

Abstract
The mechanism of the electron transfer reaction, QA−⋅QB → QAQB−⋅, was studied in isolated reaction centers from the photosynthetic bacterium Rhodobacter sphaeroides by replacing the native Q10 in the QA binding site with quinones having different redox potentials. These substitutions are expected to change the intrinsic electron transfer rate by changing the redox free energy (i.e., driving force) for electron transfer without affecting other events that may be associated with the electron transfer (e.g., protein dynamics or protonation). The electron transfer from QA−⋅ to QB was measured by three independent methods: a functional assay involving cytochrome c2 to measure the rate of QA−⋅ oxidation, optical kinetic spectroscopy to measure changes in semiquinone absorption, and kinetic near-IR spectroscopy to measure electrochromic shifts that occur in response to electron transfer. The results show that the rate of the observed electron transfer from QA−⋅ to QB does not change as the redox free energy for electron transfer is varied over a range of 150 meV. The strong temperature dependence of the observed rate rules out the possibility that the reaction is activationless. We conclude, therefore, that the independence of the observed rate on the driving force for electron transfer is due to conformational gating, that is, the rate limiting step is a conformational change required before electron transfer. This change is proposed to be the movement, controlled kinetically either by protein dynamics or intermolecular interactions, of QB by ≈5 Å as observed in the x-ray studies of Stowell et al. [Stowell, M. H. B., McPhillips, T. M., Rees, D. C., Soltis, S. M., Abresch, E. & Feher, G. (1997) Science 276, 812–816].
Protein dynamics play an important role in many biological processes (1), including electron transfer reactions (2). When protein changes precede and are slower than the intrinsic rate of electron transfer, protein dynamics will control the rate of reaction. This situation has been called conformational gating (2). In this work, we study the role of protein dynamics in an electron transfer reaction of the bacterial reaction center (RC). The RC is a membrane protein that mediates the light-induced electron and proton transfer reactions in bacterial photosynthesis. (3) After photoexcitation of the primary donor, a bacteriochlorophyll dimer (D), an electron is transferred from the donor via a bacteriopheophytin (BPhe) to the tightly bound primary quinone, QA (k ≃ 1010 s−1). Subsequent electron transfer occurs from QA−⋅ to the secondary quinone, QB, with observed rate, kAB(1) ≃ 104 s−1 (4, 5), that is,
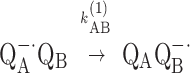 |
1 |
The observed rate (104 s−1) and activation energy (≈200 to 500 meV) measured for the electron transfer between the two quinones are within ranges that are common for reactions governed by protein dynamics. Previous studies have suggested that this reaction is accompanied by conformational changes (6–10). In this work, we examine the role of conformational gating in the observed reaction (Eq. 1).
A simplified gating model for QB reduction involves two consecutive steps:
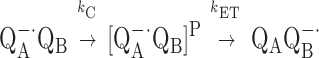 |
2 |
where QA−⋅QB represents the initial state, [QA−⋅QB]P is the state activated for electron transfer and, QAQB−⋅ is the final product state. In this model, the overall (observed) rate of the reaction depends on the values of kET and kC (2). If kC < kET, the reaction is conformationally gated. If kC > kET, the reaction is rate-limited by electron transfer. To determine which of these two cases prevails, a driving force assay was used (11). In this assay, quinones with different redox potentials (i.e., driving force) are substituted for QA, thereby changing the driving force for electron transfer and, thus, the intrinsic rate constant, kET. This will change the overall rate kAB(1) if electron transfer is rate limiting but will not change kAB(1) if the reaction is gated.
Naphthoquinones (NQ) with redox potentials different from that of native Q10 were substituted into the QA site whereas Q10 was retained in the QB site. (11–13). The substitution of QA changes the intrinsic rate of electron transfer, kET, without affecting kC since the QB site is unchanged and the structures of the substituted QA molecules are nearly identical. By using this procedure, the observed rate kAB(1) was measured as a function of the driving force for electron transfer, from which the mechanism of the kAB(1) reaction was determined. A preliminary account of this work has been published (14).
MATERIALS AND METHODS
Sample Preparation.
RCs from Rhodobacter sphaeroides strains R26.1 and 2.4.1 were isolated and purified in lauryldimethylamine-N-oxide (LDAO) as described (15). The ratios of absorbance (A280nm/A800nm) of the purified RCs were <1.25. QA and QB were removed from RCs as described (16). Reduced R. sphaeroides cytochrome (cyt) c2 was purified as described (17). Q10, MQ0, MQP, and MQ4 were obtained from Sigma. 2,3,5-trimethyl-1,4-naphthoquinone (Me3NQ) and 2,3,6,7-tetramethyl-1,4-naphthoquinone (Me4NQ) kindly were provided by J. M. Bruce (Univ. of Manchester, U.K.). They all were solubilized in ethanol before use. Q10 also was solubilized in 1% LDAO and 1% Triton X-100. Unless otherwise indicated, experiments were performed in BMK buffer, which consisted of 0.04% β–d-dodecyl-maltoside, 10 mM KCl, 0.1 mM EDTA, and 5 mM of each of the following buffers: citrate, mes, pipes, tris, ches, and caps. Reagents were of analytical grade; their sources are given in ref. 11.
