

Abstract
Botulinum neurotoxins are the most toxic proteins currently known. Based on a recently identified potent lead structure, 2,4-dichlorocinnamic acid hydroxamate, herein we report on the structure-activity relationship of a series of hydroxamate BoNT/A inhibitors. Among them, 2-bromo-4-chlorocinnamic acid hydroxamate, 2-methyl-4-chlorocinnamic acid hydroxamate and 2-trifluoromethyl-4-chlorocinnamic acid hydroxamate, displayed comparable inhibitory activity to that of the lead structure.
Botulinum neurotoxin (BoNT) is one of the most toxic proteins currently known, with a lethal dose of ∼1 ng/kg of body weight for humans.1 Although the typical BoNT poisoning by accidental food consumption is rare in modern society, serious threats have emerged with the possibility of its use as a biological weapon. C. botulinum has seven serologically distinct strains (A-G), with serotype A being the deadliest and the most threatening for potential bioterrorist attacks, given its prolonged half-life in vivo and the the ease of its production and transport.2
The active form of botulinum neurotoxin is a heterodimer consisting of a 100 kDa heavy chain (HC) coupled to a 50 kDa light chain (LC) by one or more disulfide bonds.3 Its neurotoxicity is attributed to the light chain, a Zn (II) endopeptidase that cleaves SNARE (soluble NSF-attachment protein receptor) proteins involved in neuronal synaptic vesicle function. There are three different types of SNARE proteins attacked by BoNT (VAMP, SNAP-25 and syntaxin); the target for BoNT/A is SNAP-25. The degradation of these proteins blocks the release of acetylcholine, resulting in flaccid muscle paralysis and potentially death.4
Current therapeutic options for BoNT intoxication are rather limited. An effective immunoprophylactic vaccine is available, but the development of protection is slow, with annual boosters required to produce adequate antibody titers.5 In addition, equine antitoxins can be used for the treatment of adult botulism, but this therapy also can cause severe adverse reactions including serum sickness and anaphylaxis.6 Finally, there are reports of the successful use of multiple monoclonal antibodies as BoNT/A antitoxins.7 Unfortunately, the antibodies must be administered prior to, or shortly after, BoNT exposure which makes this approach of limited therapeutic utility, particularly in case of a possible bioterrorist attack. Therefore, a pharmacological intervention that would be effective after BoNT enters neuronal cells and could be used en masse is highly desirable. In this light, small molecule inhibitors of BoNT light chain protease represent a very attractive target and several nonpeptidic, small molecule inhibitors have been reported.8
As a part of our ongoing research program focused on identifying small molecule inhibitors of BoNT/A, we reported the high-throughput screen of a library of hydroxamic acids,9 from which 4-chlorocinnamic hydroxamate, displaying an IC50 of 15 μM, resulted as a promising lead structure for further development (Fig. 1, compound 1).10 A subsequently synthesized series of compounds revealed that while replacement of the chloro substituent was not tolerated, introduction of another chloro substituent in the ortho-position (Fig. 1, compound 2) resulted in the most potent nonpeptidic BoNT inhibitor to date, with IC50 value <1 μM.10
Figure 1.
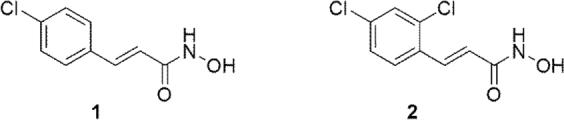
Structures of potent BoNT/A LC inhibitors, 2-chlorocinnamic acid hydroxamate (1) and 2,4-dichlorocinnamic acid hydroxamate (2).
Recently, the X-ray crystallographic structures of BoNT/A light chain with both 4-chlorocinnamic hydroxamate (1) and 2,4-dichlorocinnamic hydroxamate (2) were described.11 This study revealed the expected coordination of the hydroxyl oxygen of the hydroxamate moiety to the Zn(II) atom. In addition, the hydroxamate carbonyl oxygen forms a hydrogen bond with Tyr 366. The catalytic cleft of BoNT/A LC is lined almost entirely by hydrophobic residues and the phenyl ring of 1 and 2 binds in a tight pocket formed by Ile 161, Phe 194 and Phe 369. The increased potency of 2 compared to 1 was attributed to the dipole/electronegative contact of the additional chlorine atom with Arg 363, improving specificity and enhancing binding affinity. In addition, these structural studies revealed that the BoNT/A LC active site is capable of dramatic rearrangements in response to the electrostatic character of the substrate, which makes various structural patterns very promising in the search for new BoNT/A LC small molecule inhibitors.11 Consequently, we decided to further explore the structure-activity relationship of 2 and herein report on the synthesis and inhibitory activity of series of derivatives of the parent structure.
