H. is supported by a grant from the Boehringer Ingelheim Fonds.
ABBREVIATIONS
- CJD
Creutzfeldt-Jakob disease
- PrP
prion protein
- PrPC
cellular form of PrP
- PrPSc
scrapie form of PrP
- hPrP
human PrP
- mPrP
mouse PrP
- mPrP(121–231)
fragment of mPrP
- mPrP(23–231)
complete polypeptide of mature mPrP
- NOE
nuclear Overhauser effect
- TSE
transmissible spongiform encephalopathy
Footnotes
Data deposition: The atom coordinates of the refined mean structure of mPrP(121–231) have been deposited in the Protein Data Bank, Biology Department, Brookhaven National Laboratory, Upton, NY 11973 (PDB ID code 1AG2).
References
- 1. Prusiner S B. Trends Biochem Sci. 1996;21:482–487. doi: 10.1016/s0968-0004(96)10063-3. [DOI] [PubMed] [Google Scholar]
- 2.Weissmann C. FEBS Lett. 1996;389:3–11. doi: 10.1016/0014-5793(96)00610-2. [DOI] [PubMed] [Google Scholar]
- 3.Riesner D. Chemie in unserer Zeit. 1996;30:66–74. [Google Scholar]
- 4.Alper T, Cramp W A, Haig D A, Clarke M C. Nature (London) 1967;214:764–766. doi: 10.1038/214764a0. [DOI] [PubMed] [Google Scholar]
- 5.Griffith J S. Nature (London) 1967;215:1043–1044. doi: 10.1038/2151043a0. [DOI] [PubMed] [Google Scholar]
- 6.Prusiner S B. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 7.Büeler H, Raeber A, Sailer A, Fischer M, Aguzzi A, Weissmann C. Cell. 1993;72:1339–1347. [Google Scholar]
- 8.Wickner R B. Proc Natl Acad Sci USA. 1997;94:10012–10014. doi: 10.1073/pnas.94.19.10012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caughey B W, Dong A, Bhat K S, Ernst D, Hayes S F, Caughey W S. Biochemistry. 1991;30:7672–7680. doi: 10.1021/bi00245a003. [DOI] [PubMed] [Google Scholar]
- 10.Pan K-M, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick R J, Cohen F E, Prusiner S B. Proc Natl Acad Sci USA. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Glockshuber R, Hornemann S, Riek R, Wider G, Billeter M, Wüthrich K. In: Prions and Brain Diseases in Animals and Humans. Morrison D R O, Collinge J, editors. London: Plenum; 1998. , in press. [Google Scholar]
- 12.Stahl N, Baldwin M A, Teplow D B, Hood L, Gibson B W, Burlingame A L, Prusiner S B. Biochemistry. 1993;32:1991–2002. doi: 10.1021/bi00059a016. [DOI] [PubMed] [Google Scholar]
- 13.Riek R, Hornemann S, Wider G, Glockshuber R, Wüthrich K. FEBS Lett. 1997;413:282–288. doi: 10.1016/s0014-5793(97)00920-4. [DOI] [PubMed] [Google Scholar]
- 14.Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wüthrich K. Nature (London) 1996;382:180–182. doi: 10.1038/382180a0. [DOI] [PubMed] [Google Scholar]
- 15.Cohen F E, Pan K, Huang Z, Baldwin M, Fletterick R J, Prusiner S B. Science. 1994;264:530–531. doi: 10.1126/science.7909169. [DOI] [PubMed] [Google Scholar]
- 16.Harrison P M, Bamborough P, Daggett V, Prusiner S B, Cohen F E. Curr Opin Struct Biol. 1997;7:53–59. doi: 10.1016/s0959-440x(97)80007-3. [DOI] [PubMed] [Google Scholar]
- 17.Huang Z, Gabriel J M, Baldwin M A, Fletterick R J, Prusiner S B, Cohen F E. Proc Natl Acad Sci USA. 1994;91:7139–7143. doi: 10.1073/pnas.91.15.7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schätzl H M, Da Costa M, Taylor L, Cohen F E, Prusiner S B. J Mol Biol. 1995;245:362–374. doi: 10.1006/jmbi.1994.0030. [DOI] [PubMed] [Google Scholar]
- 19.Hornemann S, Glockhuber R. J Mol Biol. 1996;262:614–619. doi: 10.1006/jmbi.1996.0487. [DOI] [PubMed] [Google Scholar]
- 20.Güntert P, Dötsch V, Wider G, Wüthrich K. J Biomol NMR. 1992;2:619–629. [Google Scholar]
