

Abstract
To investigate the apparent genomic complexity of long-genome bacteriophages, we have sequenced the 218,948-bp genome (6479 bp terminal repeat), and identified the virion proteins (55), of Bacillus thuringiensis bacteriophage 0305φ8-36. Phage 0305φ8-36 is an atypical myovirus with three large curly tail fibers. An accurate mode of DNA pyrosequencing was used to sequence the genome and mass spectrometry was used to accomplish the comprehensive virion protein survey. Advanced informatic techniques were used to identify classical morphogenesis genes. The 0305φ8-36 genes were highly diverged; 19% of 247 closely spaced genes have similarity to proteins with known functions. Genes for virion-associated, apparently fibrous proteins in a new class were found, in addition to strong candidates for the curly fiber genes. Phage 0305φ8-36 has twice the virion protein coding sequence of T4. Based on its genomic isolation, 0305φ8-36 is a resource for future studies of vertical gene transmission.
Keywords: myovirus, Bacillus thuringiensis, pyrosequencing, virion protein, mass spectrometry
Introduction
Tailed bacteriophages are remarkably numerous (Brüssow and Kutter, 2005; Wommack and Colwell, 2000), displaying diversity in the range of hosts they infect and in the different environments from which they can be isolated (Chibani-Chennoufi et al., 2004a; Sharp, 2001). Consequently, phage genomes exhibit marked divergence, to the extent that for a newly-sequenced phage, typically 50% or more of the open reading frames (orfs) are novel (Rohwer, 2003). Phage genomes range in size from less than 20 kb to greater than 200 kb (Ackermann, 2000). However, only a small proportion of the phages in the environment with long genomes (>200 kb) have been isolated (Claverie et al., 2006). Currently, six sequenced phage genomes greater than 200 kb are in GenBank: Aeromonas phage Aeh1 (Nolan et al., 2006), Pseudomonas phage SDM-1 (Kwan et al., 2006), cyanophage PSSM-2 (Sullivan et al., 2005), Vibrio phage KVP40 (Miller et al., 2003a), and the Pseudomonas phages phiKZ (Mesyanzhinov et al., 2002) and EL (Hertveldt et al., 2005). Two other phages, Aeromonas phage 65 and Vibrio phage nt-1, also have genomes longer than 200 kb (Petrov et al., 2006). All of these phages, except SDM-1, are myoviruses (i.e., they have an icosahedral-shaped head and a contractile tail) and five have T4-like morphology (Petrov et al., 2006).
No >200 kb genome phages infective for Gram-positive bacteria are deposited in GenBank (as of May 2007), although phages in this category exist. The longest genome of all known phages is that of the myovirus Bacillus megaterium phage G (ca. 500 kb) (Claverie et al., 2006; Fangman, 1978). Sequencing of additional long-genome phages with Gram-positive hosts is vital for the determination of the extent of phage diversity. It is also critical for the addition of more members to phage protein families, which will lead to a clearer portrayal of the mechanisms by which phages evolve (Serwer et al., 2004). Studies of phages with long genomes will also provide insight into why and how such long phage genomes exist.
Bacteriophage genomic diversity derives, in part, from diversity of structure, evidence of which has existed for many years (Ackermann and DuBow, 1987; Bradley, 1967; Slopek and Krzywy, 1985). This diversity is seen among the three families of phages, delineated on tail morphology: Podoviridae (short tails), Siphoviridae (long, non-contractile tails) and Myoviridae (contractile tails) (Fauquet et al., 2005). Diversity also arises from the so-called “facultative structures”: appendages, such as baseplates, collars, knobs, filaments and many types of fibers (Ackermann, 2000). However, the functional significance of many of these facultative structures is unknown (Ackermann, 2000). Facultative structures can be complex. For example, T4 requires at least 16 proteins to create its sophisticated baseplate (Coombs and Arisaka, 1994; Mesyanzhinov, 2004; Miller et al., 2003b). This indicates that morphological diversity can incur large coding requirements, thereby accounting, in part, for long genomes.
Among the long-genome phages with Gram positive hosts, phage 0305φ8-36, infective for Bacillus thuringiensis, has several unusual characteristics. These include plaque formation only in ultra-dilute gels and aggregation, as visualized by fluorescence microscopy (Serwer et al., 2007b). This phage has a 221-kb genome, as assessed by pulse-field gel analysis (Serwer et al., 2007c). The tail of 0305φ8-36 is remarkably long, 486 nm in length (Serwer et al., 2007a), making it more than three times the length of the tail of T4 (Kostyuchenko et al., 2005). However, the most notable feature of the 0305φ8-36 tail is the presence of three “curly” fibers (approximately 187 nm long and 10 nm in diameter) that are joined to the contractile tail near the baseplate (Serwer et al., 2007a). The dimensions of 0305φ8-36 are almost identical to those of the B. cereus phage Bace-11, a classified myovirus (Ackermann et al., 1995; Fauquet et al., 2005). Aside from the curly fibers, there are other notable shared morphological features of 0305φ8-36 and Bace-11, including baseplates that appear to be elaborate. Hence, the structure and function of 0305φ8-36 and BACE-11 curly fibers are likely to be homologous. However, the only experimental evidence as to what that function might be comes from phages with morphologically less similar curly fibers, such as PBS1, AR9, PBP1 and χ (Belyaeva and Azizbekyan, 1968; Eiserling, 1967; Lovett, 1972; Schade et al., 1967).
A preliminary sequence survey, performed as described (Serwer et al., 2004), revealed that 0305φ8-36 had an unusual genome encoding highly divergent proteins. We present here the complete genomic sequence of 0305φ8-36. In order to annotate the highly divergent proteins of this novel phage, we used a comprehensive set of bioinformatic procedures. Their use was critical for several proteins that otherwise would not have been assigned a function. Mass spectrometry was used to identify virion proteins and revealed a surprisingly large number of virion protein genes. These studies confirm that 0305φ8-36 represents a new genomic group.
Results
Genome sequencing: High quality data from pyrosequencing
Determination of the genomic sequence of 0305φ8-36 was initiated by obtaining extensive dideoxy terminator sequence data from random clones. This process yielded five contigs totaling over 200,000 bp high quality sequence with an average of nine-fold sequence coverage. Whole genome sequencing was then performed by pyrosequencing (Margulies et al., 2005). A single 38-fold redundancy-derived pyrosequence contig of 212,469 bp was obtained. It represented the 0305φ8-36 genome best described in a circular form and cut at an arbitrary point. Assembly of the pyrosequence contig with the previously obtained high quality capillary data showed that there were no discrepancies in the regions that overlapped. The absence of errors in the pyrosequencing data was consistent with the quality scores provided by 454 Life Sciences (Branford, CT). Co-mixed phage genomes sequenced at lower redundancy had higher error rates. Hence, the reliability of the 0305φ8-36 pyrosequence data correlated with a high redundancy of reads (not shown).
Locating the genome termini
Comparison of heated versus non-heated digests of 0305φ8-36 DNA cleaved with the restriction endonucleases, BamHI, HindIII, NaeI and SmaI, showed no differences in banding patterns. This indicated that 0305φ8-36 did not have cohesive termini. However, the restriction profiles of various enzymes were consistent with the determined circular sequence with the exception of several extra fragments that could be explained by postulating a terminal repeat. The sizes of these fragments were used to approximately locate the position of the genomic termini. To complete the sequence, the two ends of the 0305φ8-36 genome were sequenced from DNA obtained by PCR amplification of the genome ends ligated to pUC119. Identification of the exact ends of the 0305φ8-36 genome showed that it includes a blunt-ended terminal repeat of 6479 bp. Long terminal repeats have been reported for well characterized phages SPO1 and T5 (11.5 kb and 10.1 kb, respectively) (Stewart et al., 1998; Wang et al. 2005).
General features of the genome
The complete 218,948 bp genomic sequence of 0305φ8-36 has been deposited into Genbank (accession no. EF583821). The G+C content was 41.8 % and was without remarkable regional variation. No matches to any other entity were found by Blast searching at the nucleotide level. Orfs were annotated by a series of approaches including GeneMark, Blast-matching, and examination of prospective ribosome binding sequences and intergene packing. There were 247 putative orfs identified in the 0305φ8-36 sequence. Orfs were typically tightly packed such that the total fraction of the genome covered by orfs was 94.9%, a value similar to what has been reported for other phage genomes (Kwan et al., 2005; Kwan et al., 2006; Miller et al., 2003b). Eighteen percent of the predicted gene products were 100 amino acids or shorter, the two shortest being 41 residues. Putative small gene products were included in the annotation because leaving them out precludes the opportunity for a future BlastP search to match and validate them. The small gene products were also annotated because there are precedents for the existence of short proteins with known functions in other myoviruses (Miller et al., 2003b). The inclusion of small gene products was justified by the identification of two small proteins, gp174 (82 residues) and gp197 (78 residues), as virion proteins by mass spectrometry (see below). Putative start codons were 88% AUG, 6% GUG, and 6% UUG, consistent with B. subtilis start codon usage (Kunst et al., 1997).
Frame orientation was found to divide the 0305φ8-36 genome into two distinct regions. About half the orfs (orfs108 through 199) are transcribed from the plus strand (“left arm”) and the others (orfs 200 to 100) transcribed on the minus strand (“right arm”) (Fig. 1). Exceptions are orf205 and orf208 which are transcribed from the plus strand but are located in the right arm (Fig. 1). Transcriptional patterns of different phages vary greatly but symmetry of the plus and minus strands, such as seen in the 0305φ8-36 genome, is not a feature of the long-genome phages listed earlier, and is also not a feature of other T4-like phages with genomes <200 kb (http://phage/bioc.tulane.edu/).
Fig. 1.

Genome map of phage 0305φ8-36 showing the major genome regions and functional clustering of morphogenesis orfs. A. The major regions within the 0305φ8-36 genome. Black arrows indicate the direction of transcription which divides the genome into left and right arms. Orfs 108 through 199 are transcribed from the plus strand (left arm) and orfs 200 through 100 are transcribed on the minus strand (right arm). Two orfs in the right arm (orf205 and orf208) that are transcribed from the plus strand are marked. Red arrows represent the terminal repeats (orf101 through 107) and the direction in which they are transcribed. Green indicates orfs whose products were identified as virion proteins by mass spectrometry of purified particles. Blue indicates orfs with non-virion protein functions. The head, tail and baseplate modules in the major morphogenesis gene region are indicated. The orfs encoding the putative major curly fiber proteins are marked. “Other fiber” refers to a cluster of orfs downstream of the baseplate module predicted to encode fibers other than the curly fibers. tRNA-like indicates the location of the tRNAs, which are likely to be non-functional (see text). B. The head module is expanded to show the conserved gene order. The nested scaffold gene (orf123*) within the gene encoding the capsid protease is indicated with shading. Head decor. refers to the gene encoding the putative head decoration protein.
Two tRNA-like elements were reported in the 0305φ8-36 genome by tRNAscan-SE (Lowe and Eddy, 1997). These elements were predicted to lie in the right arm (Fig. 1) in a 1090-bp region containing no predicted orfs, which would be unlikely to happen by chance. However, both predictions were of low confidence and of an unconventional class ( pseudo-tRNA, cove score = 30.2, and intron-containing Arg-tRNA, cove score = 22.4). Aragorn (Laslett and Canback, 2004) did not detect any tRNAs in the 0305φ8-36 genome. Hence it is unclear at this time if these are defective tRNA genes, unconventional tRNA genes, or just false positives.
Initial informatic analysis of 0305φ8-36 putative proteins
Relatively few genes for 0305φ8-36 proteins were identified by simple BlastP or family database searches keyed with translated sequence. Additional methods were required to increase the annotation of the genome, particularly in the morphogenesis gene region. These methods included forward Psi-Blast searches to aid the identification of divergent homologues and SDS-PAGE followed by mass spectrometry to identify virion proteins. Further bioinformatics strategies, including reverse Psi-Blast searches and custom family building operations using the UCSC Sequence Alignment and Modeling System (SAM) (Hughey and Krogh, 1996; Karplus et al., 1998) were also employed to enable the identification of several 0305φ8-36 proteins. Ultimately, 21 % of the 0305φ8-36 gene products were assigned a specific putative function. However, for 131 putative gene products (including 44 proteins ≤ 100 residues in length), no homology or functional information could be obtained, except for the observation that they are probably not present in the mature virion.
Table 1 summarizes the resulting information about the 0305φ8-36 prospective gene products (gp) for which likely functions and/or homologues could be found. The homologues detected using Psi-Blast had matches ranging from 20 to 77 % amino acid identity (Table 1). Only five of the 247 gene products were found to have proteins of named phages as their best match using Psi-Blast, and typically, there was a high degree of divergence between each of these best matches (gp16, gp213, gp239, gp61 and gp88, Table 1). None of these five matches were to phage virion proteins. One 0305φ8-36 protein (gp194) had as its best match a protein (307L) of Invertebrate iridescent virus 6, whose function is unknown. Most of the best scoring homologues to 0305φ8-36 orfs originated from bacterial genomes. Notable were 20 homologues from B. thuringiensis serovar israelensis ATCC 35646 and 12 homologues from the closely related species B. weihenstephanensis KBAB4. The B. thuringiensis serovar israelensis and B. weihenstephanensis homologues, though mostly of unassigned function, were critical for the functional annotation of many 0305φ8-36 orfs because they were frequently the only matches in a BlastP search, and, as such, enabled Psi-Blast to make a profile and then match more divergent homologues.
