

Abstract
The archipelago gene (ago) encodes the F-box specificity subunit of an SCF(skp-cullin-f box) ubiquitin ligase that inhibits cell proliferation in Drosophila melanogaster and suppresses tumorigenesis in mammals. ago limits mitotic activity by targeting cell cycle and cell growth proteins for ubiquitin-dependent degradation, but the diverse developmental roles of other F-box proteins suggests that it is likely to have additional protein targets. Here we show that ago is required for the post-mitotic shaping of the Drosophila embryonic tracheal system, and that it acts in this tissue by targeting the Trachealess (Trh) protein, a conserved bHLH-PAS transcription factor. ago restricts Trh levels in vivo and antagonizes transcription of the breathless FGF receptor, a known target of Trh in the tracheal system. At a molecular level, the Ago protein binds Trh and is required for proteasome-dependent elimination of Trh in response to expression of the Dysfusion protein. ago mutations that elevate Trh levels in vivo are defective in binding forms of Trh found in Dysfusion-positive cells. These data identify a novel function for the ago ubiquitin-ligase in tracheal morphogenesis via Trh and its target breathless, and suggest that ago has distinct functions in mitotic and post-mitotic cells that influence its role in development and disease.
Introduction
The morphogenesis of branched networks of cells with a single, fused lumen plays an important role in the development of many metazoan organs, including the vertebrate lungs, vasculature, kidneys, and mammary glands. The cellular architecture of these mammalian organs is quite similar to tubular structures in simpler metazoans, suggesting that the molecular and cellular mechanisms underlying the process of branching morphogenesis are conserved. In the fruit fly Drosophila melanogaster, tubular morphogenesis underlies formation of the tracheal system, a network of interconnected tubes that duct air throughout the developing organism. The complete embryonic tracheal network is composed of approximately 1600 polarized epithelial cells that originate in early embryogenesis as 20 ectodermal placodes distributed along either side of the embryo (Samakovlis et al., 1996). Each tracheal placode contains approximately 20 cells, which undergo two rounds of cell division, exit the cell cycle, and complete the subsequent stages of invagination and tracheogenesis without further cell division or cell death. Following invagination, primary branches bud from the tracheal sac and form a continuous lumen within each placode (Lubarsky and Krasnow, 2003). The pattern of placode branching is segmentally repeated and under fixed genetic control (Samakovlis et al., 1996). Budded branches extend toward their target tissues by a process of cell migration and cell extension, and subsequent fusion between adjacent tracheal metameres at later stages of embryogenesis produces a continuous, open tubular system.
Forward genetic screens for mutations that disrupt tracheal development have revealed a central role for fibroblast growth factor (FGF) signaling in promoting the post-mitotic cell migration and extension of the embryonic tracheal arbor (reviewed in Metzger and Krasnow, 1999). The central components of the Drosophila FGF pathway are encoded by the breathless (btl) (Klambt et al., 1992) and branchless (bnl) (Sutherland et al., 1996) genes, which encode an FGF-like receptor and an FGF-like secreted ligand respectively. The role of this receptor/ligand pair in controlling tracheal outgrowth is based upon a simple model in which the restricted expression of bnl in cells outside the tracheal placode represents a directional cue for the migration of btl-expressing cells within the tracheal placode. Initial induction of btl transcription within tracheal cells depends upon the trachealess (trh) gene, which encodes a basic helix-loop-helix Per/Arnt/Sim (bHLH-PAS) domain transcription factor (Isaac and Andrew, 1996; Kuo et al., 1996; Ohshiro and Saigo, 1997; Wilk et al., 1996) most closely homologous to the mammalian NPAS1 and NPAS3 proteins (Brunskill et al., 1999; Zhou et al., 1997). Mutation of any one of these core components - btl, bnl or trh - produces a failure of tracheal branching. Phenotypic analysis of these and other tracheogenesis genes has shown that Btl/FGF signaling is used to promote successive rounds of primary and secondary tracheal branching during embryonic Drosophila development (reviewed in Ghabrial et al., 2003). A similar role has been proposed for the FGF pathway in controlling the branching morphogenesis of tubes in the mammalian lung (Min et al., 1998).
In addition to a positive role in tracheal outgrowth, experimental evidence indicates that inappropriately timed Btl/FGF signaling can also impair tracheal development. First, in the course of normal development btl expression is not constant in tracheal cells, but oscillates: initially btl mRNA rises in all placode cells at stages 10–12 preceding primary branch formation, subsequently falls during late stage 12/early stage 13, and is re-initiated in a restricted set of cells that define sites of secondary branching at late stage 13/early stage14. Unlike early btl expression, this second wave is dependent upon a bnl-dependent feedback loop (Ohshiro et al., 2002; Ohshiro and Saigo, 1997). Second, ectopic activation of Btl, either by mutational inactivation of the btl inhibitor abnormal wing disc (awd) (Dammai et al., 2003), or by constitutive expression of bnl (Sutherland et al., 1996) or btl (this study and Lee et al., 1996), interferes with directed cell migration in the trachea. Similarly, expression of activated Ras or btl perturbs the ability of tracheal cells to form proper branching patterns in larval development (Cabernard and Affolter, 2005). These observations have led to the hypothesis that spatial and quantitative restriction of Btl activity is necessary to permit normal patterns of tracheal cell migration and fusion (Lee et al., 1996). While many genes that modulate Btl/FGF signaling have been identified, it is likely that other as yet unrecognized mechanisms are required to restrict Btl/FGF signaling to specific times and places in the developing organism.
Here we identify archipelago (ago), a gene known primarily for its role in cell proliferation control, as a component of the genetic circuitry that patterns the post-mitotic development of the embryonic tracheal system. ago encodes an F-box/WD-repeat protein that recruits target proteins to an SCF-type E3 Ub-ligase for subsequent poly-ubiquitination and proteolytic destruction. ago limits the division and growth of Drosophila eye epithelial cells by targeting the G1/S regulator Cyclin E and dMyc, the fruit fly ortholog of the human c-Myc proto-oncogene, for proteasome mediated-degradation (Moberg et al., 2001; Moberg et al., 2004). A highly conserved mammalian ago ortholog (variously termed Fbw7, Fbxw7, hAgo, hCDC4 or hSel-10) also targets Cyclin E and c-Myc and is a mutational target in a rapidly expanding array of human cancers (Balakrishnan et al., 2007; Bredel et al., 2005; Calhoun et al., 2003; Hagedorn et al., 2007; Malyukova et al., 2007; Mao et al., 2004; Maser et al., 2007; Minella et al., 2007; O’Neil et al., 2007; Rajagopalan et al., 2004; Thompson et al., 2007). Our current data indicate that ago is required for tracheal morphogenesis via a previously unrecognized target, the Trachealess (Trh) transcription factor. We find that ago mutant embryos contain excess Trh protein and ectopically express the btl gene, a known Trh target. Alleles of ago exhibit strong genetic interactions with trh and other known tracheogenesis genes, and the Ago protein is able to bind the Trh protein and regulate its proteasomal turnover via a mechanism that involves a third factor, the bHLH-PAS protein Dysfusion (Dys; (Jiang and Crews, 2003). Collectively, these data reveal a previously unappreciated developmental function for the ago tumor suppressor in the embryonic tracheal system, and identify the Trh transcription factor as a target of Ago in this process.
Materials and methods
Stocks, genetics and statistics
The ago alleles ago1 and ago3 have been previously described (Moberg et al., 2001). Unless indicated, analysis was performed on ago1/ago3 trans-heterozygotes. Full-length ago and agoΔF, a version of ago lacking the core F-box domain (Moberg et al., 2004), were cloned as PCR products into the EcoRI site of the pUAST vector (Brand and Perrimon, 1993) and used to generate UAS-ago and UAS-agoΔF stocks (D. Rennie, Massachusetts General Hospital Transgenic Drosophila Core). Other alleles used in this study were: btldev1, btlEY01638, trh10512, awdj2A4, Df(3L)Exel9000 and esg-lacZ (all from Bloomington Drosophila Stock Center), 1-eve-1 (Perrimon et al., 1991), btl-Gal4 (Shiga et al., 1996), UAS-trh (Jin et al., 2001), FRT80B, UAS-CycE, UAS-dMyc. Crosses involving the temperature-sensitive UAS-Pros261 and UAS-Prosβ21 transgenes (gift of J. Belote) were performed at 21°C. Embryos were genotyped using the TM6B, P{iab-2(1.7)lacZ}6B, Tb1 and CyO, P{elav-lacZ.H}YH2 ‘blue’ balancers, or the TM3, P{Gal4-twi.G}2.3, P{UAS-2xEGFP}AH2.3, Sb1Ser1 balancer. Statistical comparisons were made using Student’s t-Test.