A detailed procedure for reconstitution of the QA binding site with naphthoquinone and QB binding site with ubiquinone has been presented (11). The occupancy of the QA and QB sites after reconstitution were determined from the amplitudes of transients associated with charge recombination, kAD (D+QA−⋅ → DQA), and kBD (D+QAQB−⋅ → DQAQB) (18). One-hundred percent QA site occupancies and 75–90% QB site occupancies were achieved at pH 7.2.
Electron Transfer Measurements.
Absorbance changes were measured at 23°C by using a single beam spectrophotometer of local design (19). Voltage output was recorded on a digital oscilloscope (LeCroy 9310M, Chestnut Ridge, NY) and was digitally filtered (LabView, National Instruments, Austin, TX) and fitted (Peakfit, Tablecurve, or Sigmaplot, Jandel, San Rafael, CA). Actinic illumination was provided by a pulsed dye laser (PhaseR DL2100c, 590 nm, ≈0.2 J/pulse, 0.4-μs full width at half maximum) or by a ruby laser (Optic Technologies Model 130, 695 nm, ≈0.5 J/pulse, ≈10-ns full width at half maximum). Laser light intensity was determined to be saturating (>98%) before and after each set of experiments.
The Electron Transfer from QA−⋅ to QB Was Measured by Three Independent Methods.
(i) Optical absorbance changes at 412 and 470 nm. The difference in extinction between ubisemiquinone and naphthosemiquinones provides a spectral signature of the electron transfer, (NQ)A−⋅QB → (NQ)AQB−⋅, in hybrid RCs. Peaks in the differential extinction occur at wavelengths of 412 and 470 nm (20, 21). In native RCs, QA and QB are the same (Q10); there is, however an absorption change at 412 nm caused by an electrochromic shift of the BPhe absorption. No electrochromic shift is observed at 470 nm.
(ii) Optical absorbance changes at 757 nm.
At this wavelength, the absorption changes associated with electron transfer, QA−⋅QB → QAQB−⋅, are caused by an electrochromic shift of the BPhe absorption (9). The observed kinetics were corrected for fluorescent artifacts by subtracting the absorbance changes when electron transfer was blocked by terbutryne.
(iii) Two-flash cyt c2 assay (22).
Optical absorbance changes at 865 nm or at 550 nm were measured after two laser flashes separated by a variable time interval, Δt, from 1 to 10,000 μs. The fraction of photoactive RCs, f(Δt) = 1 − exp[−(kAB(1)Δt)], was calculated from the absorbance change at 865 nm due to oxidation of the primary donor, D, from the relation
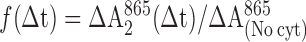 |
3 |
where ΔA2865(Δt) is the absorbance change induced by the second flash, and ΔA(No cyt)865 is the absorption change after a single flash in the absence of cyt c2. Normalization of the data required correction for several small effects, whose magnitudes were determined independently, i.e., (i) the fraction of RCs without a functional QB, (ii) the fraction of RCs without cyt c22+ bound before the first flash, (iii) the thermodynamic equilibrium between the QA−⋅QB and QAQB−⋅ states, and (iv) the exchange of cyt c23+ for cyt c22+ between flashes.
From absorption changes at 550 nm, which result from cyt c22+ oxidation, the fraction of photoactive RCs was obtained from
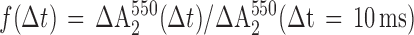 |
4 |
where ΔA2550(Δt) is the absorbance change induced by the second flash given Δt seconds after the first and ΔA2550(Δt = 10 ms) is the absorbance change induced by the second flash given 10 ms after the first. Because at 10 ms the reaction is complete, ΔA2550(Δt = 10 ms) is the maximum possible change. Normalization to ΔA2550(Δt = 10 ms), instead of the amplitude change induced by the first flash, avoids the need to apply corrections i, ii, and iii.
RESULTS
Hybrid RCs [(NQ)(A), Q10(B)].
Hybrid RCs containing different naphthoquinones in the QA site and Q10 in the QB site [(NQ)(A), Q10(B)] were prepared and assayed as described in Materials and Methods. The fraction of hybrid RCs, [(NQ)(A), Q10(B)]/{[(NQ)(A), Q10(B)] + [Q10(A), Q10(B)]}, ranged from 0.45 to 0.80 for samples containing MQP, MQ4, Me3NQ, and Me4NQ. A lower value of 0.35 was determined for samples with MQ0. From measurements of the recombination rates kAD and kBD, the in situ free-energy changes relative to Q10, δΔG0 of the substituted quinones were obtained (see Table 1) (11–13, 23, 24).
Table 1.
