

Abstract
The G-protein gated inward rectifier K+ channel (GIRK) is activated in vivo by the Gβγ subunits liberated upon Gi-coupled receptor activation. We have recapitulated the acute desensitization of receptor-activated GIRK currents in heterologous systems and shown that it is a membrane-delimited process. Its kinetics depends on the guanine nucleotide species available and could be accounted for by the nucleotide exchange and hydrolysis cycle of G proteins. Indeed, acute desensitization is abolished by nonhydrolyzable GTP analogues. Whereas regulators of G-protein signaling (RGS) proteins by their GTPase-activating protein activities are regarded as negative regulators, a positive regulatory function of RGS4 is uncovered in our study; the opposing effects allow RGS4 to potentiate acute desensitization without compromising GIRK activation.
Stimulation of Gi-coupled receptors in the heart and the brain activates the G-protein gated inward rectifier K+ channels (GIRK) (1, 2), leading to hyperpolarization and reduction of membrane excitability. Although the receptor-activated GIRK currents vary according to the receptor subtypes stimulated and the cell types examined, they generally are biphasic, with a rapid activation followed by a slower desensitization (3, 4).
Desensitization of receptor-mediated responses provides the basis for cellular adaptation to external inputs. Moreover, different receptors that activate the same signaling pathway can generate distinct temporal signals because of the differences in their desensitization kinetics. For GIRK current desensitization, two phases can be resolved. The slower one takes several minutes and probably is mediated by G-protein receptor kinases (GRKs, also known as βARKs) (5, 6). As for the acute desensitization that occurs within a minute, dephosphorylation of KACh channels in atrial myocytes has been implicated because it is augmented by cytoplasmic ATP (4). Given that ATP can serve as a substrate for various ATPases and kinases, however, the basis for the acute desensitization remains an open question.
We approached this problem by defining a minimal system for acute desensitization, by using excised inside-out membrane patches exposed to cytoplasmic solutions of known composition. Like the receptor-mediated activation of GIRK current (7), acute desensitization of receptor-induced GIRK current is also a membrane-delimited process (4). Moreover, guanine nucleotides have profound effects on the GIRK current desensitization, which persists in the absence of ATP. The regulators of G-protein signaling (RGS) proteins, which speed up the activation-deactivation kinetics of GIRK currents (8, 9), also accelerate the desensitization kinetics. In addition to RGS4’s ability to promote GTP hydrolysis of Gα subunits, we found that RGS4 coexpression increased the G-protein pool available for GIRK activation, thereby allowing acceleration of current kinetics without compromising current amplitudes. Rates of GIRK current activation and desensitization under various experimental conditions can be simulated by a model that assumes no intrinsic channel desensitization. We propose, therefore, that the nucleotide exchange and hydrolysis cycle of G proteins is sufficient to give rise to the acute desensitization in the receptor-mediated activation of GIRK channels.
MATERIALS AND METHODS
Cell Culture and Heterologous Expression.
For whole-cell recording, plasmids containing the μ-opioid receptor gene were supertransfected into a HEK cell line expressing m4AChR, GIRK1, and GIRK4 or another cell line expressing β2 adrenergic receptor, GIRK1 and GIRK2, by using Lipofectamine. Whole-cell recordings were carried out 36–48 hr after transfection.
Excised patch recordings were performed in the Xenopus oocyte expression system. To acquire a very high level of expression, 10–15 ng of μ-receptor cRNA per oocyte was injected. Otherwise, 0.3–0.5 ng per oocyte is enough for a modest amount of receptor expression to generate the acute desensitization in the patch membranes.
Electrophysiology.
For whole-cell recording, cells were bathed in a solution containing 10 mM Hepes, 20 mM KCl, 120 mM NaCl, 1 mM MgCl2, 1 mM CaCl2, and 5 mM NaOH, pH 7.4. The pipette solution consisted of 9 mM Hepes, 4.5 mM EGTA, 0.5 mM CaCl2, 1.8 mM MgCl2, 100 mM KCl, 4.5 mM K2HPO4, 27 mM NaCl, and 0.2 mM Na2GTP, pH 7.2. To measure the basal currents, the perfusion solution was switched from K+ free solution to 20 mM K+. Receptor-induced GIRK currents were measured by holding the membrane at −60 mV.