Table 1.
Identifying information of 0305φ8-36 gene products.
| Gp | aa | Identifying information1 | Homologues2 |
|
|---|---|---|---|---|
| Organism, protein identifier (% identity, E value from Psi-Blast) or (method used to detect homologue) | Paralogues | |||
| 112 | 242 | B. thuringiensis RBTH_06519 (40% over 116, 9e-15) | ||
| 113 | 194 | STR. | ||
| 114 | 248 | STR. | ||
| 116 | 181 | STR. | ||
| 117 | 635 | terminase, large subunit | B. thuringiensis RBTH_06375 (31% over 570, 1e-58) | |
| 118 | 428 | STR. | ||
| 119 | 401 | MAJOR STR., putative curly fiber | ||
| 121 | 100 | 3 predicted transmembrane helices | ||
| 122 | 648 | STR., portal |
B. thuringiensis RBTH_06377 (43% over 465, 1e-102)
Halovirus HF1 GI:32346486 (23% over 320, 1e-07) |
|
| 123 | 487 | protease (SAM) with nested scaffold (gp123*, 257 aa) | B. thuringiensis RBTH_06378 (27% over 406, 2e-29) | |
| 124 | 364 | MAJOR STR., head decoration | B. thuringiensis RBTH_06380 (27% over 420, 7e-16) | |
| 125 | 393 | MAJOR STR, HK97-like major capsid | B. thuringiensis RBTH_06381 (40% over 334, 1e-69) | |
| 127 | 331 | STR. | ||
| 128 | 143 | STR. | ||
| 129 | 989 | MAJOR STR., putative curly fiber | Clostridium tetani GI:28210805 (41% over 131, 5e-18) | |
| 130 | 1073 | STR. | ||
| 131 | 119 | MAJOR STR., putative curly fiber | ||
| 132 | 369 | STR. | ||
| 133 | 900 | STR. | gp152, gp154, gp155, gp135 | |
| 134 | 123 | STR. | gp157 | |
| 135 | 288 | STR. | gp155, gp133, gp154, gp152 | |
| 136 | 177 | STR. | ||
| 137 | 529 | STR. | B. thuringiensis RBTH_08838 (26% over 164, 7e-05) | gp139 |
| 138 | 226 | STR. | B. thuringiensis RBTH_06388 (31% over 196, 7e-14) | |
| 139 | 717 | MAJOR STR., tail sheath | B. thuringiensis RBTH_08838 (29% over 269, 5e-23) | gp137 |
| 140 | 260 | MAJOR STR., putative tail tube | B. thuringiensis RBTH_07699 (34% over 232, 3e-25) | gp141 |
| 141 | 502 | STR. | B. thuringiensis RBTH_07698 (39% over 151, 8e-23) | gp140 |
| 142 | 558 | STR. | B. thuringiensis RBTH_07697 (40% over 111, 3e-14) | gp209 |
| 143 | 218 | B. thuringiensis RBTH_07696 (40% over 114, 6e-16) | gp144 | |
| 144 | 262 | B. thuringiensis RBTH_07695 (29% over 158, 7e-11) | gp143 | |
| 145 | 1930 | STR. | B. thuringiensis RBTH_07694 (32% over 673, 9e-86) | |
| 146 | 2536 | STR., putative tape measure | ||
| 147 | 1903 | T4 gp27, baseplate hub-like (SAM) | B. thuringiensis RBTH_07688 (28% over 515, 2e-40) | |
| 148 | 127 | putative baseplate assembly | P2 gpV-like (SAM), Mu gp45-like (SAM) | |
| 149 | 187 | B. thuringiensis RBTH_07683 | ||
| 150 | 141 | B. thuringiensis RBTH_07682 (40% over 95, 1e-08) | ||
| 151 | 270 | STR., P2 gpJ-like baseplate | B. thuringiensis RBTH_07681 (33% over 261, 1e-31) | |
| 152 | 265 | STR. | gp133, gp154, gp155, gp135, gp197 | |
| 153 | 383 | STR. | ||
| 154 | 1212 | STR., beta glucosidase3 (R-Psi) | Saccharophagus degradans 2–40 GI:90020793 (30% over 107, 0.025) | gp152, gp155, gp133, gp135, gp197 |
| 155 | 936 | STR. | gp154, gp133, gp152, gp135 | |
| 156 | 204 | STR. | ||
| 157 | 127 | STR. | gp134 | |
| 158 | 293 | STR. | gp159, gp160 | |
| 159 | 270 | STR. | gp158, gp160 | |
| 160 | 267 | STR. | gp158, gp159 | |
| 161 | 114 | STR. | ||
| 162 | 372 | STR.3 | B. thuringiensis RBTH_07680 (33% over 212, 7e-21) | |
| 163 | 2143 | STR., 4 x FN3 | Mus musculus GI:90403603 (24% over 390, 6e-10) | |
| 164 | 1062 | STR., Von Willebrand’s domain | B. thuringiensis RBTH_07673 (33% over 238, 1e-29); RBTH_08837 (26% over 250, 3e-10) | |
| 165 | 421 | STR., 3 x FN3 | gp166 | |
| 166 | 406 | STR., 3 x FN3 | gp165 | |
| 167 | 591 | STR., 4 x FN3 | B. cereus GI:29896472 (28% over 198, 5e-11) | gp199 |
| 168 | 693 | STR. | ||
| 171 | 755 | STR. | B. thuringiensis RBTH_07661 (26% over 389, 1e-23); RBTH_07663 (22% over 246, 1e-04); RBTH_07662 (25% over 90, 0.29) | |
| 172 | 591 | STR. | B. thuringiensis RBTH_07660 (22% over 565, 6e-15) | |
| 173 | 686 | STR. | B. thuringiensis RBTH_07659 (20% over 614, 3e-17) | |
| 174 | 82 | STR. | ||
| 175 | 398 | STR. | B. thuringiensis RBTH_07657 (25% over 221, 1e-09) | |
| 180 | 229 | B. thuringiensis RBTH_07119 (33% over 150, 7e-16) | ||
| 181 | 316 | Archaeal primase | B. thuringiensis RBTH_07120 (31% over 291, 1e-40) | |
| 182 | 346 | dnaB helicase | B. thuringiensis RBTH_07121 (42% over 323, 1e-69) | |
| 190 | 269 | RecB | B. thuringiensis RBTH_07113 (26% over 273, 4e-24) | |
| 192 | 177 | RuvC, Holliday junction resolvase | Caldicellulosiruptor saccharolyticus GI:82501051 (28% over 169, 1e-05) | |
| 193 | 287 | C. acetobutylicum, GI:15004860 (23% over 263, 1e-12) | ||
| 194 | 170 | homing nuclease | Invertebrate iridescent virus 6, GI:15079019 (40% over 84, 1e-09) | |
| 196 | 185 | thymidine kinase | Bacillus sp. GI:89095598 (41% over 177, 2e-30) | |
| 197 | 78 | STR.3 | ||
| 198 | 135 | STR., FN3 | cd00063, FN3 (24% over 85, 2e-04) | gp167 |
| 199 | 486 | STR., 4X FN3 | Solibacter usitatus GI:67929848 (40% over 92, 2e-08) | |
| 201 | 802 | Silicibacter pomeroyi GI:56677853 (22% over 310, 2e-09) | ||
| 205 | 171 | STR. | ||
| 207 | 330 | mreB-like rod determination protein | Symbiobacterium thermophilum GI:51858110 (25% over336, 1e-16) | |
| 208 | 143 | transcription factor | C. acetobutylicum GI:15023852 (35% over 111, 7e-08) | |
| 209 | 585 | STR. | B. licheniformis GI:52349990 (30% over 245, 2e-22) | gp142 |
| 210 | 220 | HsdM | COG0286 (20% over 174, 8e-05) | |
| 213 | 139 | homing nuclease | phiKZ ORF296 GI:29135232 (51% over 54, 2e-04) | |
| 215 | 255 | dCMP deamidase | Lactobacillus gasseri GI:23003434 (62% over 122, 2e-39) | |
| 223 | 159 | Flavodoxin | Exiguobacterium sibiricum GI:68056239 (37% over 124, 2e-17) | |
| 224 | 352 | nrd beta subunit | E. sibiricum GI:68056370 (46% over 344, 2e-82) | |
| 225 | 767 | nrd alpha subunit | Geobacillus kaustophilus GI:56379288 (51% over 774, 0) | |
| 227 | 206 | B. thuringiensis RBTH_07176 (40% over 123, 1e-14) | ||
| 228 | 179 | B. thuringiensis RBTH_07176 (23% over 152, 0.025) | ||
| 232 | 145 | membrane-bound metal-dependent hydrolase | Desulfotomaculum reducens GI:88944587 (31% over 129, 2e-07) | |
| 234 | 155 | COG3236 | Escherichia coli GI:16128766 (41% over 153, 3e-29) | |
| 236 | 424 | dnaG primase | Prosthecochloris vibrioformis GI:71481478 (29% over365, 2e-33) | |
| 237 | 368 | lysin | B. cereus GI:89199806 (68% over 197, 3e-76) | |
| 5 | 236 | B. halodurans GI:10174999 (42% over 184, 1e-31) | ||
| 8 | 769 | rec. exo | C. thermocellum GI:67876361 (28% over 492, 4e-49) | |
| 9 | 182 | Mesorhizobium sp. GI:68190552 (42% over 71, 2e-07) | gp10 | |
| 10 | 244 | B. thuringiensis RBTH_02290 (35% over 128, 1e-08) | gp9 | |
| 15 | 250 | serine/threonine phosphatase | Lactococcus lactis GI:76574849 (30% over 221, 7e-17) | |
| 16 | 117 | MazG | Mx8, p26 GI:15320596 (52% over 113, 9e-21) | |
| 23 | 657 | PcrA helicase | Moorella thermoacetica GI:83590844 (42% over 642, 1e-141) | |
| 241 | 457 | recA (nb. in two segments as gene interrupted by orf29) | Mycobacterium peregrinum (33% over 374, 3e-46) | |
| 29 | 266 | mobile intron | M. vanbaalenii GI:90205066 (35% over 53, 0.004) | |
| 33 | 111 | DNA-binding protein HU | B. cereus GI:29895202 (77% over 90, 1e-26) | |
| 240 | 1348 | DNA polymerase III alpha subunit (nb. in two segments as gene interrupted by orf36) | C. thermocellum GI:67873315 (30% over 1296, 4e-157) | |
| 36 | 261 | mobile intron | B. anthracis GI:47503129, (30% over 214, 8e-15) | |
| 239 | 176 | mobile nuclease | Cyanophage P-SSM2, GI:61806163 (35% over 80, 1e-08) | |
| 39 | 361 | DNA polymerase III beta subunit | C. perfringens GI:18143659 (28% over 311, 2e-23) | |
| 41 | 104 | GroES | Anabaena variabilis GI:75705561 (41% over 92, 3e-12) | |
| 43 | 207 | Uracil-DNA glycosylase | Thermoanaerobacter tengcongensis GI:20515053 (30% over 155, 2e-10) | |
| 44 | 352 | C-terminal region of bacteriolytic enzyme | B. clausii GI:38603523 (26% over 224, 3e-05) | |
| 46 | 318 | N. farcinica GI:54019290 (25% over 330, 1e-14) | ||
| 53 | 166 | HTH type 11 transcriptional regulator | Vibrio vulnificus GI:37200665 (37% o over 69, 6e-04) | |
| 61 | 248 | B. cereus phage phBC6A51, | phBC6A51 GI:31415774 (37% over 79, 2e-06) | gp88 |
| 62 | 235 | enterotoxin/cell-wall binding protein | B. cereus GI:42784405 (69% over 110, 1e-37) | |
| 63 | 553 | metalloprotein chaperonin subunit | N. farcinica GI:54019291 (27% over 528, 2e-33) | |
| 66 | 471 | metalloprotein chaperonin subunit | Listeria monocytogenes GI:47092621 (38% over 288, 2e-43) | |
| 70 | 624 | RecQ helicase | B. thuringiensis RBTH_07170 (26% over 624, 5e-52) | |
| 79 | 577 | RecJ | B. halodurans GI:10173856 (34% over 562, 8e-95) | |
| 81 | 85 | MAJOR STR. | ||
| 88 | 154 | B. cereus phage phBC6A51 | phBC6A51 GI:31415774 (44% over 74, 9e-09) | gp61 |
| 97 | 188 | B. thuringiensis RBTH_06730 (34% over 167, 6e-23) | ||
| 99 | 1245 | putative RNA polymerase | Arabidopsis thaliana GI:24935275 (23% over 475, 2e-04) | |
Putative functions were assigned based on homologies to proteins detected using Psi-Blast, unless stated otherwise.
