

Abstract
Nitroalkenes, the nitration products of unsaturated fatty acids formed via NO-dependent oxidative reactions, have been demonstrated to exert strong biological actions in endothelial cells and monocytes/macrophages; however, little is known about their effects on vascular smooth muscle cells (VSMCs). The present study examined the role of nitro-linoleic acid (LNO2) in the regulation of VSMC proliferation. We observed that LNO2 inhibited VSMC proliferation in a dose-dependent manner. In addition, LNO2 induced growth arrest of VSMCs in the G1/S phase of the cell cycle with an upregulation of the cyclin-dependent kinase inhibitor p27kip1. Furthermore, LNO2 triggered nuclear factor-erythroid 2-related factor 2 (Nrf2) nuclear translocation and activation of the antioxidant-responsive element-driven transcriptional activity via impairing Kelch-like ECH-associating protein 1 (Keap1)-mediated negative control of Nrf2 activity in VSMCs. LNO2 upregulated the expression of Nrf2 protein levels, but not mRNA levels, in VSMCs. A forced activation of Nrf2 led to an upregulation of p27kip1 and growth inhibition of VSMCs. In contrast, knock down of Nrf2 using an Nrf2 siRNA approach reversed the LNO2-induced upregulation of p27kip1 and inhibition of cellular proliferation in VSMCs. These studies provide the first evidence that nitroalkene LNO2 inhibits VSMC proliferation through activation of the Keap1/Nrf2 signaling pathway, suggesting an important role of nitroalkenes in vascular biology.
Keywords: nitroalkenes, nuclear factor-erythroid 2-related factor 2, Kelch-like ECH-associating protein 1
Recently, increasing evidence has revealed that nitroalkenes, the nitration products of unsaturated fatty acids formed via NO-dependent oxidative reactions (38, 43, 44), are a novel class of signaling molecules in the vasculature. This notion has been emerging since our initial observations that nitro-linoleicacid (LNO2) mediates pluripotent cell signaling actions, since it induces relaxation of phenylephrine-preconstricted rat aortic rings, inhibits aggregation of human platelets, and attenuates human neutrophil superoxide generation, degranulation, and integrin expression (14, 15, 33). Subsequent data from our laboratories has shown that there is a class of nitrated unsaturated fatty acids present in healthy human circulation with levels of LNO2 and nitro-oleic acid (OA-NO2) exceeding 1μM (3, 4). Also, LNO2 and OA-NO2 can activate the nuclear transcription factor peroxisome proliferator-activated receptor-γ (PPARγ; see Refs. 3 and 45). More recently, we have demonstrated that LNO2 and OA-NO2 appear to be potent electrophiles that mediate reversible nitroalkylation reactions both in vitro and in vivo (6). Accordingly, we observed that both LNO2 and OA-NO2 repress nuclear factor (NF)-κ B activity by direct nitroalkylation, inhibiting inflammatory responses in vascular cells, including monocytes/macrophages and endothelial cells (16). In addition, LNO2 stimulates a dramatic induction of heme oxygenase-1 (HO-1) in endothelial cells; however, the putative mediators are still unknown (49). To date, it is unclear whether LNO2 acts as an electrophile to induce HO-1 expression in endothelial cells. Also, the role of nitroalkenes in the regulation of vascular smooth muscle cell (VSMC) biology remains to be explored.
Nuclear factor-erythroid 2-related factor 2 (Nrf2) belongs to the Cap “n” Collar family of basic leucine zipper transcription factors that include NF-E2, Nrf1-3, and Bach1–2 (11, 13, 26, 28, 32). Nrf2 is a pleiotropic protein that binds to a cis-acting enhancer sequence known as the antioxidant response element (ARE), also referred to as the electrophile response element with a core nucleotide sequence of 5′-RTGACNNNGC-3′ to control the basal and inducible expression of a battery of antioxidant genes and other cytoprotective phase II detoxifying enzymes, such as HO-1, ferritin, Cu/Zn superoxide dismutase, glutathione peroxidase, glutathione-S-transferases, NAD(P)H-quinone oxidoreductase (NQO) 1, NQO2, γ-glutamylcysteine synthase, and glucuronosyltransferase. Nrf2 activity is principally governed by Kelch-like ECH-associating protein 1 (Keap1). Although the precise molecular mechanisms regarding regulation of Keap 1-Nrf2 homeostasis are still unclear, growing evidence supports the following working hypothesis: Nrf2 interaction with the actin-anchored Keap1 rapidly degrades Nrf2, thereby maintaining low basal expression of Nrf2-regulated genes under normal physiological conditions. However, upon recognition of chemical signals imparted by oxidative and eletrophilic molecules or other Nrf2-activating signals, Nrf2 escapes from Keap1-mediated proteasomal degradation, translocates to the nucleus, and transactivates the expression of several cytoprotective genes.
The beneficial effects of HO-1 have been well documented in the cardiovascular system (22, 36, 40); however, the contribution of Nrf2-operated HO-1 expression in cardiovascular remodeling under pathological status remains unknown (1). Interestingly, it has been recently demonstrated that Nrf2 might play important roles in the antioxidant defense or inflammation resolution in endothelial cells (8, 10, 12, 21) and macrophages (23, 24, 25, 31, 46) and the regulation of redox status, cellular survival, or proliferation in VSMCs (2, 23, 29, 34). These results strongly support the implication of Nrf2/ARE signaling in the pathogenesis of cardiovascular disease; however, the role of Nrf2-operated signaling in the pathophysiological responses in the vasculature has been less explored.
To our knowledge, the present study is the first to provide evidence that LNO2 activates the Keap1/Nrf2 signaling pathway, resulting in upregulation of p27kip1 and eventual growth arrest of VSMCs. Our results suggest that nitroalkenes might orchestrate Keap1/Nrf2 signaling to participate in the pathogenesis of cardiovascular disease.