For oocyte giant patch recording, the pipette solution contained 10 mM Hepes, 140 mM KCl, 5 mM KOH, 1 mM MgCl2, and 1 mM CaCl2, pH 7.4. Agonists were included in the pipette solutions except for measurement of the basal activity of receptors. The concentrations of agonists used in the experiments were as follows: 50 nM [d-Ala2,N-MePhe4,Gly5-ol]enkephalin (DAMGO), or 50 nM nalorphine for μ-receptors, 10 μM acetylcholine for m4AChR, m2AChR, and m2RΔ, 100 nM (±)-epinephrine for α2A adrenergic receptors, and 50 nM d-Trp8-somatostatin-14 for SSTR-2. The bath solution was composed of 10 mM Hepes, 110 mM KCl, 5 mM K2HPO4, 30 mM NaCl, 5 mM EGTA, 0.55 mM CaCl2, and 2 mM MgCl2, pH 7.2. Guanine nucleotides were prepared as 100 or 200 mM stocks and stored in small aliquots. Unless otherwise indicated, the working concentration of guanine nucleotides was 200 μM. The membrane patch was held at −60 mV for continuous sampling of currents. The eletrophysiological data were sampled at 5 kHz but digitized at only 0.1–0.2 kHz for the analysis of macroscopic currents. Unpaired Student’s t tests were performed to determine the statistical significance.
RESULTS
Acute Desensitization of GIRK Channel Activation by G Protein-Coupled Receptors in Heterologous Expression Systems.
In HEK 293 cells transfected with cDNAs for G protein-coupled receptors and GIRK channels, receptor-activated GIRK currents typically comprise a rapid rising phase followed by desensitization (Fig. 1A). Acute desensitization also was observed in perforated patch recording of HEK cells and in oocytes recorded by the two-electrode voltage clamp method (Fig. 1B). The activation kinetics of GIRK current varied with the receptor type (Fig. 1A) and with the agonists. For instance, the amplitudes of μOR-induced currents in the same cell always followed the rank order DAMGO > morphine > methadone > nalorphine as reported before (10). The stronger the agonist, the more likely it will cause acute desensitization.
Figure 1.

Receptor-induced activation of GIRK currents. (A) m4R and μ-opioid receptor (μOR) activated GIRK current in the same cell with different kinetics of activation and desensitization. The concentrations (in μM) of agonists used are shown in parentheses. (B) Acute desensitization also was observed via perforated patch recording of HEK 293 cells (m4R, Upper) or two-electrode voltage clamp in Xenopus oocytes (α2A adrenergic receptor, Lower). (C) Activation of GIRK current by GTP in the excised inside-out patches from oocytes is sensitive to pertussis toxin (PTX). (D) Larger activated currents and acute desensitization were observed in the patches excised from cells expressing higher levels of receptors. (E) In a patch membrane with a high level of μOR even the partial agonist nalorphine (50 nM) could evoke acute desensitization of GIRK current when the solution was changed from nucleotide free to one containing GTP. (F) In a patch expressing μOR, GIRK1, GIRK2, and RGS4, similar perfusion protocol revealed acute desensitization.
Similar to the GIRK current activation in native cells (3, 4), GIRK channels in the inside-out membrane patch excised from oocytes were activated by application of GTP to the patch in a membrane-delimited fashion. This channel activation is induced by receptor stimulation, because it requires the presence of agonists for the relevant receptors in the pipette solutions and is abolished by pertussis toxin treatment (Fig. 1C). This was true for all of the Gi-coupled receptors we examined, including μ-opioid receptor, α2A adrenergic receptor, m4AChR, m2AChR, and the somatostatin receptor SSTR2.