Embryo immunohistochemistry and antibodies
Embryos were staged and fixed in 37% formaldehyde-saturated heptane, devitellinized in methanol and stored in ethanol at 4°C. These samples were rehydrated & washed in PBS with .05% Triton-X 100 (PBSTx), blocked in 5% milk powder/5% NGS in PBSTx, and incubated with the following primary antibodies: mouse anti-Tango (1:5, Developmental Studies Hybridoma Bank; DSHB), rat anti-Trh (1:200) a gift of D. Andrew (Henderson et al., 1999), mouse mAb2A12 (1:5; DSHB), rabbit anti-β-Gal (1:300; Cappel), guinea pig anti-full length Ago (1:2500; Pocono Rabbit Farm & Laboratory), rat anti-Dys (1:200) and rabbit anti-Dys (1:800) both a gift of S. Crews (Jiang and Crews, 2003). Secondary antibodies conjugated to HRP, AP, Cy3 and Cy5 were used as recommended (Jackson ImmunoResearch). Embryos from w1118 and ago1/TM3, P{Gal4-twi.G}2.3, P{UAS-2xEGFP}AH2.3, Sb1Ser1 strains were collected at stages 13/14 and sorted by absence of GFP fluorescence. Extracts were prepared in sample buffer containing DTT and resolved on 7.5% SDS-PAGE prior to Western blotting with rat anti-Trh (1:2000), or anti-β-tubulin (1:2000; Santa Cruz Biotechnology). Anti-HA and anti-Flag antibodies (Sigma) were used according to manufacturer’s instructions.
RNA analysis
RNA in situ hybridization was performed as described (Tautz, 2000). Briefly, embryos were fixed in 1.5% formaldehyde (in 0.1 M Hepes, pH 6.9, 2 mM MgSO4, 1 mM EGTA)-saturated heptane, devitellinized and stored in methanol at −20°C. Embryos were washed in PBS with 0.1% Tween 20, treated with 15ug/ml Proteinase K for 2 minutes and post-fixed with 4% paraformaldehyde. Riboprobe hybridization and subsequent immunohistochemistry were performed as described (Tautz, 2000). Sense and anti-sense digoxigenin (DIG)-labeled riboprobes were synthesized from a 1 kb PCR fragment amplified from btl cDNA and visualized by the anti-DIG-AP antibody (1:2000; Roche).
Plasmids and molecular biology
pMT-HA-ago expression plasmids have been described previously (Moberg et al., 2004). pMT-Flag-trh was generated by cloning the full-length trh ORF (Open Biosystems) into the pMT vector as an N-terminally Flag-tagged PCR product. The pACT-HA-dys plasmid (gift of S. Crews) contains an N-terminally tagged version of the dys ORF under the control of a constitutively active fragment of the actin promoter. Transfected S2 cells were induced by the addition of 0.5mM CuSO4 for 6hrs prior lysis in buffer containing 0.5M KCl, 0.1% NP-40, 35% glycerol, 10mM HEPES pH 7.0, 5mM MgCl2, 0.5mM EDTA pH 8.0, 25mM NaF, 1mM Na2VO4, 1mM DTT supplemented with protease inhibitors (Complete™ Protease Inhibitor Cocktail; Roche). Where indicated, 50uM MG132 (Calbiochem) was added 6 hrs prior to harvesting cells. Lysates were analyzed directly by immunoblot (IB), or diluted in immunoprecipitation (IP) buffer and subject to IP/IB analysis as described previously (Moberg et al., 2004).
Results
ago has role in embryonic tracheal development
Analysis of the growth restrictive role of ago in proliferating larval imaginal disc cells has provided an excellent model to understand the tumor suppressive properties of ago mammalian orthologs. However, the Ago protein is widely expressed in the early embryo (Figure 1) and mutations that disrupt it lead to embryonic death (Moberg et al., 2001). Intercrossing the ago1 or ago 3alleles (respectively encoding a C-terminal truncation in the WD-repeat/substrate-binding domain and a G1131E missense mutation in the 4th WD repeat; (Moberg et al., 2001) produces trans-heterozygous mutant embryos that develop through all embryonic stages but fail to hatch as L1 larvae (data not shown), suggesting lethality occurs during late embryogenesis. The severity of this and other phenotypes (see below) in homozygous or heteroallelic mutant animals is indistinguishable from those carrying either allele in trans to a chromosomal deficiency that removes the ago locus (Table 1; Df(3L)Exel9000), indicating that ago1 and ago3 are either null or strong loss-of-function alleles. Considered together, these observations indicate that ago may have as yet unappreciated roles in early development.
Figure 1. Ago is widely expressed during embryogenesis.
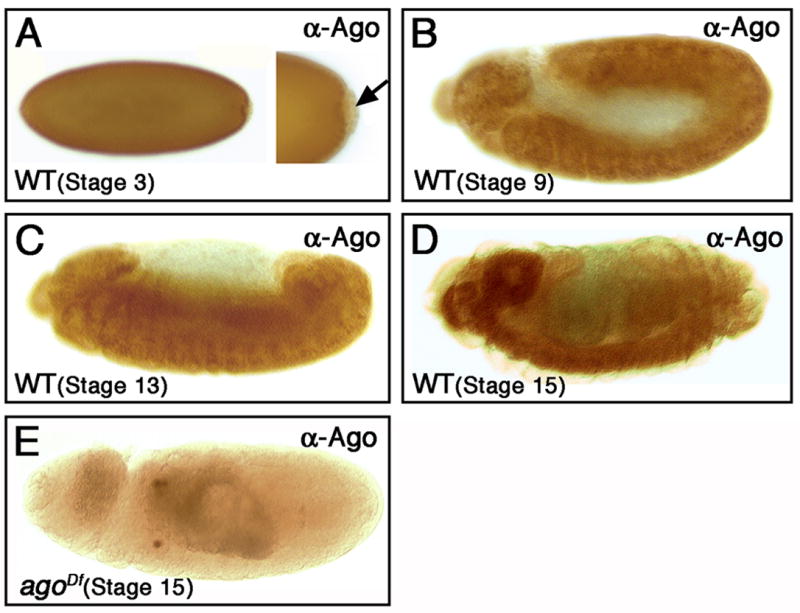
Lateral views of FRT80B control embryos stained with Ago-specific antiserum. Ago expression is detected uniformly during the cellularization (A), and germ-band extended (B) and retracted (C) stages, but is not detectable in pole cells (arrow in A). Ago is concentrated in the CNS during late embryogenesis (D). In this and all further figures, embryos are shown anterior left and dorsal up (unless indicated). Anti-Ago staining in an embryo homozygous for the Df(3L)Exel9000 deletion, which removes the ago gene (E).
Table 1. ago1 and ago3 behave as null alleles in tracheogenesis.
Percent penetrance of the dorsal trunk ‘break’ phenotype in embryos of the indicated genotypes.
| Genotype | DT Breaks (%) | n= |
|---|---|---|
| FRT80B | 3** | 61 |
| ago1/ago3 | 46 | 85 |
| ago1/Exel9000 | 39 | 57 |
| ago3/Exel9000 | 38 | 42 |
p < 0.01 compared to ago1/3.
The earliest morphological defects in ago zygotic mutant embryos are observed in the developing trachea visualized with the anti-lumen antibody mAb2A12 (Figure 2C–F). This analysis reveals two major defects in tracheal branching patterns of ago embryos. The first of these is interruptions, or ‘breaks’, in the continuity of the tracheal lumen (Figure 2C–E). The breaks occur throughout the tracheal system and are prominent in the dorsal trunk (DT; Figure 2C,D, arrowheads), the lateral truck (LT; Figure 2C, arrows), and between dorsal branches of opposing tracheal placodes (DB; Figure 2E, arrow). What appears to be a misrouting phenotype is also observed in the dorsal branches (Figure 2E, arrowhead) and ganglionic branches (data not shown). Approximately 70% of all ago mutant embryos show combinations of these break or misrouting effects. The second prominent tracheal phenotype (occurring in ~25% of all ago mutant embryos) is excess lumen convolution throughout the primary and secondary branches (Figure 2F). This combined spectrum of breaks, apparent misrouting, and convolution indicates that ago is required for normal development of the tracheal system. Moreover, the absence of any accompanying defects in the major morphogenetic events of embryogenesis (NTM and KHM, unpub.) suggests that these phenotypes reflect a requirement for ago in the trachea, rather than as a secondary effect of a more general developmental role outside this tissue.
Figure 2. ago is required for tracheogenesis.