METHODS
Cell culture and materials
Rat aortic smooth muscle cells (RASMCs) were prepared in thoracic aortic media of adult male rats by the explant methods, as originally described by Ross (42). Briefly, male Sprague-Dawley rats (Harlan, Indianapolis, IN) between the ages of 8 and 10 wk were anesthetized with pentobarbital sodium and killed by an overdose of the anesthetic. The aorta was excised rapidly, immersed in DMEM, and stripped of the adventitia with sterile forceps. The isolated aorta was longitudinally cut open, and the endothelium was removed by gently rubbing the intimal surface with sharp scissors. Thereafter, the aortic media was carefully removed under a dissecting microscope. Segments of aortic media were cut into 1-mm squares, placed on a T-25 tissue culture bottle, and grown in DMEM containing 5 mM glucose, 10% FBS, and antibiotics. The growth medium was changed every 2 days. RASMCs grew out from the explants 7–10 days after the tissue was removed. The subcultures of RASMCs from passages 7–8 were used in all experiments. The VSMCs prepared from the rats were controlled and not contaminated with fibroblasts or endothelial cells as evidenced by >99% positive immunostaining of smooth muscle-α-actin with a fluorescein isothiocyanate-conjugated-actin antibody (Sigma Chemical). Subconfluent VSMCs were incubated with Opti-MEM I (Invitrogen) for 48 h to induce a quiescent status. All animal experiments were performed according to National Institutes of Health guidelines and were approved by the Animal Care and Use Committee at the University of Michigan. Purification and quantitation of LNO2 were performed as described previously (4). Other reagents were purchased from Sigma (St. Louis, MO) unless specified.
Cell proliferation assay
VSMC proliferation was determined by different methods: 1) direct counting of the whole population of VSMCs by using a Coulter Counter (model ZF; Coulter Electronics, Hialeah, FL). For these experiments, the quiescent RASMCs cultured in six-well plates were switched to DMEM-F-12 containing 10% FCS as indicated and thereafter were detached with trypsin (0.025%)/EDTA (1 mM) for cell number determination; 2) using the cell proliferation reagent WST-1 (Roche), which measures the metabolic activity of viable cells by the formation of a formazan dye in the microplate with an ELISA plate reader. The absorbance at 450 nm directly correlates with the cell number. For these experiments, quiescent RASMCs cultured in 96-well plates were restimulated with DMEM-F-12 containing 2% FCS for 48 h. WST-1 was added for the last 4 h, and the absorbance was measured according to the manufacturer’s procedure; 3) DNA synthesis. Quiescent RASMCs were infected with the corresponding adenovirus for 24 h and replenished with DMEM-F-12 containing 10% FBS medium for an additional 48 h. The cells were pulsed with [3H]thymidine (1 μCi/ml; Sigma) for the last 4 h, washed three times with ice-cold PBS, and fixed with 10% TCA for 30 min at 4°C. The DNA was extracted with 0.2% SDS/0.2 N NaOH. Radioactivity was determined by scintillation counting.
FACS analysis
RASMCs (106 cells) were growth arrested with 0.5% FBS medium for 48 h. After serum deprivation, cells were treated with 2.5μ LNO2 or linoleic acid (LA) and then restimulated with medium containing 2% FBS for 24 h. RASMCs were collected by trypsinization, centrifuged (500 g × 5 min), and washed two times with PBS. Cell pellets were resuspended in 500 μl PBS, fixed in 1 ml ethanol, and stored at 4°C until analysis. Fixed cells were centrifuged (500 g × 5 min) and washed two times with PBS, resuspended in 500 μl staining solution (1 ml PBS, 50 ng propidium iodide, and 10 ng RNase A), incubated in the dark for 15 min at room temperature, and stored on ice. Flow cytometry studies were performed by using a FACScan flow cytometer (Becton Dickinson). The percentage of cells in different phases of the cell cycle was determined by using the ModFit LT V3.1 (Verity Software House) program.
Construction of adenovirus
Recombinant adenoviruses containing the human Nrf2 cDNA or Keap1 cDNA were prepared by the AdEasy system as previously described (18). A 6xMet-tag was added at the NH2-terminal region of the human Keap1 cDNA. The green fluorescent protein adenovirus (Ad-GFP) was used as the control in this study.
Transfection and reporter gene assay
RASMCs at ~85% confluence in 24-well plates were transiently cotransfected using LipofectAMINE 2000 (Invitrogen, Carlsbad, CA) with a plasmid containing the luciferase gene under the control of three tandem AREs in the pGL3 basic vector (Promega, Madison, WI), as described previously (12). The pRL-TK vector was used as an internal control for transfection. After transfection (24 h), cells were cultured for 4 h in Opti-MEM I and then treated with various stimuli as indicated for an additional 24 h. Each transfection was performed in triplicate at least three times. Reporter luciferase assay kits (Promega) were used to measure the luciferase activity of cells according to the manufacturer’s instructions with a luminometer (Victor II; Perkin-Elmer, Wellesley, MA).
Knock down of Nrf2
The small-interfering RNA (siRNA) sequences utilized targeted the following rat Nrf2 coding sequences: 5′-ACGCAGGAGAGGGAAGAATAAAGTT-3′, 5′-TGGAGCAA-GACTTGGGCCACTTAAA-3′, and 5′-GGAAACCTTACTCTCCC-AGTGAGTA-3′, corresponding to nucleotides at positions starting at +1,548, +1,619, and +1,763, respectively, of the rat Nrf2 mRNA. All siRNA duplexes were synthesized by Stealth RNAi (Invitrogen). RASMC were transfected with 100 nmol of each siRNA duplex using Lipofectamine 2000 and Opti-MEM for 24 h according to the manufacturer’s recommendations. Control experiments were performed using equivalent amounts of the StealthTM RNAi Negative Control Med GC (Invitrogen).
Immunochemical staining and Western blot analysis
RASMCs were cultured in a four-chamber glass slide (BD Falcon, Bedford, MA) and infected with Ad-GFP and Ad-Nrf2 for 48 h in Opti-MEM. Next, the cells were washed with PBS and fixed with 4% paraformaldehyde-PBS (20 min), permeabilized with 0.5% Triton X-100 in PBS-0.5% BSA (20 min), and blocked with PBS-1% BSA (1 h). Thereafter, cells were incubated with Nrf2 antibody, diluted 1:200 in PBS-1% BSA at 4°C overnight followed by an Alexa Fluor 594-labeled goat anti-rabbit IgG (Molecular Probes, Eugene, OR), and diluted 1:1,000 in PBS for 1 h. Nuclei were stained with Hoechst 33342 (10 μg/ml; Molecular Probes) for 10 min in PBS. Cells were washed with PBS and mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA). Immunofluorescence was visualized with a Nikon Eclipse TE-2000 inverted microscope (Melville, NY) and analyzed with a MetaMorph Imaging system (Molecular Devices, Downingtown, PA). Western blotting of whole cell lysates was performed as previously described (16). Cytoplasmic and nuclear fractions were obtained using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce) following the manufacturer’s instructions. Primary antibodies of Nrf2 (sc-13032; 1:200), p27kip1 (sc-528; 1:200), cyclin D1 (sc-718; 1:200), cyclin E (sc-481; 1:200), CDK4 (sc-260; 1:200), and lamin B1 (sc-20682; 1:200) were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies of β-tubulin (1:1,000) were from Upstate Biotechnology.