Closest homologues detected by Psi-Blast are listed with other matches included, where pertinent. The organism and protein global identifier for homologues are provided, except for B. thuringiensis homologues for which the protein identifiers, beginning RBTH_, are given. B. thuringiensis refers to Bacillus thuringiensis serovar israelensis ATCC 35646. The Psi-Blast matches of 0305φ8-36 proteins to B. thuringiensis or B. weihenstephanensis homologues were very similar. Only the results for the B. thuringiensis homologues are included in this table. Several 0305φ8-36 proteins had more than one B. thuringiensis homologue, in which instance protein identifiers of the additional homologues are included in parenthesis. 0305φ8-36 paralogues are listed.
, single peptide detected by mass spectrometry with decreased confidence (see Table S1).
Abbreviations used: aa, amino acids; FN3, Fibronectin type 3 domain; MAJOR STR., one of the eight most densely stained bands as detected by SDS-PAGE (Figure 2), copy number for each protein was estimated to be >100 copies per virion; SAM, homologous relationship determined using a SAM model; STR., virion structural protein identified by mass spectrometry with a protein identification probability of 98% or greater, unless noted (see Table S1).
Homologues to 0305φ8-36 gene products included proteins with functions associated with DNA replication, recombination and repair and nucleotide metabolism (Table 1). Proteins with these functions are commonly found in phages with long-genomes (Hertveldt et al., 2005; Mesyanzhinov et al., 2002; Miller et al., 2003a; Miller et al., 2003b; Nolan et al., 2006; Petrov et al., 2006; Sullivan et al., 2005). The genes encoding the DNA polymerase and a RecA-like protein, orf240 and 241, respectively, are interrupted by mobile introns, elements also frequently identified in phage genomes [e.g., the DNA polymerase gene of SPO1 and SPO1-like phages (Goodrich-Blair and David, 1994; Goodrich-Blair et al., 1990)]. No lysis module was identified in the 0305φ8-36 genome. However, gp237 has homology to endolysins, and gp121 is a candidate for a type I holin based on its length (100 amino acids) and three predicted transmembrane regions (Young et al., 2000). No integrase, excisionase or other proteins expected of a temperate phage were identified in 0305φ8-36.
An unexpected finding was the number of 0305φ8-36 gene products that were paralogues of other 0305φ8-36 proteins. Paralogues are homologous proteins generated by gene duplication and then retained in the same genome. The 0305φ8-36 paralogues were identified using local Psi-Blast searches of a database including 0305φ8-36 orfs. Many of the 0305φ8-36 paralogues were proteins that had no homologues from other genomes (Table 1). The genes of most paralogues were located in the virion protein and morphogenesis gene region (see below), with the exceptions of orf9, orf10 and orf209.
Identification of homologues with virion protein and morphogenesis-related functions
Psi-Blast searches found only five 0305φ8-36 proteins that had homologues with phage protein and morphogenesis-related functions. These proteins were: gp117 (terminase large subunit), gp122 (portal), gp125 (main head protein), gp139 (main tail sheath protein) and gp151 (baseplate protein) (Table 1). However, the best match to each of these proteins was not a protein from a described phage or prophage, but to a protein encoded by B. thuringiensis serovar israelensis. Notably, even the best matches were not close matches, as judged by the percent identities that ranged from 31% to 43%, highlighting the uniqueness of the 0305φ8-36 proteins. For example, the distinctiveness of the 0305φ8-36 terminase protein is such that it intersects a global terminase tree for this protein only at the center and forms the first member of a new class of DNA packaging ATPases (Serwer et al., 2007a). Intriguingly, despite the divergence of these five 0305φ8-36 structure and morphogenesis proteins, the placement of their respective genes was similar to other phages. The ordering of these genes suggested that 0305φ8-36 has functional clusters of orfs (modules) in the common order of head, tail and baseplate modules (Fig. 1). The presence of such modules was further supported by the identification of additional orfs within these modules with functions appropriate to their particular modules (see below).
Mass spectral analysis of virion proteins
SDS-PAGE followed by capillary HPLC-electrospray tandem mass spectrometry (HPLC-ESI-MS/MS) were used to directly identify proteins assembled in mature 0305φ8-36 particles (Fig. 2 and Table 1). Fifty-five such proteins were identified by this approach (Table 1). Two strategies were used to analyze 0305φ8-36 virion proteins in a phage sample purified by two CsCl gradients. (1) Proteins were separated by SDS-PAGE on a gel that was run to completion, bands were visualized by staining with Coomassie Brilliant Blue, individual gel bands were excised and digested in situ with trypsin, and the resulting peptides were analyzed by HPLC-ESI-MS/MS. This resulted in the identification of 35 virion proteins. (2) In a parallel determination, an SDS-PAGE gel was run for 20 min. The 1.5-cm region of the gel that contained the partially separated proteins was excised into seven slices followed by in-gel digestion and MS analysis. This approach yielded identification of 50 virion proteins, 20 of which were not identified by the first method. The second approach permitted identification of proteins present at low levels for which defined bands were not visualized by the first method. Five proteins identified by the first approach (gp145, gp146, gp163, gp168 and gp172) were not identified by the second. An explanation for three of these proteins, gp145, gp146 and gp163, not being detected by the second approach may be that the slice sampling did not extend high enough up the gel to include proteins with high molecular weights (each of these proteins has a molecular weight >200 kDa). All 0305φ8-36 proteins identified by mass spectrometry conclusively matched 0305φ8-36 orfs.
Fig. 2.
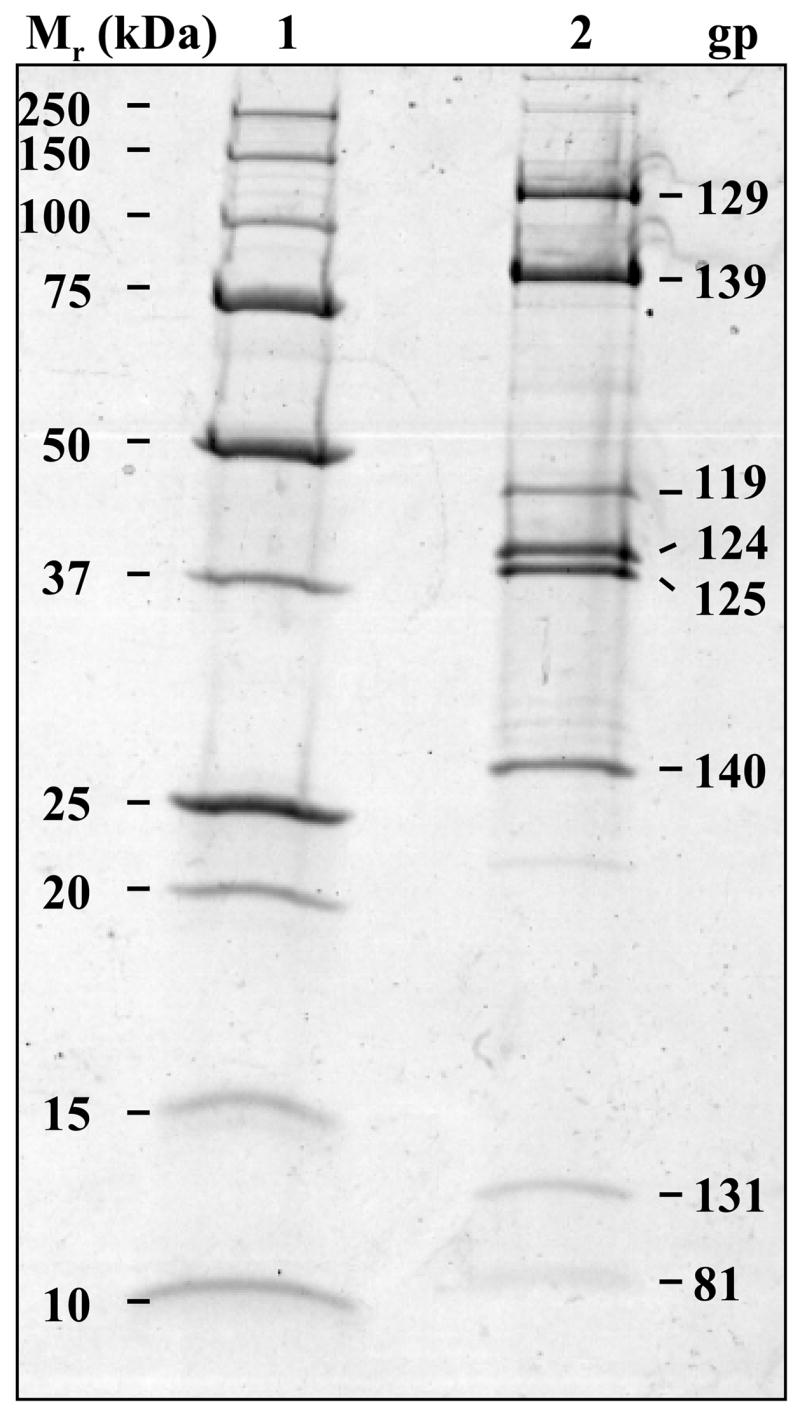
Virion proteins of 0305φ8-36 separated by SDS-PAGE. A Bio-Rad Tris-HCl gradient gel (8 to 16% polyacrylamide) was employed, and proteins were visualized by staining with Coomassie Brilliant Blue; lane 1, Precision Plus Protein Standard (Bio-Rad); lane 2, purified 0305φ8-36. The identities of the proteins in the eight most intense bands are indicated. The putative functions of these proteins are: gp125, major head protein; gp124, head decoration protein); gp139, tail sheath protein; gp140, tail tube protein; gp119, gp129 and gp131, candidates for curly fiber components; and gp81, unassigned.
The details of the searches of the tandem mass spectral analyses of 0305φ8-36 proteins against a Swiss-Prot database supplemented with all putative 0305φ8-36 protein sequences are provided in the Supplementary material (Table S.1). The majority of proteins (49) were identified with 100% probability.
Identification of the 0305φ8-36 virion proteins by mass spectrometry enabled the positions of the encoding genes in the 0305φ8-36 genome to be determined (Fig. 1, green regions; Table 1). The genes of 49 of the 55 0305φ8-36 virion proteins mapped to one region of the genome, delineating the morphogenesis gene region (Fig. 1). The genes of six virion proteins mapped outside of the main morphogenesis gene region (gp81, gp197, gp198, gp199, gp205 and gp209).
The identification of the virion proteins of 0305φ8-36 led to the recognition that there were new phage-like entities in the genomes of B. thuringiensis and B. weihenstephanensis, based on our observation that five 0305φ8-36 morphogenesis proteins had closest homology to proteins of B. thuringiensis and not to proteins of known phages (see above). In addition, 21 other 0305φ8-36 virion proteins (gp122 through gp175, Table 1) had homology to hypothetical proteins of B. thuringiensis and/or B. weihenstephanensis detected using Psi-Blast. The genes for these B. thuringiensis and B. weihenstephanensis proteins in most instances are in the same order in their respective genomes as their 0305φ8-36 matches. Even though the genomes of both B. thuringiensis and B. weihenstephanensis are in draft status and not fully assembled at the time of this writing, we were able to gain insight into the order of the genes of interest because all of the B. thuringiensis homologues to 0305φ8-36 structure and morphogenesis proteins were encoded on the 128761 base pair contig, sq1939 (NZ_AAJM01000001). Most of the B. weihenstephanensis homologues to 0305φ8-36 proteins were located on the 403024 base pair contig, ctg266 (NZ_AAOY01000001). The B. thuringiensis and B. weihenstephanensis homologues are, therefore, likely to be from the genome of either a prophage or some kind of phage relic, although contamination by infective phage can not be ruled out. That the B. thuringiensis phage-like entity may have a prophage origin is supported for by an integrase (RBTH_07144) annotated on B. thuringiensis contig sq1939. In this discussion, the B. thuringiensis phage-like region will be referred to as BtI1 and the similar, but not as extensive, phage-like region in B. weihenstephanensis will be referred to as BwK1.
Determination of proteins present in more than 100 copies per virion
In SDS-PAGE analysis of 0305φ8-36 virions, intense bands corresponding to six major proteins were detected, indicating that there was a high copy number of each of these proteins per virion (Fig. 2; gp129, gp139, gp119, gp124, gp125 and gp140). These proteins were expected to include the major head protein, the tail sheath and tail tube proteins, typically the major virion proteins of a myovirus. The main head and tail sheath proteins, gp125 and gp139, respectively, had been identified by homology. The copy numbers of the major proteins were estimated based on the intensities of their gel bands relative to that of the tail sheath protein. The copy number of the tail sheath protein (Table 2) was estimated based on the following: (1) The 0305φ8-36 sheath protein (78.2 kDa) is similar in molecular weight to the T4 tail sheath protein (gp18, 71.3 kDa); (2) the 26-nm diameter of the contracted 0305φ8-36 sheath is comparable to that of the contracted sheath of T4 (Kostyuchenko et al., 2005) and other myoviruses (Ackermann, 2000; Admiraal and Mellema, 1976; Chibani-Chennoufi et al., 2004b; Parker and Eiserling, 1983); (3) the uncontracted state of the sheath protein in 0305φ8-36 was assumed to be the same as that of T4 gp18, i.e., it was assumed that the copy number of tail sheath protein per virion varies in proportion to the tail length. Using this approach, we deduced that there were 695 ± 174 molecules per virion of the 0305φ8-36 tail sheath protein, based on 138 copies for T4 gp18 (Kostyuchenko et al., 2005). Seven other proteins were deduced to be present in over 100 copies per 0305φ8-36 virion: gp129, gp119, gp124, gp125, gp140 and gp131 and gp81 (Table 2). If the assumption in (3) above is not correct, the absolute values for the copy numbers for these eight proteins present in over 100 copies would not be accurate. However, the relative stoichiometry of these proteins would still be as estimated.
