

Abstract
The presence of a pulmonary artery aneurysm, major aortopulmonary and coronary–pulmonary collateral vessels, and severe pulmonary hypertension in an adult with unilateral pulmonary artery agenesis and previous patent ductus arteriosus ligation is very rare. A 34-year-old man experienced these conditions. When he was 10 years old, catheterization and angiography revealed right pulmonary artery agenesis, dilation of the main pulmonary artery, multiple collateral vessels extending from the aorta to the right pulmonary system, and a patent ductus arteriosus (shunt ratio, 3.57) that was then ligated; the other conditions were not corrected.
This adult patient was in New York Heart Association functional class II; mild central cyanosis was detected only during exercise. The right pulmonary arterial system was seen only at the right hilar area via collateral vessels from the subclavian, bronchial, internal mammary, and intercostal arteries. Angiography revealed collateral vessels from the right and circumflex coronary arteries to the right pulmonary system. The right intraparenchymal pulmonary arterial systems were patent but of small diameter (pulmonary artery pressure, 85 mmHg; ratio of peak right-to-left ventricular pressure, 0.94; peak pulmonary pressure unresponsive to 100% oxygen). Pulmonary vascular resistance was not estimated because of the risk of aneurysmal rupture.
We concluded that irreversible pulmonary hypertension had developed (delayed by the patent ductus arteriosus ligation in childhood) and that the patient's only chance for survival was heart-lung transplantation. To sustain the patient until surgery, we administered sildenafil. Herein, we describe the vascular conditions that accompany unilateral absence of the pulmonary artery, and therapeutic methods.
Key words: Adult; cardiovascular abnormalities; collateral circulation/physiology; coronary vessel anomalies/complications/diagnosis/physiopathology; diagnosis, differential; heart defects, congenital; hypertension, pulmonary/diagnosis/drug therapy/etiology; pulmonary artery/abnormalities/anatomy & histology/physiopathology; sildenafil; vasodilator agents/therapeutic use
Congenital unilateral absence of the pulmonary artery (UAPA) is a rare condition that is a result of embryologic defects and may be accompanied by cardiovascular, skeletal, and other anomalies. Absence of the left pulmonary artery (PA) is commonly seen in patients who have tetralogy of Fallot or truncus arteriosus; absence of the right PA is often an isolated finding. The embryologic basis for UAPA is malformation of the 6th aortic arch on the affected side.1,2 Persons with this anomaly have a normal pulmonary trunk but a unilateral absence of the branch PA. Some authors prefer to use the term “proximal interruption of a pulmonary artery,”3,4 because the more distal parenchymal pulmonary vessels are generally intact and patent, although of small diameter.
Herein, we describe the clinical and laboratory features of an adult whose UAPA and patent ductus arteriosus (PDA) were diagnosed in childhood but who at that time underwent only PDA ligation. We describe the vascular conditions that accompany UAPA, and methods of therapy.
Case Report
In June 2006, a 34-year-old man was admitted to our hospital with a history of exertional dyspnea since childhood and, for a few months, of cyanosis during heavy effort. The patient had undergone a PDA ligation at 10 years of age. He had no history of hemoptysis or of frequent pulmonary infection.
Physical examination revealed a man in outwardly good condition but in New York Heart Association functional class II. His blood pressure was 95/65 mmHg; pulse, 96 beats/min; respiratory rate, 21 breaths/min; and temperature, 36 °C. The popliteal systolic and diastolic blood pressure readings were the same as those in the arms. Chest wall excursions and breath sounds were equal in both sides of the chest. The patient's jugular venous pressure was elevated (10 cm H2O), with prominent a waves. There was a prominent right ventricular impulse, and a large area of pulsatility in the pulmonary region. Auscultation revealed a pulmonary ejection click followed by a short systolic ejection murmur, a widely split 2nd sound, and a long, diastolic, pulmonary-regurgitation murmur. Clubbing, telangiectasia, and edema were not noted. Mild mucosal and peripheral cyanosis was seen when he exercised moderately. The liver and spleen were of normal size and location. Results of tests for complete blood count, blood urea nitrogen levels, electrolyte concentrations, and C-reactive protein concentration were all normal. Protein C, protein S, and antithrombin III readings were within normal limits. The results of rheumatologic tests were negative.