Statistical analysis
All data were evaluated with a two-tailed, unpaired Student’s t-test or compared by one-way ANOVA followed by a Fisher t-test. Values are expressed as mean ± SD. A value of P < 0.05 was considered statistically significant.
RESULTS
LNO2 inhibits VSMC proliferation with cell cycle arrest of G1/S phase and induction of the p27kip1 protein expression
As an initial step to define the role of LNO2 in VSMCs, we analyzed the effect of LNO2 treatment on proliferation of cultured RASMCs. As shown is Fig. 1A, LNO2 inhibited FBS-induced VSMC proliferation in a dose-dependent manner. The antiproliferative effect of LNO2 in VSMCs is specific of the nitroalkene moiety, since equivalent concentrations of LA did not inhibit serum-mediated VSMC proliferation. Detailed analysis of cell-cycle protein expression indicated that LNO2 dose dependently upregulated the cyclin-dependent kinase inhibitor p27kip1 without affecting expression levels of cyclin D1 and E or cyclin-dependent kinase 4 (Fig. 1B). No change was observed in LA-treated cells.
Fig. 1.

Nitro-linoleic acid (LNO2) induces growth arrest of vascular smooth muscle cells (VSMCs) via upregulation of cyclin-dependent kinase inhibitor (CDKI) p27kip1. A: quiescent rat aortic smooth muscle cells (RASMCs) were stimulated as indicated for 48 h and then subjected to WST-1 assay as described in METHODS. Values are means ± SD (n = 6 experiments). *P < 0.05 vs. 2% FBS alone. B: quiescent RASMCs were treated in growth medium (2% FBS) with LNO2, linoleic acid (LA), and ethanol control (0.1%) for 24 h at the concentrations indicated and then subjected to Western blot analysis with the antibodies indicated. Equal protein loading was determined by probing the membrane with an anti-β-tubulin antibody. The results were representative of three separate experiments. C, control. C: RASMC were treated as indicated, and cell cycle distribution was determined by FACS analysis as described in METHODS. Values are means ± SD (n = 6). *P < 0.05 vs. control.
In addition, the effect of LNO2 on cell cycle progression of RASMCs was determined using flow cytometry analysis. As shown in Fig. 1C, LNO2 (2.5 μM)-treated RASMCs exhibited a higher G1 phase population but a decreased S phase population compared with control cells. The S phase population was significantly lower in the LNO2-treated cells than in the control cells 24 h after serum restimulation (2% FBS). Conversely, the G1 phase population was lower in control cells than in LNO2-treated cells. LA treatment did not alter cell cycle population compared with control cells. No significant sub-G1 population or apoptotic cells were observed.
LNO2 activates Keap1/Nrf2 pathway in VSMCs
To further elucidate the underlying molecular mechanisms, we focused on the Keap1/Nrf2 pathway because, as aforementioned, 1) the biochemical properties of nitroalkenes as electrophiles indicate that LNO2 may activate Nrf2 via nitroalkylation of Keap1 and 2) Nrf2 overexpression inhibits VSMC proliferation. In RASMCs, we observed that LNO2 triggered Nrf2 nuclear translocation in a dose-dependent manner with the activation of ARE-driven transcriptional activity, whereas LA had no effect (Fig. 2, A and B). The potency of LNO2 on activation of ARE transcriptional activity was comparable with sulforaphane, a well-established inducer of ARE transcription (data not shown; see Ref. 35). These results indicate that LNO2 activates Nrf2/ARE signaling in VSMCs. Next, we examined whether LNO2 drives Nrf2/ARE signaling by interfering with the Keap1-mediated inhibition of Nrf2 in RASMCs. The data in Fig. 2C show that adenoviral overexpression of Keap1 resulted in decreased basal ARE transcriptional activity in RASMCs that could be partially restored by LNO2. Consistent with these results, LNO2 was still able to induce Nrf2 nuclear translocation in RASMCs overexpressing Keap1 (Fig. 2D). However, in the Keap1 overexpressed cells, both LNO2-induced Nrf2 nuclear translocation and ARE activity are substantially lower compared with control GFP-expressing cells (Fig. 2, C and D). These results demonstrate that Nrf2-driven ARE signaling is under negative control by Keap1 in VSMCs. LNO2 triggers Nrf2 nuclear translocation and then activates ARE-response gene transcription by impairing the ability of Keap1 to inhibit Nrf2.
Fig. 2.
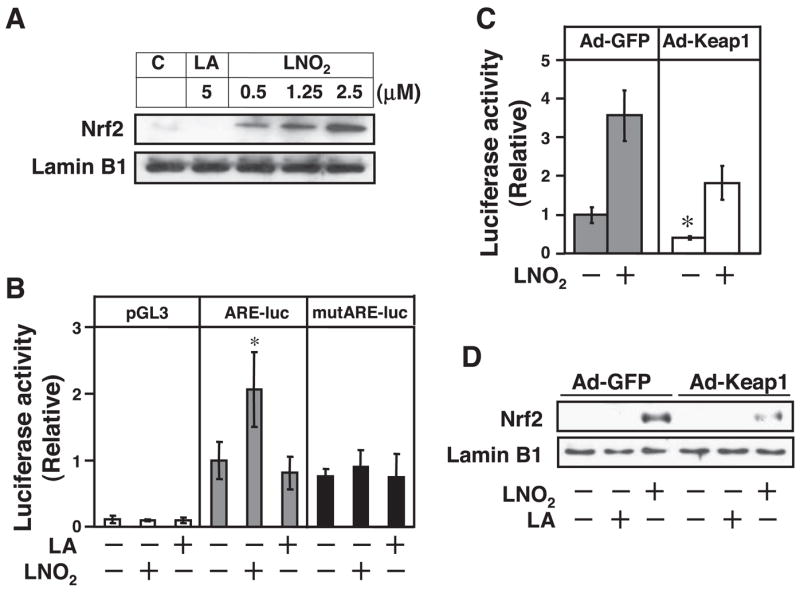
LNO2 activates Kelch-like ECH-associating protein 1 (Keap1)/nuclear factor-erythroid 2-related factor 2 (Nrf2) pathway in VSMCs. A: quiescent RASMCs were stimulated with LNO2 (2.5 μM), LA (2.5 μM), or ethanol (0.1%) as control solvent for 3 h, and then nuclear translocation of Nrf2 was analyzed as described in METHODS. B: 3× antioxidant response element (ARE)-luc and 3× mutARE-luc reporter vectors were transiently transfected into RASMCs. Cells were then treated with LNO2 (2.5 μM), LA (2.5 μM), or ethanol (0.1%) as control solvent for 24 h, and luciferase activity was measured. Values are means ± SD (n = 6). *P < 0.05 vs. control (−) in the same experimental groups. C: RASMCs were infected with green fluorescent protein adenovirus [Ad-GFP; 10 plaque-forming units (pfu)/cell] and Ad-Keap1 (10 pfu/cell) and then transiently transfected with 3× ARE-Luc. After the transfection (24 h), the cells were treated with or without LNO2 (2.5 μmol/l) for 24 h, and luciferase activity was measured. Values are means ± SD (n = 6). *P < 0.05 vs. Ad-GFP (−). D: RASMCs infected with Ad-GFP (10 pfu/cell) or Ad-Keap1 (10 pfu/cell) were stimulated with LNO2 (2.5 μM), LA (2.5 μM), or ethanol (0.1%) for 1 h, and Nrf2 nuclear protein levels were determined by Western blot. The result is representative of three separate experiments.