The kinetics and amplitudes of the GTP-induced currents were affected by the receptor activity, which is determined by the number of receptors and the agonist used to stimulate the receptors. With low-level receptor expression, the activation was slow even when the efficacious agonist DAMGO was used for stimulation (Fig. 1C). Elevated receptor expression results in faster kinetics and larger current amplitudes of the induced currents, exhibiting rapid activation and a subsequent decay phase (Fig. 1D), and recapitulating the acute desensitization observed in the whole-cell recording from the HEK293 cells. When nalorphine, a much less efficacious agonist than DAMGO, was used to stimulate the receptors, we had to express the receptors to an even higher level to observe a comparable desensitization in the excised patches (Fig. 1E, Right). This high level of receptor expression led to detectable basal activity; application of GTP to inside-out patches elicited some GIRK current, even when no agonists were included in the pipette solution. Similar dependence of acute desensitization on receptor expression level also was observed for m2AChR or a deletion mutant of m2AChR (m2RΔ) (11) that removes the βARK phosphorylation sites in the oocyte expression system. These observations indicate that an excised inside-out patch contains the basic components for generating acute desensitization, provided a sufficiently high receptor activity is achieved.
Desensitization in the Excised Patches Is Affected by Guanine Nucleotides.
Because the receptors in an excised inside-out patch are constantly stimulated by agonists in the pipette, G proteins should become activated upon exposure of the cytoplasmic face of the membrane patch to a solution containing GTP, causing dissociation of βγ dimers to activate the GIRK channels (12–14). The size of induced currents would reflect the amount of Gβγ generated by the activated receptors. The extent of acute desensitization was much more prominent if we switched from a solution without GDP to one containing GTP, as compared with what we observed upon switching from GDP- to GTP-containing solution (Fig. 1 E and F, see also Fig. 3A). The following model could account for these observations.
Figure 3.

Expression of RGS4 alters the kinetics of channel activation. (A) From a batch of oocytes, those oocytes coexpressing RGS4 yield faster channel activation and deactivation and more prominent desensitization, as evident from inside-out patch recording. (B) Peak and steady-state GIRK currents induced by switching from the nucleotide-free solution to one containing GTP (n = 10 for each group) (Left). DAMGO (50 nM) was used to activate μ-opioid receptors. The fraction of residual current normalized to the peak in the group coexpressing RGS4 is smaller, thus showing larger apparent desensitization (Right, P < 0.05). (C) A low receptor activity, because of either low level of receptor expression (Left) or the use of an inefficacious agonist to stimulate the receptors (Right), resulted in small steady-state currents. Subsequent application of GppNHp provoked larger nondesensitizing currents, presumably by disrupting the G-protein cycle.
The observed dependence of acute desensitization on guanine nucleotides indicates that the kinetics of GIRK current desensitization are influenced by the G-protein turnover cycle. Fig. 2A illustrates a simplified version of this cycle. Before receptor activation, most of the G proteins are in their GDP-bound trimeric state. Nucleotide exchange upon receptor activation leads to a transient state of the trimeric G protein with the guanine nucleotide binding pocket empty (αEβγ). Such an empty state is readily quenched by binding of either GTP or GDP. Because of the relative abundance of GTP than GDP, most of the empty-state G proteins become GTP bound, which allows the dissociation of α- and βγ-subunits to activate their respective effectors. The intrinsic GTPase activity of Gα causes the hydrolysis the GTP molecule and regenerates α-GDP, which will bind βγ-subunit to form trimeric G protein to reinitiate the cycle.
Figure 2.

A hypothetical G-protein turnover cycle is sufficient for the acute desensitization. (A) αEβγ stands for a trimeric G protein with an empty nucleotide binding site. P1, P2, or P3 indicates the fraction of G proteins in each state. The kinetic constants of the G-protein nucleotide exchange-hydrolysis cycle could account for the kinetics of GTP-activated GIRK currents. (B) Slower channel activation is expected when GTP application follows GDP, as compared with following a nucleotide free solution, whereas the steady-state level should be independent of the starting conditions. (C) An increase in the receptor activity speeds up and enhances the activation. (D) The GAP activity alone will contribute to more prominent acute desensitization. (E) Disruption of the G-protein cycle by nonhydrolyzable GTP analogues will lead to greater channel activation and the abolition of acute desensitization.