Arterial blood gases with the patient on room air showed a pH of 7.4, arterial carbon dioxide partial pressure of 32.1 mmHg, and arterial oxygen partial pressure of 82.8 mmHg; the bicarbonate level was 40.3 mmol/L, and oxygen saturation was 86%. Bilateral lower-extremity venous Doppler ultrasonography showed normal flow patterns. Whole-abdominal Doppler ultrasonography showed no abnormalities. Electrocardiography (Fig. 1) showed sinus rhythm with right ventricular hypertrophy, right-axis deviation, and right ventricular strain pattern (T-wave inversion in the right precordial leads and ST-segment depression). Echocardiography showed normal left ventricular size and function, right ventricular hypertrophy and mild dilation, flattening of the interventricular septum, right atrial dilation, moderate tricuspid regurgitation, and systolic PA pressure of 95 mmHg. Posteroanterior chest radiography (Fig. 2) revealed a loss of volume in the right lung, the absence of a PA shadow in the right hilar region, grossly diminished right pulmonary vascular markings, right hemidiaphragm elevation, contralateral lung hyperinflation and herniation beyond the midline, and apparent left PA dilation.

Fig. 1 Electrocardiogram shows right ventricular hypertrophy, right-axis deviation, and right ventricular strain pattern.
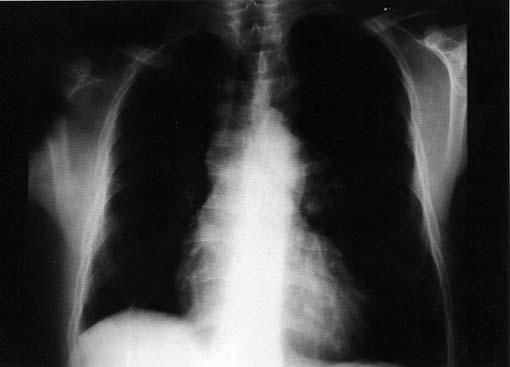
Fig. 2 Typical posteroanterior chest radiograph in our patient with unilateral absence of the right pulmonary artery.
Computed tomography (CT) showed that the ascending aorta, aortic arch, and thoracic aorta were of normal size. The widest PA diameter was 61 mm. The left main PA diameter was 28 mm. The intraparenchymal left pulmonary system was patent and normal. The right PA (diameter, 9 mm) was visible at the right hilar region because of CT contrast material spread via collateral vessels that extended from the hypertrophic bronchial arteries, the right internal mammary artery, and the intercostal arteries. The intercostal arteries had established large collateral vessels to the right intraparenchymal PA systems in the subpleural areas. There was no thrombus or dissection in either the main pulmonary trunk or the left PA system. The collateral vasculature from the intercostal arteries was retrogradely supplying the right-lung PA system in the subpleural region. The vessels of the right intraparenchymal arterial system were patent but of small diameter. The total volume of the right lung was less than that of the left (Figs. 3A and 3B).
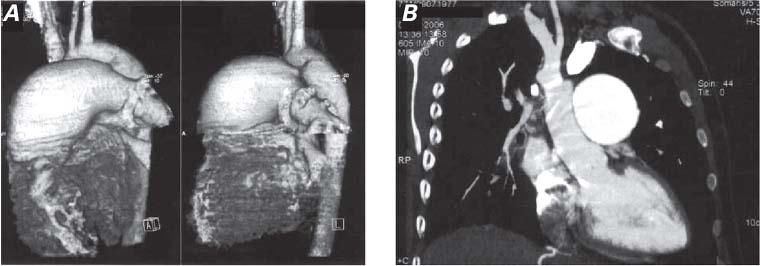
Fig. 3 Contrast-enhanced computed tomographic sections of the thorax, in 3-dimensional view (A and in 0.75-mm crosscut reconstructed view (B), show unilateral absence of the right pulmonary artery, associated vascular pathogenesis, and a total volume of the right lung less than that of the left.