Parameters describing the characteristic time of electron transfer, QA−⋅QB → QAQB−, in hybrid and native RCs
| QA | δΔG0, meV | Two-flash assay
|
Optical absorption
change assays
|
|||
|---|---|---|---|---|---|---|
| 865 and 550 nm | 412 nm | 470 nm | 757 nm | 1/kAB(1), μs (avg) | ||
| Q10 | 0 | 47 μs (37%) | 120 μs (−27%) | NA | 27 μs (26%) | 110 ± 3 |
| 200 μs (57%) | 250 μs (74%) | |||||
| MQ0 | 15 ± 5 | ND | 42 μs (36%) | 48 μs (25%) | 76 μs (47%) | 150 ± 20 |
| 340 μs (27%) | 310 μs (58%) | 320 μs (53%) | ||||
| MQP | −35 ± 9 | ND | 51 μs (27%) | 32 μs (44%) | 64 μs (29%) | 140 ± 30 |
| 220 μs (43%) | 230 μs (45%) | 260 μs (71%) | ||||
| MQ4 | −46 ± 9 | ND | 47 μs (18%) | 50 μs (13%) | 51 μs (28%) | 150 ± 20 |
| 240 μs (42%) | 210 μs (77%) | 170 μs (72%) | ||||
| Me3 NQ | −95 | ND | 52 μs (15%) | 44 μs (23%) | 33 μs (35%) | 190 ± 30 |
| 270 μs (48%) | 340 μs (63%) | 230 μs (65%) | ||||
| Me4 NQ | −120 ± 10 | 46 μs (48%) | 50 μs (24%) | NA | 50 μs (42%) | 140 ± 20 |
| 200 μs (49%) | 320 μs (34%) | 320 μs (58%) | ||||
Data were fitted with two exponentials. The characteristic times are given in microseconds together with their relative contributions in parenthesis (757 nm). The percent amplitude relative to the steady-state change (NQA−⋅ minus Q10−(13) is given in parentheses for 412 and 470 nm. δΔG0 is given relative to Q10, which is defined as 0. The tabulated values in the seventh column represent the average 1/e time of the three methods. Errors were calculated as the SD of the mean. NA, not applicable; ND, not determined.
Determination of Electron Transfer Rates, QA−⋅QB → QAQB−⋅, in Hybrid [(NQ)(A), Q10(B)] and Native (Q10(A), Q10(B)) RCs.
The electron transfer rates kAB(1) were determined by three independent methods described in Materials and Methods. The use of several methods helped to guard against possible artifacts caused by transient signals not associated with electron transfer. Of particular importance is the two-flash functional assay that directly measures electron transfer. The decay kinetics were fitted by one or two exponential rate constants (see Table 1). The fitted values should be viewed as empirical parameters and are not intended to imply a mechanism. Biexponential decays were observed even with Q10 in both binding sites and, therefore, are not related to the mixture of hybrid and native RCs. The results of the kinetic measurements are described below.
Transient absorbance changes at 412 nm after a laser flash are shown for hybrid and native RCs in Fig. 1. The solid lines represent fits to a biexponential function. Despite the change in electron driving force, the decay rates of the two hybrid samples were essentially the same. The amplitudes were normalized by using the steady-state semiquinone absorption spectra (with Δɛ412 nm(NQ−−Q10−) = 4.3 mM−1 cm−1) (11, 20, 21). In native RCs, the absorbance change is caused by an electrochromic shift of the BPhe absorption. The same shift is assumed to be present also in hybrid RCs. To estimate the fraction of hybrid RCs displaying the measured electron transfer, the electrochromic shift observed for native RCs must be added to the absorbance changes of hybrid RCs. This results in an amplitude for the (NQ)A−⋅ transient decay that is 90 ± 5% of the steady-state (i.e., expected) semiquinone decay.
Figure 1.

Optical absorbance changes at 412 nm associated with electron transfer from QA−⋅ to QB in hybrid (Me3NQ−⋅ or MQP−⋅ in the QA site and Q10 in the QB site) and native (Q10 in both QA and QB sites) RCs. In native RCs, the absorbance change is due to an electrochromic shift of the BPhe absorption. In hybrid RCs, the main effect is caused by the difference in extinction coefficient between the naphthosemiquinones (NQ−⋅ in the QA site and Q10−⋅ in the QB site). The observed transients in hybrid RCs are reduced by the electrochromic shift, which was assumed to be the same as that observed in native RCs. The data for the hybrid RCs were normalized to the expected absorption determined from the known difference in extinction coefficients between (NQ)A−⋅ and Q10−⋅ (11, 20, 21). A shallow slope determined by monitoring the recombination reactions (D+QA−⋅ → DQA and D+QAQB−⋅ → DQAQB) was subtracted for clarity. Addition of terbutryne, which inhibits electron transfer to QB, eliminated the transient kinetic phases. [Conditions: 5 μM 2.4.1RCs, BMK buffer (pH = 7.2), 0.03% LDAO. Spectral bandwidth, 10 nm].
At 470 nm, no electrochromic shifts are observed, and, therefore, no transient signals are obtained in native RCs. The amplitudes of the observed transients in hybrid RCs caused by (NQ)A−⋅ absorption decay amounted to 89 ± 5% of the expected steady-state signal (with Δɛ(NQ−470 nm−Q10−) = 2.5 mM−1 cm−1 for MQ0, MQP, and MQ4 (20, 21) and 5.0 mM−1 cm−1 for Me3NQ) (11). The characteristic times associated with the observed transients at 470 nm were, within experimental error, the same as those obtained at 412 nm (see Table 1).
The transient absorption changes at 757 nm that arise from an electrochromic shift associated with kAB(1) (9, 21, 25, 26) were measured in native as well as in all hybrid RCs. The data were fitted with a biexponential function and were in general agreement with the results obtained by the other methods. Replacement of Q10 with Q2 and Q0 in the QB site had no effect on the observed decays rates.