Table 2.
Estimated copy number of 0305φ8-36 major structural proteins.
| Gene | Functional Assignment | Mr (kDa) | Copy Numbera |
|---|---|---|---|
| Major tail components | |||
| orf139 | tail sheath | 78.2 | 695 |
| orf140 | tail tube | 27.9 | 475 |
| Major head components | |||
| orf125 | major head protein | 37.8b | 744 |
| orf124 | head decoration | 40.1 | 704 |
| Proposed major curly fiber components | |||
| orf129 | 110.8 | 212 | |
| orf119c | 43.2 | 209 | |
| orf131 | 13.2 | 187 | |
| Unknown | |||
| orf 81 | 7.5 | 309 | |
Copy number per virion was measured relative to 695 for tail sheath assuming the same number of sheath molecules per tail length as in T4. Standard errors <= 25%. There may be an additional unknown systematic error if there is differential staining of the different proteins.
Processed.
Tentative assignment.
Links between the major head protein and gp5 of HK97
The major head protein (gp125) had divergent homology (17% identity) to gp5, the well-characterized head protein of HK97 and to numerous HK97 gp5-like proteins. The gp125 match with HK97 gp5 was from end-to-end, including the N-terminal delta region of gp5 (Helgstrand et al., 2003). In view of the match between 0305φ8-36 gp125 and HK97 gp5, we searched for other elements of an HK97-like head morphogenesis system in 0305φ8-36. HK97 gp5 and some of its homologues are covalently cross-linked during maturation to form protein chain mail (Hendrix, 2005; Popa et al., 1991). However, other HK97 homologues are not cross-linked (Baker et al., 2005; Effantin et al., 2006; Fokine et al., 2005b). 0305φ8-36 gp125 is not cross-linked as indicated by the identification of this protein as the dominant component of a single intensely-stained SDS-PAGE band with an apparent molecular weight that was close to that predicted for gp125 (Fig. 2; Table 2). This conclusion is further supported by the absence of indications associated with cross-linked heads in the 0305φ8-36 SDS-PAGE profile. Others have noted that when there are cross-linked heads, SDS PAGE analysis shows two distinct high molecular weight proteins, such as reported for HK97 (Popa et al., 1991), L5 (Hatfull and Sarkis, 1993) and D29 (Ford et al., 1998), and that there are proteins that are either unable to enter, or are trapped in the stacking gel (Popa et al., 1991; Thomas, 2005).
The covalent cross-linking of HK97 gp5 plays an important role in head stability (Ross et al., 2005; Wikoff et al., 2000). It has been suggested that the presence of decoration proteins and/or extra protein domains add stability to heads are not cross-linked (Fokine et al., 2005b). The absence of cross-linking in the 0305φ8-36 head suggests a requirement for a decoration protein, a role proposed below for gp124.
Many phage head proteins are proteolytically cleaved during maturation of the head, including gp5 of HK97 (Wikoff et al., 2000). Hence, the SDS-PAGE migration of gp125 of 0305φ8-36 to a molecular weight ~10% lower than predicted was an indication that this protein might have been processed during maturation. This possibility was supported by the fact that the most N-terminal peptide of gp125 detected by mass spectrometry was FMATPSAQILIPR and that the preceding residue in the predicted sequence is an E, and not a K or an R (i.e. it was not produced by tryptic cleavage at both ends). These results indicate that 72 amino acids had been removed from the N-terminus of gp125.
Putative conserved maturation cleavage site
Within a few residues of the mature N-terminus of gp125 there was only one residue the same between gp125 and its BtI1 homologue, RBTH_06381. Fourteen residues upstream, there was a potential conserved maturation cleavage site (K^MM) in gp125 and RBTH_06381 (Fig. 4). Further support for K^MM being the maturation cleavage site is that it agreed with the consensus cleavage site, K^x[L or M] of HK97 gp5 and homologues in a SAM alignment. [It should be noted that to find the cleavage site in RBTH_06381 required extending its N-terminus to a start codon further upstream than the N-terminus annotated in the GenBank entry. The extended RBTH_06381 frame also introduced a recognizable ribosome binding sequence at the new start position (not shown)]. This raises the question of how the mature gp125 lost the remaining 14 residues. Assuming that gp125 has the conformation of its HK97 homologue, the X-ray diffraction-based structure of the HK97 head (Helgstrand et al., 2003) indicates that the N-terminus of 0305φ8-36 gp125 is exposed to the virion exterior. In this structure, the missing 14 residues are in a position to be removed by nonspecific proteolysis. The location of the predicted cleavage site of gp125 and the existence of a protease, see below, are both consistent with 0305φ8-36 having an ancestral relationship with the HK97 system.
Fig. 4.

Comparison of the secondary structure of the T4 tail tube protein (gp19) with that of the putative 0305φ8-36 tail tube protein (gp140). The regions of 0305φ8-36 gp140 with similarity to gp141, as determined by Psi-Blast, are marked with grey boxes. The indel in gp140 thus defined is removed from the comparison with T4gp19.
A putative protease containing a nested scaffold protein
The observation that gp125 is processed indicated the existence of a phage-encoded protease and led us to seek such a protein. However, initial Blast searches did not identify any protein with protease homology. To search for more divergent proteases, SAM HMM models were developed starting from the proteases P2 gpO and HK97 gp4. These models identified a potential maturation protease in the N-terminal 230 residues of 0305φ8-36 gp123. The C-terminal domain of gp123 was strongly predicted by COILS (Lupas et al., 1991) to contain an 80-residue coiled coil region. In analogy with other phages such as φ (Ziegelhoffer et al., 1992) and Mu (Morgan et al., 2002), the head protease orf of 0305φ8-36 also encodes a putative nested scaffold gene, orf123*. The potential internal start site within gp123 for the scaffold protein (gp123*) is residue 231. This start site would produce a scaffold protein of 256 residues. There is a good upstream ribosomal binding site for orf123*. Consequently, we project that, as in other phages, the sequence encoding the nested scaffold gene does double duty by encoding the C-terminal domain of the protease protein and a separate scaffold protein.
A putative head decoration protein analogous to λ gpD
The argument for a head decoration protein is supported by the presence of gp124. Although no homologues with known functions to gp124 were found by homology searches, gp124 is a good candidate for a head decoration protein. Orf124 is located between orfs for the major head protein and scaffold proteins, in the same relative position as the gene encoding the λ decoration protein (gpD) (Fig. 1). Similarly, the gene encoding the BtI1 homologue to gp124 holds the same position in the BtI1 head module as orf124 holds in the 0305φ8-36 head module. Phages often have a functional clustering of head genes (Casjens, 2003). Also, as is the case for λ gpD and λ main capsid protein (gpE), there is a 1:1 stoichiometry for 0305φ8-36 gp124 and the main head protein, gp125 (Table 2).
Triangulation number of the 0305φ8-36 head
The major head protein of phage 0305φ8-36, gp125, was estimated to be present at 744 ± 186 copies per virion. Thus the most likely T numbers for the 0305φ8-36 head are 12 and 13, based on the series of T numbers (T=1, 3, 4, 7, 9, 12, 13, 16…) that defines the possible ways in which an icosahedron can be triangulated (Casjens, 1985). No evidence of a mirror plane, such as occurs for T=12 and T=16 lattices, has been seen in electron micrographs of the 0305φ8-36 head. Thus, if icosahedral, the T number for the 0305φ8-36 head is most probably 13. Mutants of T4 with isometric heads have a T=13 lattice (Iwasaki et al., 2000; Olson et al., 2001).
Identification of tail sheath and tube proteins
The tail sheath protein (gp139) was initially identified using Psi-Blast and is a divergent homologue of phage HF2 p095, that matches the pfam04984, tail_sheath1 family (Table 1). Confirmation was obtained by a reverse Psi-Blast search starting from the tail sheath protein (gp18) of Aeh1, a T4-like phage. The search for the tail tube protein was not as straightforward because no 0305φ8-36 gene product matched a known tail tube protein in a Blast search—not even using a Blast-two-sequence strategy at very high E value. Of the orfs encoding high copy number virion proteins, orf140 was the leading tail tube gene candidate because it holds the same relative position to the tail sheath gene as do the tail tube genes of various other myoviruses, including T4 (Miller et al., 2003b), T4-like phages (Hambly et al., 2001; Tétart et al., 2001), P2 (Temple et al., 1991), Mu (Takedo et al., 1998) and Mu-like phages (Morgan et al., 2002). However, gp140 is 97 residues longer than the T4 tail tube protein (T4 gp19), raising the question of how the extra mass could be accommodated inside the tail sheath.
Some help in structurally correlating gp140 with T4 gp19 came through first analyzing paralogues. Gp140 has a paralogue in the N-terminal domain of the adjacent gp141. The paralogue domain is smaller than gp140, having a length close to the length of T4 gp19. Inspection of the alignment between gp140 to gp141 located the extra residues in gp140 to a specific position (residues 83 to 171). With the extra residues in gp140 thus located and removed from consideration, the predicted secondary structures of gp140 and T4 tail tube align well (Fig. 4).
Finally, a possible explanation for the extra mass of 0305φ8-36 gp140 versus the T4 tail tube protein was found via the observations that (1) the molar ratio of T4 tail sheath to tail tube is 1.0, but (2) the same ratio for 0305φ8-36 is 0.7 (Table 2). Based on these observations, an explanation is that each tube-forming disk of subunits is thicker in 0305φ8-36 than in T4, therefore, the 0305φ8-36 tube would require fewer disks to cover its tail length than if its tail tube protein was of a similar mass to T4 gp19. In support, the mass ratio of T4 gp19 to 0305φ8-36 is 18.5 kDa/27.9 kDa (0.7:1).
Identification of the tape measure protein
The tape measure protein (TMP) regulates tail length during assembly and fills the tail lumen (Abuladze et al., 1994; Casjens and Hendrix, 1988; Katsura, 1987; Popa et al., 1991). Identified TMPs exhibit poor sequence conservation but are predicted to have highly α-helical structures (Casjens and Henrix, 1988; Katsura and Hendrix, 1984). As such, TMPs are usually recognized by the position of their gene in addition to their length and secondary structure rather than by sequence homology (Pedulla et al., 2003). The TMP candidates in 0305φ8-36 were gp145, gp146 and gp147, based on high molecular weight and the position of the corresponding orf. Orf145, orf146 and orf147 are all located in a position appropriate for a TMP gene (downstream of the main tail sheath and tube genes) when compared to TMPs in other phage genomes with functional clustering of genes (Xu et al., 2004). Secondary structure predictions for the TMP candidates, found that the predicted percentage of residues in α-helices in gp146 was 58%, higher than the percentages predicted for gp145 and 147 (45% and 31%, respectively) (Rost, 1996; Rost and Sandler, 1993). Based on our EM measurements described above, the length of the 0305φ8-36 tail is 3.2 times the length of the phage λ. Also, the molecular weight of gp146 is 3.1 times the molecular weight of the TMP of λ (gpH). Thus, assuming that the structure of the 0305φ8-36 TMP is the same as the structure of the λ TMP, gp146 best matches the criteria for a TMP based on its molecular weight and secondary structure.
Identification of proteins associated with the baseplate
Three 0305φ8-36 proteins, gp147, gp148 and gp151, were assigned as baseplate proteins based on their homology to the baseplate proteins of other phages (Table 1). The only component of the baseplate that was functionally assigned by Psi-Blast and family database searches was gp151, which is homologous to P2 gpJ, located on the edge of the small P2 baseplate structure (Haggard-Ljungquist et al., 1995). To detect further baseplate genes, we employed SAM HMM models built with homologues of other P2 virion proteins. The SAM HMM models assigned the following: (1) gp147 as a homologue of gp27, the hub protein of T4 and gpD of P2; (2) gp148 as a homologue of gpV, the tail spike protein of P2 and gp45, a baseplate protein of Mu baseplate protein. However, gp147 and gp148 were not detected by the mass spectrometry analysis. The likely reason is that hub and tail spike proteins had been ejected from the virion by tail contraction during purification. Electron microscopy revealed that all tails in this preparation were contracted (not shown). Contracted tails were also found previously for purified 0305φ8-36 (Serwer et al., 2007a). There was also decrease in the titer of the phage sample after purification (see Methods) as would be expected if components of the baseplate had been ejected. As a consequence, functional assignment of gp147 and gp148 was made on the basis of sequence recognition alone. Given the degree of functional clustering found in the 0305φ8-36 genome, the products of the adjacent two small orfs, orf149 and orf150 (neither a virion protein detected by mass spectrometry) may also be virion components ejected during tail contraction (Table 1). Since gp147 and gp151 belong to myovirus specific families, their identification marks 0305φ8-36 as a myovirus, consistent with the EM examination.