In experiments as shown in Fig. 1 E and F, the exposure of the cytoplasmic side of the inside-out membrane patch to a nucleotide free solution causes G proteins to be arrested in the empty state because neither GDP nor GTP is available for nucleotide binding upon the receptor-catalyzed dissociation of GDP from the trimeric G proteins. Subsequent application of a solution containing GTP permits immediate binding of GTP to these empty-state G proteins and starts the entire cycle. This initially synchronized release of Gβγ leads to a transient increase of free Gβγ concentration, followed by a relaxation to its new steady-state level (Fig. 1 E and F, Right). To mimic the nucleotide exchange under more physiological conditions, where G proteins switch from the GDP- to the GTP-bound state, we exposed the patch to a solution with 200 μM GDP to favor binding of GDP to form inactive trimeric G proteins despite the continuous exposure of the receptors to their ligands. Subsequent application of GTP induced less synchronized activation of G proteins and consequently a GIRK current with slower activation and much reduced desensitization (Fig. 1 E and F, Left).
GIRK Activation Predicted from the G-Protein Hydrolysis Cycle.
To qualitatively predict the receptor-induced GIRK current under various experimental manipulation, we simulate the kinetics of induced current by the following set of differential equations:
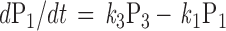 |
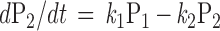 |
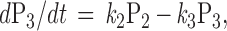 |
where Pi = [Pi]/[total G protein] represents the fraction of G proteins in a specific state. The solution will be the sum of two exponential functions. The rate constants for the two exponential functions, λ1 and λ2, are the roots of the characteristic quadratic form and satisfy:
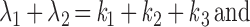 |
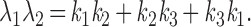 |
Like the values for λ1 and λ2, the steady-state values of P1, P2, and P3 are determined exclusively by the kinetic constants k1, k2, and k3. As long as the kinetic parameters of the cycle remain unchanged, the system should reach the same steady state when the generation of active G-protein subunits by receptors is balanced by their clearance via GTP hydrolysis and reassociation regardless of the initial conditions (Figs. 1F and 2A). This steady state can be derived by letting the right side of the differential equation equal zero.
If k2 is much larger than either k1 or k3, as in the case of a modest expression of receptors and limited hydrolysis rate of the α-GTP, the equations above can be approximated by:
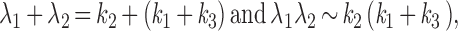 |
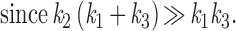 |
The roots of this approximation are therefore k2 and (k1 + k3). The physical meaning of λ1 and λ2 is readily appreciated in the exchange experiment with solutions switched from nucleotide free to GTP containing. Rapid activation and desensitization take place. The activation rate will be decided by k2, and the desensitization rate is reflected by (k1 + k3), i.e., the sum of the exchange and the hydrolysis rates. Because of the positive cooperativity of βγ activation of GIRK channels (15, 16), GIRK activation and desensitization rates are not simply the biochemically determined rates for G-protein cycle (see Discussion). This model predicts that an increase in either k1 or k3 should lead to acceleration of the desensitization time constant. It follows that acute desensitization of receptor-induced GIRK current is more apparent if receptor expression levels are higher (Figs. 1 C and D and 2C), more potent agonists are used to stimulate the receptors, or if the GTPase activity of Gα subunits is accelerated by GTPase activating proteins (GAP) such as RGS4 (Fig. 2D and Fig. 3).
If one uses nonhydrolyzable GTP analogues to disrupt the cycle, the differential equations are reduced to:
 |
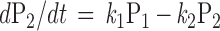 |
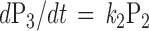 |
since k3 equals zero if the GTP analogues are absolutely hydrolysis resistant. The generation of free βγ by applying these nonhydrolyzable analogues will be biexponential with the two rate constants equal to k1 and k2. The steady state will be the maximal activation of G proteins (Figs. 2E and 3C). The nonhydrolyzable GTP analogues therefore will be useful for assessing the rates of nucleotide exchange and free βγ generation.
The Ability of RGS Protein to Accelerate GTP Hydrolysis Modifies the G-Protein Turnover Cycle.