Digital subtraction angiography showed UAPA and an aneurysm of the main and left PA (Fig. 4). The main pulmonary trunk was greatly dilated at 6.3 cm. The left peripheral PA system was diminished in luminal diameter, which suggested elevated PA pressure. A complete hemodynamic study and an oximetric study were performed by means of cardiac catheterization. Because of the risk of aneurysmal rupture, pulmonary vascular resistance was not estimated. Peak PA pressure was 85 mmHg. The ratio of peak right ventricular pressure to left ventricular pressure was 0.94, and peak pulmonary pressure did not decrease in response to 100% oxygen. Selective coronary angiography (Fig. 5) showed some collateral vessels extending from the right and circumflex coronary arteries to the right PA. An aortogram of the descending aorta (Fig. 6) showed multiple large collateral vessels that originated from the ascending aorta, aortic arch, and descending aorta and extended to the right PA system. Right renal artery angiography (Fig. 7) showed collateral vessels arising from a major branch of this artery and extending to the lower lobes of the right lung.
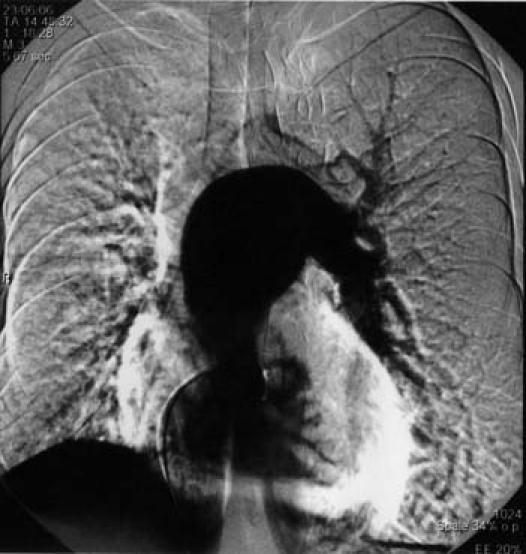
Fig. 4 Digital subtraction angiography in our patient with unilateral absence of the right pulmonary artery shows proximal interruption of the right pulmonary artery and an aneurysm of the main and left pulmonary arteries.
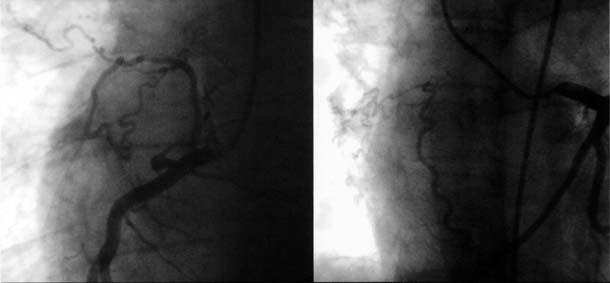
Fig. 5 Selective coronary angiography shows collateral vessels extending from the right and circumflex coronary arteries to the right pulmonary arterial system.
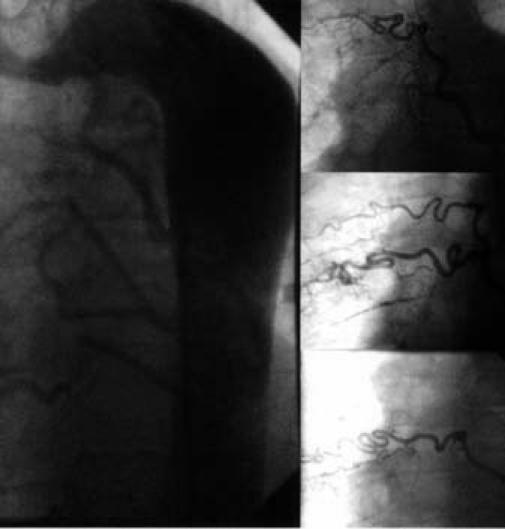
Fig. 6 Aortogram of the descending aorta shows multiple large collateral vessels extending to the right pulmonary arterial system, and selective injections of collateral vessels.
