

SUMMARY
Protrusion of the leading edge of migrating epithelial cells requires precise regulation of two actin filament (F-actin) networks, the lamellipodium and lamella. Cofilin is a downstream target of Rho-GTPase signaling that promotes F-actin cycling through its F-actin nucleating, severing and depolymerizing activity. However, its function in modulating lamellipodium and lamella dynamics, and their implications for protrusion efficiency have been unclear. Using quantitative fluorescent speckle microscopy, immunofluorescence, and electron microscopy, we establish that the Rac1/Pak1/LIMK1 signaling pathway controls cofilin activity within the lamellipodium. Enhancement of cofilin activity accelerates F-actin turnover and retrograde flow, resulting in widening of the lamellipodium. This is accompanied by increased spatial overlap of the lamellipodium and lamella networks and reduced cell edge protrusion efficiency. We propose that cofilin functions as a regulator of cell protrusion by modulating the spatial interaction of lamellipodium and lamella in response to upstream signals.
INTRODUCTION
Detailed analysis of F-actin dynamics in migrating epithelial cells by quantitative fluorescent speckle microscopy (qFSM) previously revealed two actin modules with distinct dynamic and molecular properties mediating cell protrusion (Ponti et al., 2004): the lamellipodium (Lp) and the lamella (Lm). The Lp is an actin network within 1–3 μm of the leading edge characterized by fast retrograde flow and adjacent zones of actin polymerization and depolymerization. The Lm extends from near the leading edge about 15 μm towards the cell interior, with slower retrograde flow, and randomly distributed spots of cyclic actin assembly and disassembly (Ponti et al., 2005). Molecular components associate specifically with either the Lp or the Lm: the Arp2/3 complex and cofilin, which promote F-actin treadmilling, are concentrated in the Lp (Svitkina and Borisy, 1999; Welch et al., 1997), while proteins regulating the contractile machinery, myosin II and tropomyosin, localize in the Lm and are excluded from the Lp (Gupton et al., 2005; Ponti et al., 2004). Despite the differences between the two F-actin modules, the Lp network may overlap in space with a portion of the Lm (Ponti et al., 2004). The mechanisms by which the distinct properties of Lp and Lm are established and maintained, how their interaction affects cell protrusion, and which signals are involved in co-regulating the dynamics of the two modules is largely unknown. Resolving these questions is essential to understanding the processes of F-actin-mediated cell protrusion and motility.
Due to its concentrated localization at the base of the Lp and its F-actin severing and depolymerizing activity (Bamburg, 1999), cofilin represents an excellent candidate effector for regulating the interaction between the Lp and Lm networks. Its ability to bind and depolymerize F-actin is inhibited by phosphorylation at serine 3 by the LIM and TES family kinases (Toshima et al., 2001; Yang et al., 1998). LIM kinases (LIMK) are activated by phosphorylation of Thr 508/505 (LIMK 1/2) through several Rho GTPase-mediated pathways, in particular Rac/Cdc42 acting through the p21-activated kinase Pak1 (Edwards et al., 1999) and RhoA through ROCK (Rho-associated coiled-coil containing kinase) (Maekawa et al., 1999). Conversely, slingshot (SSH) and chronophin (CIN) have been shown to act as activating phosphatases for cofilin (Huang et al., 2006).
The consequences of cofilin’s phospho-cycling between active (non-phosphorylated) and inactive (phosphorylated) forms for F-actin dynamics and its downstream effects on cell morphology can be complex. On the one hand, Rho-family GTPases promote F-actin polymerization by activating the Arp2/3 complex (Eden et al., 2002) and/or members of the formin family (Wallar and Alberts, 2003), and by inhibiting F-actin depolymerization by cofilin. On the other hand, active cofilin stimulates F-actin severing, thereby initiating the formation of new filament barbed ends that serve as sites for additional F-actin polymerization (Ichetovkin et al., 2002). Active cofilin has also been found to nucleate filaments de novo (Andrianantoandro and Pollard, 2006), and the depolymerizing function of cofilin is thought to replenish the pool of actin monomers required for further F-actin polymerization (Kiuchi et al., 2007; Pollard and Borisy, 2003). How both cofilin activation and inhibition translate into spatially controlled F-actin dynamics in cells remains unclear.
The relationships between cofilin-modulated F-actin activity and the resulting cell morphological responses are similarly complex. Localized activation of cofilin has been shown to promote local edge advancement (Ghosh et al., 2004), and pathways have been identified which link cofilin activation to growth factor stimulation in chemotactic protrusion (Chan et al., 2000; Zebda et al., 2000). This behavior has been explained by cofilin’s functions both in generating growing barbed ends and in replenishing the pool of actin monomers. However, the depolymerizing and severing activities of cofilin may also weaken the structure of the F-actin networks, leading to destabilization of the links between Lp, Lm, and/or the cytoplasmic domain of adhesion complexes. Since these links are required to convert the work of growing filaments in the Lp into edge movement, increases in cofilin activity might also be expected to reduce cell protrusivity.
In the present study, we analyzed how cofilin function regulates cell protrusion efficiency by differential control of F-actin dynamics in Lp and Lm. We perturbed signaling molecules downstream of Rac1, in particular Pak1 and LIMK1, and utilized constructs of constitutively active cofilin to gradually increase cofilin activity from baseline to very high levels. Using immunofluorescence to track changes in molecular components of the Lp and Lm, combined with quantitative fluorescent speckle microscopy of F-actin dynamics, computational tracking of cell edge movements, and electron microscopy, our data show that cofilin is a spatial organizer of Lp and Lm interaction, and by this mechanism regulates the rates of cellular leading edge protrusion.
RESULTS
Pak1, but not ROCK, controls cofilin phosphorylation at the cell leading edge
We examined the localization of inactive phosphorylated cofilin (pcofilin) to determine the contributions of the Rac1-Pak1-LIMK and RhoA-ROCK-LIMK pathways to the regulation of cofilin activity at the leading edge. These studies utilized PtK1 cells, a marsupial kidney epithelial cell line in which F-actin organization, kinetics and kinematics have been extensively characterized (Gupton et al., 2005; Gupton and Waterman-Storer, 2006; Ponti et al., 2004; Ponti et al., 2005; Wittmann et al., 2003). In control cells, pcofilin localized in diffuse punctae throughout protrusions at the cell edge and within the cell body (Fig. 1A). Expression of a constitutively active Rac1 mutant (Rac1Q61L, referred to as RacQL) induces the formation of a uniform protrusion all around the unattached cell edge (Wittmann et al., 2003) and increases the level of inactive pcofilin in the first 1.5 μm from the leading edge (up 17 % vs. control cells, Fig. 1A,B). Expression of the Pak1 (auto)inhibitory domain (PID) or a kinase-defective, dominant negative LIMK1 D460N (LIMK DN) mutant downstream of RacQL decreased pcofilin levels at the leading edge by 33 % and 30 % respectively (Fig. 1A,B).
Fig. 1. Pak1, but not ROCK, regulates cofilin phosphorylation at the cell leading edge.
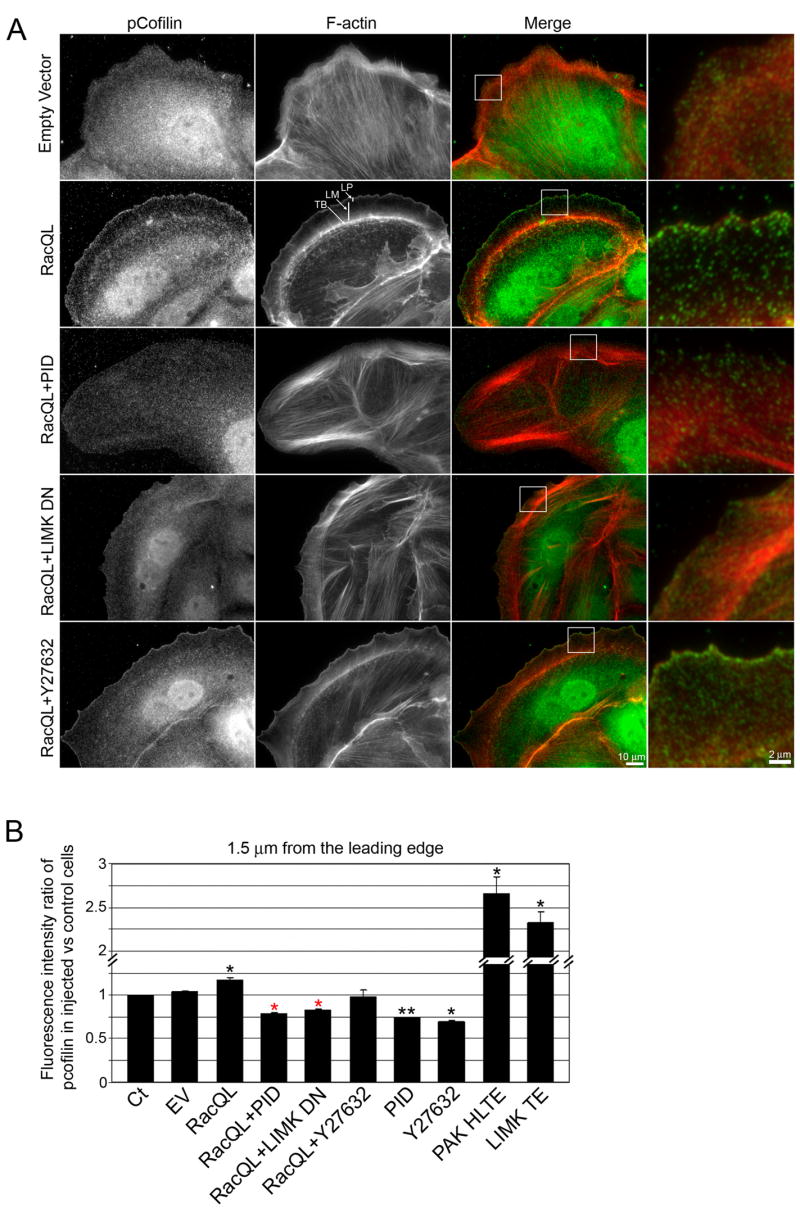
(A) Immunolocalization of phosphorylated-cofilin (pcofilin, green) and F-actin phalloidin staining (red) in control cells (empty vector), active Rac1-expressing cells (RacQL) alone or in combination with Pak1 inhibitory domain (RacQL+PID), dominant negative LIMK (RacQL+LIMK DN) or ROCK inhibitor (Y27632). Bar = 10 μm. Boxed regions are magnified to the right of the ‘Merge’ column. Bar = 2 μm. LP: Lp, LM: Lm, TB: transverse bundles.