Given that the α-GDP generated by the intrinsic GTPase activity of α-GTP will associate with and inactivate βγ dimers, proteins that accelerate the GTPase activity of α-GTP may alter the kinetics and steady-state production of free βγ dimers. A family of RGS has been characterized based on their ability to interact with certain G-protein α-subunits and accelerate their GTP hydrolysis (8, 17–19). The GAP activity of RGS proteins should speed up the deactivation of GIRK channels upon agonists removal, but cannot explain how GIRK activation can become accelerated without a reduction in the current amplitude.
We found that RGS4 also caused faster activation and deactivation of GTP-induced currents in the excised patches exposed to μ-agonists (for DAMGO, activation time, 1.63 ± 0.4 sec vs. 4.66 ± 2.15 sec, P < 0.05; deactivation time constant t1/e 3.39 ± 0.34 sec vs. 35.0 ± 8.9 sec, P < 0.05) (Fig. 3A). In addition, the amplitudes of GTP-induced currents were slightly larger at the peak and slightly smaller 30 sec after application, thereby showing more prominent desensitization (Fig. 3B). When the receptor number was low or when a partial agonist was used for the receptor stimulation, RGS4 precipitated the development of desensitization so much that the steady-state current amplitudes were rather small (Fig. 3C). Irreversible activation of G proteins by the nonhydrolyzable GTP analogue GppNHp induced much larger GIRK currents without desensitization (Fig. 3C), indicating that the RGS4 protein enhanced GIRK current desensitization without suppressing the ability of G-protein subunits to activate GIRK channels.
RGS Proteins Also Enhance G-Protein Activation.
A shortened lifetime of free α-GTP and βγ because of GAP activity of RGS4 is expected to reduce GIRK activation. However, the amplitude of GTP-induced GIRK current was not significantly altered by RGS4 coexpression. It thus appeared that RGS4 did not significantly reduce the amount of βγ dimers at the steady state, in spite of its GAP activity. To look for other RGS4 functions besides its GAP activity, we tested for the effect of RGS4 coexpression on GIRK current activation by the nonhydrolyzable GTP analogues.
When an inside-out membrane patch excised from an oocyte expressing GIRK channels but no receptors was exposed to GTPγS, the GIRK channels were activated slowly to reach a plateau within 3–4 min (Fig. 4A). The current induced by GTPγS was much larger if the oocyte also expressed RGS4 (1.08 ± 0.14 nA vs. 0.26 ± 0.03 nA, P < 0.05) (Fig. 4A). When the μ-receptors were expressed with the channels and agonists were included in the pipette to activate receptors, RGS4 coexpression led to larger current induced by replacing GDP with GTPγS (for DAMGO, 5.54 ± 0.36 nA vs. 0.41 ± 0.04 nA, P < 0.05; for nalorphine, 4.83 ± 0.99 nA vs. 0.63 ± 0.10 nA, P < 0.05), whereas the time required to reach maximal activation was not much affected by RGS4 coexpression (Fig. 4 B and C). This experiment revealed that RGS4 coexpression caused a larger population of G proteins to be available for receptor-mediated channel activation. Catalyzing nucleotide exchange of this expanded G-protein pool in oocytes coexpressing RGS4, the receptors also appeared to activate more G protein in a given amount of time. As a result, RGS proteins speed up G-protein signaling without much compromise in the steady-state amplitude of the response.
Figure 4.

RGS4 increased the amplitude of GTPγS-induced GIRK current. (A) In the absence of any exogenous receptors, RGS4 coexpression caused a larger GIRK current induced by GTPγS. (B and C) When μ-receptors were expressed and stimulated by DAMGO (B) or nalorphine (C), RGS4 coexpression drastically increased the amplitudes of the induced current but not the time to reach maximal activation.
DISCUSSION
In this study, we examine how G-protein turnover affects the kinetics of GIRK channel activation by receptors in heterologous expression systems. Recorded from both mammalian cells and excised inside-out oocyte membrane patches, these receptor-activated GIRK currents displayed acute desensitization, even in the absence of cytoplasmic ATP. Nonhydrolyzable GTP analogues maximally activated GIRK channels and abolished acute desensitization. Both activation and desensitization of GIRK current were accelerated by coexpressing RGS4, which not only enhances the GTPase activity of Gαi’s but also increases the pool of G proteins that can mediate GIRK activation by receptors. These observations thus uncovered an additional function of RGS proteins and suggest that the nucleotide exchange and hydrolysis cycle of G proteins is sufficient to cause acute desensitization.