The functional two-flash assay was performed at two wavelengths (865 and 550 nm) for hybrid and for native RCs. The result of an 865 nm assay is shown in Fig. 2. After one laser flash, the charged species, D+QA−⋅QB, was formed. Cyt c22+ reduced the primary donor to form DQA−⋅QB (τ = 0.8 μs in RCs with a bound cyt c22+). A small residual absorbance (ΔA1) remained due to RCs without cyt c22+ bound before the first flash. For RCs in which a second flash was given before the electron left QA−⋅, no charge separation on the timescale of our experiments and consequently no optical transients were observed. In RCs in which electron transfer had occurred before the second flash, the photoactive state, DQAQB−⋅, could undergo charge separation (to form D+QA−⋅ QB−⋅) with a concomitant optical absorbance change observable at 865 nmΔA2. The absorbance change after the second flash is directly related to the rate of electron transfer (see Eq. 3). Similar measurements of optical absorbance changes at 550 nm (not shown) caused by oxidation of cyt c22+ by D+ after a second flash also were made (see Eq. 4).
Figure 2.

Optical absorbance changes at 865 nm for native RCs in the presence of cyt c22+ after two laser flashes spaced Δt apart. Laser flash artifacts are truncated. The ratio of ΔA2 to ΔANo cyt gives the fraction of photoactive RCs at the time of the second flash (see Eq. 3). ΔA1 is due to the fraction of RCs that do not have a bound cyt c22+. Addition of terbutryne to inhibit electron transfer to QB eliminated the absorbance change in response to the second laser flash. (Conditions: 3 μM 2.4.1 RCs with Q10 reconstituted into the QA and QB sites by addition of Q10 in LDAO. After reconstitution, LDAO was 0.03%. 100 μM cyt c22+, 5 μM ascorbate, 1 mM Tris, and 0.04% maltoside; spectral bandwidth, 10 nm.)
The applicability of the measurements at 865 nm is limited to the time interval 1 μs < Δt < 100 μs. The lower limit is given by the electron transfer time from cyt c22+ to D+ (≈0.8 μs). The upper limit is given by the cyt c23+/cyt c22+ exchange time (0.5–0.7 ms): If cyt c23+/cyt c22+ exchange takes place between flashes, D+ will be reduced by cyt c22+. Accurate measurements at 550 nm were obtained only for time intervals Δt > 40 μs because of the relatively poor signal-to-noise ratio associated with the small difference in extinction between oxidized and reduced cyt c2 (≈21 mM−1 cm−1). For time 40 μs < Δt < 100 μs, measurements were performed at both wavelengths; the two sets of measurements agreed within experimental error.
The normalized fraction of photoactive RCs (calculated by using Eqs. 3 and 4) is plotted in Fig. 3 as a function of the time interval between flashes for RCs having either Q10 or Me4NQ in the QA site. The solid line represents a biexponential fit to the data (see Table 1). The results in Fig. 3 show that the rate of electron transfer is the same for both quinones. RCs from the strain 2.4.1 containing carotenoid were used for the studies shown in Fig. 3 to eliminate a small absorption change at very short times in carotenoid-free RCs (R26.1). Control experiments (not shown) confirmed that 2.4.1 and R26.1 RCs display the same electron transfer rates.
Figure 3.

The fraction of photoactive RCs as a function of the time interval between two laser flashes for native (•), and hybrid (□) RCs. The solid line represents a least squares biexponential fit to the data. A minimum of 20 min dark recovery was allowed between sets of flashes. (865 nm assay conditions given in Fig. 2; at 550 nm, cyt c22+ concentration was reduced to ≈30 μM; spectral bandwidth, 4 nm.)
The results determined from the three independent methods are consistent with one another (Table 1). A model independent way to compare the data from samples with different QA molecules is to compare the time required to decay to 1/e of the initial state. The 1/e times, averaged over the three methods, for RCs with various quinones in the QA sites are similar and show no correlation with the redox potential of QA (see Table 1). The rates of each of the two components of the biexponetial fit derived from the two-exponential analysis also do not vary systematically with driving force.
pH Dependence of Electron Transfer in Hybrid (Me3NQ(A), Q10(B)) and Native (Q10(A), Q10(B)) RCs.
The rate kAB(1) was measured at 470 and 757 nm over the pH range of 4.7 to 10.3 for hybrid RCs with Me3NQ in the QA site and for native RCs. The rate was independent of the driving force throughout the entire range. The hybrid RCs retained the pH dependence of native RCs; that is, the rate was pH-independent below pH 8 and decreased by a factor of ≈10 per pH unit above pH 8 (19, 25, 27).
Effect of Exogenous Quinone Concentration [Q10] on Electron Transfer.
We measured the effect of quinone concentration [Q10] on kAB(1) in native RCs. One phase of the biexponential rate was unaffected by [Q10] and remained ≈7,000 s−1 whereas the second rate depended strongly on [Q10] (e.g., at 5 μM [Q10], k2 = 1,300 s−1 and, at 85 μM [Q10], k2 = 25,000 s−1). These experiments are still in progress.