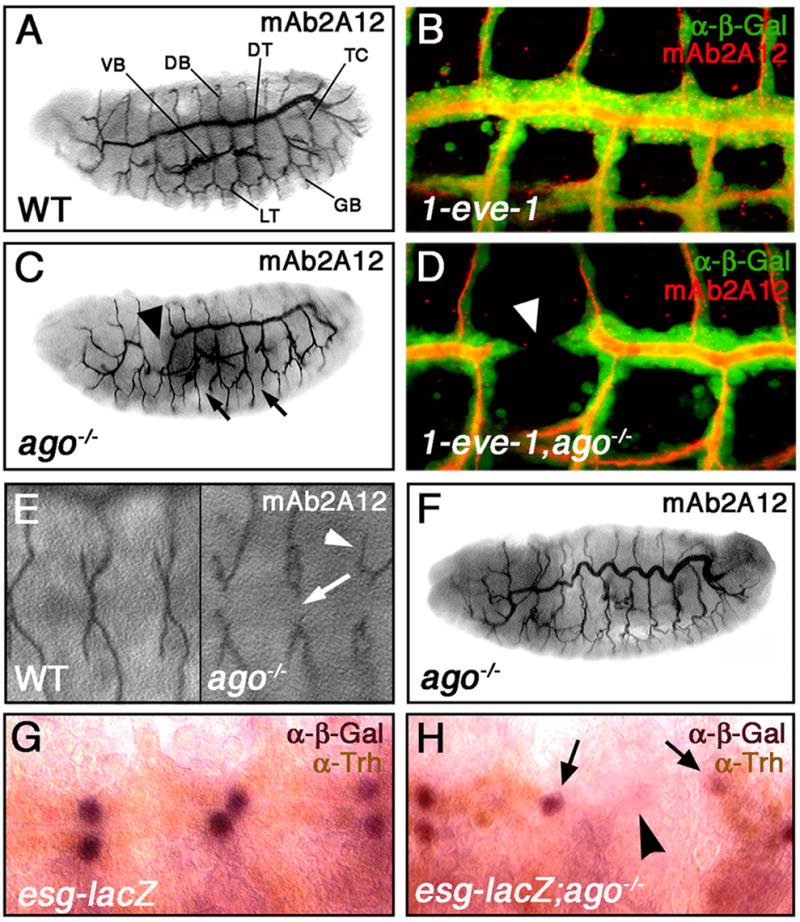
Lateral views of FRT80B (A) or ago embryos (C,F) stained with mAb2A12 to visualize the tracheal. Major tracheal branches are indicated in (A): dorsal trunk (DT), lateral trunk (LT), transverse connectives (TC), dorsal branches (DB), ganglionic branches (GB) and visceral branches (VB). ago mutants display interruptions in the lumen of the DT (arrowhead in C) and LT (arrows in C). 1-eve-1 (B) or 1-eve-1, ago (D) embryos stained with anti-β-Gal (green) to mark tracheal cells and mAb2A12 to mark the lumen (red). The DT lumen interruptions in ago mutant embryos correspond to physical breaks in the DT (arrowhead in D). Dorsal views of FRT80B (E, left panel) and ago (E, right panel) embryos stained with mAb2A12. ago mutant embryos show defects in DB fusion (arrow) as well as DB misrouting (arrowhead). The DT lumens in ago mutant embryos display a convolution defect (F). Staining of esg-lacZ (G) and esg-lacZ;ago (H) embryos with anti-Trh (brown) and anti-β-Gal (purple). Arrows in H mark fusion cells in adjacent placodes. Arrowhead indicates a break in the dorsal trunk. Panels B and D are reconstructions of serial sections.
The DT break phenotype occurs in almost half of all ago mutant embryos (46%, n=85) with an average of 1.2±0.09 breaks among those that show the phenotype (n=39). On a per fusion event basis, this corresponds to a greater than 15-fold increase in the rate of defective interplacode fusion events in ago mutants compared to control embryos (Supplemental Figure 1). Due to the prevalence of this phenotype, DT morphogenesis was selected as a system in which to characterize the role of ago in the developing tracheal system. To investigate the cellular architecture of DT breaks in ago mutant embryos, the 1-eve-1 enhancer trap line, which carries a lacZ insertion in the trh gene and expresses β-galactosidase (β-gal) in every tracheal cell (Perrimon et al., 1991; Wilk et al., 1996), was placed in the wild type and ago mutant backgrounds (Figure 2B,D). Anti-β-gal staining of these embryos reveals that lumenal gaps detected with the mAb2A12 antibody reflect physical gaps between cells of adjacent tracheal placodes (Figure 2D, arrowhead). Interplacode fusion along the DT is normally dependent upon specialized ‘fusion cells’, which are specified 2 per placode by a Notch-dependent process (Ikeya and Hayashi, 1999). At the appropriate stage, one migrates anteriorly and the other posteriorly to meet with fusion cells coming from adjacent placodes. Thus, the gaps between adjacent DT placodes in ago mutant embryos suggest either that ago is necessary for proper fusion cell specification, or that ago mutations impair fusion cell migration and/or fusion. To test these hypotheses, DT fusion cells were visualized in wild type and ago mutant stage 15 embryos (Figure 2G,H) using the fusion cell-specific enhancer trap escargot (esg)-lacZ (Tanaka-Matakatsu et al., 1996). Analysis of wild type embryos in which the process of DT fusion is complete shows esg-lacZ expression (purple nuclei; Figure 2G) in a pair of adjacent fusion cell nuclei. In ago mutant embryos, pairs of esg-lacZ positive fusion cells flank regions of DT breaks (Figure 2H; arrows mark fusion cell nuclei; arrowhead marks gap in DT). Analysis of a second fusion cell marker, the Dysfusion protein (Jiang and Crews, 2003), gives similar results (data not shown). Thus ago DT breaks are not associated with altered fusion cell numbers but rather a failure of ago mutant DT fusion cells to fuse with adjacent placodes. This differs significantly from tracheal phenotypes elicited by btlGal4-driven overexpression the putative Ago target Notch (Fryer et al., 2004; Gupta-Rossi et al., 2001; Oberg et al., 2001; Tsunematsu et al., 2004; Wu et al., 2001), which results in a complete loss of fusion cells (Ikeya and Hayashi, 1999). The lack of an effect on fusion cell number also contrasts with ago proliferative phenotypes in imaginal discs (Moberg et al., 2001; Moberg et al., 2004). To further confirm this, we measured total cell number in wild type and ago mutant DTs using an anti-Tango antiserum (Sonnenfeld et al., 1997). This analysis shows that these genotypes have approximately equal numbers of nuclei per DT segment (wt=16.7±0.4, n=6; ago=17.0±0.7, n=9). Thus the primary effect of ago loss in the DT is not on cell number, but rather on the ability of cells to follow the normal developmental program of cell migration and fusion.
Because Ago protein is expressed uniformly in tracheal and non-tracheal cells (Supplemental Figure 2), the requirement for ago in the DT could be indicative of a cell-autonomous role for ago in tracheal cells or it might reflect a non-cell autonomous role for ago outside the developing placode in, for example, the mesodermal ‘bridge cells’ necessary for DT fusion (Wolf and Schuh, 2000). To determine the site of ago action, a wild type UAS-ago transgene and a dominant-negative UAS-agoΔF transgene were expressed specifically in the developing tracheal system using the btl-Gal4 driver (Shiga et al., 1996). btl-Gal4 driven expression of wildtype ago in an ago1/ago3 mutant background significantly reduces the frequency of DT breaks (Figure 3E; compare yellow and blue bars) and the other observed tracheal phenotypes in ago mutant embryos (data not shown). Moreover, btl-Gal4 driven expression of the agoΔF dominant-negative transgene in otherwise wild type tracheal cells is sufficient to produce DT breaks at similar penetrance as zygotic deficiency for ago (Table 2 and Figure 3A,D). The agoΔF allele carries an internal deletion of the core F-box domain (Moberg et al., 2004), and is defective in ubiquitin-dependent protein degradation. While these data do not rule out a secondary role for ago in non-tracheal cells, they do argue that ago plays a required role in a protein degradation pathway that is active in embryonic tracheal cells.
Figure 3. ago acts within tracheal cells and interacts with Btl/FGF signaling components.

mAb2A12 staining of stage 15 embryos (A–C). btl-Gal4 driven expression of agoΔF (A), btl (B) or trh (C) phenocopies the ago DT break phenotype (arrowheads). (D,E) Quantitative summary of the DT break phenotype in stage 15 embryos of the indicated genotypes. (D) btl-Gal4 driven expression of agoΔF (yellow), btl (red) or trh (green) phenocopies ago mutations (blue). (E) The ago mutant phenotype is suppressed by expression of wild type ago in the btl-Gal4 domain (yellow), or by reducing the genetic dosage of either btl (red) or trh (green). * p<0.05 and ** p<0.01 compared to FRT80B (D) or ago (E).
Table 2. ago is phenocopied by ectopic expression of FGF pathway members, but not the Ago targets Cyclin E and dMyc.