Receptor Number and Agonist Efficacy Affect GIRK Channel Activation.
When agonists of different efficacies are used to stimulate the receptors, different critical numbers of receptors are required for the nucleotide exchange activity to exceed the GTPase activity to produce sufficient active G-protein subunits for effector activation. For instance, morphine is a more efficacious μ-receptor agonist than methadone. Whereas the same cell always responded to morphine with stronger GIRK activation, it was not uncommon for the methadone-activated GIRK current in one cell to be larger than the morphine-activated current in another cell, probably because of a higher number of μ-receptors in the former cell. Similarly, morphine activates the μ-opioid receptor of neurons in brain slices (20) but not of the acutely dissociated neurons that have been enzymatically treated (21). This finding might be accounted for by a reduction of functional receptors in the acutely dissociated neurons.
Receptor activity can be altered by phosphorylation. Phosphorylated by βARK, receptors are less efficient in catalyzing the nucleotide exchange to generate Gβγ and induce smaller GIRK currents. Protein kinase C-dependent receptor phosphorylation also has been reported to uncouple receptors from the effectors (22, 23). Receptor phosphorylation, however, is not necessary for the generation of acute desensitization in our experiments. The mutant m2 receptor (m2RΔ) (11) without the βARK phosphorylation sites was still capable of activating currents that exhibited acute desensitization. Moreover, in the excised patch experiments, ATP was not included in the cytoplasmic solution and yet we still observed acute desensitization. The recovery of GIRK current from acute desensitization in the absence of ATP also argues against dephosphorylation of any signaling component as the primary cause of acute desensitization.
Guanine Nucleotide Composition Is a Critical Determinant of the Acute Desensitization.
GIRK channels are activated by βγ dimers. Thus, acute desensitization of GIRK currents may reflect the second-to-second changes in the amount of free βγ. As predicted from the G-protein turnover model, acute desensitization was more readily detected when the membrane patch was exposed to a nucleotide-free solution before encountering GTP, whereas it was less pronounced if the excised inside-out patch was first exposed to GDP then to GTP (Figs. 1 E and F, and 2B).
The receptor activity is another factor that influences the distribution of G proteins in different states before they are exposed to GTP. For instance, acute desensitization was not always observed by switching from the nucleotide-free solution to one containing GTP (Fig. 1C). This situation could arise if not enough receptors were activated to release GDP from all of the trimeric G proteins before GTP application, so that some G proteins were empty whereas others remained GDP bound and gave rise to GIRK activation with two components of different rates. On the other hand, when the receptor activity is high, some G proteins might remain in the empty state even when they are exposed to an excess of GDP, because of continuous dissociation of GDP catalyzed by receptors. The distribution between the GDP-bound and empty states is thus a dynamic equilibrium governed by the receptors.
It has been suggested that activation of GIRK channels by βγ exhibits positive cooperativity (15, 16), which may make GIRK channels more sensitive to small changes in the level of free Gβγ when free [βγ] is close to its binding affinity for the channels. Moreover, phosphatidylinositol bisphosphate (PIP2) may enhance the GIRK channel activation by Gβγ (24, 25) and hence affect the extent of acute desensitization of GIRK current. This hypothesis may explain the observation that cytoplasmic ATP potentiated the acute desensitization of GIRK currents in the excised patch membranes from atrial myocytes (4); PIP2 could be generated by lipid kinases to sensitize GIRK channel activation by Gβγ.
Nonhydrolyzable Analogues of GTP Eliminate Acute Desensitization by Disrupting the Cycle.
To test whether acute desensitization arises largely from the kinetic behavior of G-protein cycle, we disrupted the cycle by nonhydrolyzable analogues of GTP. If the acute desensitization were mediated by any second messengers downstream of G proteins, we might expect to observe even more robust desensitization by these nonhydrolyzable analogues. In contrast, we found that application of GppNHp produced larger GIRK currents than application of GTP, but without noticeable acute desensitization (Fig. 4C). Our results differ from the reported suppression of muscarinic potassium currents by GTPγS (4), which could arise from additional mechanisms in the atrial myocyte membranes. Nonetheless, the elimination of GIRK desensitization by nonhydrolyzable GTP analogues applied to excised oocyte membrane patches indicates that the G-protein exchange-hydrolysis cycle could account for acute desensitization.