DISCUSSION
In this work, we tested whether conformational changes or intrinsic electron transfer determines the kinetics of the observed reaction, QA−⋅QB → QAQB−⋅, in RCs from R. sphaeroides. This was accomplished by substituting naphthoquinone molecules with redox potentials different from that of native Q10 into the QA binding site while retaining Q10 in the QB site. The experimental results showed that the observed reaction rates were independent of the electron transfer driving force.
Analysis of the Driving Force Assay.
For a typical electron transfer reaction, the observed rate depends on the free energy for electron transfer as given by the Marcus theory (28):
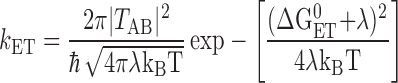 |
5 |
where kET is the intrinsic electron transfer rate constant, ΔGET0 is the free energy for electron transfer (defined as the energy of the final minus the initial state), λ is the reorganization energy, TAB is the tunneling matrix element, and kBT is the thermal energy. In principle, the independence of kAB(1) on ΔGET0 could be caused by the reaction being activationless (−ΔGET0 = λ). However, this is not the case for kAB(1), for which a large activation energy is observed (9, 19, 29, 30). Values for λ and ΔGET0 have been determined to be ≈1.1 eV and ≈−0.1 eV, respectively (13, 19, 31, 32). For these values, kET is expected to vary by a factor of 10 over the range of free energy spanned in this study* (see dashed line in Fig. 4). This is in contrast to the observed invariance of the rate with electron transfer driving force shown in Fig. 4. A fit of the data to a biexponential function (see Table 1) shows that neither component of the decay depends on electron transfer driving force. These results show that kAB(1) is not limited by electron transfer.
Figure 4.

The electron transfer rate kAB(1) as a function of redox free energy (driving force) for electron transfer (data from the second and seventh columns of Table 1). The solid line gives a best fit to the data. The dashed line represents the dependence predicted by the Marcus theory for an electron transfer limited reaction using the measured free energy difference between the initial, QA−⋅QB, and final, QAQB−⋅, states of 60 meV (19) and 1.1 eV for the reorganization energy (13, 31). Error bars represent one SD of the mean. Quinones incorporated into the QA site listed in order of decreasing redox potential were: 1, MQ0; 2, Q10; 3, MQP; 4, MQ4; 5, Me3NQ; and 6, Me4NQ. Redox energies are quoted with respect to Q10 (see • without error bars).
Conformational Gating.
The independence of kAB(1) on driving force can be explained by a conformational gating mechanism (2). Fig. 5 depicts a kinetic scheme for an electron transfer reaction coupled with a conformational change. The protein can exist in two different states indicated by different shapes in Fig. 5: the D (oval) and P (rectangle) states, for Distal and Proximal, as will be discussed below. In the D state, QB is in an inactive conformation, and electron transfer is impaired; in the P state, QB is in the active configuration, and electron transfer can proceed. The rate of equilibration between the D and P states in the dark is kc′. The gating model requires that, in the dark, the RC is predominantly in the D state. Light excitation results in formation of D+QA−⋅QB, with QB inactive. Conversion to the QB active configuration occurs with the conformational gating rate kC. Once the RC is in the QB active configuration, electron transfer can occur with a rate kET (given by Eq. 5). Thus, for the conformational gating model the observed kAB(1) ≃ 104 s−1 = kC. The intrinsic electron transfer rate must be much greater than the observed rate (i.e., kAB(1) ≪ kET). This is consistent with recent calculations suggesting that the intrinsic electron transfer rate for this reaction is >>104 s−1 (33). The model is also consistent with the work of Li et al. (30). These authors reported an additional fast phase (in the 105−106 s−1 range) in the kAB(1) kinetics measured at 398 nm and attributed this phase to electron transfer in a population of RCs active for electron transfer in the dark (i.e., our P state in the dark; see Fig. 5).
Figure 5.

Proposed model for conformational gating of electron transfer from QA−⋅ to QB. Two conformational states are shown, distal (D; oval), which is inactive, and proximal (P; rectangle), which is active in electron transfer. The main sequence of steps in the reaction are (1) excitation of RCs in the distal state, (2) conversion to the proximal state, and (3) electron transfer. A fraction of RCs in the proximal state before light excitation may give rise to a faster observed rate through reactions 1′ and 3.
The gating model accounts for the observation by Kleinfeld et al. (6) that electron transfer from QA−⋅ to QB proceeds in RCs cooled to cryogenic temperature under illumination but does not proceed in RCs cooled in the dark. RCs frozen in the dark are in the inactive conformation (D state). At low temperature, there is insufficient thermal energy for conformational interconversion. RCs cooled under illumination are frozen in the active P state, which explains the observed electron transfer in these RCs.
Molecular Basis of the Gating Mechanism.