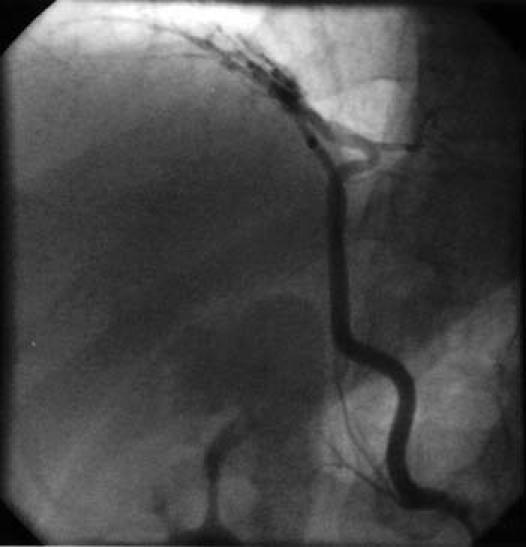
Fig. 7 Arterial angiography shows a major branch of the right renal artery supplying collateral vessels to segments of the lower lobe of the right lung.
The patient experienced episodes of cyanosis, and he had echocardiographically detected right ventricular dilation; accordingly, his UAPA was considered inoperable. Several cardiac surgeons declined to operate because of the high risk of surgery in a patient who had severe pulmonary hypertension. Eventual heart-lung transplantation seemed the best option, but, until that surgery could occur, the only remaining alternative was conservative treatment of the pulmonary hypertension by pharmacologic vasodilation. We decided to treat the patient with sildenafil citrate at a daily dosage that would be gradually increased from 75 mg to 150 mg daily.
After discharge from the hospital, the patient was scheduled for regular follow-up by clinical examination and telephone interview. He was well at 1 and 3 months. He remained stable on sildenafil at 10 months, but he reported no improvement in his exercise capacity and quality of life. On catheterization, peak PA pressure was the same as it had been 10 months before.
In August 2007, we learned that the patient, who was still awaiting heart-lung transplantation, had experienced sudden cardiac death at home.
Discussion
Unilateral absence of the PA is difficult to diagnose, and it may occur as an isolated condition or in association with other congenital heart defects. Cardiovascular anomalies associated with UAPA are tetralogy of Fallot, right aortic arch, septal defects, and PDA.5–7 Most cases of UAPA are diagnosed and surgically treated during the patient's 1st year of life.2,8 Some patients with isolated UAPA have a benign clinical course, and the diagnosis may not be made until they are adults. Patients who survive to adulthood usually present with few symptoms.6,9,10 Clinically, UAPA is characterized by increased pulmonary blood flow to the existing PA, by the presence of aortopulmonary collateral vessels extending to the lung that lacks the PA branch, and by pulmonary hypertension, right ventricular failure, and cyanosis.