LNO2 upregulates the expression of Nrf2 protein levels in VSMCs
Because the Keap1-dependent negative regulation of Nrf2 activity mainly occurs at the posttranscriptional level of Nrf2, we examined the effect of LNO2 on Nrf2 expression in VSMCs. The expression of Nrf2 protein was barely detectable in RASMCs; however, LNO2 increased the expression of Nrf2 protein time dependently, reaching a peak at 1 h and declining thereafter until 16 h (Fig. 3). LA did not affect the protein level of Nrf2 in RASMCs (Fig. 3). The mRNA level of Nrf2 was not changed by either LNO2 or LA in RASMCs (data not shown). These results demonstrate a posttranscriptional mechanism of Nrf2 regulation by LNO2 in VSMCs. It is conceivable that LNO2 enables Nrf2 to escape Keap1-dependent degradation, leading to stabilization of Nrf2 and eventual activation of Nrf2/ARE signaling in VSMCs.
Fig. 3.

LNO2 increases Nrf2 protein expression in VSMCs. RASMCs were serum deprived for 24 h and subsequently treated with LNO2 (2.5 μM) or LA (2.5 μM) as indicated. Nrf2 protein expression was determined by Western blot analysis. The same membranes were reproved for β-tubulin to confirm equal loading. The results were representative of three separate experiments. SF16, serum free medium at 16 h used as a negative control; Ad-Nrf2, positive control using cell lysate from adenoviral Nrf2-infected RASMCs.
Activation of Nrf2 upregulates p27kip1 expression that is under a negative control of Keap1 in VSMCs
It is well-established that p27kip1 is a potent inhibitor of VSMC growth (9, 47). Thus we determined whether LNO2-activated Nrf2 upregulates p27kip1, leading to growth arrest in VSMCs. Forced activation of Nrf2, a process that triggers its nuclear translocation and subsequent transcriptional activity, was achieved by overexpression of Nrf2 (27). Accordingly, we generated an activated status of Nrf2 in RASMCs through adenoviral overexpression, which was evident because of enhanced protein expression levels and nuclear translocation of Nrf2 (Fig. 4A). Indeed, overexpression of Nrf2 by Ad-Nrf2 (10 plaque-forming units/cell) upregulated p27kip1 protein levels, which were compatible to the levels induced by LNO2 (2.5 μM) in RASMCs (Fig. 4B). The activation of Nrf2 time dependently upregulated p27kip1 protein levels in RASMCs (Fig. 4C). In contrast, ectopic expression of Keap1 attenuated the upregulation of p27kip1 by Nrf2 in a dose-dependent manner (Fig. 4D). These results demonstrate that Keap1/Nrf2 signaling regulates p27kip1 expression in VSMCs, suggesting that Nrf2 activation by LNO2 upregulates p27kip1 in VSMCs.
Fig. 4.

Keap1-Nrf2 interaction is linked with p27kip1 expression in VSMCs. A: top, RASMCs infected with Ad-GFP (10 pfu/cell) and Ad-Nrf2 (10 pfu/cell) for 48 h were subjected to immunochemical staining with an anti-Nrf2 antibody and anti-rabbit IgG conjugated with an Alexa Fluor 594. The images are representative of three separate experiments. Bottom: representative result of Nrf2 protein expression in RASMCs infected with Ad-Nrf2. B: RASMCs were infected with Ad-GFP (10 pfu/cell) or Ad-Nrf2 (10 pfu/cell) for 48 h. The Ad-GFP-infected cells were treated with or without LNO2 (2.5 μM). Expression of p27kip1 was determined by Western blot. C: RASMCs were infected with Ad-GFP (10 pfu/cell) or Ad-Nrf2 (10 pfu/cell) for 48 h. Expression of p27kip1 was determined by Western blot. D: RASMCs were infected with Ad-Nrf2 (10 pfu/cell) or Ad-GFP (10 pfu/cell), and with Ad-Keap1 (10 and 30 pfu/cell, respectively) for 48 h. The cell lysates were subjected to analysis of p27kip1 protein expression by Western blot. The results were representative of three separate experiments.
Nrf2 is crucial for LNO2-induced growth inhibition of VSMCs
To address the functional consequence of LNO2-mediated Nrf2 activation in VSMC proliferation, we examined the effects of a forced activation of Nrf2 on cellular growth in RASMCs. Forced Nrf2 activation led to inhibition of VSMC growth as determined by [3H]thymidine incorporation (Fig. 5A) and a total cell count (Fig. 3B). Consistent with these results, Levonen et al. (29) have recently shown that Nrf2 overexpression inhibits proliferation of cultured human and rabbit VSMCs. To further determine the role of Nrf2 in LNO2-mediated inhibition of VSMC proliferation, we applied an Nrf2 loss-of-function approach using Nrf2 siRNA in RASMCs. Because we have observed that the basal protein expression level of Nrf2 is very low (Fig. 3), the efficacy of the Nrf2 siRNA approach was determined in the RASMCs with an upregulation of Nrf2 protein levels by LNO2. As shown in Fig. 6A, the LNO2-induced upregulation of Nrf2 was almost blocked by transfection with Nrf2 siRNA duplexes targeting the rat Nrf2 mRNA in RASMCs. Accordingly, LNO2-mediated upregulation of p27kip1 and inhibition of VSMC proliferation were abrogated by using an Nrf2 siRNA approach (Fig. 6, B and C). These data demonstrate that LNO2 activates Nrf2 to inhibit VSMC growth.