Structural changes responsible for the gating. Structural changes involved in the gating process were elucidated recently by comparing the x-ray crystal structure of the RC cooled to cryogenic (90 K) temperatures under illumination (light structure, D+QAQB−⋅) with the structure of the RC cooled in the dark (dark structure, DQAQB) (10). In the light structure, QB had moved closer to QA and had undergone a 180° propeller twist about the isoprenoid chain (see Fig. 6). The movement of QB is expected to affect both the rate and thermodynamic equilibrium of the electron transfer reaction. The more direct pathway (and shorter distance) between QA−⋅ and QB in the light structure increases the rate of electron transfer. The greater number of hydrogen bonds to QB, compared with the dark structure, stabilizes QAQB−⋅ with respect to QA−⋅QB, thereby increasing the driving force for electron transfer. The position of QB in the light structure, hydrogen bonded to Ser-L223 and to His-L190, corresponds to the active P (proximal) conformation in Figs. 5 and 6. The position of QB in the dark structure, hydrogen-bonded only to the amide hydrogen of Ile-L224, corresponds to the inactive D (distal) conformation in Figs. 5 and 6. Thus, the conformational gating step involves the movement of QB by ≈5 Å from the distal to the proximal site.
Figure 6.

Comparison of the binding positions for QB determined from the light (D+QA QB−⋅) and dark (DQAQB) x-ray crystal structures of the RC (10). Movement from an inactive–distal (black) to an active–proximal (gray) binding site is proposed as the major structural change involved with conformational gating of electron transfer from QA−⋅ to QB. Hydrogen bonding partners are connected by dotted lines. Modified from ref. 10.
What controls the kinetics of the gating process?
The unhindered movement and rotation of the quinone in solution would proceed on the nanosecond timescale (34, 35), that is, 4–5 orders of magnitude faster than the observed value of 1/kAB(1). Consequently, the rate of QB movement from the distal to the proximal site must be controlled by a process other than diffusion. What is the rate limiting step for the QB movement? Several possibilities can be envisioned; three of these are discussed below.
The most likely gating process is associated with protein dynamics that allow the quinone to move into the proximal position. Support for gating by protein dynamics comes from experiments that showed (i) light-induced proton binding changes on the timescale of kAB(1) that occur in the absence of electron transfer from QA−⋅ to QB (36), (ii) light-induced electrogenic transients in the absence of electron transfer from QA−⋅ to QB with the same rate and pH dependence as kAB(1) in native and EQ(L212) mutant RCs (8), and (iii) the importance of protein fluctuations in electron transfer reactions of the RC (37, 38). Structural changes associated with protein dynamics should, in principle, be observable in the x-ray crystal structures. However, no large changes between protein structures of light and dark adapted RCs were reported (10). The largest change was the loss of electron density for Glu-H173 (close to QB) in the light structure, which was attributed to greater disorder. The absence of large changes can be explained if the gating is due to protein “breathing” modes not detected at the cryogenic temperatures at which the structures were determined or if the gating motion involves only small movements of many parts of the protein or changes in water structure around the quinone region, not sufficiently well resolved in the x-ray crystal structures.
Another possibility is that conformational changes at the QB site are triggered by QA reduction. In the gating scheme shown in Fig. 5, a triggering mechanism implies that coupling between the QA and QB sites exists and that kC > kC′. Evidence for coupling comes from experiments which show (i) an altered recombination rate (D+QA−⋅ → DQA) and EPR spectrum of QA−⋅ when inhibitors are present in the QB site (19, 39) and (ii) proton binding by amino acids near QB in response to QA reduction (32, 40, 41). Electrostatic calculations also suggest coupling between the QA and QB binding sites (42, 43).
A third possibility is that the rate-limiting step involves the breaking of the intermolecular interactions between QB in the distal state and the protein. The result reported in this work and by McComb et al. (44), that the rates of kAB(1) for RCs with Q10, Q2, or Q0 in the QB site are the same, excludes interactions associated with the isoprenoid tail as rate limiting. However, interactions of the quinone head group, for example, π-stacking with Phe-L216 or hydrogen-bonding between the O-4 carbonyl of QB and the amide hydrogen of Ile-L224, may limit the rate of movement between the D and P states.
Non-Exponential Rates.
The kinetics of kAB(1) observed in this study were nonexponential and could be fitted with a biexponential function. Biphasic rates have been reported by several groups (9, 30, 45). In this work one component of the rate was observed to depend on the exogenous quinone concentration. We postulate that the origin of this component of the kinetics is associated with the fraction of RCs that do not have a bound quinone in the QB site. In these RCs, the rate limiting (gating) step is associated with quinone binding. This may explain both the variability and the discrepancy between rates reported by different groups. The discussion of the molecular basis of the gating mechanism presented in the previous section refers to the invariant part of the kinetics associated with those RCs that have a bound quinone.
Conformational Gating in Other Systems.
Electron transfer reactions gated by ligand binding or redox-state dependent protein changes occur in other biological systems (46, 47). It is likely that some of the electron transfer reactions of energy transducing proteins, including cytochrome oxidase, cyt b6f/cyt bc1, complex I, and others are conformationally gated. For example, crystallographic data of the bc1 complex show an ≈10 Å movement of the Fe-S subunit between two sites, one for reduction of the Fe-S center by ubiquinol and another for oxidation of the Fe-S center by cytochrome c (48, 49). To understand protein-catalyzed electron transfer reactions in general, it is necessary to establish whether the intrinsic reaction step or the associated conformational changes are rate limiting. The technique described in this work, whereby the free energy for electron transfer is varied by cofactor substitution, illustrates a general approach to investigate reaction mechanisms and energetics.