The principal differential diagnosis is Swyer-James (or Macleod's) syndrome, in which the affected hemithorax may or may not be small. The differential diagnostic criteria of UAPA for this syndrome have been described.6,9,11 Anomalous origin of the PA should also be considered. In this condition, one of the PAs originates directly from the ascending aorta or from the transverse aortic arch. Either or both of the PAs can originate directly from the aorta; in almost 80% of cases, it is the right PA, and in approximately 75% of cases, a PDA is present. The clinical manifestations usually occur in infants (or, more rarely, in newborns) as respiratory distress or congestive heart failure due to increased pulmonary resistance. Unilateral absence of the PA is completely different from anomalous origin of the PA, and angiocardiography distinguishes between these conditions.12–14
The clinical presentation of UAPA is variable; approximately 30% of patients remain asymptomatic throughout their lives.2 There may be mild dyspnea on exertion,15 and up to 40% of patients experience recurrent pulmonary infections.10 Pool and colleagues1 reviewed 98 cases and found a 10% incidence of hemoptysis, but other researchers have documented a higher frequency of hemoptysis as the presenting symptom, especially in adults.10 When hemoptysis occurs, it is most often inconsequential and arises from hypertrophic collateral vessels.1,3,10
In adult patients, chronic thromboembolic occlusion of a PA can be mistaken for UAPA.16 Reaching a differential diagnosis can be difficult because of shared features seen on chest radiography, pulmonary angiography, CT, and magnetic resonance imaging. Because the respective methods of surgical correction vary substantially, accurate diagnosis is important. Chronic pulmonary embolism can manifest itself on CT scan in particular as complete occlusive disease in vessels that are smaller than adjacent patent vessels. Other pulmonary CT angiographic findings that characterize and distinguish chronic pulmonary embolism from UAPA include evidence of recanalization, webs or flaps in the PAs, and partial filling defects that form obtuse angles with the vessel wall.17
Pulmonary blood supply via major aortopulmonary collateral arteries is commonly associated with pulmonary atresia with ventricular septal defects (PA/VSD) and occasionally with tetralogy of Fallot. Only very rarely have these major collateral arteries been described in the presence of pulmonary atresia with intact ventricular septum.18 Pulmonary atresia with intact ventricular septum is an uncommon condition, and postnatal survival is dependent on the patency of the ductus arteriosus, the patency, presence, and development of major aortopulmonary collateral arteries, and intra-atrial communications.19 Two unusual cases of muscular pulmonary atresia with an intact septum have been reported. In both, the pulmonary blood supply came entirely from aortopulmonary collateral vessels. These observations suggest right ventricular outflow tract obstruction early in fetal development, with involution of the pulmonary trunk, 6th-arch derivatives, and persistence of primitive aortopulmonary connections. Pulmonary atresia with intact septum and PA/VSD are 2 separate pathologic entities that occur at different stages in fetal development.20
In early embryonic development, transient systemic-to-pulmonary collateral arteries may arise during 2 extended periods. During maldevelopment of the pulmonary outflow tract, these transient connections may persist as systemic-to-pulmonary collateral arteries. The timing and the extent of pulmonary outflow tract maldevelopment may determine the origin and distribution of the collateral arteries. When pulmonary obstruction occurs at a very late stage of fetal development or after birth, bronchial arteries can develop into systemic-to-pulmonary collateral arteries.21 Fadel and associates22 showed that PA occlusion stimulates angiogenesis in the systemic circulation of the ipsilateral lung and increases systemic-to-pulmonary blood flow. After a period of pulmonary occlusion, revascularization normalizes the systemic blood flow to the lung and induces a partial loss of collateral vessels. It has been shown that, in congenital heart disease, certain aortopulmonary collateral arteries have a marked histologic similarity to the ductus arteriosus.23 It is argued that these connections can persist in human beings as aortopulmonary collateral vessels, in certain cases with abnormalities in the pulmonary part of the cardiac outflow tract.23
In 2002, Ten Harkel and colleagues24 searched the literature for cases of isolated UAPA and determined that pulmonary hypertension was present in 44% of the 108 cases so identified. Surgery was performed in 17% of the 108 patients because of recurrent hemoptysis, intractable pulmonary infections, severe pulmonary hypertension, cyanosis, and congestive heart failure. The primary procedures were pneumonectomy, lobectomy, ligation and embolotherapy of arteriovenous malformations, and revascularization or reconstruction of the PA. The overall mortality rate was 7%.