Opposing Actions of RGS4 Result in Accentuated GIRK Desensitization.
We found that RGS4 promoted the acute desensitization in the membrane patches because of its GAP activity. The peak current amplitude was not reduced despite the GAP activity (Fig. 3B), thanks to the ability of RGS4 to enhance the receptor-mediated activation of G proteins. GTPγS-induced GIRK current was 5- to 10-fold larger when RGS4 was coexpressed while the rate of this GIRK activation depended on receptor activity, indicating that RGS4 increased the G-protein pool available for GIRK channel activation (Fig. 4 B and C). Is this enlarged G-protein pool a consequence of the GAP activity of RGS4 or up-regulation of G-protein expression? Does RGS4 accelerate the nucleotide exchange as well as increase the G-protein pool? Further study of this RGS function may shed light on these issues. The opposing effects of RGS4 may account for its ability to alter the kinetics but not the steady-state amplitudes of the GIRK currents.
An Additional State of the G Protein Implicated by the Acute Desensitization of Whole-Cell Current.
The exchange-hydrolysis cycle of G proteins in Fig. 2A can explain the acute desensitization in the excised inside-out patches but not in the whole-cell configuration, because the hypothetical empty state should not exist when abundant guanine nucleotides are present in the cytoplasm. Nonetheless, acute desensitization of GIRK current is also accentuated by RGS4 in the whole-cell recording (not shown), indicating that it arises from the G-protein cycle. Instead of including the empty state in the G-protein state as in Fig. 2A, one could envision a state (αGDPβγ*) that determines the readiness of G proteins for activation as soon as receptors are stimulated by agonists. The G proteins in such a state still will be GDP bound hence inactive for effector coupling but would have undergone some translocation or conformation change so that GDP will dissociate immediately after agonist application (Fig. 5). The inclusion of this G-protein state would lead to acute desensitization of whole-cell currents. The kinetics indicating the transition from αGDPβγ* to αGTP+βγ in the whole-cell configuration is expected to be slower than the transition from αEβγ to αGTP+βγ in the excised patch. It will be interesting to determine what the αGDPβγ* state represents physically, and whether RGS4 exerts its positive regulation on this state.
Figure 5.

A modified model to account for the acute desensitization in the whole-cell configuration. A “readiness” state (αGDPβγ*) needs to be introduced to explain the observed acute desensitization in the whole-cell recording. The corresponding cycle in the patch experiment is included on the left for comparison. R* indicates an agonist-bound active receptor.
Acknowledgments
We are grateful to Drs. Mark von Zastrow and Jennifer Whistler for providing the μ-receptor gene and for stimulating discussions throughout the course of this study. We thank our lab colleagues for comments on the manuscript. This study was supported by National Institutes of Health Grant NS15963. Y.N.J and L.Y.J. are Howard Hughes Medical Institute Investigators.