Acknowledgments
We thank J. M. Bruce for the Me3NQ and Me4NQ, H. L. Axelrod for purified cytochrome c2, E. C. Abresch for reaction centers, and M. L. Paddock and M. R. Gunner for helpful discussions. This work was supported by National Institutes of Health Grants 2 RO1GM 14637 and GM 13191 and National Science Foundation Grant MCB 94-16652.
ABBREVIATIONS
- RC
bacterial reaction center
- NQ
naphthoquinone
- Me3NQ
2,3,5-trimethyl-1,4-naphthoquinone
- Me4NQ
2,3,6,7-tetramethyl-1,4-naphthoquinone
- LDAO
lauryldimethylamine-N-oxide
- cyt c2
R. sphaeroides cytochrome c2
- BPhe
bacteriopheophytin
- MQ0
menadione
- MQP
vitamin k3
- MQ4
menatetronone
Footnotes
References
- 1. McCammon J A, Harvey S C. Dynamics of Proteins and Nucleic Acids. New York: Cambridge Univ. Press; 1987. [Google Scholar]
- 2.Hoffman B M, Ratner M A. J Am Chem Soc. 1987;109:6237–6242. [Google Scholar]
- 3.Feher G, Allen J P, Okamura M Y, Rees D C. Nature (London) 1989;339:111–116. [Google Scholar]
- 4.Okamura M Y, Feher G. Annu Rev Biochem. 1992;61:861–896. doi: 10.1146/annurev.bi.61.070192.004241. [DOI] [PubMed] [Google Scholar]
- 5.Shinkarev V P, Wraight C A. In: The Photosynthetic Reaction Center. Deisenhofer J, Norris J R, editors. Vol. 1. New York: Academic; 1993. pp. 193–255. [Google Scholar]
- 6.Kleinfeld D, Feher G, Okamura M Y. Biochemistry. 1984;23:5780–5786. doi: 10.1021/bi00319a017. [DOI] [PubMed] [Google Scholar]
- 7.Debus R J, Feher G, Okamura M Y. Biochemistry. 1986;25:2276–2287. doi: 10.1021/bi00356a064. [DOI] [PubMed] [Google Scholar]
- 8.Brzezinski P, Okamura M Y, Feher G. In: The Photosynthetic Bacterial Reaction Center II. Breton J, Vermeglio A, editors. New York: Plenum; 1992. pp. 321–330. [Google Scholar]
- 9.Tiede D M, Vazquez J, Cordova J, Marone P A. Biochemistry. 1996;35:10763–10775. doi: 10.1021/bi9605907. [DOI] [PubMed] [Google Scholar]
- 10.Stowell M H B, McPhillips T M, Rees D C, Soltis S M, Abresch E, Feher G. Science. 1997;276:812–816. doi: 10.1126/science.276.5313.812. [DOI] [PubMed] [Google Scholar]
- 11.Graige M S, Paddock M L, Bruce J M, Feher G, Okamura M Y. J Am Chem Soc. 1996;118:9005–9016. [Google Scholar]
- 12.Gunner M R, Tiede D M, Prince R C, Dutton P L. In: Functions of Quinones in Energy Conserving Systems. Trumpower B L, editor. New York: Academic; 1982. pp. 265–269. [Google Scholar]
- 13.Labahn A, Bruce J M, Okamura M Y, Feher G. Chem Phys. 1995;197:355–366. [Google Scholar]
- 14.Graige M S, Paddock M L, Feher G, Okamura M Y. Biophys J. 1996;70:A11. [Google Scholar]
- 15.Paddock M L, Rongey S H, Abresch E C, Feher G, Okamura M Y. Photosynth Res. 1988;17:75–96. doi: 10.1007/BF00047682. [DOI] [PubMed] [Google Scholar]
- 16.Okamura M Y, Isaacson R A, Feher G. Proc Natl Acad Sci USA. 1975;72:3491–3495. doi: 10.1073/pnas.72.9.3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Axelrod H L, Feher G, Allen J P, Chirino A J, Day M W, Hsu B T, Rees D C. Acta Crystallogr D. 1994;50:596–602. doi: 10.1107/S0907444994001319. [DOI] [PubMed] [Google Scholar]
- 18.Okamura M Y, Debus R J, Kleinfeld D, Feher G. In: Functions of Quinones in Energy Conserving Systems. Trumpower B L, editor. New York: Academic; 1982. pp. 299–317. [Google Scholar]
- 19.Kleinfeld D, Okamura M Y, Feher G. Biochim Biophys Acta. 1984;766:126–140. doi: 10.1016/0005-2728(84)90224-x. [DOI] [PubMed] [Google Scholar]
- 20.Verméglio A. Biochim Biophys Acta. 1977;459:516–524. doi: 10.1016/0005-2728(77)90050-0. [DOI] [PubMed] [Google Scholar]
- 21.Shopes R J, Wraight C A. Biochim Biophys Acta. 1985;806:348–356. doi: 10.1016/0005-2728(85)90242-7. [DOI] [PubMed] [Google Scholar]
- 22.Parson W W. Biochim Biophys Acta. 1969;189:384–396. doi: 10.1016/0005-2728(69)90169-8. [DOI] [PubMed] [Google Scholar]