Candidates for major components of the curly fibers
The curly fibers of 0305φ8-36 are unique among sequenced bacteriophages. Not surprisingly, homology searches were unable to predict which 0305φ8-36 proteins were curly fiber components. Candidates for the major components of these fibers were identified in the following way. The curly fiber protein(s) were assumed not to be encoded by a gene with another identified function. Omitting the major head and tail proteins left four unassigned high-copy-number proteins (gp119, gp129, gp131, and gp81). Orf119, orf129 and orf131 occur within the main morphogenesis gene region (Fig. 1) but orf81 is outside this region. Notably, despite the extensive numbers of BtI1 homologues to 0305φ8-36 virion proteins, there were no BtI1 homologues to gp119, gp129 or gp131 (Table 1), hence the orfs encoding components of the curly fibers were assumed to be in 0305φ8-36 and absent in BtI1. Finally, the copy numbers of gp119, gp129 and gp131 are all about 200, suitable to form a heterotrimer that could polymerize to form the fibers.
To further explore the feasibility of gp119, gp129 and gp131 being the major components of the curly fibers, an estimation of the total volume formed by the three fibers was made for comparison to the individual, or collective, volumes formed by the three proteins. The three fibers were estimated to occupy a total volume of 44,000 nm3, assuming each fiber was a cylinder of the measured dimensions [187 nm long and 10 nm in diameter (Serwer et al., 2007a)]. The total volume of gp119, gp129 and gp131 in one virion were also estimated, using the copy numbers calculated previously (Table 2) and an average protein partial specific volume of 0.73 cm3/g. These calculations found that gp129 would occupy 65% of the estimated curly fiber volume. Similarly, gp131 and gp129 together would occupy 72% of the estimated curly fiber volume, and gp119, gp129 and gp131 together would occupy 97% of the estimated curly fiber volume. Thus, the results are consistent with the assumption that all three proteins are curly fiber components.
Orf129 and orf131 are clustered close to one another in the 0305φ8-36 genome, separated by a single orf (orf130) encoding a low-copy-number virion protein. The proximity of orf129 to orf131 supports the assumption that their products are present in the same structure, in light of the extensive functional clustering in the 0305φ8-36 head, tail and baseplate modules. Orf119, however, is not clustered near orf129 and orf131 in the 0305φ8-36 genome. Orf119 is positioned between the head proteins and the large terminase gene (Fig. 1). The assignment of gp119 to the curly fiber is, therefore, only tentative, particularly as the 44,000 nm3 volume estimate for the curly fibers could be an overestimate. A second possible function for gp119 is that it is a head protein that associates with the major head protein(s) at a stoichiometry of less than 1:1, as has been observed for T4 hoc (Black et al., 1994; Mesyanzhinov, 2004).
Putative fiber region
Downstream of the orfs encoding homology-identified baseplate proteins (orf147 to orf151) is a region of almost 38 kb that mainly contains orfs encoding virion proteins (orf152 to orf175; Table 1). The functions of these proteins are unknown. However, their orfs are downstream of conventionally clustered modules for head, tail and baseplate formation (Casjens, 2003) (see Fig. 1). In other phage genomes that follow the conventional clustering of orfs, this position would typically be occupied by orfs related to fiber formation (Casjens, 2003). This suggests that at least some 0305φ8-36 orfs between orf152 and orf175 encode fibers not yet observed by electron microscopy. There are few Blast matches to gp152 through gp175, and no specific function can be assigned to any of these proteins. However, several of these proteins have domains with similarity to frequently observed folding domains, most typically fibronectin type III folds (FN3) (gp163, gp165, gp166, gp167; Table 1). Also, gp164 contains a von Willebrand factor (VWA) domain (Table 1) including region 1 of a metal ion dependent adhesion site, or MIDAS motif (DXSXS, where X is any amino acid) (Whittaker and Hynes, 2002), starting at position 405.
Virion protein-based coding complexity of 0305φ8-36
The number of virion proteins in myoviruses varies. For example, P2 has 16 virion proteins (GenBank accession no. AF063097), while T4 has 36 virion proteins (Mesyanzhinov, 2004; a similar list is provided in Miller et al., 2003b). To quantitatively compare the virion protein-based coding complexity of 0305φ8-36 to P2 and T4, we defined this complexity to be the length of DNA required to encode all the proteins in the mature virion. By this definition, the virion protein-based coding complexity for phage T4 is three-times higher than it is for P2 (Table 3). The increased virion protein-based coding complexity of T4 compared to P2 results from increased numbers of different proteins in its baseplate and tail fibers, not from substantially increased numbers of different proteins in its head or tail (Table 3). In this respect, T4 has been considered to be the most complex phage studied to date (Mesyanzhinov, 2004; Miller et al., 2003b). However, 0305φ8-36 is twice as complex as T4 and six-times as complex as P2. The virion protein-based coding complexity of 0305φ8-36 is so high that 42% of its 219 kb genome is required to encode all the virion proteins.
Table 3.
Comparison of the types and lengths of proteins identified in the mature virion of the myoviruses, P2, T4 and 0305φ8-36.
| P21 | T41 | 0305φ8-361 |
|---|---|---|
| capsid proteins | ||
|
| ||
| gpQ (344), gpN (357), gpL (169) | gp20 (524), gp23 (521), gp24 (427), soc (80), hoc (376), alt (682) | gp122 (648), gp125 (393), gp124 (364) |
|
| ||
| tail proteins | ||
| FI (396), FII (172), gpX (67), gpR (155), gpS (150), gpE (91), gpU (159), gpD (387), gpT (815) | gp18 (659), gp19 (138), gp3 (176), gp15 (272), gp29 (590) | gp139 (717), gp140 (260), gp146 (2536) |
|
| ||
| baseplate proteins | ||
| gpV (211), gpW (115), gpJ (302) | gp53 (196), gp5 (575), gp6 (660), gp7 (1032), gp8 (334), gp9 (288), gp10 (602), gp11 (219), gp25 (132), gp26 (208), gp27 (391), gp28 (177), gp48 (364), gp54 (320), td (286), frd (193) | gp147 (1903), gp148 (127), gp151 (270) |
|
| ||
| fiber/facultative structure/unknown proteins | ||
| gpH (669) | gp12 (527), gp13 (309), gp14 (256), wac (487), gp34 (1289), gp35 (372), gp36 (221), gp37 (1026), gp38 (183) | gp113 (194), gp114 (248), gp116 (181), gp118 (428), gp119 (401), gp127 (331), gp128 (143), gp129 (989), gp130 (1073), gp131 (119), gp132 (369), gp133 (900), gp134 (123), gp135 (288), gp136 (177), gp137 (529), gp138 (226), gp141 (502), gp142 (558), gp145 (1930), gp152 (265), gp153 (383), gp154 (1212), gp155 (936), gp156 (204), gp157 (127), gp158 (293), gp159 (270), gp160 (267), gp161 (114), gp162 (372), gp163 (2143), gp164 (1062), gp165 (421), gp166 (406), gp167 (591), gp168 (693), gp171 (755), gp172 (591), gp173 (686), gp174 (82), gp175 (398), gp197 (78), gp198 (135), gp199 (486), gp205 (171), gp209 (585), gp81 (85) |
|
| ||
| Sum of protein lengths (Total length of DNA required in bp) | ||
| 4559 (13677) | 15092 (45276) | 30738 (92214) |
Protein lengths in amino acids are provided in parenthesis
Discussion
Phage 0305φ8-36 was previously shown to have unusual qualities, including unusual growth characteristics and atypical morphology, the most prominent feature being its three large curly fibers that join to the upper aspect of its baseplate (Serwer et al., 2007a; Serwer et al., 2007c). The results presented here indicate that these unusual characteristics of 0305φ8-36 are accompanied by an unusual 218,948 bp (6479 bp terminal repeat) genome, particularly notable for its extensive virion protein-based coding complexity. Of the 247 closely packed putative 0305φ8-36 gene products, only 34% had Psi-Blast matches. This observation is not unusual for a newly sequenced phage (Rohwer, 2003). However, the percentage of virion proteins identified by Psi-Blast (7%) is particularly low. Furthermore, the degree of divergence in these matches indicated no recent ancestry with any known phage, including those with similarly sized genomes. Thus, we propose that 0305φ8-36 be considered as the first member of a new phage genomic group.
Two technologies not routinely applied to the characterization of phages were used to characterize the genome and proteins of 0305φ8-36. Pyrosequencing was used to help sequence the genome (apparently the first use for a whole bacteriophage genome). The pyrosequencing produced data of quality higher than expected based on previous studies (Goldberg et al., 2006; Margulies et al., 2005). The higher redundancy used here (38-fold) is the only known explanation for this difference. The cost per base to obtain the high quality 0305φ8-36 sequence was substantially lower than it was using dideoxy terminator capillary DNA sequencing, an advantage of pyrosequencing reported previously at lower redundancy (Goldberg et al., 2006; Margulies et al., 2005).
In addition to pyrosequencing, SDS-PAGE/mass spectrometry has been utilized here to obtain a more comprehensive identification of the virion proteins. In parallel with identification of proteins in discrete gel bands, we also used short gel separations (1.5 cm) and excised unstained slices prior to in-gel digestion and HPLC-ESI-MS/MS analysis. This approach (often termed gel-LCMS) is somewhat analogous to MudPIT (Washburn et al., 2001) in which the first separation is based on strong cation exchange chromatography rather than SDS-PAGE. Characterization of replicate samples by two complementary approaches is important for maximizing the information content of the analyses. While mass spectrometry has been used previously to identify phage structural proteins (Chibani-Chennoufi et al., 2004b; Lavigne et al., 2006; Mann et al., 2005; Naryshkina et al., 2006), to our knowledge, the simple, but effective method used here (gel-LCMS) has not previously been applied to phages.
Phage 0305φ8-36 has the classical morphogenesis genes grouped by function into clusters, plus three novel genes whose products are strong candidates for components of the curly fibers. In addition, 0305φ8-36 has genes for many other novel virion proteins of unknown function. These 0305φ8-36 “extra” genes are organized in several modules in the morphogenesis region of the genome, suggesting several functions and several locations in the virion. Although the locations of the extra proteins within the 0305φ8-36 virion are not known from direct observation, both the genomic location of orfs 163 through 167 and the nature of the FN3 or VWA domain(s) they encode suggest that these proteins have a role in the formation of fibers that are external. Specifically, FN3 (pfam00041) and VWA (pfam00092) are domains initially characterized in fibronectin and von Willebrand’s factor, respectively. They are widely distributed in nature and generally carry out protein-protein or protein-polysaccharide binding functions (Chi-Rosso et al., 1997; Colombatti et al., 1993; Potts and Campbell, 1996; Whittaker and Hynes, 2002). These functions of FN3 and VWA domains support the location of gp163, 164, 165, 166 and 167 in an external virion fiber or fibers. An important focus for future studies is analysis of 0305φ8-36 interactions, including the extensive phage-phage interactions that are known to occur during propagation of 0305φ8-36 (Serwer et al., 2007a). All the putative fiber proteins are possible sources of these latter interactions.
Previous studies indicate that direct identification of 0305φ8-36 fibers is likely to involve complications. “Extra” or “contraction” fibers have been observed on phages PBS1 and AR9, in addition to their curly fibers (Belyaeva and Azizbekyan, 1968; Eiserling, 1967). Notably, these fibers of PBS1 and AR9 are not visible on every particle, possibly because they are in conformations that make them difficult to resolve (such as aligned with the tail sheath) (Eiserling, 1967). Alternatively, some of the contraction fibers may have been lost from virions during purification, storage or negative staining. Loss of fibers can result from any of these processes (Ackermann and DuBow, 1987; Bradley, 1965; Thomas, 2005). That 0305φ8-36 particles are unstable after purification was discussed earlier.