- 21.Bartels C, Xia T, Billeter M, Güntert P, Wüthrich K. J Biomol NMR. 1995;6:1–10. doi: 10.1007/BF00417486. [DOI] [PubMed] [Google Scholar]
- 22.Neri D, Szyperski T, Otting G, Senn H, Wüthrich K. Biochemistry. 1989;28:7510–7516. doi: 10.1021/bi00445a003. [DOI] [PubMed] [Google Scholar]
- 23.Ikura M, Kay L E, Tschudin R, Bax A. J Magn Reson. 1990;86:204–209. doi: 10.1016/j.jmr.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 24.Fesik S W, Zuiderweg E R P. J Magn Reson. 1988;78:588–593. [Google Scholar]
- 25.Kumar A, Ernst R R, Wüthrich K. Biochem Biophys Res Comm. 1980;95:1–6. doi: 10.1016/0006-291x(80)90695-6. [DOI] [PubMed] [Google Scholar]
- 26.Güntert P, Mumenthaler C, Wüthrich K. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 27.Wüthrich K, Billeter M, Braun W. J Mol Biol. 1983;169:949–961. doi: 10.1016/s0022-2836(83)80144-2. [DOI] [PubMed] [Google Scholar]
- 28.Luginbühl P, Szyperski T, Wüthrich K. J Magn Reson. 1995;109:229–233. [Google Scholar]
- 29.Güntert P, Braun W, Billeter M, Wüthrich K. J Am Chem Soc. 1989;111:3997–4004. [Google Scholar]
- 30.Williamson M P, Havel T F, Wüthrich K. J Mol Biol. 1985;182:295–315. doi: 10.1016/0022-2836(85)90347-x. [DOI] [PubMed] [Google Scholar]
- 31.Güntert P, Berndt K D, Wüthrich K. J Biomol NMR. 1993;3:601–606. doi: 10.1007/BF00178259. [DOI] [PubMed] [Google Scholar]
- 32.Weiner P K, Kollman P A, Nguyen D T, Case D A. J Comp Chem. 1986;7:230–252. doi: 10.1002/jcc.540070216. [DOI] [PubMed] [Google Scholar]
- 33.Luginbühl P, Güntert P, Billeter M, Wüthrich K. J Biomol NMR. 1996;8:136–146. doi: 10.1007/BF00211160. [DOI] [PubMed] [Google Scholar]
- 34.Hornemann S, Korth C, Oesch B, Riek R, Wider G, Wüthrich K, Glockshuber R. FEBS Lett. 1997;413:277–281. doi: 10.1016/s0014-5793(97)00921-6. [DOI] [PubMed] [Google Scholar]
- 35.Billeter M, Riek R, Wider G, Hornemann S, Glockshuber R, Wüthrich K. Proc Natl Acad Sci USA. 1997;94:7281–7285. doi: 10.1073/pnas.94.14.7281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wüthrich K. NMR of Proteins and Nucleic Acids. New York: Wiley; 1986. [Google Scholar]
- 37.Palmer M S, Dryden A J, Hughes J T, Collinge J. Nature (London) 1991;352:340–342. doi: 10.1038/352340a0. [DOI] [PubMed] [Google Scholar]
- 38.Spera S, Bax A. J Am Chem Soc. 1991;113:5490–5492. [Google Scholar]
- 39.James T L, Liu H, Ulyanov N B, Farr-Jones S, Zhang H, Donne D G, Kaneko K, Groth D, Mehlhorn I, Prusiner S B, Cohen F E. Proc Natl Acad Sci USA. 1997;94:10086–10091. doi: 10.1073/pnas.94.19.10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Donne G D, Viles J H, Groth D, Mehlhorn I, James T L, Cohen F E, Prusiner S B, Wright P E, Dyson H J. Proc Natl Acad Sci USA. 1997;94:13452–13457. doi: 10.1073/pnas.94.25.13452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harper E T, Rose G D. Biochemistry. 1993;32:7605–7609. doi: 10.1021/bi00081a001. [DOI] [PubMed] [Google Scholar]
- 42.Priola S A, Chesebro B A. J Virol. 1995;69:7754–7758. doi: 10.1128/jvi.69.12.7754-7758.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prusiner S B. Phil Trans R Soc London B. 1994;343:447–463. doi: 10.1098/rstb.1994.0043. [DOI] [PubMed] [Google Scholar]
- 44.Goldfarb L G, Petersen R B, Tabaton M, Brown P, LeBlanc A C, Montagna P, Cortelli P, Julien J, Vital C, Pendelbury W W, et al. Science. 1992;258:806–808. doi: 10.1126/science.1439789. [DOI] [PubMed] [Google Scholar]
- 45.Koradi R, Billeter M, Wüthrich K. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]