Pulmonary hypertension is characterized by a progressive increase in pulmonary vascular resistance and has a poor prognosis. Current management includes supportive therapy and specific drugs that regulate endothelial function. Sildenafil citrate, a highly selective inhibitor of phosphodiesterase type 5, induces pulmonary vasodilation. Animal models and human studies have shown that sildenafil is an effective agent in treating pulmonary hypertension.25 The effect of sildenafil on the pulmonary vasculature appears to be independent of the underlying cause. Although the reported results in pulmonary hypertension are promising, sildenafil is unproven in treating congenital heart disease, and randomized controlled studies will be required to validate its safety, efficacy, and dosage.25
We believe that, depending on the species of animal, the ductus arteriosus and other collateral arteries connect systemic and pulmonary circuits and may close permanently or attain a capacity to close and reopen, depending on physiologic need. In our patient, the ductus arteriosus and systemic-to-pulmonary collateral vessels probably persisted in the right lung. The prognosis of patients with UAPA can depend largely on the coexisting cardiac anomaly (left-to-right shunt) and on the presence or absence of pulmonary hypertension. When pulmonary hypertension develops, UAPA has a worse prognosis, because PA aneurysmal dissection or rupture may occur. These complications are rare and are usually lethal.
In our patient, UAPA was present with a PDA and large aortopulmonary and coronary artery-to-PA collateral vessels. The PDA ligation in childhood probably delayed the development of his pulmonary hypertension. A patient with UAPA and a high-pressure pulmonary aneurysm can survive for a relatively long time, even without corrective surgery, and can experience prolonged periods of few symptoms. Although sildenafil had no favorable effects on our patient's high PA pressure and exercise tolerance, progressive clinical and hemodynamic deterioration did not detectably occur for months. Such a time of stability should be regarded as an opportunity for the physician to sustain the patient who is awaiting heart-lung transplantation.
Footnotes
Address for reprints: Gulumser Heper, MD, 32. sok 5/13, 06490 Bahcelievler, Ankara, Turkey. E-mail: heperg@hotmail.com
References
- 1.Pool PE, Vogel JH, Blount SG Jr. Congenital unilateral absence of a pulmonary artery. The importance of flow in pulmonary hypertension. Am J Cardiol 1962;10:706–32. [DOI] [PubMed]
- 2.Toews WH, Pappas G. Surgical management of absent right pulmonary artery with associated pulmonary hypertension. Chest 1983;84:497–9. [DOI] [PubMed]
- 3.Anderson RC, Char F, Adams P Jr. Proximal interruption of a pulmonary arch (absence of one pulmonary artery); case report and a new embryologic interpretation. Dis Chest 1958; 34:73–86. [DOI] [PubMed]
- 4.Kieffer SA, Amplatz K, Anderson RC, Lillehei CW. Proximal interruption of a pulmonary artery. Am J Roentgenol Radium Ther Nucl Med 1965;95:592–7. [DOI] [PubMed]
- 5.Werber J, Ramilo JL, London R, Harris VJ. Unilateral absence of a pulmonary artery. Chest 1983;84:729–32. [DOI] [PubMed]
- 6.Gluck MC, Moser KM. Pulmonary artery agenesis diagnosis with ventilation and perfusion scintiphotography. Circulation 1970;41:859–67. [DOI] [PubMed]
- 7.Borgeson E, Vogel JH. Congenital unilateral absence of the left pulmonary artery. Echocardiography in reverse. Chest 1980;77:106–7. [DOI] [PubMed]
- 8.Presbitero P, Bull C, Haworth SG, de Leval MR. Absent or occult pulmonary artery. Br Heart J 1984;52:178–85. [DOI] [PMC free article] [PubMed]
- 9.Wyman SM. Congenital absence of pulmonary artery: its demonstration by roentgenography. Radiology 1954;62:321–8. [DOI] [PubMed]
- 10.Cogswell TL, Singh S. Agenesis of the left pulmonary artery as a cause of hemoptysis. Angiology 1986;37(3 Pt 1):154–9. [DOI] [PubMed]
- 11.Grum CM, Yarnal JR, Cook SA, Cordasco EM, Tomashefski JF. Unilateral hyperlucent lung. Non-invasive diagnosis of pulmonary artery agenesis. Angiology 1981;32:194–207. [DOI] [PubMed]
- 12.Keane JF, Maltz D, Bernhard WF, Corwin RD, Nadas AS. Anomalous origin of one pulmonary artery from the ascending aorta. Diagnostic, physiological and surgical considerations. Circulation 1974;50:588–94. [DOI] [PubMed]
- 13.Penkoske PA, Castaneda AR, Fyler DC, Van Praagh R. Origin of pulmonary artery branch from ascending aorta. Primary surgical repair in infancy. J Thorac Cardiovasc Surg 1983; 85:537–45. [PubMed]
- 14.Kirklin JW, Barratt-Boyes BG. Origin of the right or left pulmonary artery from the ascending aorta. In: Cardiac surgery: morphology, diagnostic criteria, natural history, techniques, results, and indications. New York: Churchill Livingstone Inc.; 1986. p. 939–44.