Percent penetrance of the dorsal trunk ‘break’ phenotype in embryos of the indicated genotypes.
| Genotype | DT Breaks (%) | n= |
|---|---|---|
| FRT80B | 3** | 61 |
| ago1/ago3 | 46 | 85 |
| btl-Gal4:UAS-agoΔF | 39 | 56 |
| btl-Gal4:UAS-trh | 28 | 56 |
| btl-Gal4:btlEY01638 | 31 | 53 |
| btl-Gal4:UAS-CycE | 18** | 64 |
| btl-Gal4:UAS-dMyc | 3** | 66 |
p < 0.01 compared to ago1/3.
ago acts upstream of trachealess and breathless
Because loss of ago leads to excess activity of proteins normally regulated by Ago-dependent degradation (Moberg et al., 2001; Moberg et al., 2004), known Ago target proteins were tested for their ability to reproduce ago mutant tracheal phenotypes when expressed in the btl-Gal4 domain. Expression of cycE and dMyc, two targets of Ago in imaginal disc cells, do not reproduce the ago mutant tracheal phenotype (Table 2), suggesting that ago controls tracheal morphogenesis via a novel target. Significantly, btl-Gal4 driven expression of either trh or its downstream target btl produces DT breaks of similar penetrance and expressivity as ago mutations (Figure 3B–D), and also leads to lumen convolution (data not shown). Moreover, loss-of-function alleles of trh (trh10512; (Isaac and Andrew, 1996) or btl (btldev1; (Kennison and Tamkun, 1988) are each able to dominantly suppress ago tracheal phenotypes (Figure 3E, green and red bars respectively). The sensitivity of ago phenotypes to the genetic dosage of trh and btl suggests that ago may inhibit Trh in the developing trachea. To test the effect ago mutations on Trh protein levels, wild type and ago mutant embryos were stained with a Trh-specific antiserum (Figure 4). Compared to control embryos, ago mutant embryos contain significantly higher levels of Trh in all tracheal cells (Figure 4A,B). This effect is also evident in immunoblot blot analysis of Trh levels in ago and control embryos (Figure 4C). Trh protein is normally detected in the nuclei of all tracheal cells, but is specifically eliminated from fusion cells, including those of the dorsal trunk, by a mechanism that requires fusion-cell specific expression of the Dysfusion (Dys) bHLH-PAS domain transcription factor (Jiang and Crews, 2003). In addition to limiting the steady-state levels of Trh in all tracheal cells, ago is also required for the Dys-stimulated elimination of Trh that occurs specifically in fusion cells (Figure 4D, E). Expression of Dys occurs normally in ago embryos (see middle panel, Figure 4E), but in these same cells Trh levels do not decline, and DT fusion cells with high levels of both Dys and Trh are readily observed (Figure 4E, arrows). Thus, the antagonistic genetic interaction between ago and trh in the tracheal system appears to have its basis in a requirement for ago in limiting the steady-state levels of Trh in all tracheal cells, and in the specific elimination of Trh from fusion cells.
Figure 4. ago regulates Trh levels and is required for Trh elimination in tracheal fusion cells.

Anti-Trh staining in ago/TM6B, P{iab-2(1.7)lacZ}6B, Tb1 (A) and ago1/ago3 (B) embryos. (C) Anti-Trh (top) and anti-β-tub (bottom; loading control) Western blot analysis of stage 13/14 control and ago mutant embryos (10 embryos/lane). FRT80B (D) and ago (E) embryos stained with anti-Trh (green, right panel) and anti-Dys (red, center panel) to mark fusion cells. Trh levels are reduced in Dys-positive wild type fusion cells (arrows in D), but not in Dys-positive ago mutant fusion cells (arrows in E).
The effect of ago alleles on Trh levels, and the genetic interaction between ago and the Trh target gene btl, indicates that ectopic Trh-driven transcription of btl may contribute to the ago phenotype. The developmental regulation of the btl promoter is complex and involves inputs from a number of factors other than Trh, including the transcriptional activators pointed (Klambt, 1993; Ohshiro et al., 2002) and ventral veinless (Anderson et al., 1996), and the transcriptional repressors anterior open (Ohshiro et al., 2002) and spalt (Kuhnlein and Schuh, 1996). To test whether the failure to down-regulate Trh in ago mutant cells is sufficient to cause mis-expression of btl, RNA in situ hybridization analysis was performed using a btl-specific anti-sense RNA probe. In stage 15 control embryos, levels of btl mRNA expression in the DT do not rise above the background signal obtained with btl anti-sense (Figure 5A) or sense (data not shown) probes. In contrast, pairs of btl-positive cells are evident along the DT of all ago mutant embryos (Figure 5B; see arrows). The location and pattern of this ectopic btl expression suggest that these paired cells correspond to fusion cells, and that failure to eliminate Trh in these cells leads to ectopic btl transcription. The lack of a similar effect in the remaining ago mutant tracheal cells suggests that excess Trh that must collaborate with fusion-cell specific factors in order to drive ectopic btl transcription.
Figure 5. btl transcription is deregulated in ago mutant embryos.

RNA in situ hybridization of FRT80B (A) and ago (B) stage 16 embryos with btl anti-sense probe. Arrows in B denote btl-expressing cells. (C) Lateral view of a stage 15 ago;awd−/+ embryo stained with mAb2A12 showing enhanced expressivity of the ago mutant phenotype. (D) Quantitative summary of the DT break phenotype in stage 15 embryos of the indicated genotypes showing interactions between ago and awd.
In view of the requirement for btl in the ago phenotype (see Table 3 and Figure 3B) and the finding that excess Btl activity disrupts DT architecture (see Figure 3B and Dammai et al., 2003; Lee et al., 1996; Ohshiro et al., 2002), the effect of ago on btl suggests that Trh-driven transcriptional deregulation of btl is an important element of ago DT phenotype. However, while the molecular effect of ago inactivation on Trh and btl is fully penetrant, the resultant morphological defects in tracheal structure are not. This might be expected if the elevated levels of Trh and btl reach a threshold that is sufficient to perturb tracheal development in a portion of embryos, but that other pathways acting redundantly to ago are able to enforce the developmental down-regulation of the Btl pathway. If so, then ago mutations might be predicted to sensitize the tracheal system to other mutations that increase Btl activity by non-transcriptional mechanisms. To test this, a loss-of-function allele of the abnormal wing discs gene (awdj2A4; (Spradling et al., 1999), which encodes a factor that promotes the endocytic down-regulation of the Btl receptor in embryonic tracheal cells (Dammai et al., 2003), was assayed for its ability to enhance the ago tracheal phenotype (Figure 5C,D). Reducing awd gene dosage by half in ago mutant animals strongly enhances both the penetrance (from 46% of embryos to greater than 80% of embryos; see Figure 5D, Table 4, and Supplemental Figure 1) of the DT break phenotype and its expressivity among affected embryos (from 1.2±0.09 breaks/embryo, n=39 to 6.1±0.95 breaks/embryo, n=41). These ago/ago,awd/+ embryos also show a much more severe disruption of the entire tracheal system than do ago mutants alone (compare Figure 5C to 2C). Moreover, a significant fraction of ago/+,awd/+ trans-heterozygous embryos show DT breaks. In view of the data indicating that ago and awd are both upstream of btl via transcriptional and endocytic mechanisms respectively, these strong synthetic effects suggest that ago and awd mutations collaborate to deregulate Btl in the developing tracheal system.
Table 3. The ago phenotype is dominantly supressed by alleles of FGF pathway members, and is rescued by tracheal cell specific expression of Ago.
Percent penetrance of the dorsal trunk ‘break’ phenotype in embryos of the indicated genotypes.
| Genotype | DT Breaks (%) | n= |
|---|---|---|
| FRT80B | 3** | 61 |
| ago1/ago3 | 46 | 85 |
| ago1/ago3;btl-Gal4:UAS-ago | 15** | 73 |
| ago1/ago3;trh10512/+ | 31* | 135 |
| ago1/ago3;btldev1/+ | 26** | 99 |
p < 0.05 and
p < 0.01 compared to ago1/3
Table 4. ago dominantly interacts with an allele of the Btl antagonist awd.
Penetrance of indicated phenotypes in embryos of the indicated genotypes.
| Genotype | DT Breaks (%) | N= |
|---|---|---|
| FRT80B | 3 | 61 |
| ago3/+ | 7 | 56 |
| awdj2A4/+ | 17* | 42 |
| ago3,awdj2A4/+ | 30** | 33 |
| ago3,awdj2A4/ago1 | 83** | 52 |
p < 0.05 and
p < 0.01 compared to FRT80B.