ABBREVIATIONS
- GIRK
G-protein gated inward rectifier K+ channel
- RGS
regulators of G-protein signaling
- GAP
GTPase-activating protein
- DAMGO
[d-Ala2,N-MePhe4,Gly5-ol]enkephalin
References
- 1. Lüscher C, Jan L Y, Stoffel M, Malenka R C, Nicoll R A. Neuron. 1997;19:687–695. doi: 10.1016/s0896-6273(00)80381-5. [DOI] [PubMed] [Google Scholar]
- 2.Wickman K D, Nemec J, Gendler S J, Clapham D E. Neuron. 1998;20:103–114. doi: 10.1016/s0896-6273(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 3.Kurachi Y, Nakajima T, Sugimoto T. Pflügers Arch. 1987;410:227–233. doi: 10.1007/BF00580270. [DOI] [PubMed] [Google Scholar]
- 4.Shui Z, Boyett M R, Zang W J. J Physiol (London) 1997;505:77–93. doi: 10.1111/j.1469-7793.1997.077bc.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shui Z, Boyett M R, Zang W J, Haga T, Kameyama K. J Physiol (London) 1995;487:359–366. doi: 10.1113/jphysiol.1995.sp020885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shui Z, Khan I A, Tsuga H, Haga T, Boyett M R. J Physiol (London) 1998;507:325–334. doi: 10.1111/j.1469-7793.1998.325bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soejima M, Noma A. Pflügers Arch. 1984;400:424–431. doi: 10.1007/BF00587544. [DOI] [PubMed] [Google Scholar]
- 8.Saitoh O, Kubo Y, Miyatani Y, Asano T, Nakata H. Nature (London) 1997;390:525–529. doi: 10.1038/37385. [DOI] [PubMed] [Google Scholar]
- 9.Doupnik C A, Davidson N, Lester H A, Kofuji P. Proc Natl Acad Sci USA. 1997;94:10461–10466. doi: 10.1073/pnas.94.19.10461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu Y, Zhang L, Yin X, Sun H, Uhl G R, Wang J B. J Biol Chem. 1997;272:28869–28874. doi: 10.1074/jbc.272.46.28869. [DOI] [PubMed] [Google Scholar]
- 11.Pals-Rylaarsdam R, Xu Y, Witt-Enderby P, Benovic J L, Hosey M M. J Biol Chem. 1995;270:29004–29011. doi: 10.1074/jbc.270.48.29004. [DOI] [PubMed] [Google Scholar]
- 12.Logothetis D E, Kurachi Y, Galper J, Neer E J, Clapham D E. Nature (London) 1987;325:321–326. doi: 10.1038/325321a0. [DOI] [PubMed] [Google Scholar]
- 13.Wickman K, Clapham D E. Physiol Rev. 1995;75:865–885. doi: 10.1152/physrev.1995.75.4.865. [DOI] [PubMed] [Google Scholar]
- 14.Reuveny E, Slesinger P A, Inglese J, Morales J M, Iniguez-Lluhi J A, Lefkowitz R J, Bourne H R, Jan Y N, Jan L Y. Nature (London) 1994;370:143–146. doi: 10.1038/370143a0. [DOI] [PubMed] [Google Scholar]
- 15.Kurachi Y, Ito H, Sugimoto T. Pflügers Arch. 1990;416:216–218. doi: 10.1007/BF00370247. [DOI] [PubMed] [Google Scholar]
- 16.Hosoya Y, Kurachi Y. In: Potassium Channels: Molecular Structure, Function, and Diseases. Kurachi Y, Jan L, Lazdunski M, editors. San Diego: Academic; 1998. , in press. [Google Scholar]
- 17.Berman D M, Wilkie T M, Gilman A G. Cell. 1996;86:445–452. doi: 10.1016/s0092-8674(00)80117-8. [DOI] [PubMed] [Google Scholar]
- 18.Dohlman H G, Apaniesk D, Chen Y, Song J P, Nusskern D. Mol Cell Biol. 1995;15:3635–3643. doi: 10.1128/mcb.15.7.3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koelle M R, Horvitz H R. Cell. 1996;84:115–125. doi: 10.1016/s0092-8674(00)80998-8. [DOI] [PubMed] [Google Scholar]
- 20.North R A, Williams J T. J Physiol (London) 1985;364:265–280. doi: 10.1113/jphysiol.1985.sp015743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ingram S, Wilding T J, McCleskey E W, Williams J T. Mol Pharmacol. 1997;52:136–143. doi: 10.1124/mol.52.1.136. [DOI] [PubMed] [Google Scholar]
- 22.Garcia D E, Brown S, Hille B, Mackie K. J Neurosci. 1998;18:2834–2841. doi: 10.1523/JNEUROSCI.18-08-02834.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gereau R W T, Heinemann S F. Neuron. 1998;20:143–151. doi: 10.1016/s0896-6273(00)80442-0. [DOI] [PubMed] [Google Scholar]
- 24.Huang C L, Feng S, Hilgemann D W. Nature (London) 1998;391:803–806. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- 25.Sui J L, Petit-Jacques J, Logothetis D E. Proc Natl Acad Sci USA. 1998;95:1307–1312. doi: 10.1073/pnas.95.3.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]