- 15.Bouros D, Pare P, Panagou P, Tsintiris K, Siafakas N. The varied manifestation of pulmonary artery agenesis in adulthood. Chest 1995;108:670–6. [DOI] [PubMed]
- 16.Moser KM, Olson LK, Schlusselberg M, Daily PO, Dembitsky WP. Chronic thromboembolic occlusion in the adult can mimic pulmonary artery agenesis. Chest 1989;95:503–8. [DOI] [PubMed]
- 17.Wittram C, Maher MM, Yoo AJ, Kalra MK, Shepard JA, McLoud TC. CT angiography of pulmonary embolism: diagnostic criteria and causes of misdiagnosis. Radiographics 2004;24:1219–38. [DOI] [PubMed]
- 18.Freedom RM, Burrows PE, Smallhorn JF. Pulmonary atresia and intact ventricular septum. In: Freedom RM, Benson LN, Smallhorn JF, editors. Neonatal heart disease. London: Springer-Verlag; 1992. p. 285–307.
- 19.Ishizawa E, Horiuchi T, Tadokoro M, Suzuki Y, Yamaki S. Surgical management of pulmonary atresia, and critical pulmonary stenosis with intact ventricular septum. Tohoku J Exp Med 1986;150:135–44. [DOI] [PubMed]
- 20.Mildner RJ, Kiraly L, Sreeram N. Pulmonary atresia, “intact ventricular septum”, and aortopulmonary collateral arteries. Heart 1997;77:173–5. [DOI] [PMC free article] [PubMed]
- 21.DeRuiter MC, Gittenberger-de Groot AC, Poelmann RE, VanIperen L, Mentink MM. Development of the pharyngeal arch system related to the pulmonary and bronchial vessels in the avian embryo. With a concept on systemic-pulmonary collateral artery formation. Circulation 1993;87:1306–19. [DOI] [PubMed]
- 22.Fadel E, Wijtenburg E, Michel R, Mazoit JX, Bernatchez R, Decante B, et al. Regression of the systemic vasculature to the lung after removal of pulmonary artery obstruction. Am J Respir Crit Care Med 2006;173:345–9. [DOI] [PubMed]
- 23.de Ruiter MC, Gittenberger-de Groot AC, Rammos S, Poelmann RE. The special status of the pulmonary arch artery in the branchial arch system of the rat. Anat Embryol (Berl) 1989;179:319–25. [DOI] [PubMed]
- 24.Ten Harkel AD, Blom NA, Ottenkamp J. Isolated unilateral absence of a pulmonary artery: a case report and review of the literature. Chest 2002;122:1471–7. [DOI] [PubMed]
- 25.Leibovitch L, Matok I, Paret G. Therapeutic applications of sildenafil citrate in the management of paediatric pulmonary hypertension. Drugs 2007;67:57–73. [DOI] [PubMed]