Ago binds Trh and restricts Trh levels in cells
The ability of ago mutations to elevate Trh levels in vivo and to uncouple Trh levels from the rise in Dys indicates that Ago acts downstream of Dys as part of the mechanism that eliminates Trh protein in fusion cells. To examine interactions between Ago and Trh more closely, epitope tagged versions of these proteins were co-expressed in S2 cells (Figure 6A,B). Analysis shows that while Trh accumulates to high levels when expressed in S2 cells, co-expression of Ago results in a subtle but reproducible reduction (~20% by density quantitation; Photoshop) in Trh levels (Figure 6A, top panel, lanes 1–2). This effect appears to parallel the negative effect ago has on Trh levels in non-fusion cells that do not normally express Dys (see Figure 4B). However, when Ago and Trh are co-expressed with Dys, the in vivo trigger of Trh down-regulation, Trh levels drop dramatically (Figure 6A, top panel, lane 3) in much the same way Trh levels drop in response to Dys expression in vivo (see Figure 4D and in (Jiang and Crews, 2003)). The parallel decline in the level of Ago is consistent with findings that other F-box proteins are destabilized by reductions in the level of their substrates (Li et al., 2004) and mimics the relationship between dMyc and Ago stability in S2 cells (Moberg et al., 2004). Examination of the effect of Dys on Trh reveals that expression of dys is sufficient to decrease Trh levels in the absence of exogenous Ago (Figure 6B, lane3). However, as is observed in embryonic tracheal cells (Figure 4E), this effect of Dys in S2 cells still requires endogenous Ago since RNAi-knockdown of ago is sufficient to block it (Figure 6B, lane 4). Addition of the proteasome inhibitor MG132 also blocks the Ago/Dys-stimulated decline in Trh levels in S2 cells (Figure 6B, lane 5). In parallel to these in vitro effects, btl-Gal4 mediated expression of dominant-negative alleles of the proteasome subunits Pros26 and Prosβ2 (Belote and Fortier, 2002) is sufficient to phenocopy ago alleles and block down-regulation of Trh in embryonic fusion cells (Figure 6C). From these data, it appears that Trh is a target of a proteasomal degradation pathway in embryonic tracheal cells and in cultured S2 cells, and that in both cases the mechanism underlying this effect requires ago and can be greatly potentiated by co-expression of dys.
Figure 6. Ago regulates Trh levels in S2 cells and during embryogenesis.
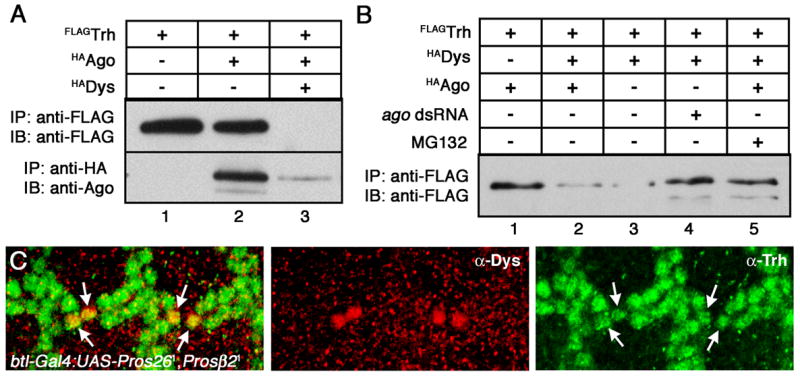
Quantification of the steady-state levels of either Trh and Ago by immunoprecipitation-immunoblot (IP/IB) from S2 cells transfected with the indicated plasmids. (A) Trh accumulates to detectable levels in S2 cells (lane 1), are slightly reduced in the presence of Ago (lane 2), and are greatly reduced after co-expression of Ago and Dys (lane 3). (B) The Dys-mediated reduction in Trh levels occurs in the absence of transfected Ago (lane 3) but is blocked by ago dsRNA treatment (lane 4) or by blocking proteasome function with MG-132 (lane 5). (C) Inhibition of proteasome function also stabilizes Trh levels in vivo. btl-Gal4:UAS-Pros261,UAS-Prosβ21 embryos stained with anti-Trh (green, right panel) and anti-Dys (red, center panel) show a failure to down-regulate Trh levels in fusion cells (arrows).
The genetic and functional interactions between ago and trh suggest that their encoded products may physically interact in cells. To test this hypothesis, Flag-tagged Trh and HA-tagged versions of either wild type Ago, the AgoΔF dominant negative protein, or the truncated Ago1 protein (lacking an intact WD domain) were expressed in S2 cells and analyzed by co-immunoprecipiation (Figure 7). Wild type Ago, Ago1, and AgoΔF are all recovered in anti-Flag immunoprecipitates (Figure 7A, lanes 2–4) and reciprocally, Flag-Trh is readily detected in anti-HA immunoprecipitates from cells expressing HA-AgoΔF (Figure 7B). Thus, in the absence of exogenous Dys, Trh is able to interact with all three forms of Ago in S2 cells, including the Ago1 WD-truncation mutant that is defective in Trh regulation in vivo. Co-expression of Dys with these combinations of Ago and Trh, and simultaneously blocking proteasome function so as to stabilize the Ago-Trh complex, induces a significant change the nature of the Ago-Trh interaction (Figure 7C). The form of Trh protein that accumulates in MG132-treated, Dys-expressing cells remains competent to bind wild type Ago but fails to interact with the WD-truncation mutant Ago1 (Figure 7C, top panel, compare lanes 3 and 5). Thus, an ago mutation that deregulates Trh levels in vivo is defective in binding to the form of Trh normally targeted for proteasome-dependent elimination in Dys-expressing fusion cells. Considered together, these data show that Ago and Trh can bind specifically to one another, and that this interaction is required to limit Trh levels in vivo.
Figure 7. Physical interaction between Ago and Trh.

IP/IB analysis from S2 cells expressing the indicated cDNAs shows that wild type Ago, AgoΔF, and Ago1 are recovered in anti-Flag precipitates (A) and that Trh is reciprocally recovered in anti-HA precipitates from cells expressing AgoΔF (B). IP/IB analysis of MG132-treated cells expressing the indicated cDNAs (C) shows that Trh is bound equally well by Ago (top panel, lane 2) and Ago1 (top panel, lane 4) in the absence of Dys, but that in Dys-positive cells Trh is bound only by wild type Ago (top panel, lane 3) but not the Ago1 mutant (top panel, lane 5) that is defective in Trh regulation in vivo. Loading controls for Ago (middle panel) and Trh (bottom panel) are shown.
Discussion
The biological properties of individual F-box proteins are to a large degree determined by their repertoire of target proteins. In the case of the Drosophila Ago F-box protein, failure to degrade these targets promotes excess proliferation of imaginal disc cells. This observation has led to the identification of Cyclin E and Myc proteins as ago targets (Moberg et al., 2001; Moberg et al., 2004). However the broad pattern of Ago expression in the embryo suggests that it might regulate distinct processes and targets in other cell types. In view of the rapidly growing body of work showing that inactivation of human ago/Fbw7 is a common event in a variety of cancers (e.g. (Malyukova et al., 2007; O’Neil et al., 2007; Thompson et al., 2007), identification of these targets may provide important insight into the biology of cancers lacking ago function.
Here we show that Drosophila Ago is required for the post-mitotic morphogenesis of the embryonic tracheal system and that this requirement is due, at least in part, to the ability of Ago to bind directly to a previously unrecognized target, the Trh transcription factor, and stimulate its proteasomal degradation. This ago degradation mechanism appears to fulfill different regulatory roles in different populations of tracheal cells. In non-fusion tracheal cells, ago is required to limit overall levels of Trh, which is normally expressed at moderate levels throughout the tracheal system. In tracheal fusion cells the ago degradation mechanism appears to be strongly potentiated by an unidentified signal generated by Dys, such that Trh is completely eliminated from Dys-expressing cells. At a genetic level, the dependence of ago tracheal phenotypes on trh gene dosage argues that that elevated Trh levels are primarily responsible for branching defects that occur in ago zygotic mutant embryos. In support of this, persistent Trh expression is also observed in ago mutant fusion cells in other tracheal branches (NTM and KHM, unpublished observation). This novel role for Ago in tracheal development is supported by the independent finding that homozygosity for a genomic deletion containing the ago locus is associated with cell migration defects in embryonic tracheal metameres (Myat et al., 2005).
Many important developmental events are controlled by multiple mechanisms that collaborate to regulate a key step in the process. This somewhat redundant control insulates the process from defects in any single pathway, such that major defects only occur when multiple control mechanisms are blocked. The observation that the effect of ago mutations on Trh and btl levels is completely penetrant, but the resulting morphological defects are not, suggests that another pathway acts redundantly to ago to control tracheal development. The strong, dominant enhancement of the ago phenotype by a mutation in the awd gene fits very well into a model in which multiple pathways are responsible for the precisely timed down-regulation of the Breathless/FGF pathway: ago attenuates btl transcription by degrading Trh, awd lowers levels of Btl protein on the cell surface by promoting its endocytic internalization (Dammai et al., 2003), and other pathways act independently to control expression of the FGF ligand branchless in non-tracheal cells (Merabet et al., 2005; Sutherland et al., 1996). Thus, the incomplete penetrance of the ago phenotype is not indicative of an insignificant role for the gene in tracheal development, but rather may indicate that the tracheal system uses multiple mechanisms to redundantly control a key step in its development.