Fig. 5.
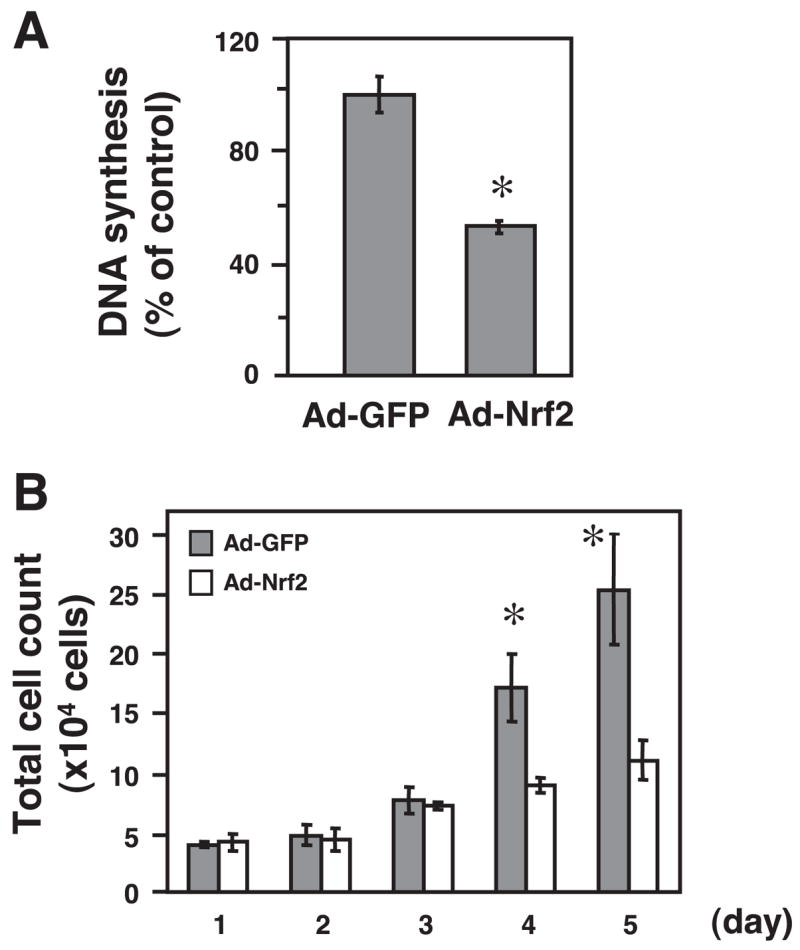
Nrf2 overexpression inhibits VSMC proliferation. The infected RASMCs (10 pfu/cell) were subjected to cellular proliferation assays. A: cell numbers were measured by Coulter counter after indicated periods. Values are means ± SD (n = 6). *P < 0.05 vs. Ad-Nrf2 in the same group. B: 1 day after the adenoviral infection, DNA synthesis in these cells was assessed by [3H]thymidine incorporation. Values are means ± SD (n = 3). *P < 0.05 vs. Ad-GFP.
Fig. 6.

Knock down of Nrf2 abolishes LNO2-mediated growth inhibition in VSMCs. A: RASMCs were transiently transfected with Nrf2 small-interfering RNA (siRNA) duplexes for 24 h and then were treated with LNO2 (2.5 μM) or LA (2.5 μM) for 1 h. Cell lysates were subjected to analysis of Nrf2 protein expression by Western blot. B: RASMCs with knock down of Nrf2 as described above were stimulated as indicated for 48 h. Expression of p27kip1 was analyzed by Western blot. The results are representative of two separate experiments. C: RASMCs with knock down of Nrf2 as described above were stimulated as indicated for 48 h. RASMC proliferation was assessed by WST-1 assay as described in METHODS. Values are means ± SD (n = 6). *P < 0.05 vs. 2% FBS-treated cells.
In summary, our results have demonstrated that the nitroalkene derivative of LNO2 inhibits VSMC proliferation through upregulation of p27kip1. We have provided evidence that LNO2 impairs Keap1-mediated inhibition of Nrf2 and stabilizes Nrf2 protein expression, which therefore leads to enhancement of Nrf2 translocation to the nucleus. Furthermore, we demonstrate that Nrf2 activation upregulates p27kip1 and inhibits cellular proliferation in VSMCs. Nrf2 is crucial for the LNO2-induced growth inhibition of VSMCs. Taken together, these data indicate that LNO2 drives the Keap1/Nrf2 pathway to upregulate p27kip1, leading to growth arrest in VSMCs.
DISCUSSION
It is firmly established that protein tyrosine nitration under diseased conditions represents a shift from the signal-transducing physiological actions of NO to oxidative and potentially pathogenic pathways (5, 41); however, the production of nitrated lipids under biological conditions, structural isomer distribution, and biological signaling actions are either not considered or incompletely characterized (19, 20, 37, 48). A series of studies from our laboratories have demonstrated that nitroalkene derivatives of LNO2 and OA-NO2 are present in healthy human circulation with levels exceeding 1 μM. These levels are sufficient to inhibit the activation of platelets, oxidative stress in leukocytes, proinflammmatory cytokine secretion in macrophages, and the inflammatory tumor necrosis factor-α-mediated monocyte adhesion to endothelial cells. In the present study, we demonstrate for the first time that physiological levels of LNO2 inhibit VSMC proliferation. Because all of these nitroalkene-mediated actions in vascular cells are beneficial for ameliorating the pathophysiological process of various vascular diseases (17, 30, 39), it is highly appreciable that nitroalkene derivatives of LNO2 and OA-NO2 are novel endogenous modulators that maintain functional integrity of the different cells in the vasculature, thereby regulating cardiovascular homeostasis. Further analysis of the underlying mechanisms and pathophysiological relevance of these nitroalkene-mediated actions will provide new insight into not only biological functions of nitroalkenes but also development of novel therapeutic strategies for cardiovascular disease.