For analysis of the evolution of the extra virion proteins of 0305φ8-36, the question arises: How similar are the functions of the extra genes in the various long-genome bacteriophages? It appears that the answer is that the function differs among phages. For example, the data indicate that the functions of the extra proteins of phage phiKZ are not involved in assembling a virion, but rather in duplicating host functions (Fokine et al., 2005a). Also, several members of the T4 superfamily have genomes substantially longer than the T4 genome, and also do not appear to use their extra DNA for encoding proteins involved in assembly (Comeau et al., 2007). For example, T4-like cyanophages, such as S-PM2, use their extra DNA to encode proteins that assist the photosynthetic metabolism specific to their hosts (Mann et al., 2005). In contrast to these phages, the use by 0305φ8-36 of its extra DNA for virion proteins suggests an evolutionary response to selective forces applied during the extracellular phase of its life cycle. Although the details of protein function during this evolution are not known, possibilities include broadening host range (Chibani-Chennoufi et al., 2004a; Claverie et al., 2006; Miller et al., 2003a), or “sensing” the environment to allow host infection to occur only in suitable conditions [e.g., wac fibritin whiskers in T4, (Letarov et al., 2005)]. Alternatively, some of the extra proteins may function to facilitate flagella-dependent adsorption via the curly fibers (Ackermann and DuBow, 1987; Lindberg, 1973). Other tailed phages with long, curly fibers (e.g., PBS1, AR9, PBP1 and χ) use these fibers to adsorb reversibly to host flagella as a primary receptor before adsorbing to a secondary receptor on the cell wall (Ackermann and DuBow, 1987; Lindberg, 1973; Raimondo et al., 1968; Samuel et al., 1999; Schade et al., 1967). Additional possible functions of the extra 0305φ8-36 proteins are environmental interactions with genetic programming to enhance long-term virion viability in the absence of viable host [e.g., phage-phage interactions (Serwer et al., 2007a) or phage-clay interactions (Vettori et al., 1999)].
Thus, 0305φ8-36 is a resource for further studies to elucidate the relationship between complex structure and both host and environmental selective forces that have influenced its evolution. In addition, the high divergence of all 0305φ8-36 genes indicates that 0305φ8-36 is a member of an anciently branched group of phages that have been relatively isolated from horizontal exchange with other known phage groups. Assuming comparable isolation from all groups, 0305φ8-36 is also a resource for future studies of vertical descent in phage genomes. Hence, 0305φ8-36 may provide a valuable link to defining the ancestral myovirus.
Materials and Methods
Sequencing the 0305φ8-36 genome
Bacillus thuringiensis phage 0305φ8-36 was propagated as previously described (Serwer et al., 2007a). Phage DNA was extracted as described previously (Serwer et al., 2007c), with the exception that the freeze-thawing step prior to enzymatic degradation of host RNA and DNA was omitted. Two approaches were used in the sequencing of the 0305φ8-36 genome. First, phage 0305φ8-36 DNA was shotgun cloned in pUC119 and subjected to dideoxysequencing using a Beckman-Coulter Biomek 3000 robot and CEQ 8000 capillary sequencer according to the manufacturer’s directions. Instruction files used for the robot are found at http://biochem.uthscsa.edu/~hs_lab/scripting/Biomek_3000_Methods.html. Second, when the dideoxysequencing reached 9-fold redundancy, the 0305φ8-36 DNA was included in a mixture of four other phage genomes totaling 0.8 Mb and sequenced by pyrosequencing (Margulies et al., 2005) by 454 Life Sciences (Branford, CT). The five phage genomes had been subjected to at least a genomic survey, of the type previously described (Serwer et al., 2004). Contigs returned by 454 Life Sciences were assigned to their respective genomes by matching them to previously obtained dideoxy shotgun sequence data. The 0305φ8-36 contig sequence was converted to a single phd file using the program fastaq2phd (available at the Informatics at the University of Oklahoma Advanced Center for Genome Technology website; http://www.genome.ou.edu/informatics.html) and combined with the dideoxy shotgun data through use of the programs Phrap (Ewing and Green, 1998; Ewing et al., 1998) and Consed (Gordon et al., 1998) compiled for use with long reads as described in the documentation for the programs. All quality values in the final sequence were greater than 64. The longest homopolymer runs found in the 0305φ8-36 sequence were 9xA, 6xT, 5xC and 5xG.
Locating the genome termini
The approximate positions of the 0305φ8-36 genome termini were located using restriction enzyme analysis. The locations of the exact left and right ends of the genome were determined by sequencing PCR-amplified ligation products. These products were created by amplifying from intact phage DNA after ligation to pUC119 that had been cleaved with HincII, to give it blunt-ends.
Genome analyses
Frame prediction of the 0305φ8-36 genome was obtained with GeneMark (Lukashin and Borodovsky, 1998), Heuristic GeneMark (Besemer and Borodovsky, 1999), and frame-by-frame GeneMark (Shmatkov et al., 1999) implemented at the Borodovsky Bioinformatics www site (http://opal.biology.gatech.edu/GeneMark/). If multiple translation initiation sites for a frame were nominated by these methods, the site selected for inclusion in the GenBank submission was chosen based on the quality of its ribosome binding site and/or close packing with the nearest upstream feature. The presence of open reading frames in selected regions, such as those where there were no predictions by GeneMark, were also explored using ORF finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html). The frames were numb ered in the order of discovery.
Searches for tRNAs were conducted using tRNAscan-SE, version 1.23 (Lowe and Eddy, 1997) implemented at the Lowe laboratory web site (http://lowelab.ucsc.edu/tRNAscan-SE/) and ARAGORN, versions 1.1 and 1.2 (Laslett and Canback, 2004) available at the Lund Swegene Bioinformatics Facility website (https://pcmbioekol-bioinf2.mbioekol.lu.se/ARAGORN1.1/HTML/aragornA.html).
Homologies to 0305φ8-36 orfs were investigated using a locally implemented version of Psi-Blast (Altschul et al., 1997) with the entire NCBI nr plus env_nr databases. Homologies were also explored using rpsblast (McGinnis and Madden, 2004) and HMMER (Eddy, 1998) with the Pfam database (Finn et al., 2006). Matches of borderline significance were investigated by a reverse Psi-Blast search against a database in which all 0305φ8-36 frames had been included, starting from a member of the putative related family. SAM (Hughey and Krogh, 1996; Karplus et al., 1998) was obtained from Richard Hughey and implemented locally. SAM was used to create local Hidden Markov Models of several protein families when the relationship of a 0305φ8-36 orf was otherwise unclear. These orfs are described in the Results section. The SAM family building procedure followed the target2k strategy (Hughey et al., 2003) except that the Blast prefilter was replaced either with a Psi-Blast prefilter or scoring of the entire NCBI nr plus env_nr database. The SAM models were subjected to a tuneup operation (Hughey et al., 2003) and then used to screen a library of 0305φ8-36 frames.
All of the 0305φ8-36 frames were also subjected to the following analytical routines: secondary structure prediction by PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/); coiled coil prediction using the COILS server (Lupas et al., 1991) available at EMBnet (http://www.ch.embnet.org/software/COILS_form.html); transmembrane helix and other distributional data prediction by the Statistical Analyses of Protein Sequences package (SAPS)(Brendel et al., 1992), implemented at EMBnet (http://www.ch.embnet.org/software/SAPS_form.html); transmembrane helix prediction by TMHMM2.0 (Krogh et al., 2001) implemented at the Centre of Biological Sequence Analysis at the Technical University of Denmark (http://www.cbs.dtu.dk/services/TMHMM/).
Nucleotide sequence accession number
The 0305φ8-36 phage genome has been deposited in GenBank under the accession no. EF583821.
Phage purification
Two CsCl step gradients were used to purify phage 0305φ8-36. Stocks of phage 0305φ8-36 were prepared as previously described (Serwer et al., 2007a). The agarose-containing overlay was almost liquid and was harvested without the addition of buffer. This suspension was centrifuged (5000 rpm, 6 min, 4 °C) in a JA rotor in Beckman Avanti J-25 centrifuge. The resulting supernatant was decanted (titer was ~4 × 1011 pfu/ml) and incubated in the presence of DNAase (final concentration 100 μg/ml) for 1 h at 30 °C. A CsCl step gradient was constructed using a buffer composed of 0.1 M Tris-HCl (pH 7.4), 0.05 M MgSO4 and 0.5 M NaCl with CsCl that was added to achieve the following buoyant densities (and volumes), in order from the top of the gradient to the bottom: 1.59 g/ml (0.75 ml), 1.52 g/ml (0.75 ml), 1.41 g/ml (1.2 ml), 1.30 g/ml (1.5 ml) and 1.21 g/ml (1.8 ml). Phage suspension (5.8 ml) was loaded onto the top of this gradient and spun at 33,000 rpm for 1.5 h at 18 °C in an SW41 rotor in a Beckman Coulter Optima LE-80K ultracentrifuge. The phage bands from six tubes were harvested and combined (2.5 ml). The titer was ~3 × 1010 pfu/ml. The buoyant density of the purified phage suspension was 1.41 g/ml. This suspension was further purified by placement between two layers of buffer containing CsCl, the lower layer having a buoyant density of 1.52 g/ml (1.2 ml) and the upper having a buoyant density of 1.36 g/ml (1.3 ml). The sample was centrifuged for 2 h at 42,000 rpm and 4 °C in a SW55Ti rotor. The resulting harvested phage band had a buoyant density of 1.42 g/ml. The phage suspension was dialysed against three changes of 0.2 M NaCl, 0.01 M Tris-HCl, 0.05M MgCl2 at 4 °C. The titer was ~7 × 109 pfu/ml. The second step-gradient centrifugation was used instead of buoyant density centrifugation to reduce the time needed for centrifugation and to minimize the loss of particles.
Analyses of virion proteins
The proteins of the purified phage particles were subjected to SDS-PAGE according to the method of Laemmli (1970). Phage samples diluted in sample buffer were heated at 95 °C for 2 min and then loaded onto Tris-HCl Ready Gels (Bio-Rad). Electrophoresis was performed using a Criterion electrophoresis unit (Bio-Rad) according to the manufacturer’s directions. Proteins were stained with Coomassie Brilliant Blue R250 (Bio-Rad). Protein markers (Bio-Rad) that were run in an outside lane were used for estimation of molecular weights.
Mass Spectrometry
Coomassie-stained gel bands and slices were manually excised and digested in situ with trypsin (Promega modified) in 40 mM NH4HCO3 at 37 °C for 4 h. The digests were analyzed by mass spectrometry without further purification. Capillary HPLC-electrospray ionization tandem mass spectra (HPLC-ESI-MS/MS) were acquired on a Thermo Fisher LTQ linear ion trap mass spectrometer fitted with a New Objective PicoView 550 nanospray interface. On-line HPLC separation of the digests was accomplished with an Eksigent NanoLC micro HPLC: column, PicoFrit™ (New Objective; 75 μm i.d.) packed to 10 cm with C18 adsorbent (Vydac; 218MS 5 μm, 300 Å); mobile phase A, 0.5% acetic acid (HAc)/0.005% trifluoroacetic acid (TFA); mobile phase B, 90% acetonitrile/0.5% HAc/0.005% TFA; gradient 2 to 42% B in 30 min; flow rate, 0.4 μl/min. MS conditions were: ESI voltage, 2.9 kV; isolation window for MS/MS, 3; relative collision energy, 35%; scan strategy, survey scan followed by acquisition of data dependent collision-induced dissociation (CID) spectra of the seven most intense ions in the survey scan above a set threshold. The uninterpreted CID spectra were searched against the Swiss-Prot database supplemented with all putative 0305φ8-36 protein sequences using Mascot (Matrix Science; London, UK). Methionine oxidation was considered as a variable modification for all searches. Cross correlation of the Mascot results with X! Tandem and determination of protein identity probabilities were accomplished by Scaffold (Proteome Software). Searches considering “semi-tryptic” cleavages were used to detect proteolytically processed N-terminal sequences for the high copy-number proteins.
For some phage virion proteins, only one peptide was identified by the search of the combined 0305φ8-36_Swiss-Prot database. As such, the potential exists for these to be false positive assignments. However, since all proteins, except one (gp197) identified on the basis of a single peptide mapped to the main morphogenesis gene region of the genome with high probability, it is highly likely that even the single peptide assignments represent true components of the virion.
Quantification of phage virion proteins
To quantify proteins after SDS-PAGE, gel patterns obtained with four different dilutions of total phage protein were digitalized using a Bio-Rad GS-800 Imaging Densitometer. The images were then converted to chromatograms using ImageJ (available at: http://rsb.info.nih.gov/ij/download.html) after which the peaks were integrated. The integrated intensity of the tail sheath protein at each of four dilutions was used to generate a standard curve with which to correct for signal saturation effects. For each of the other proteins, a concentration relative to the tail sheath protein was calculated at each of three different dilutions taking saturation and relative mass into account. The standard errors of the three determinations for each protein were within 25%. No correction was attempted for any systematic differential staining of the respective proteins that might occur.
Supplementary Material
Fig. 3.

Alignment of the N-terminus of 0305φ8-36 major head protein gp125 with BtI1 homologue RBTH_06381. The N-terminus of RBTH_06381 extends beyond the site annotated in the GenBank entry. This extension is shown in bold. The N-terminal fragment of gp125 detected by mass spectrometry is indicated in blue. The predicted conserved protease cleavage site of gp125 is underlined. The first 41 predicted residues of gp125 are not shown. Alignment was by BlastP.