In an attempt to improve the potency of our initial lead structure, we chose two structural modifications: a) fusing an additional aromatic ring to the benzene moiety of the parent structure (3a-b and 4a-d) and b) replacing the ortho-Cl atom with substituents of varying electronegativity and steric requirements (5a-k) (Fig. 2).
Figure 2.
Structural modifications of the lead structure.
In the case of the ring fused derivatives, we rationalized that “freezing out” the conformational dynamics could contribute to increased stability of the enzyme-inhibitor complex.12 Furthermore, we assumed that the additional aromatic ring might stack with the Tyr 366 residue, improving the binding affinity. Due to the mostly hydrophobic character of (1) and (2), there are very few hydrogen bonds formed between the inhibitor and the residues in the catalytic cleft.11 In order to provide greater potential for these interactions, we designed a series of fused ring derivatives with varying hydrogen bond capability (4a-d). (Fig. 2) Compounds (5a-k) were designed to examine the boundaries and flexibility of the interaction with the Arg 363 residue in the active site of the enzyme. We envisaged that due to the dipole/electronegative character of this interaction, introduction of a substituent with higher electronegativity would increase the inhibitory activity.11 To test this hypothesis, we designed a series of compounds bearing substituents with varying stereoelectronic requirements (5a-k). (Fig. 2)
All the ring fused derivatives were obtained by standard synthetic procedures from commercially available starting materials13 (Scheme 1). The crucial intermediates in the synthesis of the ortho-modified derivatives, aldehydes 12a-j, were either prepared (12b-e; Scheme 2A),14 or purchased (12a,f-j; Scheme 2B). The desired hydroxamate moiety was typically introduced by activation of the corresponding carboxylic acid with ethyl chloroformate followed by treatment with hydroxylamine,15 or by treatment of the appropriate methyl ester with 50% aqueous hydroxylamine in the presence of catalytic amount of potassium cyanide or stoichiometric amount of KOH.16
Scheme 1.

Synthesis of ring-fused hydroxamates. Reagents and conditions: (i) CuI, CuCl, DMF, 150 °C, 24h, 63%; (ii) 1. ethyl chloroformate, Et3N, THF, 0 °C to rt, 15min, 2. NH2OH, MeOH, rt, 18h, 2 steps, 47- 60%; (iii) 1. NaBH4, MeOH, 0 °C to rt, 1h, 2. TsOH, toluene, 160 °C, 2h, 2 steps 81%; (iv) oxalyl dibromide, 110 °C, 5h, 11%; (v) 1. (trimethylsilyl)diazomethane, toluene/MeOH, rt, 3h, 2. KCN (cat.), 50% NH2OH (aq.), THF/MeOH, rt, 18h, 2 steps 36−43%; (vi) methyl thioglycolate, NaH, DMSO, rt, 5min, 24%; (vii) KCN (cat.), 50% NH2OH (aq.), THF/MeOH, rt, 18h, 15−37%. (viii) MeI, K2CO3, DMF, 60°C, 24 h, 72%.
Scheme 2.

Synthesis of ortho-modified hydroxamates. Reagents and conditions: (vii) KCN (cat.), 50% NH2OH (aq.), THF/MeOH, rt, 18h, 15−37%. (ix) 1. BH3-DMS, THF, rt to 70 °C, 3 h, 2. MnO2, CH2Cl2, rt, 18 h, 2 steps 34%; (x) MeI, K2CO3, DMF, 75 °C, 18 h, 95%; (xi) 1. LiBH4, THF, rt to 60 °C, 2 h, 2. MnO2, CH2Cl2, rt, 18 h, 2 steps, 94%; (xii) NaSMe, DMF, rt, 1 h, 91%; (xiii) Fe, HCl, EtOH/AcOH/H2O, 100°C, 15 min., rt, 40 min. (64%); (xiv) methyl diethylphosphonoacetate, NaH, DMF, rt, 18h, 78−99%; (xv) Zn(CN)2, Pd(PPh3)4, DMF, 160°C, 10 min., microwave, (50%); (xvi) Oxone®, MeOH-H2O, 0°C to rt, 5 h, 60%; (xvii) 50% NH2OH (aq.), 1M KOH/MeOH, THF, 0°C, 2−4, 12−58%.