(B) Fluorescence intensity ratio of pcofilin in injected/control cells at the cell edge (± SEM). The experiment was repeated at least three times; n = 20 to 50 cells for each condition. Red *, P < 0.05 vs. RacQL expressing cells. Black *, P < 0.05 and **, P < 0.01 vs. control cells.
Consistent with these observations, expression of the PID or LIMK DN also modified RacQL-induced actin organization. RacQL-expressing cells present a dense F-actin meshwork in the Lp and a dense transverse actin bundle at the base of the Lm (Fig. 1A, F-actin middle panel, arrows). In contrast, an increase in active cofilin downstream of RacQL due to PID or LIMK DN expression induced the formation of a dense F-actin network both in the Lp and the Lm, and of prominent actin bundles in the cell body. Inhibition of ROCK did not significantly decrease leading edge cofilin phosphorylation in RacQL-expressing cells (12 % decrease vs. uninhibited RacQL cells, P = 0.544, Fig. 1A,B). However, inhibition of Pak1 or ROCK in PtK1 control cells decreased pcofilin levels at the cell edge by 26 % and by 31 %, respectively. As a positive control, expression of active Pak1 (Pak H83,86L T423E) or active LIMK (LIMK T508EE) induced an increase of 127 % and of 99 %, respectively, in pcofilin (Fig. s1B and S1). These results indicate that Pak1, but not ROCK, controls cofilin phosphorylation at the leading edge of active Rac1-expressing cells, and that regulation of cofilin via the Pak1-LIMK1 signaling pathway is a required component of Rac1-induced leading edge actin organization.
Increase of active cofilin affects F-actin kinematics at the cell edge and induces the formation of a region of fast actin retrograde flow
We used fluorescent speckle microscopy (FSM) to define the effects of Pak1-regulated cofilin activity on actin dynamics at the leading edge (Videos 1–7; Fig. 2A,B). In control cells (Video 1), F-actin underwent a fast retrograde flow in the Lp and a slow retrograde flow in the Lm. Expression of RacQL (Video 2) did not significantly alter the flow velocity in the Lp (P = 0.992 vs. control cells) but induced a widening of the Lp (Fig. 2A,B).
Fig. 2. Increase of active cofilin induces the formation of a protrusion with fast actin retrograde flow.

(A) Single frames of actin fluorescent speckle time-lapse series of PtK1 cell expressing GFP (empty vector), RacQL alone or in combination with PID, LIMK DN, CIN WT, CFL SA, or CFL SA alone. Bar = 5 μm. White arrows indicate the lines used to generate kymographs.
(B) Kymograph analysis of actin retrograde flow in the cells depicted in A. White lines highlight speckle translocation used to calculate flow velocities; steeper streaks = faster flow rates. Time bar (t) = 2 min; scale bar (d) = 2 μm.
(C–D) Average F-actin flow rates measured at the leading edge (C) and 5 μm from the leading edge (D) of injected cells (± SEM). EV = empty vector control. n = 4 to 25 cells for each condition, with a minimum of 100 measurements per condition. *, P < 0.0001 vs. control cells; **, P < 0.0001 vs. RacQL-expressing cells. The experiment was repeated at least three times.
Inhibition of Pak1 downstream of RacQL (Video 3) reduced the width of the Lp compared to RacQL alone, and enhanced the rate of F-actin retrograde flow in the Lp. Pak1 inhibition was also associated with a two-fold decrease in the rate of F-actin retrograde flow in the Lm, as previously described (Wittmann et al., 2003). Importantly, more direct enhancement of cofilin activity downstream of Pak1 by expressing either LIMK DN (Video 4), an active chronophin phosphatase (CIN, Video 5), or a non-phosphorylatable active cofilin (CFL S3A, referred to as CFL SA, Video 6) increased the rate of F-actin retrograde flow not only at the leading edge (Fig. 2C) but throughout the entire protrusion (Fig. 2D). Similar blurring of the gradient in retrograde flow that distinguishes the Lp and Lm (Fig. 2B) was observed with constitutively active cofilin S3A in the absence of a RacQL background (Fig. 2B–D;Video 7). This further supports the conclusion that the effects on leading edge dynamics observed are an intrinsic result of cofilin activation.
The region of enhanced F-actin retrograde flow at the leading edge results from widening of the Lp
In comparison to control cells, cells with enhanced active cofilin displayed a wider region at the cell edge characterized by rapid actin retrograde flow. This effect could be due to (i) a widening of the Lp, (ii) an inhibition of Lp formation accompanied by an increase of the rate of F-actin retrograde flow in the remaining Lm (Gupton et al., 2005) or (iii) a fusion of the Lp/Lm networks. To distinguish between these possibilities, we used the definition of Lp and Lm as possessing myosin II-insensitive and -sensitive F-actin flows, respectively (Gupton et al., 2005; Ponti et al., 2004).
Treatment with blebbistatin, a nonmuscle myosin II ATPase inhibitor (Straight et al., 2003), reduced Lm retrograde flow by two fold, but did not affect Lp flow in control cells (empty vector EV, Fig. 3A–C, Video 8). Similar results were obtained in cells expressing RacQL and in cells with enhanced cofilin activity (RacQL and RacQL+CFL SA, Fig. 3A–C, Video 9,10). Spatially resolved maps of F-actin flow speed confirmed that blebbistatin treatment did not affect the characteristic gradient of fast flow at the leading edge to slower flow in the Lm region (Fig. 3D). Thus, the broad region of fast F-actin flow in cells with active cofilin is myosin II-independent, supporting its definition as a Lp.
Fig. 3. Retrograde flow in cells with increased active cofilin is myosin II-independent.

(A) Kymograph analysis of actin retrograde flow in a control cell expressing GFP (empty vector) and cells treated with 100 μM blebbistatin expressing GFP, RacQL alone or RacQL+CFL SA. White lines highlight speckle translocation used to calculate flow velocities. t = 1.5 min; d = 1.5 μm.
(B–C) Average F-actin flow rates measured at the leading edge (B) and 5 μm from the leading edge (C) of injected cells (± SEM). EV = empty vector control. **, P < 0.01 compared to EV + blebbistatin. n ≥ 9 cells for each condition. The experiment was repeated three times.
(D) F-actin flow maps computed from qFSM analysis of time-lapse movies of a control cell expressing GFP and cells treated with 100 μM blebbistatin expressing GFP, RacQL alone or RacQL+CFL SA. Flow rates are color coded, ranging from fast flow in red to slow flow in blue. Flow maps have been averaged over 60 frames, i.e. 5 min, and have been created with the same color-coded speed scale in order to allow comparison of cells under different conditions.
To verify the widening of the Lp in cells with enhanced cofilin activity, we characterized the molecular composition of regions subadjacent to the leading edge. Immunofluorescence localization of high molecular weight isoforms of tropomyosin established that tropomyosin was reduced near the leading edge of control and active Rac1-expressing cells. Increase of active cofilin downstream of Rac1 induced a major depletion of tropomyosin in the entire protrusion (Fig. S2A). These observations were confirmed by quantification of the fluorescence intensity ratio of tropomyosin/F-actin from the leading edge into the cell center (Fig. S2B–D). Tropomyosin was depleted in the first 4 μm adjacent to the cell edge in active Rac1-expressing cells, a distance corresponding to the size of the Lp (Fig. 2A,B). Cells with enhanced cofilin activity (RacQL+PID, RacQL+LIMK DN and RacQL+CFL SA) had a two-fold decreased level of tropomyosin in the first 7.5 μm of the protrusion compared to active Rac1 alone.
Similarly, myosin II was absent in the Lp of active Rac1-expressing cells and, interestingly, was depleted much further from the cell edge in the protrusions of cells with enhanced cofilin activity, as compared to controls (Fig. S3A–D). These results confirm that activation of cofilin downstream of Rac1 causes an expansion of the Lp from the leading edge to extend throughout large parts of the protrusion.
Active cofilin modifies F-actin kinetics and increases polymerization-competent free barbed ends at the cell edge in an Arp2/3 independent fashion
F-actin flow analysis, coupled with changes in localization of signature molecules, suggested that enhancement of cofilin activity induces the formation of a broad Lp in the protrusion. To verify this hypothesis, we analyzed the spatial organization of F-actin assembly/disassembly rates in these cells (Fig. 4A). Actin turnover maps, indicated that control cells had a ~2 μm wide band of strong polymerization along the leading edge (red punctae) juxtaposed to a similarly narrow band of depolymerization (green punctae). Together, these reflect the spatial organization of assembly and disassembly in a treadmilling Lp. The Lm is represented by a region with random foci of weaker polymerization/depolymerization (Fig. 4A). Expression of active Rac1 induced actin polymerization in a narrower (~1 μm), yet more homogeneous band along the leading edge. Increases of active cofilin markedly modified RacQL-induced F-actin kinetics: expression of PID, LIMK DN, CIN, or CFL SA all induced polymerization extending from the leading edge to deeper within the protrusion (Fig. 4A). Behind the wider polymerization band, cells expressing PID still presented a region characteristic of Lm turnover. However, in cells expressing kinase-defective LIMK, CIN, or active CFL, no characteristic pattern of the Lm was observed, and instead the region mainly displayed depolymerization events. Again, a similar phenotype was obtained when CFL SA alone was expressed in the absence of active RacQL (Fig. 4A).