Our previous studies have demonstrated that LNO2 or/and OA-NO2 directly activate PPARγ, induce HO-1 expression, and inhibit NF-κB activity (3, 16, 45 49). They also appear to be potent electrophiles that mediate reversible nitroalkylation reactions with target proteins (6). However, it is likely the electrophilic nature of LNO2- and OA-NO2-mediated alkylation of NF-κB, rather than PPARγ activation or HO-1 induction, contributes to their anti-inflammatory actions in monocyte/macrophages and endothelial cells (16). It has been demonstrated that the elevated cysteine content of Keap1 is highly reactive to electrophiles (26, 28), and such a direct interaction between electrophiles with Keap1 impairs the Keap1-mediated inhibition of Nrf2/ARE signaling. In the present study, we have shown that LNO2 might employ similar mechanisms to activate Nrf2 in VSMCs. LNO2 triggered Nrf2 nuclear translocation via bypassing the Keap1 negative control gate. Consequently, ARE transcriptional activity was increased, resulting in the upregulation of p27kip1 and eventual VSMC growth arrest. As aforementioned, HO-1 is one of the Nrf2/ARE response genes. The finding of LNO2-mediated activation of Keap1/Nrf2/ARE signaling provides, at least in part, the molecular basis for the antioxidative properties of nitroalkenes in our previous observations (49). These results suggest that the biological effects of nitroalkenes in vascular cells may be partially due to their electrophilic-dependent nitroalkylation of target proteins, such as NF-κB and Keap1.
In addition, Nrf2/ARE signaling is regulated by interaction with other factors, such as the small Maf protein that is used for ARE binding. The recruitment of cofactors for transcriptional activation of target genes may be critical for generating cross-talk with other signaling cascades involving CCAAT/enhancer-binding protein β and PPARγ (13, 26, 28, 32). These interactions might lead to a special activation of downstream events resulting in differential biological consequences. For example, there is a functional codependence between Nrf2/HO-1 and phosphatidylinositol 3-kinase/protein kinase B in mediating cytoprotection against oxidative stress-induced cell death in VSMCs (7). Thus a careful study of nitroalkene-mediated Keap1/Nrf2/ARE signaling in VSMCs is recommended in future investigations.
The levels of nitroalkenes appear to be increased under pathophysiological conditions via a complex NO-dependent redox signaling mechanism, thus terminating the pathogenic chain propagation reactions of lipid radicals (43, 44). It is likely that nitroalkene levels might be elevated during vascular remodeling associated with various disorders. In addition, preliminary results from a separate experiment have revealed that Nrf2 expression is upregulated in rat carotid arteries after balloon injury (data not shown). Therefore, it is possible that endogenous nitroalkenes operate Keap1/Nrf2/ARE signaling to protect the vasculature from damage.
Acknowledgments
GRANTS
This work was supported by American Diabetes Association Grant JFA 7-05-JF-12 (T. Cui) and National Heart, Lung, and Blood Institute Grants HL-068878 and HL-075397 (Y. E. Chen) and HL-58115 and HL-64937 (B. A. Freeman). L. Villacorta and M. T. Garcia-Barrio were supported by a postdoctoral fellowship (0525495B) and a Beginning Grant-in-Aid (0465202B) from the American Heart Association Southeast Affiliate, respectively.
References
- 1.Alam J, Cook JL. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am J Respir Cell Mol Biol. 2007;36:166–174. doi: 10.1165/rcmb.2006-0340TR. [DOI] [PubMed] [Google Scholar]
- 2.Anwar AA, Li FY, Leake DS, Ishii T, Mann GE, Siow RC. Induction of heme oxygenase 1 by moderately oxidized low-density lipoproteins in human vascular smooth muscle cells: role of mitogen-activated protein kinases and Nrf2. Free Radic Biol Med. 2005;39:227–236. doi: 10.1016/j.freeradbiomed.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 3.Baker PR, Lin Y, Schopfer FJ, Woodcock SR, Groeger AL, Batthyany C, Sweeney S, Long MH, Iles KE, Baker LM, Branchaud BP, Chen YE, Freeman BA. Fatty acid transduction of nitric oxide signaling: multiple nitrated unsaturated fatty acid derivatives exist in human blood and urine and serve as endogenous peroxisome proliferator-activated receptor ligands. J Biol Chem. 2005;280:42464–42475. doi: 10.1074/jbc.M504212200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker PR, Schopfer FJ, Sweeney S, Freeman BA. Red cell membrane and plasma linoleic acid nitration products: synthesis, clinical identification, and quantitation. Proc Natl Acad Sci USA. 2004;101:11577–11582. doi: 10.1073/pnas.0402587101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baldus S, Eiserich JP, Brennan ML, Jackson RM, Alexander CB, Freeman BA. Spatial mapping of pulmonary and vascular nitrotyrosine reveals the pivotal role of myeloperoxidase as a catalyst for tyrosine nitration in inflammatory diseases (Abstract) Free Radic Biol Med. 2002;33:1010. doi: 10.1016/s0891-5849(02)00993-0. [DOI] [PubMed] [Google Scholar]
- 6.Batthyany C, Schopfer FJ, Baker PR, Duran R, Baker LM, Huang Y, Cervenansky C, Branchaud BP, Freeman BA. Reversible posttranslational modification of proteins by nitrated fatty acids in vivo. J Biol Chem. 2006;281:20450–20463. doi: 10.1074/jbc.M602814200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brunt KR, Fenrich KK, Kiani G, Tse MY, Pang SC, Ward CA, Melo LG. Protection of human vascular smooth muscle cells from H2O2-induced apoptosis through functional codependence between HO-1 and AKT. Arterioscler Thromb Vasc Biol. 2006;26:2027–2034. doi: 10.1161/01.ATV.0000236204.37119.8d. [DOI] [PubMed] [Google Scholar]