The Ago WD repeat region binds Cyclin E and dMyc, and the current work demonstrates that it also binds Trh. Broadly, the Ago-Trh interaction is quite similar to interactions with Cyclin E and dMyc: it is required for the down-regulation of substrate levels in vivo, and its disruption elevates levels of substrate that then drive downstream phenotypes. For substrates like Myc, site-specific phosphorylation generates a motif that binds to the Ago WD-region and stimulates rapid, SCF-mediated protein turnover of the target protein (reviewed in Minella and Clurman, 2005). In contrast, the data here suggest that Trh can physically interact with Ago in two distinct configurations: one that does not require an intact WD-domain and a second WD-dependent mode of binding. The observation that the Ago1 allele can participate in the first complex but not the second and is defective in Trh regulation in vivo, suggests that like other Ago targets, WD-dependent binding is associated with rapid Trh turnover. Expression of Dys appears to shift the balance in favor of this second mode of binding. Combined with the genetic and phenotypic data implicating ago as an in vivo regulator of Trh activity, these molecular data support a model in which Ago can bind to Trh in the absence of Dys and inefficiently stimulate Trh turnover by a WD-dependent mechanism. This inefficient mechanism may be responsible for the fairly mild increase in Trh levels observed in all ago mutant dorsal trunk cells. However, in the presence of Dys, the efficiency of Trh turnover in DT fusion cells is enhanced to the degree that the entire pool of Trh is rapidly eliminated. Interestingly, the correlate of this hypothesis - that ectopic expression of dys in non-fusion cells should be sufficient to trigger down-regulation of endogenous Trh -was confirmed in a recent study (Jiang and Crews, 2006).
The nature of the Dys-generated signal responsible for this effect is not currently known. Precedent with other Ago targets suggests that it may involve Trh phosphorylation (Fryer et al., 2004; Koepp et al., 2001; Nateri et al., 2004; Sundqvist et al., 2005; Wei et al., 2005; Welcker et al., 2004; Welcker et al., 2003; Yada et al., 2004; Ye et al., 2004). Recent work on the mammalian ago ortholog Fbw7 has shown that interactions with substrates can also be modulated by interaction with accessory factors (Punga et al., 2006), or by conformational changes in the substrate driven by the isomerization of proline residues within the Ago/Fbw7 binding motif (van Drogen et al., 2006). Proline isomerization has been implicated in the degradation of mammalian c-Myc (Yeh et al., 2004), but such mechanisms are not currently known to play a role in the degradation of either Myc or bHLH-PAS proteins in Drosophila. An important goal of future studies will be to determine if any of these types of mechanisms are involved in Dys-induced Trh degradation in tracheal cells.
The requirement for ago in tracheal cells suggests that the consequences of ago loss vary considerably depending on the proliferative state of the cells, their location within the organism, and their developmental stage. ago mutant clones in the mitotically active larval eye disc show no evidence of excessive Trh levels or deregulated Btl/FGF signaling (NTM and KHM, unpub.) and conversely, ago zygotic mutant trachea do not display ‘extra cell’ defects similar to those observed in the eye. The origins of this tissue specificity are currently not clear, although it might simply reflect the differential expression patterns of Ago targets in various mitotic and post-mitotic cell populations. There is currently no evidence that the mammalian Trh homologs NPAS-1 and NPAS-3 are degraded by an Ago/Fbw7 dependent mechanism in mammalian cells. However the finding that Fbw7 knock-out mouse embryos display defects in vascular development (Tetzlaff et al., 2004; Tsunematsu et al., 2004) seems to indicate that the Fbw7 ligase may also target proteins involved in tubular morphogenesis, and intriguingly both NPAS1 and NPAS3 have been linked either to this process (Levesque et al., 2007) or to the transcriptional control of FGFR genes (Pieper et al., 2005). It has been suggested that the Fbw7 vascular defects arise due to Notch misregulation. However since FGF signaling is known to control branching morphogenesis of the mammalian vasculature and lung (Min et al., 1998; Walgenbach et al., 1995), the data presented here raise the possibility that vascular phenotypes in Fbw7 knock-out mouse embryos may also reflect the deregulation of developmental pathways that control branching morphogenesis via mammalian homologs of trh and btl.
Supplementary Material
Supplemental Figure 1. Quantitation of the penetrance of ago and ago;awd DT ‘breaks’ per fusion event.
Supplemental Figure 2. Stage 15 1-eve-1 embryos stained with anti-β-Gal (green) to show tracheal cells and anti-Ago (red) showing nuclear ago expression in all cells in the field.
Acknowledgments
We apologize to those whose work could not be cited due to space constraints. We thank D. Andrew, S. Crews, S. Bray, J. Belote, and T. Hsu for gifts of fly stocks and reagents. Where noted, stocks and antibodies were also obtained from BDGP and DSHB. We are grateful to S. Burdick for technical assistance, and to M.M. Gilbert, C. Krisel, A. Vrailas, D. Marenda, J. Mitchell, D. Zarnescu, and K. Moses for helpful comment and discussion. NTM is funded by an Emory Woodruff Fellowship and a Predoctoral Fellowship from the American Heart Association. KHM is funded by RO1 GM079242-01.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson MG, Certel SJ, Certel K, Lee T, Montell DJ, Johnson WA. Function of the Drosophila POU domain transcription factor drifter as an upstream regulator of breathless receptor tyrosine kinase expression in developing trachea. Development. 1996;122:4169–78. doi: 10.1242/dev.122.12.4169. [DOI] [PubMed] [Google Scholar]
- Balakrishnan A, Bleeker FE, Lamba S, Rodolfo M, Daniotti M, Scarpa A, van Tilborg AA, Leenstra S, Zanon C, Bardelli A. Novel somatic and germline mutations in cancer candidate genes in glioblastoma, melanoma, and pancreatic carcinoma. Cancer Res. 2007;67:3545–50. doi: 10.1158/0008-5472.CAN-07-0065. [DOI] [PubMed] [Google Scholar]
- Belote JM, Fortier E. Targeted expression of dominant negative proteasome mutants in Drosophila melanogaster. Genesis. 2002;34:80–2. doi: 10.1002/gene.10131. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–15. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Bredel M, Bredel C, Juric D, Harsh GR, Vogel H, Recht LD, Sikic BI. Functional network analysis reveals extended gliomagenesis pathway maps and three novel MYC-interacting genes in human gliomas. Cancer Res. 2005;65:8679–89. doi: 10.1158/0008-5472.CAN-05-1204. [DOI] [PubMed] [Google Scholar]
- Brunskill EW, Witte DP, Shreiner AB, Potter SS. Characterization of npas3, a novel basic helix-loop-helix PAS gene expressed in the developing mouse nervous system. Mech Dev. 1999;88:237–41. doi: 10.1016/s0925-4773(99)00182-3. [DOI] [PubMed] [Google Scholar]
- Cabernard C, Affolter M. Distinct roles for two receptor tyrosine kinases in epithelial branching morphogenesis in Drosophila. Dev Cell. 2005;9:831–42. doi: 10.1016/j.devcel.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Calhoun ES, Jones JB, Ashfaq R, Adsay V, Baker SJ, Valentine V, Hempen PM, Hilgers W, Yeo CJ, Hruban RH, Kern SE. BRAF and FBXW7 (CDC4, FBW7, AGO, SEL10) mutations in distinct subsets of pancreatic cancer: potential therapeutic targets. Am J Pathol. 2003;163:1255–60. doi: 10.1016/S0002-9440(10)63485-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dammai V, Adryan B, Lavenburg KR, Hsu T. Drosophila awd, the homolog of human nm23, regulates FGF receptor levels and functions synergistically with shi/dynamin during tracheal development. Genes Dev. 2003;17:2812–24. doi: 10.1101/gad.1096903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer CJ, White JB, Jones KA. Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol Cell. 2004;16:509–20. doi: 10.1016/j.molcel.2004.10.014. [DOI] [PubMed] [Google Scholar]