Fig. 4. Active cofilin increases F-actin treadmilling and induces formation of polymerization-competent free barbed filament ends at the cell edge.
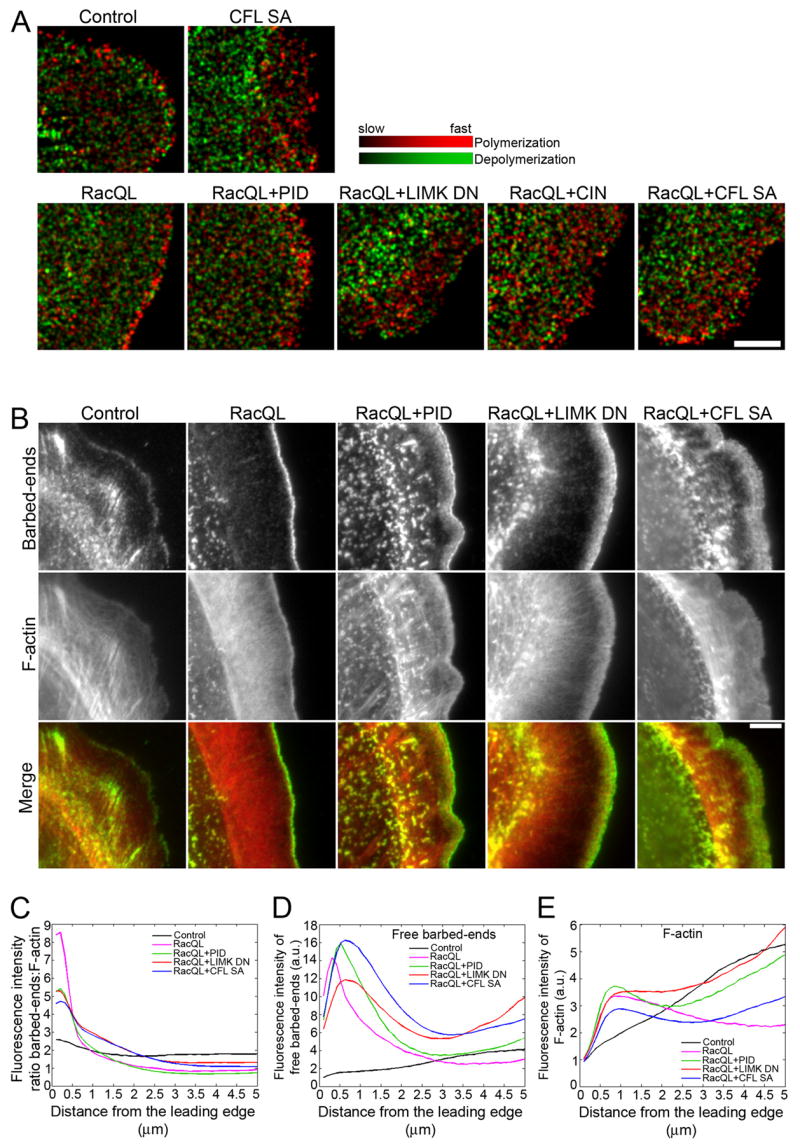
(A) F-actin turnover maps computed from qFSM analysis of time-lapse movies of cells expressing GFP (empty vector), CFL SA, RacQL alone or in combination with PID, LIMK DN, CIN or CFL SA. Turnover maps depict F-actin polymerization (red) and depolymerization (green) rates. Maps have been averaged over 24 frames, i.e. 2 min. Bar = 5 μm. n ≥ 4 cells analyzed for each condition.
(B) Free barbed-end actin incorporation (green) and F-actin phalloidin staining (red) in control cells, cells expressing RacQL alone or in combination with PID, LIMK DN or CFL SA. Bar = 5 μm.
(C) Fluorescence intensity ratio of actin incorporation marking free barbed-ends relative to F-actin in control (black), RacQL (pink), RacQL+PID (green), RacQL+LIMK DN (red) and RacQL+CFL SA (blue), measured from the cell edge (0 μm) into the cell center (5μm).
(D–E) Fluorescence intensity of free barbed-end actin incorporation (D) and F-actin (E) in injected cells (control: black, RacQL: pink, RacQL+PID: green, RacQL+LIMK DN: red, RacQL+CFL SA: blue), measured from the cell edge (0 μm) into the cell center (5 μm). In C–E, data shown represent one experiment, and are averaged from n ≥ 7 cells for each condition. The experiment was repeated at least three times, with similar results.
Since cofilin activities include severing/depolymerization (Carlier et al., 1997) as well as indirect promotion of barbed end formation and polymerization of F-actin (Condeelis, 2001; Ghosh et al., 2004), and may even directly mediate de novo nucleation of filaments (Andrianantoandro and Pollard, 2006), we further analyzed the localization and density of polymerization-competent free barbed filament ends. In control cells, free barbed ends were distributed in a narrow rim along the leading edge and at the end of F-actin bundles inside the protrusion (Fig. 4B). Expression of active Rac1 increased the concentration of free barbed ends that homogeneously localized along the leading edge, in agreement with the narrow band of polymerization measured by qFSM (Fig. 4B). In contrast, free barbed filament ends spread widely from the leading edge inside the protrusion of cells with enhanced cofilin activity (Fig. 4B: see RacQL+PID, RacQL+LIMK DN and RacQL+CFL SA). Quantitation of the barbed end fluorescence intensity to F-actin intensity ratio (Fig. 4C–E) confirmed that whereas polymerization-competent free barbed filament ends localized in a 0.5–1 μm wide band along the leading edge in RacQL-expressing cells (pink), this band widened in the presence of PID (~ 1.5 μm wide band, green), LIMK DN (~ 2 μm wide band, red) or CFL SA (~ 2.5–3 μm wide band, blue).
Several studies suggest a synergy between cofilin and Arp2/3 pathways, whereby the severing activity of cofilin can amplify the branching activity of the Arp2/3 complex (DesMarais et al., 2004; Ichetovkin et al., 2002). Thus, we examined whether increased cofilin activity would broaden the area of Arp2/3 localization in the Lp. Arp2/3 is distributed throughout the PtK1 cell, with the highest concentration within 1–2 μm from the leading edge (Fig. 5A). RacQL-expression induced spreading of Arp2/3 into the 2–4 μm region corresponding to the widened Lp (Fig. 5A) and also increased its apparent density (Fig. 5B–D, pink). Enhancement of active cofilin downstream of Rac1 reduced the width of the band of Arp2/3 localization (Fig.s 5A: see RacQL+PID, RacQL+LIMK DN and RacQL+CFL SA). Quantitation of the fluorescence intensity from the leading edge towards the cell center confirmed that Arp2/3 increased in density but remained localized in the first ~ 1–1.5 μm adjacent to the leading edge in cells with enhanced cofilin activity, similar to its localization in control cells (Fig. 5B–D). Thus, enhancement of cofilin activity downstream of Rac1 increases the width of the actin treadmilling array and the density of polymerization competent filament barbed ends independently of the localization of Arp2/3.
Fig. 5. Arp3 does not extend into protrusions of cells with increased active cofilin.
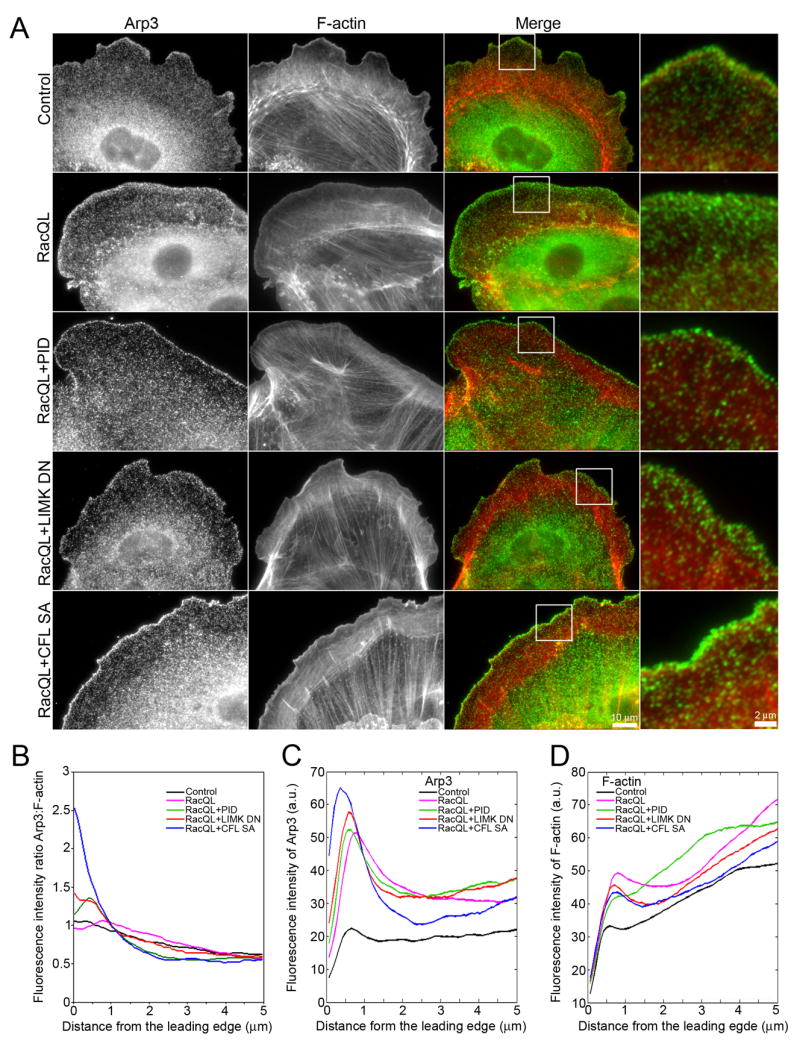
(A) Immunolocalization of Arp3 (green) and F-actin phalloidin staining (red) in control cell, or cell expressing RacQL alone or in combination with PID, LIMK DN or CFL SA. Bar = 10 μm. Boxed regions are magnified to the right of the ‘Merge’ column. Bar = 2 μm.