- 8.Buckley BJ, Marshall ZM, Whorton AR. Nitric oxide stimulates Nrf2 nuclear translocation in vascular endothelium. Biochem Biophys Res Commun. 2003;307:973–979. doi: 10.1016/s0006-291x(03)01308-1. [DOI] [PubMed] [Google Scholar]
- 9.Chen D, Krasinski K, Sylvester A, Chen J, Nisen PD, Andres V. Downregulation of cyclin-dependent kinase 2 activity and cyclin A promoter activity in vascular smooth muscle cells by p27(KIP1), an inhibitor of neointima formation in the rat carotid artery. J Clin Invest. 1997;99:2334–2341. doi: 10.1172/JCI119414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen XL, Dodd G, Thomas S, Zhang X, Wasserman MA, Rovin BH, Kunsch C. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am J Physiol Heart Circ Physiol. 2006;290:H1862–H1870. doi: 10.1152/ajpheart.00651.2005. [DOI] [PubMed] [Google Scholar]
- 11.Chen XL, Kunsch C. Induction of cytoprotective genes through Nrf2/antioxidant response element pathway: a new therapeutic approach for the treatment of inflammatory diseases. Curr Pharm Des. 2004;10:879–891. doi: 10.2174/1381612043452901. [DOI] [PubMed] [Google Scholar]
- 12.Chen XL, Varner SE, Rao AS, Grey JY, Thomas S, Cook CK, Wasserman MA, Medford RM, Jaiswal AK, Kunsch C. Laminar flow induction of antioxidant response element-mediated genes in endothelial cells. A novel anti-inflammatory mechanism. J Biol Chem. 2003;278:703–711. doi: 10.1074/jbc.M203161200. [DOI] [PubMed] [Google Scholar]
- 13.Cho HY, Reddy SP, Kleeberger SR. Nrf2 defends the lung from oxidative stress. Antioxid Redox Signal. 2006;8:76–87. doi: 10.1089/ars.2006.8.76. [DOI] [PubMed] [Google Scholar]
- 14.Coles B, Bloodsworth A, Clark SR, Lewis MJ, Cross AR, Freeman BA, O’Donnell VB. Nitrolinoleate inhibits superoxide generation, degranulation, and integrin expression by human neutrophils: novel antiinflammatory properties of nitric oxide-derived reactive species in vascular cells. Circ Res. 2002;91:375–381. doi: 10.1161/01.res.0000032114.68919.ef. [DOI] [PubMed] [Google Scholar]
- 15.Coles B, Bloodsworth A, Eiserich JP, Coffey MJ, McLoughlin RM, Giddings JC, Lewis MJ, Haslam RJ, Freeman BA, O’Donnell VB. Nitrolinoleate inhibits platelet activation by attenuating calcium mobilization and inducing phosphorylation of vasodilator-stimulated phospho-protein through elevation of cAMP. J Biol Chem. 2002;277:5832–5840. doi: 10.1074/jbc.M105209200. [DOI] [PubMed] [Google Scholar]
- 16.Cui T, Schopfer FJ, Zhang J, Chen K, Ichikawa T, Baker PR, Batthyany C, Chacko BK, Feng X, Patel RP, Agarwal A, Freeman BA, Chen YE. Nitrated fatty acids: endogenous anti-inflammatory signaling mediators. J Biol Chem. 2006;281:35686–35698. doi: 10.1074/jbc.M603357200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dzau VJ, Braun-Dullaeus RC, Sedding DG. Vascular proliferation and atherosclerosis: new perspectives and therapeutic strategies. Nat Med. 2002;8:1249–1256. doi: 10.1038/nm1102-1249. [DOI] [PubMed] [Google Scholar]
- 18.Fu M, Zhang J, Tseng YH, Cui T, Zhu X, Xiao Y, Mou Y, De Leon H, Chang MM, Hamamori Y, Kahn CR, Chen YE. Rad GTPase attenuates vascular lesion formation by inhibition of vascular smooth muscle cell migration. Circulation. 2005;111:1071–1077. doi: 10.1161/01.CIR.0000156439.55349.AD. [DOI] [PubMed] [Google Scholar]
- 19.Goodwin DC, Landino LM, Marnett LJ. Effects of nitric oxide and nitric oxide-derived species on prostaglandin endoperoxide synthase and prostaglandin biosynthesis. FASEB J. 1999;13:1121–1136. doi: 10.1096/fasebj.13.10.1121. [DOI] [PubMed] [Google Scholar]
- 20.Goodwin DC, Landino LM, Marnett LJ. Reactions of prostaglandin endoperoxide synthase with nitric oxide and peroxynitrite. Drug Metab Rev. 1999;31:273–294. doi: 10.1081/dmr-100101918. [DOI] [PubMed] [Google Scholar]
- 21.Hosoya T, Maruyama A, Kang MI, Kawatani Y, Shibata T, Uchida K, Warabi E, Noguchi N, Itoh K, Yamamoto M. Differential responses of the Nrf2-Keap1 system to laminar and oscillatory shear stresses in endothelial cells. J Biol Chem. 2005;280:27244–27250. doi: 10.1074/jbc.M502551200. [DOI] [PubMed] [Google Scholar]
- 22.Immenschuh S, Schroder H. Heme oxygenase-1 and cardiovascular disease. Histol Histopathol. 2006;21:679–685. doi: 10.14670/HH-21.679. [DOI] [PubMed] [Google Scholar]
- 23.Ishii T, Itoh K, Ruiz E, Leake DS, Unoki H, Yamamoto M, Mann GE. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: activation by oxidatively modified LDL and 4-hydroxynonenal. Circ Res. 2004;94:609–616. doi: 10.1161/01.RES.0000119171.44657.45. [DOI] [PubMed] [Google Scholar]
- 24.Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, Yamamoto M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem. 2000;275:16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- 25.Itoh K, Mochizuki M, Ishii Y, Ishii T, Shibata T, Kawamoto Y, Kelly V, Sekizawa K, Uchida K, Yamamoto M. Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-deoxy-delta(12,14)-prostaglandin j(2) Mol Cell Biol. 2004;24:36–45. doi: 10.1128/MCB.24.1.36-45.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2006;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 27.Kobayashi A, Kang MI, Watai Y, Tong KI, Shibata T, Uchida K, Yamamoto M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol Cell Biol. 2006;26:221–229. doi: 10.1128/MCB.26.1.221-229.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kobayashi M, Yamamoto M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv Enzyme Regul. 2006;46:113–140. doi: 10.1016/j.advenzreg.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 29.Levonen AL, Inkala M, Heikura T, Jauhiainen S, Jyrkkanen HK, Kansanen E, Maatta K, Romppanen E, Turunen P, Rutanen J, Yla-Herttuala S. Nrf2 gene transfer induces antioxidant enzymes and suppresses smooth muscle cell growth in vitro and reduces oxidative stress in rabbit aorta in vivo. Arterioscler Thromb Vasc Biol. 2007;27:741–747. doi: 10.1161/01.ATV.0000258868.80079.4d. [DOI] [PubMed] [Google Scholar]