Acknowledgments
Mass spectral analyses were performed in the UTHSCSA Institutional Mass Spectrometry Laboratory. We would like to thank Dr. Borries Demeler and Jeremy Mann in the UTHSCSA Bioinformatics Center for assistance with computational aspects of the project, and Brano Djenic and Kyle Wallace for technical assistance. This research was supported by grants from The Robert J. Kleberg, Jr. and Helen C. Kleberg Foundation, the Welch Foundation (AQ-764) and the National Institutes of Health (GM24365).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abuladze NK, Gingery M, Tsai J, Eiserling FA. Tail length determination in bacteriophage T4. Virology. 1994 Mar;199(2):301–10. doi: 10.1006/viro.1994.1128. [DOI] [PubMed] [Google Scholar]
- Ackermann HW. Tailed bacteriophages: The order Caudovirales. Adv Virus Res. 2000;51:135–201. doi: 10.1016/S0065-3527(08)60785-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackermann H-W, DuBow MS. Viruses of Prokaryotes. 1 & 2. CRC Press; Boca Raton, Florida: 1987. [Google Scholar]
- Ackermann HW, Yoshino S, Ogata S. A Bacillus phage that is a living fossil. Can J Microbiol. 1995;41:294–297. [Google Scholar]
- Admiraal G, Mellema JE. The structure of the contractile sheath of bacteriophage Mu. J Ultrastruc Res. 1976;56:48–64. doi: 10.1016/s0022-5320(76)80140-2. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker ML, Jiang W, Rixon FJ, Chiu W. Common ancestry of herpesviruses and tailed DNA bacteriophages. J Virol. 2005;79:14967–14970. doi: 10.1128/JVI.79.23.14967-14970.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyaeva NN, Azizbekyan RR. Fine structure of new Bacillus subtilis phage AR9 with complex morphology. Virology. 1968;34:176–179. doi: 10.1016/0042-6822(68)90023-8. [DOI] [PubMed] [Google Scholar]
- Besemer J, Borodovsky M. Heuristic approach to deriving models for gene finding. Nucleic Acids Res. 1999;27:3911–3920. doi: 10.1093/nar/27.19.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black LW, Showe MK, Steven AC. Morphogenesis of the bacteriophage T4 head. In: Karam JD, editor. Molecular Biology of T4. American Society for Microbiology; Washington, D.C: 1994. [Google Scholar]
- Bradley DE. The morphology and physiology of bacteriophages as revealed by the electron microscope. J R Microsc Soc. 1965;84:257–316. [PubMed] [Google Scholar]
- Bradley DE. Ultrastructure of bacteriophages and bacteriocins. Bacteriol Rev. 1967;31:230–314. doi: 10.1128/br.31.4.230-314.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendel V, Bucher P, Nourbakhsh I, Blaisdell BE, Karlin S. Methods and algorithms for statistical analysis of protein sequences. Proc Natl Acad Sci USA. 1992;89:2002–2006. doi: 10.1073/pnas.89.6.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüssow H, Kutter E. Phage Ecology. In: Kutter E, Sulakvelidze A, editors. Bacteriophages: Biology and Applications. CRC Press; Boca Raton: 2005. pp. 129–163. [Google Scholar]
- Casjens S. An introduction to virus structure and assembly. In: Casjens S, editor. Virus Structure and Assembly. Jones & Bartlett; Boston: 1985. pp. 1–28. [Google Scholar]
- Casjens S. Prophages and bacterial genomics: what have we learned so far? Mol Microbiol. 2003;49:277–300. doi: 10.1046/j.1365-2958.2003.03580.x. [DOI] [PubMed] [Google Scholar]
- Casjens S, Hendrix R. Control mechanisms in dsDNA bacteriophage assembly. In: Calender R, editor. The Bacteriophages. Vol. 1. Plenum Press; New York: 1988. pp. 15–91. [Google Scholar]
- Chibani-Chennoufi S, Bruttin A, Dillmann M, Brussow H. Phage-host interaction: an ecological perspective. J Bacteriol. 2004a;186:3677–3686. doi: 10.1128/JB.186.12.3677-3686.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chibani-Chennoufi S, Dillmann ML, Marvin-Guy L, Rami-Shojaei S, Brussow H. Lactobacillus plantarum bacteriophage LP65: A new member of the SPO1-like genus of the family Myoviridae. J Bacteriol. 2004b;186:7069–7083. doi: 10.1128/JB.186.21.7069-7083.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi-Rosso G, Gotwals PJ, Yang J, Ling L, Jiang K, Chao B, Baker DP, Burkly LC, Fawell SE, Koteliansky VE. Fibronectin Type III Repeats Mediate RGD-independent Adhesion and Signaling through Activated beta 1 Integrins. J Biol Chem. 1997;272:31447–31452. doi: 10.1074/jbc.272.50.31447. [DOI] [PubMed] [Google Scholar]
- Claverie JM, Ogata H, Audic S, Abergel C, Suhre K, Fournier P. Mimivirus and the emerging concept of “giant” virus. Virus Res. 2006;117:133–144. doi: 10.1016/j.virusres.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Colombatti A, Bonaldo P, Doliana R. A common feature appears to be involvement in multiprotein complexes. Proteins that incorporate vWF domains participate in numerous biological events. Matrix. 1993;13:297–306. doi: 10.1016/s0934-8832(11)80025-9. [DOI] [PubMed] [Google Scholar]
- Comeau A, Bertrand C, Letarov A, Tétart F, Krisch H. Modular architecture of the T4 phage superfamily: A conserved core genome and a plastic periphery. Virology. 2007 doi: 10.1016/j.virol.2006.12.031. [DOI] [PubMed] [Google Scholar]
- Coombs DH, Arisaka F. T4 tail structure and function. In: Karam JD, editor. Molecular Biology of T4. American Society for Microbiology; Washington, D.C: 1994. [Google Scholar]
- Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- Effantin G, Boulanger P, Neumann E, Letellier L, Conway JF. Bacteriophage T5 structure reveals similarities with HK97 and T4 suggesting evolutionary relationships. J Mol Biol. 2006;361:993–1002. doi: 10.1016/j.jmb.2006.06.081. [DOI] [PubMed] [Google Scholar]
- Eiserling FA. The structure of Bacillus subtilis bacteriophage PBS1. J Ultrastruct Res. 1967;17:342–347. doi: 10.1016/s0022-5320(67)80053-4. [DOI] [PubMed] [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using Phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using Phred. I. Accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Fangman WL. Separation of very large DNA molecules by gel electrophoresis. Nucleic Acids Res. 1978;5:653–665. doi: 10.1093/nar/5.3.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA, editors. Virus Taxonomy. VIIIth Report of the International Committee on Taxonomy of Viruses; Oxford: Elsevier/Academic Press. 2005. [Google Scholar]
- Finn RD, Mistry J, Schuster-Bockler B, Griffiths-Jones S, Hollich V, Lassmann T, Moxon S, Marshall M, Khanna A, Durbin R, Eddy SR, Sonnhammer EL, et al. Pfam: clans, web tools and services. Nucleic Acids Res. 2006;34(Database issue):D247–D251. doi: 10.1093/nar/gkj149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fokine A, Kostyuchenko VA, Efimov AV, Kurochkina LP, Sykilinda NN, Robben J, Volckaert G, Hoenger A, Chipman PR, Battisti AJ, Rossmann MG, Mesyanzhinov VV. A three-dimensional cryo-electron microscopy structure of the bacteriophage phiKZ head. J Mol Biol. 2005a;352:117–124. doi: 10.1016/j.jmb.2005.07.018. [DOI] [PubMed] [Google Scholar]
- Fokine A, Leiman PG, Shneider MM, Ahvazi B, Boeshans KM, Steven AC, Black LW, Mesyanzhinov VV, Rossmann MG. Structural and functional similarities between the capsid proteins of bacteriophages T4 and HK97 point to a common ancestry. Proc Natl Acad Sci. 2005b;102:7163–7168. doi: 10.1073/pnas.0502164102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford ME, Sarkis GJ, Belanger AE, Hendrix RW, Hatfull GF. Genome structure of mycobacteriophage D29: implications for phage evolution. J Mol Biol. 1998;279:143–164. doi: 10.1006/jmbi.1997.1610. [DOI] [PubMed] [Google Scholar]
- Goldberg SMD, Johnson J, Busam D, Feldblyum T, Ferriera S, Friedman R, Halpern A, Khouri H, Kravitz SA, Lauro FM, Li K, Rogers YH, et al. A Sanger/pyrosequencing hybrid approach for the generation of high-quality draft assemblies of marine microbial genomes. Proc Natl Acad Sci. 2006;103:11240–11245. doi: 10.1073/pnas.0604351103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich-Blair H, David AS. The DNA polymerase genes of several HMU-bacteriophages have similar group I introns with highly divergent open reading frames. Nucleic Acids Research. 1994;22:3715–3721. doi: 10.1093/nar/22.18.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich-Blair H, Scarlato V, Gott JM, Xu MQ, Shub DA. A self-splicing group I intron in the DNA polymerase gene of Bacillus subtilis bacteriophage SPO1. Cell. 1990;63:417–424. doi: 10.1016/0092-8674(90)90174-d. [DOI] [PubMed] [Google Scholar]
- Gordon D, Abajian C, Green P. Consed: A graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- Haggard-Ljungquist E, Jacobsen E, Rishovd S, Six EW, Nilssen O, Sunshine MG, Lindqvist BH, Kim KJ, Barreiro V, Koonin EV. Bacteriophage P2: genes involved in baseplate assembly. Virology. 1995;213:109–121. doi: 10.1006/viro.1995.1551. [DOI] [PubMed] [Google Scholar]
- Hambly E, Tétart F, Desplats C, Wilson WH, Krisch HM, Mann NH. A conserved genetic module that encodes the major virion components in both the coliphage T4 and the marine cyanophage S-PM2. Proc Natl Acad Sci. 2001;98:11411–11416. doi: 10.1073/pnas.191174498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatfull GF, Sarkis GJ. DNA sequence, structure and gene expression of mycobacteriophage L5: a phage system for mycobacterial genetics. Mol Microbiol. 1993;7:395–405. doi: 10.1111/j.1365-2958.1993.tb01131.x. [DOI] [PubMed] [Google Scholar]
- Helgstrand C, Wikoff WR, Duda RL, Hendrix RW, Johnson JE, Liljas L. The refined structure of a protein catenane: The HK97 bacteriophage capsid at 3.44 A resolution. J Mol Biol. 2003;334:885–899. doi: 10.1016/j.jmb.2003.09.035. [DOI] [PubMed] [Google Scholar]
- Hendrix RW. Bacteriophage HK97: Assembly of the capsid and evolutionary connections. Adv Virus Res. 2005;64:1–14. doi: 10.1016/S0065-3527(05)64001-8. [DOI] [PubMed] [Google Scholar]
- Hertveldt K, Lavigne R, Pleteneva E, Sernova N, Kurochkina L, Korchevskii R, Robben J, Mesyanzhinov V, Krylov VN, Volckaert G. Genome comparison of Pseudomonas aeruginosa large phages. J Mol Biol. 2005;354:536–45. doi: 10.1016/j.jmb.2005.08.075. [DOI] [PubMed] [Google Scholar]
- Hughey R, Karplus K, Krogh A. University of California; Santa Cruz, CA: 2003. SAM: Sequence alignment and modeling software system. Technical Report UCSC-CRL-99-11. online at http://www.soe.ucsc.edu/research/compbio/papers/sam_doc/sam_doc.html. [Google Scholar]
- Hughey R, Krogh A. Hidden Markov models for sequence analysis: Extension and analysis of the basic method. Comput Appl Biosci. 1996;12:95–107. doi: 10.1093/bioinformatics/12.2.95. [DOI] [PubMed] [Google Scholar]
- Iwasaki K, Trus BL, Wingfield PT, Cheng N, Campusano G, Rao VB, Steven AC. Molecular architecture of bacteriophage T4 capsid: Vertex structure and bimodal binding of the stabilizing accessory protein, soc. Virology. 2000;271:321–333. doi: 10.1006/viro.2000.0321. [DOI] [PubMed] [Google Scholar]
- Karplus K, Barrett C, Hughey R. Hidden markov models for detecting remote protein homologies. Bioinformatics. 1998;14:846–856. doi: 10.1093/bioinformatics/14.10.846. [DOI] [PubMed] [Google Scholar]
- Katsura I. Determination of bacteriophage lambda tail length by a protein ruler. Nature. 1987;327:73–75. doi: 10.1038/327073a0. [DOI] [PubMed] [Google Scholar]
- Katsura I, Hendrix RW. Length determination in bacteriophage Lambda tails. Cell. 1984;39:691–698. doi: 10.1016/0092-8674(84)90476-8. [DOI] [PubMed] [Google Scholar]
- Kostyuchenko VA, Chipman PR, Lieman PG, Arisaka F, Mesyanzhinov VV, Rossmann MG. The tail structure of T4 and its mechanism of contraction. Nat Struct Mol Biol. 2005;12:810–813. doi: 10.1038/nsmb975. [DOI] [PubMed] [Google Scholar]
- Krogh A, Larsson B, Von Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Kunst F, Ogasawara N, Moszer I, Albertini AM, Alloni G, Azevedo V, Bertero MG, Bessieres P, Bolotin A, Borchert S, Borriss R, Boursier L, et al. The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature. 1997;390:249–256. doi: 10.1038/36786. [DOI] [PubMed] [Google Scholar]