(B) Arp3/F-actin fluorescence intensity ratio in control (black), RacQL- (pink), RacQL+PID- (green), RacQL+LIMK DN- (red) and RacQL+CFL SA- (blue) expressing cells, measured from the cell edge (0 μm) into the cell center (5 μm).
(C-D) Fluorescence intensity of Arp3 (C) and F-actin (D) in injected cells (control: black, RacQL: pink, RacQL+PID: green, RacQL+LIMK DN: red, RacQL+CFL SA: blue), measured from the cell edge (0 μm) into the cell center (5 μm). In B–D, data shown represent one experiment, and are averaged from n ≥ 9 cells for each condition. The experiment was repeated at least three times, with similar results.
Increased cofilin activity deregulates coordinated edge protrusion and retraction
To evaluate the role of cofilin in cell protrusion, we investigated the effect of active cofilin on leading edge dynamics. In control cells, protrusion events propagate as transverse waves of positive velocities along the cell edge (visible in kymographs as diagonal red stripes), intercepted by retraction events (visible as diagonal blue stripes; Fig. 6A; wave pattern highlighted by white lines). In cells expressing RacQL, protrusion of the entire cell edge alternates with retraction of the entire cell edge (visible in kymographs as vertical red and blue stripes; Fig. 6A, dashed black lines). In cells expressing RacQL+LIMK DN, RacQL+CIN, RacQL+CFL SA, or CFL SA alone (Fig. 6A), such patterns of coordinated edge movement are dramatically reduced, suggesting that increased active cofilin disrupts the spatiotemporal coordination of leading edge movements.
Fig. 6. Coordination of leading edge dynamics is lost in cells with increased active cofilin.

(A) Kymographs of protrusion and retraction dynamics of the leading edge of a migrating PtK1 control cell, or a cell expressing RacQL alone, RacQL + LIMK DN, RacQL + CFL SA, or CFL SA alone. Edge displacements are encoded with warm color (red) for protrusion and cold color (blue) for retraction. t = 30 sec; d = 5 μm.
(B) Average velocity of a 40 μm leading edge section over 10 min (blue bars, ± SEM). *, P < 0.05 compared to zero average velocity. Average of the absolute velocity of a 40 μm leading edge section over 10 min (purple bars, ± SEM). **, P < 0.05 vs. control cells. Levels of active cofilin as percentage of the level in cells expressing LIMK TE (red line).
(C) Protrusion efficiency. Black *, P < 0.05 compared to a protrusion efficiency of one. n = 4 to 11 cells for each condition.
We calculated the average net protrusion velocity over multiple cycles to examine how the loss of coordinated movement affected productive edge advancement (Fig. 6B, blue bars; red line indicates increasing active cofilin levels, see Methods). Only control cells advanced with a net velocity significantly different from 0 (P = 0.003) in comparison to cells expressing RacQL, RacQL+LIMK DN, RacQL+CIN, RacQL+CFL SA or CFL SA alone (P > 0.12 for all conditions). In contrast, the average instantaneous edge velocity was significantly different from 0 for all conditions (Fig. 6B, purple bars). Thus, the rate of productive advancement of the leading edge depends primarily on how much time the cell edge spends in a protruding or retracting state, and only secondarily on how fast the edge moves in absolute terms.
To test this conclusion further, we defined protrusion efficiency as the ratio between the distances the edge travels in the protruding and the retracting states (Fig. 6C). A ratio equal to 1 indicates that protrusion and retraction events cancel one another, retaining a constant average position of the cell edge, while greater than 1 indicates net advancement of the entire leading edge. With the exception of the ratio for control cells, none of the protrusion efficiency scores in the other conditions were statistically different from 1 (P = 0.08 for RacQL, P > 0.50 for all other conditions; Fig. 6C). Together, these data show that control cells spend more time in a protruding than in a retracting state, whereas cells expressing RacQL+LIMK DN, RacQL+CFL SA and CFL SA do not significantly protrude on average, despite similar or even higher instantaneous edge velocities compared to control cells. This suggests that cells with increased cofilin activity lose the balance between local protrusion and retraction events along the edge. In contrast, the insignificant edge advancement of RacQL cells is primarily related to the lower instantaneous edge velocity (Fig. 6B).
Cofilin spatially reorganizes Lp and Lm
To identify the origin of the decrease in protrusion efficiency associated with increased active cofilin levels, we examined the spatial organization of F-actin dynamics behind the leading edge using single speckle classification of speckle life time and speed. As described in (Ponti et al., 2004), in migrating epithelial cells under control conditions, fast-moving and short-lived speckles are markers of a fast treadmilling Lp network whose rapid retrograde flow is powered by F-actin polymerization. Slow-moving and long-lived speckles are markers of the more stable Lm network whose slower retrograde flow is driven by actomyosin contraction.
Here, we applied the same analysis to test whether variations in cofilin activity would affect the spatial distribution of Lp and Lm speckles. The criterion for dividing the total population of tracked speckles into a group of fast-moving, short-lived speckles (group 1) and a group of slow-moving, long-lived speckles (group 2) was the maximal spatial separation of the two groups, i.e. the analyzed cellular area was divided into two regions and the thresholds for speed and lifetime were set such that in the region where speckles from group 1 are in majority, the number of speckles from group 2 are minimized, and vice-versa in the second region. Slow-moving, short-lived speckles were not classified. They consist of fluorophores incorporated into a mixed population of filaments with heterogeneous turnover and motion properties, thus generating an unstable image signal. Despite the relatively low percentage of classifiable speckles, each of the two groups contained at least several thousand speckles usable as stable markers of different F-actin dynamics (see Fig. 7A).
Fig. 7. Spatial reorganization of Lp and Lm in cells with increased active cofilin.
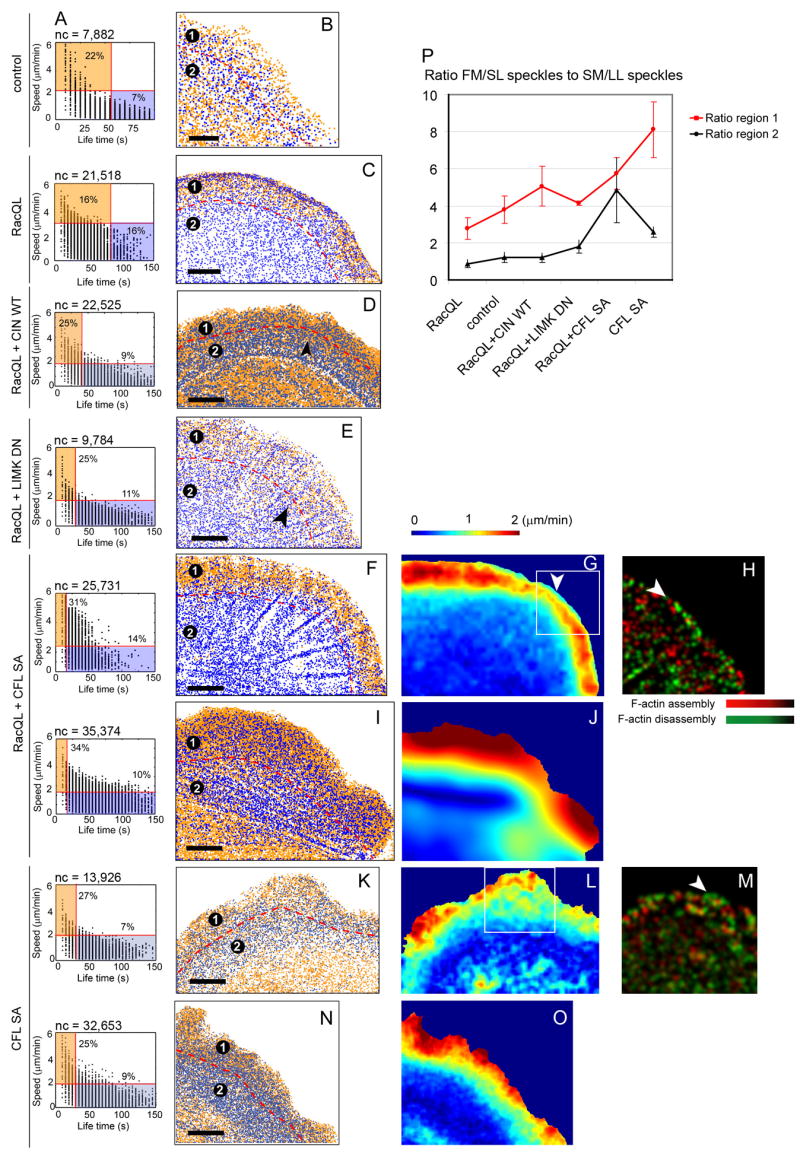
(A) Speckle lifetime and speed distributions. Red lines: speed and velocity thresholds for Lm (blue) and Lp (orange) segmentations. Percentages indicate the relative number of classified speckles compared to the total number of speckles. nc indicates the number of unambiguously classifiable speckles.
(B,C,D,E,F,I,K,N) Lm speckles (blue) and Lp speckles (orange) in control cell (B), or cells expressing RacQL alone (C), or in combination with CIN (D), LIMK DN (E), CFL SA (F and I) or cells expressing CFL SA alone (K and N). Bar = 5 μm.
(G,J,L,O) F-actin flow speed maps in cells expressing RacQL + CFL SA (G and J) or CFL SA alone (L and O). Maps are averaged over 24 frames, i.e. 2 min.