- Ghabrial A, Luschnig S, Metzstein MM, Krasnow MA. Branching morphogenesis of the Drosophila tracheal system. Annu Rev Cell Dev Biol. 2003;19:623–47. doi: 10.1146/annurev.cellbio.19.031403.160043. [DOI] [PubMed] [Google Scholar]
- Gupta-Rossi N, Le Bail O, Gonen H, Brou C, Logeat F, Six E, Ciechanover A, Israel A. Functional interaction between SEL-10, an F-box protein, and the nuclear form of activated Notch1 receptor. J Biol Chem. 2001;276:34371–8. doi: 10.1074/jbc.M101343200. [DOI] [PubMed] [Google Scholar]
- Hagedorn M, Delugin M, Abraldes I, Allain N, Belaud-Rotureau MA, Turmo M, Prigent C, Loiseau H, Bikfalvi A, Javerzat S. FBXW7/hCDC4 controls glioma cell proliferation in vitro and is a prognostic marker for survival in glioblastoma patients. Cell Div. 2007;2:9. doi: 10.1186/1747-1028-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson KD, Isaac DD, Andrew DJ. Cell fate specification in the Drosophila salivary gland: the integration of homeotic gene function with the DPP signaling cascade. Dev Biol. 1999;205:10–21. doi: 10.1006/dbio.1998.9113. [DOI] [PubMed] [Google Scholar]
- Ikeya T, Hayashi S. Interplay of Notch and FGF signaling restricts cell fate and MAPK activation in the Drosophila trachea. Development. 1999;126:4455–63. doi: 10.1242/dev.126.20.4455. [DOI] [PubMed] [Google Scholar]
- Isaac DD, Andrew DJ. Tubulogenesis in Drosophila: a requirement for the trachealess gene product. Genes Dev. 1996;10:103–17. doi: 10.1101/gad.10.1.103. [DOI] [PubMed] [Google Scholar]
- Jiang L, Crews ST. The Drosophila dysfusion basic helix-loop-helix (bHLH)-PAS gene controls tracheal fusion and levels of the trachealess bHLH-PAS protein. Mol Cell Biol. 2003;23:5625–37. doi: 10.1128/MCB.23.16.5625-5637.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Crews ST. dysfusion Transcriptional Control of Drosophila Tracheal Migration, Adhesion, and Fusion. Mol Cell Biol. 2006;26:6547–56. doi: 10.1128/MCB.00284-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Anthopoulos N, Wetsch B, Binari RC, Isaac DD, Andrew DJ, Woodgett JR, Manoukian AS. Regulation of Drosophila tracheal system development by protein kinase B. Dev Cell. 2001;1:817–27. doi: 10.1016/s1534-5807(01)00090-9. [DOI] [PubMed] [Google Scholar]
- Kennison JA, Tamkun JW. Dosage-dependent modifiers of polycomb and antennapedia mutations in Drosophila. Proc Natl Acad Sci U S A. 1988;85:8136–40. doi: 10.1073/pnas.85.21.8136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klambt C. The Drosophila gene pointed encodes two ETS-like proteins which are involved in the development of the midline glial cells. Development. 1993;117:163–76. doi: 10.1242/dev.117.1.163. [DOI] [PubMed] [Google Scholar]
- Klambt C, Glazer L, Shilo BZ. breathless, a Drosophila FGF receptor homolog, is essential for migration of tracheal and specific midline glial cells. Genes Dev. 1992;6:1668–78. doi: 10.1101/gad.6.9.1668. [DOI] [PubMed] [Google Scholar]
- Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, Elledge SJ. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science. 2001;294:173–7. doi: 10.1126/science.1065203. [DOI] [PubMed] [Google Scholar]
- Kuhnlein RP, Schuh R. Dual function of the region-specific homeotic gene spalt during Drosophila tracheal system development. Development. 1996;122:2215–23. doi: 10.1242/dev.122.7.2215. [DOI] [PubMed] [Google Scholar]
- Kuo YM, Jones N, Zhou B, Panzer S, Larson V, Beckendorf SK. Salivary duct determination in Drosophila: roles of the EGF receptor signalling pathway and the transcription factors fork head and trachealess. Development. 1996;122:1909–17. doi: 10.1242/dev.122.6.1909. [DOI] [PubMed] [Google Scholar]
- Lee T, Hacohen N, Krasnow M, Montell DJ. Regulated Breathless receptor tyrosine kinase activity required to pattern cell migration and branching in the Drosophila tracheal system. Genes Dev. 1996;10:2912–21. doi: 10.1101/gad.10.22.2912. [DOI] [PubMed] [Google Scholar]
- Levesque BM, Zhou S, Shan L, Johnston P, Kong Y, Degan S, Sunday ME. NPAS1 regulates branching morphogenesis in embryonic lung. Am J Respir Cell Mol Biol. 2007;36:427–34. doi: 10.1165/rcmb.2006-0314OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Gazdoiu S, Pan ZQ, Fuchs SY. Stability of homologue of Slimb F-box protein is regulated by availability of its substrate. J Biol Chem. 2004;279:11074–80. doi: 10.1074/jbc.M312301200. [DOI] [PubMed] [Google Scholar]
- Lubarsky B, Krasnow MA. Tube morphogenesis: making and shaping biological tubes. Cell. 2003;112:19–28. doi: 10.1016/s0092-8674(02)01283-7. [DOI] [PubMed] [Google Scholar]
- Malyukova A, Dohda T, von der Lehr N, Akhondi S, Corcoran M, Heyman M, Spruck C, Grander D, Lendahl U, Sangfelt O. The tumor suppressor gene hCDC4 is frequently mutated in human T-cell acute lymphoblastic leukemia with functional consequences for Notch signaling. Cancer Res. 2007;67:5611–6. doi: 10.1158/0008-5472.CAN-06-4381. [DOI] [PubMed] [Google Scholar]
- Mao JH, Perez-Losada J, Wu D, Delrosario R, Tsunematsu R, Nakayama KI, Brown K, Bryson S, Balmain A. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature. 2004;432:775–9. doi: 10.1038/nature03155. [DOI] [PubMed] [Google Scholar]
- Maser RS, Choudhury B, Campbell PJ, Feng B, Wong KK, Protopopov A, O’Neil J, Gutierrez A, Ivanova E, Perna I, Lin E, Mani V, Jiang S, McNamara K, Zaghlul S, Edkins S, Stevens C, Brennan C, Martin ES, Wiedemeyer R, Kabbarah O, Nogueira C, Histen G, Aster J, Mansour M, Duke V, Foroni L, Fielding AK, Goldstone AH, Rowe JM, Wang YA, Look AT, Stratton MR, Chin L, Futreal PA, DePinho RA. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature. 2007;447:966–71. doi: 10.1038/nature05886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merabet S, Ebner A, Affolter M. The Drosophila Extradenticle and Homothorax selector proteins control branchless/FGF expression in mesodermal bridge-cells. EMBO Rep. 2005;6:762–8. doi: 10.1038/sj.embor.7400462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger RJ, Krasnow MA. Genetic control of branching morphogenesis. Science. 1999;284:1635–9. doi: 10.1126/science.284.5420.1635. [DOI] [PubMed] [Google Scholar]
- Min H, Danilenko DM, Scully SA, Bolon B, Ring BD, Tarpley JE, DeRose M, Simonet WS. Fgf-10 is required for both limb and lung development and exhibits striking functional similarity to Drosophila branchless. Genes Dev. 1998;12:3156–61. doi: 10.1101/gad.12.20.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minella AC, Clurman BE. Mechanisms of tumor suppression by the SCF(Fbw7) Cell Cycle. 2005;4:1356–9. doi: 10.4161/cc.4.10.2058. [DOI] [PubMed] [Google Scholar]
- Minella AC, Grim JE, Welcker M, Clurman BE. p53 and SCF(Fbw7) cooperatively restrain cyclin E-associated genome instability. Oncogene. 2007 doi: 10.1038/sj.onc.1210518. [DOI] [PubMed] [Google Scholar]
- Moberg KH, Bell DW, Wahrer DC, Haber DA, Hariharan IK. Archipelago regulates Cyclin E levels in Drosophila and is mutated in human cancer cell lines. Nature. 2001;413:311–6. doi: 10.1038/35095068. [DOI] [PubMed] [Google Scholar]
- Moberg KH, Mukherjee A, Veraksa A, Artavanis-Tsakonas S, Hariharan IK. The Drosophila F box protein archipelago regulates dMyc protein levels in vivo. Curr Biol. 2004;14:965–74. doi: 10.1016/j.cub.2004.04.040. [DOI] [PubMed] [Google Scholar]
- Myat MM, Lightfoot H, Wang P, Andrew DJ. A molecular link between FGF and Dpp signaling in branch-specific migration of the Drosophila trachea. Dev Biol. 2005;281:38–52. doi: 10.1016/j.ydbio.2005.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nateri AS, Riera-Sans L, Da Costa C, Behrens A. The ubiquitin ligase SCFFbw7 antagonizes apoptotic JNK signaling. Science. 2004;303:1374–8. doi: 10.1126/science.1092880. [DOI] [PubMed] [Google Scholar]