Once prepared, all compounds were evaluated with recombinant BoNT/A light chain protease (LC/A) in a FRET-based assay employing SNAPtide™ (List Biological Laboratories, Inc., Campbell, CA) as the enzymatic substrate.17 While the fused ring hydroxamates (3a-b and 4a-d) showed only modest inhibition (Table 1), refuting our hypotheses, some of the ortho-substituted derivatives displayed significant inhibitory activity (Table 1). The best substituents proved to be the electron withdrawing groups, with the Br (5b), CF3 (5g) and NO2 (5j) derivatives showing low micromolar or submicromolar inhibitory activity (Table 1). Surprisingly, similar inhibitory activity was observed in case of the methyl-substituted derivative 5f (Table 1). This finding is very interesting as it sheds new light on the nature of the interaction between the substituent in ortho-position and the Arg 363 residue. Unfortunately, no clear trend was observed between inhibitory activity and electron donating/withdrawing character of the substituent (5a-k).
Table 1.
BoNT/A LC inhibitory activitya
| Compound | IC50 (μM)b |
|---|---|
| 2 | 0.9 ± 0.1 |
| 3a | 21 ± 5 |
| 3b | 71 ± 1 |
| 4a | 41 ± 0.3 |
| 4b | 45 ± 7 |
| 4c | n.a. |
| 4d | 38 ± 7 |
| 5a | 4.4 ± 0.1 |
| 5b | 0.7 ± 0.1 |
| 5c | 9 ± 2 |
| 5d | 5.1 ± 0.6 |
| 5e | 25 ± 6 |
| 5f | 0.8 ± 0.1 |
| 5g | 0.6 ± 0.1 |
| 5h | 13 ± 5 |
| 5i | 2 ± 0.6 |
| 5j | 17 ± 0.4 |
| 5k | 12 ± 3 |
Inhibitors were evaluated as previously described.17
Values are means of two experiments performed in triplicate, standard deviation is given in parentheses (n.a. = not active up to 100 μM).
In summary, we have synthesized and evaluated a series of BoNT/A LC protease inhibitors as analogs of the recently discovered potent parent structure (2). While the increased conformational constraint imported by the inclusion of an additional aromatic ring resulted in dramatic loss of inhibitory activity, introduction of bromo (5b), trifluoromethyl (5g) and methyl (5f) substituents in the ortho position led to increased inhibitory activity, yielding three new small molecule inhibitors with comparable potency to the parent molecule. In light of these data, future efforts to uncover the full extent of BoNT/A LC flexibility are warranted.
Acknowledgments
This work was supported by contract NO1-AI30050 from the National Institutes of Health and The Skaggs Institute for Chemical Biology. We wish to thank Dr. Lisa Eubanks for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
Supplementary data
Supplementary material associated with this article (experimental procedures, NMR data) can be found in the online version at doi:
References and notes
- 1.Singh BR. Nature. 2000;7:617. doi: 10.1038/77900. [DOI] [PubMed] [Google Scholar]
- 2.Hicks RP, Hartell MG, Nichols DA, Bhattacharjee AK, Von Hamont JE, Skillman DR. Curr. Med. Chem. 2005;12:667–690. doi: 10.2174/0929867053202223. [DOI] [PubMed] [Google Scholar]
- 3.Oguma K, Fujinaga Y, Inoue K. Microbiol. Immunol. 1995;39:161. doi: 10.1111/j.1348-0421.1995.tb02184.x. [DOI] [PubMed] [Google Scholar]
- 4.Simpson LL. Annu. Rev. Pharmacol. Toxicol. 2004;44:167. doi: 10.1146/annurev.pharmtox.44.101802.121554. [DOI] [PubMed] [Google Scholar]