(H,M) F-actin turnover in cell expressing RacQL + CFL SA (H) or CFL SA alone (M). F-actin polymerization: red; depolymerization green. Maps are averaged over 24 frames, i.e. 2 min.
(P) Ratio between the number of fast-moving, short-lived speckles (FM/SL) and slow-moving, long-lived speckles (SM/LL). Measured separately for the leading edge region (circle 1 in B, C, D, E, F, I, K, N; red line, ± SEM) and the region adjacent to the leading edge region (circle 2 in B, C, D, E, F, I, K, N; black line, ± SEM), respectively. n = 4 to 8 cells for each condition.
In control cells, the classification indicated a 1–2 μm wide region adjacent to the leading edge which consists mostly of fast-moving, short-lived speckles (Fig. 7B, orange, region #1) in front of a wider band of slow-moving, long-lived speckles (Fig. 7B, blue, region #2). The classification algorithm accounts only for the lifetime and speed of a speckle but not the direction of motion relative to the leading edge. Thus, a significant population of fast-moving, short-lived speckles is also found in the lower left corner of region #2, which is associated with anterograde actin flow in the cell body. The same behavior is observed in Fig. 7D, I, K, N.
In RacQL-expressing cells, the region of fast-moving, short-lived speckles gets distinctly wider (Fig. 7C), consistent with the data in Figs 2–4, suggesting that under these conditions the width of the fast treadmilling Lp network is increased. Importantly, to achieve maximal spatial separation between the two groups, it was necessary to raise the thresholds for both lifetime and velocity as compared to control cells. This reflects the changes in the absolute rates of F-actin turnover and faster flow induced by expression of RacQL, although the organization of F-actin dynamics into Lp and Lm, is preserved. In fact, in these cells thresholds could be found that excluded nearly all fast-moving, short-lived speckles from region #2, but none of the thresholds allowed exclusion of slow-moving, long-lived from region #1. Thus, in agreement with the co-localization of overlapping Lp and Lm found in (Ponti et al., 2004), we concluded that two F-actin populations with different dynamics co-exist subadjacent to the cell edge.
Upon expression of RacQL+CIN or RacQL+LIMK DN, the spatial separation between the two speckle categories became less distinct. The band of fast-moving, short-lived speckles extended further away from the cell edge (Fig. 7D and E, arrow), but we still observed spatial co-localization of the two speckle categories at the leading edge. Upon expression of RacQL+CFL SA or CFL SA alone, the trend of an increasingly denser and wider population of fast-moving, short-lived speckles overlapping a scarcer population of slow-moving, long-lived continued (Fig. 7F, I, K and L). To achieve optimal separation of the two speckle groups in cells with increasingly higher cofilin activity, the threshold for the lifetime had to be substantially lowered as compared to RacQL-expressing cells. This need for adjustment reflects the faster F-actin turnover under elevated cofilin activity. In half of all observed cells expressing RacQL+CFL SA, a rim of slow-moving, long-lived speckles at the leading edge was reflected by a narrow band of slow flow (Fig. 7G, arrow) and significant depolymerization (Fig. 7H, arrow). This was also observed with CFL-SA itself (Fig. 7L and M).
To further quantify the overlap of the two speckle groups, we computed their number ratios in regions 1 and 2 (Fig. 7P; data presented in the order of increasing levels of active cofilin). The boundary between the regions is delineated by red dashed lines (Fig. 7B, C, D, E, F, I, K, N). In control cells, the leading edge was dominated by fast-moving, short-lived speckles (region 1) whereas the remaining network was dominated by slow-moving, long-lived speckles (region 2). The fraction of fast-moving, short-lived speckles gradually increased with enhancement of cofilin activity, both within the leading edge region (region 1) and further inside the protrusion (region 2). The ratio increase for region 2 in RacQL+CFL SA cells was accompanied by large cell to cell variation. As mentioned above, in about 50 % of the cells, a rim of slow-moving, long-lived speckles was found at the very leading edge (Fig. 7F–H). In these cells, region 2 was nearly devoid of fast-moving, short-lived speckles (Fig. 7F). In the other 50 % of the cells, spatial separation of the two speckle groups was difficult to discern (Fig. 7I), resulting in a strong increase of the fraction of fast-moving, short-lived speckles in region 2. Together, these data show that global elevation of cofilin activity obfuscates the spatial separation of two speckle groups that report the distinct dynamics of Lp and Lm. This is consistent with immunofluorescence data presented in Fig.s 5, S2, and S3, as well as experiments blocking myosin II activity shown in Fig. 3, which suggested gradual dislocation of Lm-specific molecular markers by Lp-specific markers as the level of cofilin activity was increased.
Our interpretations were fully supported by electron micrographs of PtK1 cells arrested at various levels of cofilin activity (Fig.s 8 and S4). Electron microscopy (EM) allowed us to directly image the distinct morphologies of Lp (shown in yellow) and Lm (shown in red). In control cells, a homogenous, dense filament array defining the lamellipodial actin network is found at the cell edge, followed by the Lm which is morphologically characterized by transversal actin bundles (highlighted in pink) interspersed by the isotropic actin network (Gupton et al., 2005). A horizontal belt of dense actin bundles (green) delineates the Lm from the cell body (Fig. 8). A similar organization is found in RacQL-expressing cells. However, here some of the actin bundles reorient from transversal to orthogonal throughout the Lm and visibly reach the cell edge. Increased levels of active cofilin induce a widening of the isotropic actin network that defines the Lp (Fig.s 8 and S4: RacQL+CFL SA, CFL SA). To varying extents, the Lm contains transverse actin bundles (Fig.s 8 and S4: RacQL+CFL SA 1st set of panels) or orthogonal bundles reaching the leading edge (Fig.s 8 and S4: RacQL+CFL SA 2nd set of panels, CFL SA), which are superimposed by a dense, homogeneous filament array characteristic of a Lp network.
Fig. 8. Active cofilin regulates Lp and Lm network organization as imaged by electron microscopy.
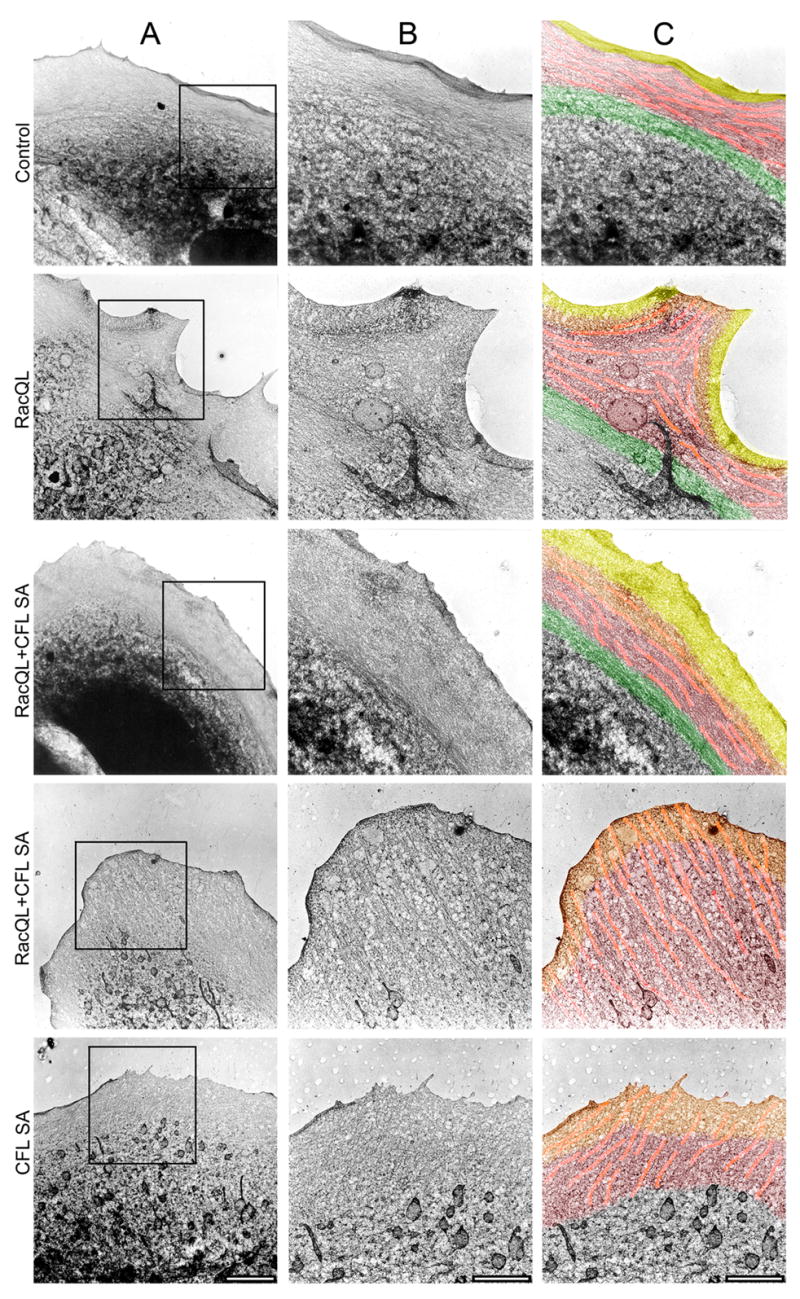
(A) Electron micrographs of control cells and cells expressing RacQL, RacQL+CFL SA or CFL SA. Bar = 5 μm.
(B and C) Higher magnification images of the inserts in column A. The Lp (yellow), defined by a dense network adjacent to the cell edge, is followed by the Lm (red) containing a large population of actin bundles (highlighted in pink). A region of transverse bundles (green) delineates the cell body from the Lm. Increased levels of active cofilin induce a widening of the Lp (RacQL+CFL SA, CFL SA) and cause Lm actin bundles to visibly extend to the leading edge (RacQL+CFL SA 2nd set of panels, CFL SA). The overlap between the Lp and the Lm is indicated in orange. White bars = 2.4 μm. Stereo pairs of these images can be seen in Fig. S4. The experiment was repeated at least three times for each condition, with similar results.
DISCUSSION
We report here the critical function of cofilin regulated via Pak1 and LIMK1 as a spatial organizer of the Lp and Lm F-actin networks during spontaneous epithelial cell protrusion. In control cells, the Lm extends 10–20 μm from the cell body in the direction of protrusion, with a narrow (1–2 μm) Lp partially overlapping the Lm at the leading edge. Increased levels of active cofilin markedly affect this organization: the Lp becomes distinctly wider, with F-actin network assembly and disassembly at significantly higher rates. At the same time the Lm extends fully to the leading edge. However, higher polymerization rates promoted by the increased number of free barbed filament ends in the Lp do not result in higher cell protrusion rates, but lead to faster polymerization-driven retrograde flow. Our results indicate a new role for cofilin as a regulator of the interaction between spatially overlapping Lp and Lm filaments, directly impacting on the efficiency with which assembly of these actin networks is locally transformed into cell edge protrusion.
From Rac1 to cofilin: a key signaling pathway to regulate actin polymerization
Expression of constitutively active Rac1 confirmed that this GTPase mediates a signaling cascade which controls cofilin activity at the leading edge that is dependent on downstream activation of the kinases Pak1 and LIMK1 (Edwards et al., 1999), but independent of ROCK, an effector kinase of RhoA that is also known to activate LIMK1 (Maekawa et al., 1999). Consistent with the participation of Pak1 and LIMK1 in a linear signaling pathway leading to suppression of cofilin activity downstream of Rac1, we observed that specific inhibition of the activity of any of these two signaling molecules resulted in changes in actin dynamics qualitatively similar to those obtained after expression of a constitutively active cofilin. Advanced live cell imaging and data modeling allowed us to quantify the gradual increase of specificity of cofilin function on a continuous scale as we stepped down the signaling pathway from Rac1 to Pak1 to LIMK1, ending with the prominent phenotype associated with expression of constitutively active cofilin. Introduction of the cofilin S3A mutant produces a stable pool of active cofilin that cannot be inactivated by phosphorylation and which out-competes endogeneous, phosphorylated forms of cofilin, thereby generating a “pure” active cofilin phenotype. In verification of this approach, we have also observed changes in cofilin-dependent actin dynamics upon siRNA-mediated depletion of CIN phosphatase in MTLn3 cells that are consistent with the results presented here (DerMardirossian, et. al, submitted).
Two region-dependent functions of Pak
Our quantitative live cell microscopy shows that cofilin inactivation in RacQL-expressing cells prolongs the lifetime of newly formed filaments at the leading edge (Fig. 7A) and, thus, widens the Lp (Fig. 2B). A similar result has been recently reported when cofilin was depleted using RNAi in Drosophila S2 cells (Iwasa and Mullins, 2007). In contrast, expression of the N-terminal Pak1 regulatory region (PID, aa 83–149), which acts as a dominant-negative inhibitor of Pak1 in vivo (Zenke et al., 1999), produces a modest increase in the steady-state levels of active cofilin, accompanied by a reduction in the width of the Lp (Fig. 2A,B). At these concentrations the severing function of cofilin dominates. In agreement with this model, increased rates of F-actin treadmilling yield faster polymerization-driven retrograde flow in the Lp (Fig.s 2B,C, and 7A).
qFSM maps of F-actin turnover show that PID-expressing cells still exhibit a region with random foci of assembly/disassembly characteristic of the Lm (Fig. 4A). However, F-actin retrograde flow is markedly reduced in this zone when Pak1 function is inhibited (Fig. 2B,D). Thus, Pak1 exhibits a region-dependent functionality in regulating F-actin dynamics. In the Lp, Pak1 promotes turnover of F-actin via regulation of cofilin phosphorylation, thereby also increasing the rate of polymerization-driven retrograde flow. In the Lm Pak1-regulated cofilin activity plays no role in mediating F-actin turnover. This is in line with the presence of long isoforms of tropomyosin, which selectively block cofilin-binding to Lm filaments (Fig. S2; (Gupton et al., 2005)). We conclude that Pak1 affects myosin II-driven F-actin flow in the Lm via yet-unidentified signaling pathways acting independently of cofilin. Our results therefore suggest that Pak1 is in a key position to coordinate the organization and interaction of Lp and Lm in response to upstream Rho GTPase signals.
Arp2/3-independent polymerization in the Lp
As a nucleator of actin polymerization, the Arp2/3 complex plays a central role in the cellular control of actin dynamics (Pollard and Borisy, 2003; Welch and Mullins, 2002). In PtK1 control cells, a narrow band of F-actin polymerization at the leading edge coincides spatially with equally thin bands of Arp2/3 and of polymerization-competent barbed ends (Fig.s 4A,B and 5A). With increased cofilin activity this correspondence is lost: whereas the bands of polymerization and free barbed ends become broader (Fig. 4A,B), the band of Arp2/3 is not affected and remains thin (Fig. 5A). Thus, in cells with high cofilin activity, Arp2/3-independent pathways must be present to stimulate Lp F-actin assembly. In agreement with (Andrianantoandro and Pollard, 2006), increased cofilin activity could contribute to de novo nucleation of F-actin filaments independent of Arp2/3 activation. We propose that this mechanism must be supported additionally by cofilin-mediated severing of existing F-actin, which also yields polymerization-competent free barbed ends throughout the Lp, and pointed end disassembly. Combined increases of assembly and disassembly enhance the overall rate of lamellipodial F-actin treadmilling. This explains the significantly increased rates of polymerization-driven retrograde flow and increased width of the Lp measured in cells in which the concentration of active cofilin exceeds the levels below which it acts as a pure severing molecule (as represented in the model, Fig. S5). Thus, our in vivo measurements suggest that the antagonizing functions of cofilin as a severer and nucleator of F-actin, identified in vitro, can overlap in a synergistic fashion.
Cofilin activity regulates the colocalization of Lm and Lp
Our high resolution analysis of F-actin dynamics at the level of single speckles, as well as structural analysis by electron microscopy, shows that cofilin functions as a spatial organizer of the Lp and Lm networks. The Lp, which in control cells exclusively occupies a narrow rim at the leading edge, is re-structured under gradually higher levels of active cofilin to become a distinctively wider region of accelerated retrograde flow that completely overlaps the Lm (see Fig. 2 and model Fig. S5).
The characteristic increase in the average flow speed calculated from the undivided speckle population at the leading edge could be due to (i) a growing proportion of Lp speckles without change in Lp and Lm flows, or (ii) a specific acceleration of Lp speckles. Our data provides more evidence for the former: Ratiometric analysis of tropomyosin:F-actin immunostaining (Fig. S2), which serves as a molecular marker for the fraction of Lm filaments at the cell edge, indicated that enhanced levels of active cofilin increase the relative number of Lp filaments. However, the tropomyosin:F-actin ratio at the leading edge is never less than ~30% of the ratio found in a Lp-free region. Thus, in all conditions Lm filaments do reach the leading edge. On the contrary, the threshold for which we obtained maximal spatial separation between the fast-moving, short-living (Lp) and the slow-moving, long-living (Lm) speckle groups was nearly constant (~2 μm/min) for all conditions (Fig. 7A). Therefore, within the limits of single speckle analysis it is unlikely that increased rates of F-actin treadmilling upon elevated cofilin activity also selectively accelerates Lp retrograde flow.
Proper organization of the Lp and Lm networks regulates cell protrusion
The markedly increased assembly rates accompanying enhanced cofilin activity do not translate into faster rates of cell edge protrusion. On the contrary, the overall protrusion is significantly impaired (Fig. 6). In epithelial cells, protrusion is a dynamic process where for any single time point certain sectors of the leading edge advance while others retract, alternating in cycles of 60–100 s (Machacek and Danuser, 2006). The cell will display net protrusion only if phases of edge advancement dominate phases of retraction. Our observations indicate that enhanced cofilin activity must therefore deregulate the local balance of edge advancement and retraction.
To translate F-actin assembly into edge advancement, propulsive forces generated by filament elongation must be counteracted by mechanical coupling of the F-actin cytoskeleton to the extracellular matrix (ECM). This is achieved by engagement of the Lm network with adhesions near the cell leading edge. Thus, one possibility for the loss of protrusivity in cells with enhanced cofilin activity would be the disruption of the normal distribution of focal adhesions at the front of the cell. However, in these cells, focal adhesions are intact and form close to the leading edge: we observe that 40 % of paxillin foci are still localized in the first 0.5 μm from the cell edge (compared to 12 % in control cells and 65 % in RacQL-expressing cells; Fig. S6). The presence of focal adhesions thus makes it likely that the Lm of cells with enhanced cofilin is still coupled to the ECM and not responsible for the loss of protrusion.
Alternatively, we propose that the decreased protrusion efficiency under increased cofilin activity results from a partial decoupling of Lp and Lm. This is supported by (i) the increase in retrograde flow rates in the Lp and (ii) the presence of a region of slow flow at the leading edge in about 50 % of these cells that appears to result from slippage of the entire Lp network relative to the Lm. In addition, high concentrations of active cofilin may affect the mechanical stability of the Lm network. From the lower ratio of tropomyosin:F-actin observed with higher levels of cofilin activity (Fig. S2), we conclude that under these conditions active cofilin displaces tropomyosin from Lm filaments, leading to increased rates of filament severing and depolymerization in the Lm. Further evidence for this proposition comes from the 50 % of cells, where enhanced cofilin activity results in a substantial extension of the Lp over the entire protrusion and the separability of distinct Lp and Lm speckle populations is lost. In these cases, high cofilin activity may disrupt the coupling of the majority of Lm filaments to adhesions which, in control conditions, contributes to the differentiation of flow speeds in the Lp and Lm. Thus, we propose that destabilization of the Lm is an additional factor in the loss of protrusion.
In our study, increased global activation of cofilin results in enhanced filament elongation, accelerated retrograde flow and reduced protrusion efficiency, suggesting that cofilin negatively regulates cell protrusion. This agrees with recent observations that constitutively active cofilin inhibits directional sensing and chemotaxis (Mouneimne et al., 2006). Our study now provides a mechanism for this result by placing cofilin at the center stage of controlling the mechanical interactions between Lp, Lm and adhesions in response to upstream signaling, a process which must be delicately balanced to transform F-actin assembly into productive cell advancement.
In view of earlier studies, where a transient and localized activation of cofilin suggested that cofilin is a positive regulator of cell protrusion (Dawe et al., 2003; Ghosh et al., 2004; Zebda et al., 2000), we propose a functional switch in the morphological responses to the local versus global regulation of cofilin activity. Local and transient activation of cofilin at the scale of microns and seconds promotes F-actin severing and depolymerization at the base of the Lp and increased F-actin assembly in proximal regions at the leading edge (Andrianantoandro and Pollard, 2006; Kiuchi et al., 2007), without affecting the mechanical integration of Lp and Lm, and the Lm and substrate. Thus, higher F-actin turnover yields, in this case, increased local cell edge protrusion. Conversely, increased steady state activity of cofilin on a time scale of minutes to hours, which reveals the function of cofilin as a nucleator of de novo filaments that are most probably weakly coupled to one another, results in a spatial reorganization and partial disintegration of Lp and Lm F-actin networks that, in turn, negatively regulates protrusion.
Together, these data indicate the requirement for spatial integrity of the Lp and Lm networks for the conversion of F-actin polymerization at the leading edge into effective cell protrusion. This competes with the requirement for F-actin disassembly to replenish the pool of G-actin necessary for filament reassembly. Cofilin defines a central switch in the balance of these two processes depending on the spatial and temporal scale of its activation. Thus, the global versus local regulation of cofilin by Pak1-mediated signals plays a critical role in determining the effect of cofilin as a positive or negative promoter of cell protrusion.
EXPERIMENTAL PROCEDURES
Cell culture and microinjection
Ptk1 cells were cultured in Ham’s F12 medium pH 7.2 (Sigma-Aldrich) containing 25 mM Hepes (GIBCO), 10% FBS (Gemini Bio-Products, CA), 100 U/ml penicillin, 0.1 mg/ml streptomycin and 0.25 μg/ml amphotericin B (GIBCO) at 37°C, 5% CO2. Two days prior to experiments, cells were plated on #1.5 coverslips. Plasmids encoding EGFP or EGFP-Rac1Q61L (RacQL) were injected in the cell nucleus, alone (150 ng/μl) or in combination with plasmids encoding Pak1 autoinhibitory domain (aa. 83–149 of human Pak1, myc-PID), kinase defective LIMK D460N (LIMK DN), chronophin wild type (CIN) or non-phosphorylatable and constitutively active cofilin S3A (HA-CFL S3A) (200 ng/μl). Plasmids encoding myc-PID, constitutively active Pak1 H83,86L T423E (myc-Pak HLTE), active LIMK T508EE (GFP-LIMK TE) or cofilin S3A (GFP-CFL SA) were injected alone (200 ng/μl). For FSM experiments, recombinant GST-PID H83L (aa 67–149 of human Pak1) was expressed and purified as described (Zenke et al., 1999), and injected (3.56 mg/ml) in PtK1 cells expressing GFP-RacQL. X-Rhodamine-conjugated actin, labeled on its lysine residues, was prepared as described (Gupton et al., 2005) and injected into cells at 1.3–1.5 mg/ml. Plasmids and fluorescent actin were co-injected into the nucleus. Protein expression was assessed by detection of the GFP from the EGFP empty vector, the GFP-RacQL or the GFP-CFL SA constructs. 3 to 6 h after injection, cells were mounted in chambers for live-cell microscopy or fixed for immunofluorescence staining. For ROCK inhibition, cells were treated with 10 μM Y-27632 (Calbiochem) in culture medium for 1h at 37°C, 5% CO2 before fixation. For myosin II inhibition, cells were mounted in chambers containing 100 μM blebbistatin (Calbiochem) in culture medium and were imaged as described below.
Immunofluorescence microscopy
Control and injected cells were fixed in cytoskeletal buffer (CB; 10 mM MES, 3 mM MgCl2, 138 mM KCl, 2 mM EGTA, pH 6.9) containing 4% paraformaldehyde, permeabilized in CB containing 0.5% Triton X-100 and blocked with 2% BSA in CB. Cells were then immunolabeled for the following: phosphorylated-cofilin (pcofilin, a gift from J. Bamburg, Colorado State University, Fort Collins, CO), long isoforms of tropomyosin (TM311, Sigma-Aldrich), myosin IIA heavy chain (Sigma-Aldrich) or Arp3 (a gift from M. Welch, University of California, Berkeley, CA) using the appropriate Alexa Fluor 568-conjugated secondary antibodies (Molecular Probes, Eugene, OR). F-actin was detected using Alexa Fluor 350-conjugated phalloidin (Molecular Probes). Cells were mounted on slides with mowiol mounting medium (Calbiochem) according to manufacturer’s instructions.
Immunofluorescence localization of free barbed filament ends was performed as described (Symons and Mitchison, 1991).
Epifluorescence images of fixed cells were acquired on an inverted microscope (Eclipse TE 2000-U, Nikon) equipped with electronically controlled shutter, filter wheels, and a 14-bit cooled CCD camera (either Orca II, Hamamatsu, or Cool SNAP HQ, Photometrics) controlled by MetaMorph® software (Universal Imaging Corp.) using 60×/1.4 NA Plan Apo DIC or 100×/1.4 NA Plan Apo Ph3 objective lens (Nikon) (Wittmann et al., 2003).
Immunofluorescence analysis
Fluorescence intensity of pcofilin was measured along the free edge in a 1.5 μm wide region extending from the leading edge into the cell center. All measurements were made from controls and injected cells plated on the same coverslip.
Quantification of the fluorescence of F-actin, free barbed-ends, tropomyosin, MHC or Arp3 as a function of the distance from the cell edge was obtained with custom software written in Matlab (Mathworks). Bands of constant distance to the cell edge were constructed and individual fluorescence intensities accumulated and averaged in each band to produce fluorescence intensities vs distance-to-the-cell-edge graphs.
Assessment of active cofilin
Data in Fig.s 6 and 7 is presented in order of increasing active cofilin levels, as indirectly gauged from pcofilin (inactive) immunofluorescence intensity (Fig.s 1 and S1). Since LIMK is the only known kinase that phosphorylates cofilin, we used cells expressing constitutively active LIMK (LIMK TE) to set the value of fluorescence intensity for maximal pcofilin concentration. The level of active (nonphosphorylated) cofilin in any other condition was estimated by the decrease of pcofilin immunofluorescence relative to LIMK TE-expressing cells. Accordingly, RacQL had the lowest levels of active cofilin (50 ± 3 %, n = 40 cells), while cofilin activity gradually increased in control cells (57 ± 2 %, n = 24 cells) and in cells expressing RacQL+CIN (63.5 ± 2 %, n = 55 cells) or RacQL+LIMK DN (64 ± 2 %, n = 22 cells) (Fig. 6B, red line).
Fluorescent speckle microscopy
Cells were prepared for live-cell microscopy as described (Gupton et al., 2005). Actin fluorescent speckle microscopy time-lapse series were acquired at 5 sec intervals for 10 min using a 100×/1.4 NA Plan Apo objective lens (Nikon) on a spinning disk confocal microscope system described in (Adams et al., 2003), using either a 14-bit Orca II or CoolSnapHQ camera.
Image analysis and quantification
F-actin flow rates at the cell leading edge were measured by kymograph analysis as described (Salmon et al., 2002). At least five randomly placed lines normal to the free cell edge were used to construct 5 kymographs of each cell and five flow rate measures were calculated for each region (Lp/Lm) in each kymograph.
FSM time lapse image series were analyzed as outlined in (Danuser and Waterman-Storer, 2006) using the fsmCenter software package written in Matlab (Mathworks) and C. Details on speckle identification, tracking and F-actin network turnover analysis can be found in (Ponti et al., 2005; Ponti et al., 2003).
Electron microscopy
Ptk1 cells were grown for two or three days on carbon coated formvar 100 mesh finder grids (EMS) (Luxenburg et al., 2007). Viewing a large number of cells on a single grid, by using the grid finders, allows localization of the exact individual cell (injected vs control) in both light and in electron microscope imaging. Control cells and injected cells were chemically fixed in CB containing 4% PFA, washed and stained with aqueous 2% OsO4 and 2% uranyl acetate. Dehydration in increasing concentrations of reagent grade ethanol (15, 20, 50, 70, 95, and 100%; 3 min per change) was followed by drying from liquid C02 by the critical-point method according to (Anderson, 1951) and (Buckley, 1975). Images were obtained under low-dose conditions using a Tecnai 12 microscope (FEI electron optics) equipped with a Lab6 filament (Denka) at 120kV. Tilt angles from ± 15 to ± 20 ° were used for the stereo-pair images (Fig. S4). Kodak SO-163 plates were developed for 13 min using D19 developer (Eastman Kodak Co., Rochester, NY).
Supplementary Material
Acknowledgments
We acknowledge the excellent technical assistance of B. Fowler. We thank J. Birkenfeld for cofilin constructs; M. Welch for antibodies to Arp3; J. Bamburg for antibodies to phosphorylated cofilin; B. Shin for help with microscopy; and J. Lim for images of PtK1 with actin alone. This work was supported by NIH grants GM39434 and GM44428 to G.M.B; GM67230 to C.W.S and G.D., and Cell Migration Consortium U54 GM64346 to G.D. V.D. is a fellow of the American Heart Association (Western States Affiliate) and M.M. is a Swiss National Science Foundation fellow. The electron microscopy portion of the work was supported by NIH Cell Migration Consortium U54 GM64346 to D.H.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams MC, Salmon WC, Gupton SL, Cohan CS, Wittmann T, Prigozhina N, Waterman-Storer CM. A high-speed multispectral spinning-disk confocal microscope system for fluorescent speckle microscopy of living cells. Methods. 2003;29:29–41. doi: 10.1016/s1046-2023(02)00282-7. [DOI] [PubMed] [Google Scholar]
- Anderson TF. Techniques for the preservation of three-dimensional structure in preparing specimens. EM TRANS NY ACAD SCI. 1951;13:130–134. [Google Scholar]
- Andrianantoandro E, Pollard TD. Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol Cell. 2006;24:13–23. doi: 10.1016/j.molcel.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Bamburg JR. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu Rev Cell Dev Biol. 1999;15:185–230. doi: 10.1146/annurev.cellbio.15.1.185. [DOI] [PubMed] [Google Scholar]
- Buckley IK, Porter KR. Electron microscopy of critical-point-dried whole cultured cells. J Microscopy. 1975;104:107–120. doi: 10.1111/j.1365-2818.1975.tb04010.x. [DOI] [PubMed] [Google Scholar]
- Carlier MF, Laurent V, Santolini J, Melki R, Didry D, Xia GX, Hong Y, Chua NH, Pantaloni D. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin-based motility. J Cell Biol. 1997;136:1307–1322. doi: 10.1083/jcb.136.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AY, Bailly M, Zebda N, Segall JE, Condeelis JS. Role of cofilin in epidermal growth factor-stimulated actin polymerization and lamellipod protrusion. J Cell Biol. 2000;148:531–542. doi: 10.1083/jcb.148.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condeelis J. How is actin polymerization nucleated in vivo? Trends Cell Biol. 2001;11:288–293. doi: 10.1016/s0962-8924(01)02008-6. [DOI] [PubMed] [Google Scholar]
- Danuser G, Waterman-Storer CM. Quantitative fluorescent speckle microscopy of cytoskeleton dynamics. Annu Rev Biophys Biomol Struct. 2006;35:361–387. doi: 10.1146/annurev.biophys.35.040405.102114. [DOI] [PubMed] [Google Scholar]
- Dawe HR, Minamide LS, Bamburg JR, Cramer LP. ADF/cofilin controls cell polarity during fibroblast migration. Curr Biol. 2003;13:252–257. doi: 10.1016/s0960-9822(03)00040-x. [DOI] [PubMed] [Google Scholar]
- DesMarais V, Macaluso F, Condeelis J, Bailly M. Synergistic interaction between the Arp2/3 complex and cofilin drives stimulated lamellipod extension. J Cell Sci. 2004;117:3499–3510. doi: 10.1242/jcs.01211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden S, Rohatgi R, Podtelejnikov AV, Mann M, Kirschner MW. Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature. 2002;418:790–793. doi: 10.1038/nature00859. [DOI] [PubMed] [Google Scholar]
- Edwards DC, Sanders LC, Bokoch GM, Gill GN. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol. 1999;1:253–259. doi: 10.1038/12963. [DOI] [PubMed] [Google Scholar]
- Ghosh M, Song X, Mouneimne G, Sidani M, Lawrence DS, Condeelis JS. Cofilin promotes actin polymerization and defines the direction of cell motility. Science. 2004;304:743–746. doi: 10.1126/science.1094561. [DOI] [PubMed] [Google Scholar]
- Gupton SL, Anderson KL, Kole TP, Fischer RS, Ponti A, Hitchcock-DeGregori SE, Danuser G, Fowler VM, Wirtz D, Hanein D, Waterman-Storer CM. Cell migration without a lamellipodium: translation of actin dynamics into cell movement mediated by tropomyosin. J Cell Biol. 2005;168:619–631. doi: 10.1083/jcb.200406063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupton SL, Waterman-Storer CM. Spatiotemporal feedback between actomyosin and focal-adhesion systems optimizes rapid cell migration. Cell. 2006;125:1361–1374. doi: 10.1016/j.cell.2006.05.029. [DOI] [PubMed] [Google Scholar]
- Huang TY, DerMardirossian C, Bokoch GM. Cofilin phosphatases and regulation of actin dynamics. Curr Opin Cell Biol. 2006;18:26–31. doi: 10.1016/j.ceb.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Ichetovkin I, Grant W, Condeelis J. Cofilin produces newly polymerized actin filaments that are preferred for dendritic nucleation by the Arp2/3 complex. Curr Biol. 2002;12:79–84. doi: 10.1016/s0960-9822(01)00629-7. [DOI] [PubMed] [Google Scholar]
- Iwasa JH, Mullins RD. Spatial and temporal relationships between actin-filament nucleation, capping, and disassembly. Curr Biol. 2007;17:395–406. doi: 10.1016/j.cub.2007.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiuchi T, Ohashi K, Kurita S, Mizuno K. Cofilin promotes stimulus-induced lamellipodium formation by generating an abundant supply of actin monomers. J Cell Biol. 2007;177:465–476. doi: 10.1083/jcb.200610005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luxenburg C, Geblinger D, Klein E, Anderson K, Hanein D, Geiger B, Addadi L. The architecture of the adhesive apparatus of cultured osteoclasts: from podosome formation to sealing zone assembly. PLoS ONE. 2007;2:e179. doi: 10.1371/journal.pone.0000179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machacek M, Danuser G. Morphodynamic profiling of protrusion phenotypes. Biophys J. 2006;90:1439–1452. doi: 10.1529/biophysj.105.070383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, Obinata T, Ohashi K, Mizuno K, Narumiya S. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 1999;285:895–898. doi: 10.1126/science.285.5429.895. [DOI] [PubMed] [Google Scholar]
- Mouneimne G, DesMarais V, Sidani M, Scemes E, Wang W, Song X, Eddy R, Condeelis J. Spatial and temporal control of cofilin activity is required for directional sensing during chemotaxis. Curr Biol. 2006;16:2193–2205. doi: 10.1016/j.cub.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- Ponti A, Machacek M, Gupton SL, Waterman-Storer CM, Danuser G. Two distinct actin networks drive the protrusion of migrating cells. Science. 2004;305:1782–1786. doi: 10.1126/science.1100533. [DOI] [PubMed] [Google Scholar]
- Ponti A, Matov A, Adams M, Gupton S, Waterman-Storer CM, Danuser G. Periodic patterns of actin turnover in lamellipodia and lamellae of migrating epithelial cells analyzed by quantitative Fluorescent Speckle Microscopy. Biophys J. 2005;89:3456–3469. doi: 10.1529/biophysj.104.058701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponti A, Vallotton P, Salmon WC, Waterman-Storer CM, Danuser G. Computational analysis of F-actin turnover in cortical actin meshworks using fluorescent speckle microscopy. Biophys J. 2003;84:3336–3352. doi: 10.1016/S0006-3495(03)70058-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon WC, Adams MC, Waterman-Storer CM. Dual-wavelength fluorescent speckle microscopy reveals coupling of microtubule and actin movements in migrating cells. J Cell Biol. 2002;158:31–37. doi: 10.1083/jcb.200203022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR, Mitchison TJ. Dissecting temporal and spatial control of cytokinesis with a myosin II Inhibitor. Science. 2003;299:1743–1747. doi: 10.1126/science.1081412. [DOI] [PubMed] [Google Scholar]
- Svitkina TM, Borisy GG. Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J Cell Biol. 1999;145:1009–1026. doi: 10.1083/jcb.145.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symons MH, Mitchison TJ. Control of actin polymerization in live and permeabilized fibroblasts. J Cell Biol. 1991;114:503–513. doi: 10.1083/jcb.114.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toshima J, Toshima JY, Amano T, Yang N, Narumiya S, Mizuno K. Cofilin phosphorylation by protein kinase testicular protein kinase 1 and its role in integrin-mediated actin reorganization and focal adhesion formation. Mol Biol Cell. 2001;12:1131–1145. doi: 10.1091/mbc.12.4.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallar BJ, Alberts AS. The formins: active scaffolds that remodel the cytoskeleton. Trends Cell Biol. 2003;13:435–446. doi: 10.1016/s0962-8924(03)00153-3. [DOI] [PubMed] [Google Scholar]
- Welch MD, DePace AH, Verma S, Iwamatsu A, Mitchison TJ. The human Arp2/3 complex is composed of evolutionarily conserved subunits and is localized to cellular regions of dynamic actin filament assembly. J Cell Biol. 1997;138:375–384. doi: 10.1083/jcb.138.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch MD, Mullins RD. Cellular control of actin nucleation. Annu Rev Cell Dev Biol. 2002;18:247–288. doi: 10.1146/annurev.cellbio.18.040202.112133. [DOI] [PubMed] [Google Scholar]
- Wittmann T, Bokoch GM, Waterman-Storer CM. Regulation of leading edge microtubule and actin dynamics downstream of Rac1. J Cell Biol. 2003;161:845–851. doi: 10.1083/jcb.200303082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, Nishida E, Mizuno K. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature. 1998;393:809–812. doi: 10.1038/31735. [DOI] [PubMed] [Google Scholar]
- Zebda N, Bernard O, Bailly M, Welti S, Lawrence DS, Condeelis JS. Phosphorylation of ADF/cofilin abolishes EGF-induced actin nucleation at the leading edge and subsequent lamellipod extension. J Cell Biol. 2000;151:1119–1128. doi: 10.1083/jcb.151.5.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenke FT, King CC, Bohl BP, Bokoch GM. Identification of a central phosphorylation site in p21-activated kinase regulating autoinhibition and kinase activity. J Biol Chem. 1999;274:32565–32573. doi: 10.1074/jbc.274.46.32565. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.