- O’Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, Hardwick J, Welcker M, Meijerink JP, Pieters R, Draetta G, Sears R, Clurman BE, Look AT. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to {gamma}-secretase inhibitors. J Exp Med. 2007 doi: 10.1084/jem.20070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberg C, Li J, Pauley A, Wolf E, Gurney M, Lendahl U. The Notch intracellular domain is ubiquitinated and negatively regulated by the mammalian Sel-10 homolog. J Biol Chem. 2001;276:35847–53. doi: 10.1074/jbc.M103992200. [DOI] [PubMed] [Google Scholar]
- Ohshiro T, Emori Y, Saigo K. Ligand-dependent activation of breathless FGF receptor gene in Drosophila developing trachea. Mech Dev. 2002;114:3–11. doi: 10.1016/s0925-4773(02)00042-4. [DOI] [PubMed] [Google Scholar]
- Ohshiro T, Saigo K. Transcriptional regulation of breathless FGF receptor gene by binding of TRACHEALESS/dARNT heterodimers to three central midline elements in Drosophila developing trachea. Development. 1997;124:3975–86. doi: 10.1242/dev.124.20.3975. [DOI] [PubMed] [Google Scholar]
- Perrimon N, Noll E, McCall K, Brand A. Generating lineage-specific markers to study Drosophila development. Dev Genet. 1991;12:238–52. doi: 10.1002/dvg.1020120309. [DOI] [PubMed] [Google Scholar]
- Pieper AA, Wu X, Han TW, Estill SJ, Dang Q, Wu LC, Reece-Fincanon S, Dudley CA, Richardson JA, Brat DJ, McKnight SL. The neuronal PAS domain protein 3 transcription factor controls FGF-mediated adult hippocampal neurogenesis in mice. Proc Natl Acad Sci U S A. 2005;102:14052–7. doi: 10.1073/pnas.0506713102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punga T, Bengoechea-Alonso MT, Ericsson J. Phosphorylation and ubiquitination of the transcription factor sterol regulatory element-binding protein-1 in response to DNA binding. J Biol Chem. 2006;281:25278–86. doi: 10.1074/jbc.M604983200. [DOI] [PubMed] [Google Scholar]
- Rajagopalan H, Jallepalli PV, Rago C, Velculescu VE, Kinzler KW, Vogelstein B, Lengauer C. Inactivation of hCDC4 can cause chromosomal instability. Nature. 2004;428:77–81. doi: 10.1038/nature02313. [DOI] [PubMed] [Google Scholar]
- Samakovlis C, Manning G, Steneberg P, Hacohen N, Cantera R, Krasnow MA. Genetic control of epithelial tube fusion during Drosophila tracheal development. Development. 1996;122:3531–6. doi: 10.1242/dev.122.11.3531. [DOI] [PubMed] [Google Scholar]
- Shiga Y, Tanaka-Matakatsu M, Hayashi S. A nuclear GFP/beta-galactosidase fusion protein as a marker for morphogenesis in living Drosophila. Dev Growth Diffn. 1996;38:99–106. [Google Scholar]
- Sonnenfeld M, Ward M, Nystrom G, Mosher J, Stahl S, Crews S. The Drosophila tango gene encodes a bHLH-PAS protein that is orthologous to mammalian Arnt and controls CNS midline and tracheal development. Development. 1997;124:4571–82. doi: 10.1242/dev.124.22.4571. [DOI] [PubMed] [Google Scholar]
- Spradling AC, Stern D, Beaton A, Rhem EJ, Laverty T, Mozden N, Misra S, Rubin GM. The Berkeley Drosophila Genome Project gene disruption project: Single P-element insertions mutating 25% of vital Drosophila genes. Genetics. 1999;153:135–77. doi: 10.1093/genetics/153.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundqvist A, Bengoechea-Alonso MT, Ye X, Lukiyanchuk V, Jin J, Harper JW, Ericsson J. Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7) Cell Metab. 2005;1:379–91. doi: 10.1016/j.cmet.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Sutherland D, Samakovlis C, Krasnow MA. branchless encodes a Drosophila FGF homolog that controls tracheal cell migration and the pattern of branching. Cell. 1996;87:1091–101. doi: 10.1016/s0092-8674(00)81803-6. [DOI] [PubMed] [Google Scholar]
- Tanaka-Matakatsu M, Uemura T, Oda H, Takeichi M, Hayashi S. Cadherin-mediated cell adhesion and cell motility in Drosophila trachea regulated by the transcription factor Escargot. Development. 1996;122:3697–705. doi: 10.1242/dev.122.12.3697. [DOI] [PubMed] [Google Scholar]
- Tautz D. Whole mount in situ hybridization for the detection of mRNA in Drosophila embryos. In: Kessler C, editor. Nonradioactive Analysis of Biomolecules. Berlin; New York: Springer; 2000. pp. 573–80. [Google Scholar]
- Tetzlaff MT, Yu W, Li M, Zhang P, Finegold M, Mahon K, Harper JW, Schwartz RJ, Elledge SJ. Defective cardiovascular development and elevated cyclin E and Notch proteins in mice lacking the Fbw7 F-box protein. Proc Natl Acad Sci U S A. 2004;101:3338–45. doi: 10.1073/pnas.0307875101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson BJ, Buonamici S, Sulis ML, Palomero T, Vilimas T, Basso G, Ferrando A, Aifantis I. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J Exp Med. 2007 doi: 10.1084/jem.20070872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunematsu R, Nakayama K, Oike Y, Nishiyama M, Ishida N, Hatakeyama S, Bessho Y, Kageyama R, Suda T, Nakayama KI. Mouse Fbw7/Sel-10/Cdc4 is required for notch degradation during vascular development. J Biol Chem. 2004;279:9417–23. doi: 10.1074/jbc.M312337200. [DOI] [PubMed] [Google Scholar]
- van Drogen F, Sangfelt O, Malyukova A, Matskova L, Yeh E, Means AR, Reed SI. Ubiquitylation of cyclin E requires the sequential function of SCF complexes containing distinct hCdc4 isoforms. Mol Cell. 2006;23:37–48. doi: 10.1016/j.molcel.2006.05.020. [DOI] [PubMed] [Google Scholar]
- Walgenbach KJ, Gratas C, Shestak KC, Becker D. Ischaemia-induced expression of bFGF in normal skeletal muscle: a potential paracrine mechanism for mediating angiogenesis in ischaemic skeletal muscle. Nat Med. 1995;1:453–9. doi: 10.1038/nm0595-453. [DOI] [PubMed] [Google Scholar]
- Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG., Jr The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell. 2005;8:25–33. doi: 10.1016/j.ccr.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Welcker M, Orian A, Jin J, Grim JA, Harper JW, Eisenman RN, Clurman BE. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci U S A. 2004;101:9085–90. doi: 10.1073/pnas.0402770101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welcker M, Singer J, Loeb KR, Grim J, Bloecher A, Gurien-West M, Clurman BE, Roberts JM. Multisite phosphorylation by Cdk2 and GSK3 controls cyclin E degradation. Mol Cell. 2003;12:381–92. doi: 10.1016/s1097-2765(03)00287-9. [DOI] [PubMed] [Google Scholar]
- Wilk R, Weizman I, Shilo BZ. trachealess encodes a bHLH-PAS protein that is an inducer of tracheal cell fates in Drosophila. Genes Dev. 1996;10:93–102. doi: 10.1101/gad.10.1.93. [DOI] [PubMed] [Google Scholar]
- Wolf C, Schuh R. Single mesodermal cells guide outgrowth of ectodermal tubular structures in Drosophila. Genes Dev. 2000;14:2140–5. doi: 10.1101/gad.180900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Lyapina S, Das I, Li J, Gurney M, Pauley A, Chui I, Deshaies RJ, Kitajewski J. SEL-10 is an inhibitor of notch signaling that targets notch for ubiquitin-mediated protein degradation. Mol Cell Biol. 2001;21:7403–15. doi: 10.1128/MCB.21.21.7403-7415.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, Nakayama KI. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. Embo J. 2004;23:2116–25. doi: 10.1038/sj.emboj.7600217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Nalepa G, Welcker M, Kessler BM, Spooner E, Qin J, Elledge SJ, Clurman BE, Harper JW. Recognition of phosphodegron motifs in human cyclin E by the SCF(Fbw7) ubiquitin ligase. J Biol Chem. 2004;279:50110–9. doi: 10.1074/jbc.M409226200. [DOI] [PubMed] [Google Scholar]
- Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, Counter CM, Nevins JR, Means AR, Sears R. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol. 2004;6:308–18. doi: 10.1038/ncb1110. [DOI] [PubMed] [Google Scholar]
- Zhou YD, Barnard M, Tian H, Li X, Ring HZ, Francke U, Shelton J, Richardson J, Russell DW, McKnight SL. Molecular characterization of two mammalian bHLH-PAS domain proteins selectively expressed in the central nervous system. Proc Natl Acad Sci U S A. 1997;94:713–8. doi: 10.1073/pnas.94.2.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Quantitation of the penetrance of ago and ago;awd DT ‘breaks’ per fusion event.
Supplemental Figure 2. Stage 15 1-eve-1 embryos stained with anti-β-Gal (green) to show tracheal cells and anti-Ago (red) showing nuclear ago expression in all cells in the field.