- 23.Woodbury N W, Parson W W, Gunner M R, Prince R C, Dutton P L. Biochim Biophys Acta. 1986;851:6–22. doi: 10.1016/0005-2728(86)90243-4. [DOI] [PubMed] [Google Scholar]
- 24.Gunner M R, Dutton P L. J Am Chem Soc. 1989;111:3400–3412. [Google Scholar]
- 25.Verméglio A, Clayton R K. Biochim Biophys Acta. 1977;461:159–165. doi: 10.1016/0005-2728(77)90078-0. [DOI] [PubMed] [Google Scholar]
- 26.Bylina E J, Kirmaier C K, McDowell L, Holten D, Youvan D C. Nature (London) 1988;336:182–184. [Google Scholar]
- 27.Wraight C A. Biochim Biophys Acta. 1979;548:309–327. doi: 10.1016/0005-2728(79)90138-5. [DOI] [PubMed] [Google Scholar]
- 28.Marcus R A, Sutin N. Biochim Biophys Acta. 1985;811:265–322. [Google Scholar]
- 29.Mancino L J, Dean D P, Blankenship R E. Biochim Biophys Acta. 1984;764:46–54. [Google Scholar]
- 30.Li J L, Gilroy D, Tiede D M, Gunner M R. Biochemistry. 1998;37:2818–2829. doi: 10.1021/bi971699x. [DOI] [PubMed] [Google Scholar]
- 31.Allen J P, Williams J C, Graige M S, Paddock M L, Labahn A, Feher G, Okamura M Y. Photosynth Res. 1998;55:227–233. [Google Scholar]
- 32.McPherson P H, Schonfeld M, Paddock M L, Okamura M Y, Feher G. Biochemistry. 1994;33:1181–1193. doi: 10.1021/bi00171a018. [DOI] [PubMed] [Google Scholar]
- 33.Page C C, Farid R S, Moser C C, Dutton P L. Biophys J. 1996;70:A343. [Google Scholar]
- 34.Chaiken I M, Freedman M H, Lyerla J R, Cohen J S. J Biol Chem. 1973;248:884–891. [PubMed] [Google Scholar]
- 35.Gupte S, Wu E S, Hoechli L, Hoechli M, Jacobson K, Sowers A E, Hackenbrock C R. Proc Natl Acad Sci, USA. 1984;81:2606–2610. doi: 10.1073/pnas.81.9.2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maroti P, Wraight C A. Biophys J. 1997;73:367–381. doi: 10.1016/S0006-3495(97)78077-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McMahon B H, Muller J D, Wraight C A, Nienhaus G U. Biophys J. 1998;74:2567–2587. doi: 10.1016/S0006-3495(98)77964-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Utschig L M, Ohigashi Y, Thurnauer M C, Tiede D M. Biochemistry. 1998;37:8278–8281. doi: 10.1021/bi980395n. [DOI] [PubMed] [Google Scholar]
- 39.Butler W F, Johnston D C, Shore H B, Fredkin D R, Okamura M Y, Feher G. Biophys J. 1980;32:967–992. doi: 10.1016/S0006-3495(80)85030-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maroti P, Hanson D K, Schiffer M, Sebban P. Nat Struct Biol. 1995;2:1057–1059. doi: 10.1038/nsb1295-1057. [DOI] [PubMed] [Google Scholar]
- 41.Miksovska J, Maroti P, Tandori J, Schiffer M, Hanson D K, Sebban P. Biochemistry. 1996;35:15411–15417. doi: 10.1021/bi961299u. [DOI] [PubMed] [Google Scholar]
- 42.Beroza P, Fredkin D R, Okamura M Y, Feher G. Biophys J. 1995;68:2233–2250. doi: 10.1016/S0006-3495(95)80406-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lancaster C R D, Michel H, Honig B, Gunner M R. Biophys J. 1996;70:2469–2492. doi: 10.1016/S0006-3495(96)79820-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McComb J C, Stein R R, Wraight C A. Biochim Biophys Acta. 1990;1015:156–171. doi: 10.1016/0005-2728(90)90227-u. [DOI] [PubMed] [Google Scholar]
- 45.Hienerwadel R, Thibodeau D, Lenz F, Nabedryk E, Breton J, Kreutz W, Mantele W. Biochemistry. 1992;31:5799–5808. doi: 10.1021/bi00140a016. [DOI] [PubMed] [Google Scholar]
- 46.Bechtold R, Kuehn C, Lepre C, Isied S S. Nature (London) 1986;322:286–288. doi: 10.1038/322286a0. [DOI] [PubMed] [Google Scholar]
- 47.Walker M C, Tollin G. Biochemistry. 1991;30:5546–5555. doi: 10.1021/bi00236a030. [DOI] [PubMed] [Google Scholar]
- 48.Xia D, Yu C A, Kim H, Xian J Z, Kachurin A M, Zhang L, Yu L, Deisenhofer J. Science. 1997;277:60–66. doi: 10.1126/science.277.5322.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Z L, Huang L S, Shulmeister V M, Chi Y I, Kim K K, Hung L W, Crofts A R, Berry E A, Kim S H. Nature (London) 1998;392:677–684. doi: 10.1038/33612. [DOI] [PubMed] [Google Scholar]