- Kwan T, Liu J, DuBow M, Gros P, Pelletier J. The complete genomes and proteomes of 27 Staphylococcus aureus bacteriophages. Proc Natl Acad Sci. 2005;102:5174–5179. doi: 10.1073/pnas.0501140102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan T, Liu J, DuBow M, Gros P, Pelletier J. Comparative genomic analysis of 18 Pseudomonas aeruginosa bacteriophages. J Bacteriol. 2006;188:1184–1187. doi: 10.1128/JB.188.3.1184-1187.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laslett D, Canback B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004;32:11–16. doi: 10.1093/nar/gkh152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavigne R, Noben JP, Hertveldt K, Ceyssens PJ, Briers Y, Dumont D, Roucourt B, Krylov VN, Mesyanzhinov VV, Robben J, Volckaert G. The structural proteome of Pseudomonas aeruginosa bacteriophage phiKMV. Microbiology. 2006;152:529–534. doi: 10.1099/mic.0.28431-0. [DOI] [PubMed] [Google Scholar]
- Letarov A, Manival X, Desplats C, Krisch HM. gpwac of the T4-type bacteriophages: Structure, function, and evolution of a segmented coiled-coil protein that controls viral infectivity. J Bacteriol. 2005;187:1055–1066. doi: 10.1128/JB.187.3.1055-1066.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg AA. Bacteriophage Receptors. Ann Rev Microbiol. 1973;27:205–241. doi: 10.1146/annurev.mi.27.100173.001225. [DOI] [PubMed] [Google Scholar]
- Lovett PS. PBP1: a flagella-specific bacteriophage mediating transduction in Bacillus pumilus. Virology. 1972;47:743–752. doi: 10.1016/0042-6822(72)90564-8. [DOI] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashin A, Borodovsky M. GeneMark hmm: new solutions for gene finding. Nucleic Acids Res. 1998;26:1107–1115. doi: 10.1093/nar/26.4.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- Mann NH, Clokie MRJ, Millard A, Cook A, Wilson WH, Wheatley PJ, Letarov A, Krisch HM. The genome of S-PM2, a “photosynthetic” T4-type bacteriophage that infects marine synechococcus strains. J Bacteriol. 2005;187:3188–3200. doi: 10.1128/JB.187.9.3188-3200.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis S, Madden TL. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004;32:W20–W25. doi: 10.1093/nar/gkh435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesyanzhinov VV, Maramorosch K, Shatkin AJ, editors. Adv Virus Res. Vol. 63. Academic Press; 2004. Bacteriophage T4: Structure, assembly, and initiation infection studied in three dimensions. [DOI] [PubMed] [Google Scholar]
- Mesyanzhinov VV, Robben J, Grymonprez B, Kostyuchenko VA, Bourkaltseva MV, Sykilinda NN, Krylov VN, Volckaert G. The genome of bacteriophage phiKZ of Pseudomonas aeruginosa. J Mol Biol. 2002;317:1–19. doi: 10.1006/jmbi.2001.5396. [DOI] [PubMed] [Google Scholar]
- Miller ES, Heidelberg JF, Eisen JA, Nelson WC, Durkin S, Ciecko A, Feldblyum TV, White O, Paulsen IT, Nierman WC, Lee J, Szczypinski B, et al. Complete genome sequence of the broad-host-range vibriophage KVP40: Comparative genomics of a T4-related bacteriophage. J Bacteriol. 2003a;185:5220–5233. doi: 10.1128/JB.185.17.5220-5233.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller ES, Kutter E, Mosig G, Arisaka F, Kunisawa T, Ruger W. Bacteriophage T4 Genome. Microbiol Mol Biol Rev. 2003b;67:86–156. doi: 10.1128/MMBR.67.1.86-156.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan GJ, Hatfull GF, Casjens S, Hendrix RW. Bacteriophage Mu genome sequence: analysis and comparison with Mu-like prophages in Haemophilus, Neisseria and Deinococcus. J Mol Biol. 2002;317:337–359. doi: 10.1006/jmbi.2002.5437. [DOI] [PubMed] [Google Scholar]
- Naryshkina T, Liu J, Florens L, Swanson SK, Pavlov ARVPN, Inman R, Minakhin L, Kozyavkin SA, Washburn M, Mushegian A, Severinov K. Thermus thermophilus bacteriophage phiYS40 genome and proteomic characterization of virions. J Mol Biol. 2006;364:667–77. doi: 10.1016/j.jmb.2006.08.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan J, Petrov V, Bertrand C, Krisch H, Karam J. Genetic diversity among five T4-like bacteriophages. Virol J. 2006;3:30. doi: 10.1186/1743-422X-3-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson NH, Gingery M, Eiserling FA, Baker TS. The structure of isometric capsids of bacteriophage T4. Virology. 2001;279:385–391. doi: 10.1006/viro.2000.0735. [DOI] [PubMed] [Google Scholar]
- Parker ML, Eiserling FA. Bacteriophage SPO1 structure and morphogenesis. I Tail structure and length regulation. J Virol. 1983;43:239–249. doi: 10.1128/jvi.46.1.239-249.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedulla ML, Ford ME, Houts J, Karthikeyan T, Wadsworth C, Lewis JA, Jacobs-Sera D, Falbo J, Gross J, Pannunzio NR, Brucker W, Kumar V, et al. Origins of highly mosaic mycobacteriophage genomes. Cell. 2003;113:171–182. doi: 10.1016/s0092-8674(03)00233-2. [DOI] [PubMed] [Google Scholar]
- Petrov VM, Nolan JM, Bertrand C, Levy D, Desplats C, Krisch HM, Karam JD. Plasticity of the gene functions for DNA replication in the T4-like phages. J Mol Biol. 2006;361:46–68. doi: 10.1016/j.jmb.2006.05.071. [DOI] [PubMed] [Google Scholar]
- Popa M, McKelvey TA, Hempel J, Hendrix RW. Bacteriophage HK97 structure: Wholesale covalent cross-linking between the major head shell subunits. J Virol. 1991;65:3227–3237. doi: 10.1128/jvi.65.6.3227-3237.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts JR, Campbell ID. Structure and function of fibronectin modules. Matrix Biol. 1996;15:313–320. doi: 10.1016/s0945-053x(96)90133-x. [DOI] [PubMed] [Google Scholar]
- Raimondo LM, Lundh NP, Martinez RJ. Primary adsorption site of phage PBS1: the flagellum of Bacillus subtilis. J Virol. 1968;2:256–264. doi: 10.1128/jvi.2.3.256-264.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohwer F. Global phage diversity. Cell. 2003;113:141. doi: 10.1016/s0092-8674(03)00276-9. [DOI] [PubMed] [Google Scholar]
- Ross PD, Cheng N, Conway JF, Firek BA, Hendrix R, Duda RL, Steven AC. Crosslinking renders bacteriophage HK97 capsid maturation irreversible and effects an essential stabilization. EMBO J. 2005;24:1352–1363. doi: 10.1038/sj.emboj.7600613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost B. PHD. Meth Enzymol. 1996;266:525–539. doi: 10.1016/s0076-6879(96)66033-9. [DOI] [PubMed] [Google Scholar]
- Rost B, Sandler C. PHDsec. J Mol Biol. 1993;232:584–599. doi: 10.1006/jmbi.1993.1413. [DOI] [PubMed] [Google Scholar]
- Samuel ADT, Pitta TP, Ryu WS, Danese PN, Leung ECW, Berg HC. Flagellar determinants of bacterial sensitivity to Chi-phage. Proc Natl Acad Sci. 1999;96:9863–9866. doi: 10.1073/pnas.96.17.9863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schade SZ, Adler J, Ris H. How bacteriophage Chi attacks motile bacteria. J Virol. 1967;1:599–609. doi: 10.1128/jvi.1.3.599-609.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serwer P, Hayes S, Thomas J, Hardies S. Propagating the missing bacteriophages: a large bacteriophage in a new class. Virol J. 2007a;4:21. doi: 10.1186/1743-422X-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serwer P, Hayes SJ, Thomas J, Demeler B, Hardies SC. Isolation of novel large and aggregating bacteriophages. In: Clokie M, Kropinski AM, editors. Bacteriophages: Methods and protocols. 2007b. in press. [DOI] [PubMed] [Google Scholar]
- Serwer P, Hayes SJ, Thomas J, Griess GA, Hardies SC. Rapid determination of genomic DNA length for new bacteriophages. Electrophoresis. 2007c doi: 10.1002/elps.200600672. in press. [DOI] [PubMed] [Google Scholar]
- Serwer P, Hayes SJ, Zaman S, Lieman K, Rolando M, Hardies SC. Improved isolation of undersampled bacteriophages: finding of distant terminase genes. Virology. 2004;329:412–424. doi: 10.1016/j.virol.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Sharp R. Bacteriophages: biology and history. J Chem Tech Biotech. 2001;76:667–672. [Google Scholar]
- Shmatkov AM, Melikyan AM, Chernousko FL, Borodovsky M. Finding prokaryotic genes by the “frame-by-frame” algorithm: targeting gene starts and overlapping genes. Bioinformatics. 1999;15:874–886. doi: 10.1093/bioinformatics/15.11.874. [DOI] [PubMed] [Google Scholar]
- Slopek S, Krzywy T. Morphology and ultrastructure of bacteriophages. An electron microscopic study. Arch Immunol Ther Exp (Warsz) 1985;33:1–217. [PubMed] [Google Scholar]
- Stewart CR, Gaslightwala I, Hinata K, Krolikowski KA, Needleman DS, Peng AS, Peterman MA, Tobias A, Wei P. Genes and regulatory sites of the ‘host-takeover module’ in the terminal redundancy of Bacillus subtilis bacteriophage SPO1. Virology. 1998;246:329–340. doi: 10.1006/viro.1998.9197. [DOI] [PubMed] [Google Scholar]
- Sullivan MB, Coleman ML, Weigele P, Rohwer F, Chisholm SW. Three Prochlorococcus cyanophage genomes: Signature features and ecological interpretations. PLoS Biol. 2005;3:e144. doi: 10.1371/journal.pbio.0030144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takedo S, Sasaki T, Ritani A, Howe M, Arisaka F. Discovery of the tail tube gene of bacteriophage Mu and sequence analysis of the sheath and tube genes. Biochim Biophys Acta. 1998;1399:88–92. doi: 10.1016/s0167-4781(98)00102-x. [DOI] [PubMed] [Google Scholar]
- Temple LM, Forsburg SL, Calendar R, Christie GE. Nucleotide sequence of the genes encoding the major tail sheath and tail tube proteins of bacteriophage P2. Virology. 1991;181:353–358. doi: 10.1016/0042-6822(91)90502-3. [DOI] [PubMed] [Google Scholar]
- Tétart F, Desplats C, Kutateladze M, Monod C, Ackermann HW, Krisch HM. Phylogeny of the major head and tail genes of the wide-ranging T4-type bacteriophages. J Bacteriol. 2001;183:358–366. doi: 10.1128/JB.183.1.358-366.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JA. PhD thesis. La Trobe University; Australia: 2005. [Google Scholar]
- Vettori C, Stotzky G, Yoder M, Gallori E. Interaction between bacteriophage PBS1 and clay minerals and transduction of Bacillus subtilis by clay-phage complexes. Environ Microbiol. 1999;1:347–355. doi: 10.1046/j.1462-2920.1999.00044.x. [DOI] [PubMed] [Google Scholar]
- Wang J, Jiang Y, Vincent M, Sun Y, Yu H, Wang J, Bao Q, Kong H, Hu S. Complete genome sequence of bacteriophage T5. Virology. 2005;332:45–65. doi: 10.1016/j.virol.2004.10.049. [DOI] [PubMed] [Google Scholar]
- Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001 Mar;19(3):242–7. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- Whittaker CA, Hynes RO. Distribution and Evolution of von Willebrand/Integrin A Domains: Widely Dispersed Domains with Roles in Cell Adhesion and Elsewhere. Mol Biol Cell. 2002;13:3369–3387. doi: 10.1091/mbc.E02-05-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikoff WR, Liljas L, Duda RL, Tsuruta H, Hendrix RW, Johnson JE. Topologically linked protein rings in the bacteriophage HK97 capsid. Science. 2000;289:2129–2133. doi: 10.1126/science.289.5487.2129. [DOI] [PubMed] [Google Scholar]
- Wommack KE, Colwell RR. Virioplankton: Viruses in aquatic ecosystems. Microbiol Mol Biol Rev. 2000;64:69–114. doi: 10.1128/mmbr.64.1.69-114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Hendrix R, Duda RL. Conserved translational frameshift in dsDNA bacteriophage tail assembly genes. Mol Cell. 2004;16:11–21. doi: 10.1016/j.molcel.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Young R, Wang IN, Roof WD. Phages will out: strategies of host cell lysis. TRENDS in Microbiology. 2000;8:120–128. doi: 10.1016/s0966-842x(00)01705-4. [DOI] [PubMed] [Google Scholar]
- Ziegelhoffer T, Yau P, Chandrasekhar G, Kochan J, Georgopoulos C, Murialdo H. The purification and properties of the scaffolding protein of bacteriophage lambda. J Biol Chem. 1992;267:455–461. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.