- 30.Ley K. Molecular mechanisms of leukocyte recruitment in the inflammatory process. Cardiovasc Res. 1996;32:733–742. [PubMed] [Google Scholar]
- 31.Li N, Alam J, Venkatesan MI, Eiguren-Fernandez A, Schmitz D, Di Stefano E, Slaughter N, Killeen E, Wang X, Huang A, Wang M, Miguel AH, Cho A, Sioutas C, Nel AE. Nrf2 is a key transcription factor that regulates antioxidant defense in macrophages and epithelial cells: protecting against the proinflammatory and oxidizing effects of diesel exhaust chemicals. J Immunol. 2004;173:3467–3481. doi: 10.4049/jimmunol.173.5.3467. [DOI] [PubMed] [Google Scholar]
- 32.Li N, Nel AE. Role of the Nrf2-mediated signaling pathway as a negative regulator of inflammation: implications for the impact of particulate pollutants on asthma. Antioxid Redox Signal. 2006;8:88–98. doi: 10.1089/ars.2006.8.88. [DOI] [PubMed] [Google Scholar]
- 33.Lima ES, Di Mascio P, Rubbo H, Abdalla DS. Characterization of linoleic acid nitration in human blood plasma by mass spectrometry. Biochemistry. 2002;41:10717–10722. doi: 10.1021/bi025504j. [DOI] [PubMed] [Google Scholar]
- 34.Liu XM, Peyton KJ, Ensenat D, Wang H, Schafer AI, Alam J, Durante W. Endoplasmic reticulum stress stimulates heme oxygenase-1 gene expression in vascular smooth muscle. Role in cell survival. J Biol Chem. 2005;280:872–877. doi: 10.1074/jbc.M410413200. [DOI] [PubMed] [Google Scholar]
- 35.Morimitsu Y, Nakagawa Y, Hayashi K, Fujii H, Kumagai T, Nakamura Y, Osawa T, Horio F, Itoh K, Iida K, Yamamoto M, Uchida K. A sulforaphane analogue that potently activates the Nrf2-dependent de-toxification pathway. J Biol Chem. 2002;277:3456–3463. doi: 10.1074/jbc.M110244200. [DOI] [PubMed] [Google Scholar]
- 36.Morita T. Heme oxygenase and atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:1786–1795. doi: 10.1161/01.ATV.0000178169.95781.49. [DOI] [PubMed] [Google Scholar]
- 37.Napolitano A, Camera E, Picardo M, d’Ishida M. Reactions of hydro(pero)xy derivatives of polyunsaturated fatty acids/esters with nitrite ions under acidic conditions. Unusual nitrosative breakdown of methyl 13-hydro(pero)xyoctadeca-9,11-dienoate to a novel 4-nitro-2-oximinoalk-3-enal product. J Org Chem. 2002;67:1125–1132. doi: 10.1021/jo015973b. [DOI] [PubMed] [Google Scholar]
- 38.O’Donnell VB, Eiserich JP, Chumley PH, Jablonsky MJ, Krishna NR, Kirk M, Barnes S, Darley-Usmar VM, Freeman BA. Nitration of unsaturated fatty acids by nitric oxide-derived reactive nitrogen species peroxynitrite, nitrous acid, nitrogen dioxide, and nitronium ion. Chem Res Toxicol. 1999;12:83–92. doi: 10.1021/tx980207u. [DOI] [PubMed] [Google Scholar]
- 39.Osterud B, Bjorklid E. Role of monocytes in atherogenesis. Physiol Rev. 2003;83:1069–1112. doi: 10.1152/physrev.00005.2003. [DOI] [PubMed] [Google Scholar]
- 40.Perrella MA, Yet SF. Role of heme oxygenase-1 in cardiovascular function. Curr Pharm Design. 2003;9:2479–2487. doi: 10.2174/1381612033453776. [DOI] [PubMed] [Google Scholar]
- 41.Radi R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc Natl Acad Sci USA. 2004;101:4003–4008. doi: 10.1073/pnas.0307446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ross R. The smooth muscle cell. II. Growth of smooth muscle in culture and formation of elastic fibers. J Cell Biol. 1971;50:172–186. doi: 10.1083/jcb.50.1.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rubbo H, Parthasarathy S, Barnes S, Kirk M, Kalyanaraman B, Freeman BA. Nitric oxide inhibition of lipoxygenase-dependent liposome and low-density lipoprotein oxidation: termination of radical chain propagation reactions and formation of nitrogen-containing oxidized lipid derivatives. Arch Biochem Biophy. 1995;324:15–25. doi: 10.1006/abbi.1995.9935. [DOI] [PubMed] [Google Scholar]
- 44.Rubbo H, Radi R, Trujillo M, Telleri R, Kalyanaraman B, Barnes S, Kirk M, Freeman BA. Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation. Formation of novel nitrogen-containing oxidized lipid derivatives. J Biol Chem. 1994;269:26066–26075. [PubMed] [Google Scholar]
- 45.Schopfer FJ, Lin Y, Baker PR, Cui T, Garcia-Barrio M, Zhang J, Chen K, Chen YE, Freeman BA. Nitrolinoleic acid: an endogenous peroxisome proliferator-activated receptor gamma ligand. Proc Natl Acad Sci USA. 2005;102:2340–2345. doi: 10.1073/pnas.0408384102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Srisook K, Cha YN. Super-induction of HO-1 in macrophages stimulated with lipopolysaccharide by prior depletion of glutathione decreases iNOS expression and NO production. Nitric Oxide. 2005;12:70–79. doi: 10.1016/j.niox.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 47.Tanner FC, Boehm M, Akyurek LM, San H, Yang ZY, Tashiro J, Nabel GJ, Nabel EG. Differential effects of the cyclin-dependent kinase inhibitors p27(Kip1), p21(Cip1), and p16(Ink4) on vascular smooth muscle cell proliferation. Circulation. 2000;101:2022–2025. doi: 10.1161/01.cir.101.17.2022. [DOI] [PubMed] [Google Scholar]
- 48.Tetsuka T, Daphna-Iken D, Srivastava SK, Baier LD, DuMaine J, Morrison AR. Cross-talk between cyclooxygenase and nitric oxide pathways: prostaglandin E2 negatively modulates induction of nitric oxide synthase by interleukin 1. Proc Natl Acad Sci USA. 1994;91:12168–12172. doi: 10.1073/pnas.91.25.12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wright MM, Schopfer FJ, Baker PR, Vidyasagar V, Powell P, Chumley P, Iles KE, Freeman BA, Agarwal A. Fatty acid transduction of nitric oxide signaling: nitrolinoleic acid potently activates endothelial heme oxygenase 1 expression. Proc Natl Acad Sci USA. 2006;103:4299–4304. doi: 10.1073/pnas.0506541103. [DOI] [PMC free article] [PubMed] [Google Scholar]