- 5.Arnon SS, Schechter R, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Hauer J, Layton M, Lillibridge S, Osterholm MT, O'Toole T, Parker G, Perl TM, Russell PK, Swerdlow DL, Tonat K. JAMA. 2001;285:1059. doi: 10.1001/jama.285.8.1059. [DOI] [PubMed] [Google Scholar]
- 6.Black RE, Gunn RA. Am. J. Med. 1980;69:567. doi: 10.1016/0002-9343(80)90469-6. [DOI] [PubMed] [Google Scholar]
- 7.a Amersdorfer P, Wong C, Smith T, Chen S, Deshpande S, Sheridan R, Marks JD. Vaccine. 2002;20:1640. doi: 10.1016/s0264-410x(01)00482-0. [DOI] [PubMed] [Google Scholar]; b Nowakowski A, Wang C, Powers DB, Amersdorfer P, Smith TJ, Montgomery VA, Sheridan R, Blake R, Smith LA, Marks JD. Proc. Natl. Acad. Sci. 2002;99:11346. doi: 10.1073/pnas.172229899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dickerson TJ, Janda KD. ACS Chem. Biol. 2006;1:359. doi: 10.1021/cb600179d. [DOI] [PubMed] [Google Scholar]
- 9.Boldt GE, Kennedy JP, Hixon MS, McAllister LA, Barbieri JT, Tzipori S, Janda KD. J.Comb. Chem. 2006;8:513. doi: 10.1021/cc060010h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boldt GE, Kennedy JP, Janda KD. Org. Lett. 2006;8:1729. doi: 10.1021/ol0603211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Silvaggi NR, Boldt GE, Hixon MS, Kennedy JP, Tzipori S, Janda KD, Allen KN. Chem. Biol. 2007;14:533. doi: 10.1016/j.chembiol.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 12.Choi S, Silverman RB. J. Med. Chem. 2002;45:4531. doi: 10.1021/jm020134i. [DOI] [PubMed] [Google Scholar]
- 13.a Jia ZJ, Wu Y, Huang W, Zhang P, Song Y, Woolfrey J, Sinha U, Arfsten AE, Edwards ST, Hutchaleelaha A, Hollennbach SJ, Lambing JL, Scarborough RM, Zhu B-Y. Bioorg. Med. Chem. Lett. 2004;14:1229. doi: 10.1016/j.bmcl.2003.12.054. [DOI] [PubMed] [Google Scholar]; b Hauser FM, Prasanna S. Synthesis. 1980:621. [Google Scholar]; c Varnavas A, Lassiani L, Valenta V, Berti F, Tontini A, Mennuni L, Makovec F. Eur. J. Med. Chem. 2004;39:85. doi: 10.1016/j.ejmech.2003.11.010. [DOI] [PubMed] [Google Scholar]; d Bridges AJ, Lee A, Maduakor EC, Schwartz CE. Tetrahedron Lett. 1992;33:7499. [Google Scholar]; e Sakamuri S, George C, Flippen-Anderson J, Kozikowski AP. Tetrahedron Lett. 2000;41:2055. [Google Scholar]
- 14.a Kasuga J-I, Hashimoto Y, Miyachi H. Bioorg. Med. Chem. Lett. 2006;16:771. doi: 10.1016/j.bmcl.2005.11.029. [DOI] [PubMed] [Google Scholar]; b Nag M, Jenks WS. J. Org. Chem. 2005;70:3458. doi: 10.1021/jo047995e. [DOI] [PubMed] [Google Scholar]; c Raitio KH, Savinainen JR, Vepsalainen J, Laitinen JT, Poso A, Jarvinen T, Nevalainen T. J. Med. Chem. 2006;49:2022. doi: 10.1021/jm050879z. [DOI] [PubMed] [Google Scholar]
- 15.Reddy AS, Kumar MS, Reddy GR. Tetrahedron Lett. 2000;41:6285. [Google Scholar]
- 16.a Ho CY, Strobel E, Ralbovsky J, Galemmo RA., Jr. J. Org. Chem. 2005;70:4873. doi: 10.1021/jo050036f. [DOI] [PubMed] [Google Scholar]; b Marson CM, Savy P, Rioja AS, Mahadevan T, Mikol C, Veerupillai A, Nsubuga E, Chahwan A, Joel SP. J. Med. Chem. 2006;49:800. doi: 10.1021/jm051010j. [DOI] [PubMed] [Google Scholar]
- 17.Eubanks LM, Hixon MS, Jin W, Hong S, Clancy CM, Tepp WH, Baldwin MR, Malizio CJ, Goodnough MC, Barbieri JT, Johnson EA, Boger DL, Dickerson TJ, Janda KD. Proc. Natl. Acad. Sci. 2007;104:2602. doi: 10.1073/pnas.0611213104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data
Supplementary material associated with this article (experimental procedures, NMR data) can be found in the online version at doi: