

Abstract
Sialic acids are important cell-surface molecules of animals in the deuterostome lineage. Although humans do not express easily detectable amounts of N-glycolylneuraminic acid (Neu5Gc, a hydroxylated form of the common sialic acid N-acetylneuraminic acid, Neu5Ac), it is a major component in great ape tissues, except in the brain. This difference correlates with lack of the hydroxylase activity that converts CMP-Neu5Ac to CMP-Neu5Gc. Here we report cloning of human and chimpanzee hydroxylase cDNAs. Although this chimpanzee cDNA is similar to the murine homologue, the human cDNA contains a 92-bp deletion resulting in a frameshift mutation. The isolated human gene also shows evidence for this deletion. Genomic PCR analysis indicates that this deletion does not occur in any of the African great apes. The gene is localized to 6p22–p23 in both humans and great apes, which does not correspond to known chromosomal rearrangements that occurred during hominoid evolution. Thus, the lineage leading to modern humans suffered a mutation sometime after the common ancestor with the chimpanzee and bonobo, potentially affecting recognition by a variety of endogenous and exogenous sialic acid-binding lectins. Also, the expression of Neu5Gc previously reported in human fetuses and tumors as well as the traces detected in some normal adult humans must be mediated by an alternate pathway.
Keywords: Pongidae/evolution/neuraminic acids/PCR/hominoids
Humans are evolutionarily related to the African great apes, Pan troglodytes (the chimpanzee), Pan paniscus (the bonobo or pygmy chimpanzee) and Gorilla gorilla (the gorilla) (1–6). Current data indicate that the last common ancestor of humans with the great apes was very likely shared with the chimpanzee and bonobo (7–16). Given the ≈99% identity at the DNA and amino acid-sequence level, it is proposed that only a few changes in gene expression or function are responsible for the morphological and functional differences between humans and these closely related primates (4). Such differences in gene regulation and function could also explain variations in susceptibility and biological response to diseases such as cancer, hepatitis, AIDS, malaria, and intestinal infections (17–22).
The sialic acids are a family of acidic sugars typically found at the outer end of the cell surface and secreted glycoconjugates of all vertebrates (23–26). The two most common forms of sialic acid found in mammalian cells are N-acetylneuraminic acid (Neu5Ac) and its hydroxylated derivative, N-glycolylneuraminic acid (Neu5Gc). The conversion from Neu5Ac to Neu5Gc can positively or negatively affect interactions involving several of the known endogenous and exogenous receptors for sialic acids such as CD22, myelin-associated glycoprotein, sialoadhesin, and the influenza A virus hemagglutinin (27–33). Interestingly, even in animals with large amounts of Neu5Gc in other tissues, its level in the brain is always extremely low (34–36). We recently reported a major biochemical difference between the great apes and humans, with the latter showing a loss of activity of the enzyme CMP-N-acetylneuraminic acid hydroxylase (37). This enzyme converts the nucleotide sugar donor CMP-Neu5Ac to CMP-Neu5Gc in other animals (38–46). Loss of hydroxylase activity provides a potential explanation for the finding that although the body fluids and tissues of all of the great apes (chimpanzee, bonobo, gorilla, and orangutan) express high levels of this sialic acid, corresponding samples from humans contain little or no detectable Neu5Gc (37). However, the regulation of this hydroxylase activity is very complex and involves multiple interacting factors (38–45). Moreover, expression of Neu5Gc has been reported in human fetal tissues (47) and in certain human cancers (47–49), and our previous study (37) noted that some human tissues had small quantities of an HPLC peak corresponding to Neu5Gc. Thus, it has been suggested that the activity of this hydroxylase may be suppressed in adult humans, rather than eliminated altogether. To explore this issue, we have cloned the human and chimpanzee cDNAs encoding the CMP-Neu5Ac hydroxylase. We find that the human lineage has suffered a genetic mutation in the coding region of this cDNA. While this manuscript was in preparation, another group reported the same deletion in a human hydroxylase cDNA (50). However, that paper reported an incomplete cDNA sequence, which did not include the initial 5′ translation start site. It also did not address the uniqueness of this mutation for humans and its implications for hominoid evolution.
MATERIALS AND METHODS
Identification and Characterization of Expressed Sequence Tag (EST) Clones.
The previously reported mouse CMP-Neu5Ac hydroxylase sequence (45) was used to carry out blast searches of the human EST database at the National Center for Biotechnology Information website (http://www.ncbi.nlm.nih.gov/dbEST/index.html). Several clones with >80% homology to the murine hydroxylase sequence were found. These were obtained from the company Research Genetics, and subjected to complete sequencing of both strands by the dideoxy chain-termination method (performed by the MacConnell Research Corporation, or in the laboratory of S. Hedrick, using an Applied Biosystems Prism Sequencer).
5′ RACE (Rapid Amplification of cDNA Ends).
RACE was performed by using primers based on the 3′ regions of the EST clones. Epstein–Barr virus (EBV)-transformed lymphocytes from chimpanzees and humans were cultured in RPMI medium 1640 supplemented with 10% fetal calf serum and 1% l-glutamine. Messenger RNA was isolated from ≈30 million cells by using a Qiagen Oligotex Direct mRNA isolation kit (Chatsworth, CA). Cells were lysed, homogenized using Qiagen’s QIAshredder, and incubated with Oligotex beads to allow mRNA to hybridize with (dT)30 oligonucleotides covalently linked to the Oligotex beads. The mRNA–Oligotex complex was then washed twice, and mRNA was eluted with a low-salt buffer. GIBCO/BRL’s 5′ RACE system was then used to perform cDNA amplification. Briefly, 2.5 μg of mRNA was reverse-transcribed with a gene-specific primer RACE1 (CTG TGT TTC CAA CAT AAA TGG G) using Superscript II reverse transcriptase. The mRNA was then degraded with RNase, and the cDNA was purified by using GlassMAX spin cartridges. After dC-tailing of the cDNA with terminal deoxynucleotidyltransferase (TdT) and dCTP, the cDNA was PCR-amplified by using the Abridged Anchor Primer (GIBCO/BRL) and a second gene-specific primer, RACE2 (ACA GCC TCT CCA GCC AAT CAG ATG), to obtain a nested product. The cDNA was then further nested by reamplifying using PCR with the Abridged Universal Amplification Primer (GIBCO/BRL) and the primer RACE3 (AAC CAC CAT CCA CGG GCA AAA GCA), and finally TA cloned by using Invitrogen’s Original TA Cloning Kit. Sequencing of the products was carried out as above.
Cloning of Full-Length cDNA for Chimpanzee and Human Hydroxylase by Reverse Transcription (RT)–PCR.
Two primers, NOTC51 (AAG CAG AGC GGC CGC CAG ACG ATG GGC AGC ATC G) and 31BAM (TGT CTT GGA TCC TTT TCT CTT CCT GTT TCC TC), were designed using 5′ RACE and the 3′ EST sequences common to human and chimpanzee. One microgram of mRNA was reverse-transcribed using 50 pmol of random hexamer, 200 units of Superscript II, and 20 units of RNase inhibitor in a 40-μl reaction with First Strand Buffer, 200 μM dNTP, and 10 mM DTT. A 2-μl portion of the reverse transcription reaction was then PCR-amplified with 20 pmol of NOTC51 primer, 20 pmol of 31BAM primer, and the Expand Long Template PCR System from Boehringer Mannheim with 1.75 mM MgCl2. The PCR products were then gel-purified by using the QIAEX Gel Extraction system and cloned by using Invitrogen’s TA Cloning Kit. Sequencing of three independent clones of each PCR product was done as described above. Final sequences are based on overlapping sequencing runs covering both strands.
Genomic PCR Analysis.
EBV-transformed lymphocytes from human, chimpanzee, bonobo, and gorilla were cultured as described above. Genomic DNA was prepared according to standard procedures (51), and PCR was carried out using primers R5 and R3, corresponding to the 5′ and 3′ ends of the 92-bp region, respectively. Twenty picomoles of R5 (GTC TGT CAG ATG CAC AAA CGA C) and 20 pmol of R3 (CCA GCT CAT CTT GAC AGA AGC) were used to amplify 1 μg of genomic DNA from each species in a 50-μl reaction with 200 μM dNTPs, 2.5 mM MgCl2, 20 mM Tris⋅Cl (pH 8.4), and 50 mM KCl. PCR conditions were as follows: 95°C for 5 min followed by 30 cycles of 94°C, 60 sec; 58°C, 60 sec; 72°C 45 sec, ending with 72°C for 10 min. Ten microliters of the PCR product was then electrophoresed on a 1.5% agarose gel and stained with ethidium bromide.
PCR Amplification of Human 3′ Untranslated Region (UTR) Sequences.
Primers 244MF and 244MR were derived from 3′ UTR sequence of EST244303 (244MF, CAC AGA GGA AAC AGG AAG AG; 244MR, TTG TTA TGC ATG TGA GCG G). PCR was carried out using 20 pmol of each primer and 200 ng of human genomic DNA in a total volume of 20 μl containing 200 μM dNTPs and 1.2 units of Taq polymerase in a buffer of 1.5 mM MgCl2, 15 mM (NH4)2SO4, 60 mM Tris⋅Cl (pH 8.5), and 5% dimethyl sulfoxide. Using a Perkin–Elmer GeneAmp PCR System 9600, the following conditions were used: denaturation at 95°C for 120 sec followed by 32 amplification cycles of 94°C for 30 sec; 58°C for 30 sec; 72°C for 60 sec; and extension at 72°C for 7 min. Electrophoresis of the PCR products through a 2% agarose gel followed by ethidium bromide staining revealed the expected 359-bp product (data not shown).
Isolation of Bacterial Artificial Chromosome (BAC) Clones.
Human genomic clones were identified by screening pools of the human BAC library release III from Research Genetics (Huntsville, AL) by PCR of 3′ UTR sequences. Primers and conditions were as described above, except 100 ng of DNA was used as template. DNA from the positive human genomic BAC clone 443N17 was purified by using a QIAGEN-tip 100 and a modified Plasmid Midi Kit midiprep protocol. Briefly, a 100-ml culture was treated as two 50-ml aliquots until addition to the column. Solutions P1, P2, and P3 were used at 2.5× volumes (compared with the standard protocol) per 50 ml of culture. The column elution buffer QF was heated to 65°C and added to the tip-100 in 1-ml aliquots.
Southern Blot Analysis of BAC Clones.
BAC DNA was prepared, restriction-digested, and analyzed using Southern hybridization performed according to standard procedures (51). BAC DNA (2 μg) was digested overnight at 37°C with BamHI or EcoRI and electrophoresed on a 0.8% agarose gel, which was then shaken for 1 hr in denaturation buffer (0.5 M NaOH/1.5 M NaCl) and neutralized 1 hr in neutralization buffer (1 M Tris⋅Cl, pH 7.5/1.5 M NaCl). The DNA was then transferred for 1 hr 45 min in 20× standard saline citrate (SSC; (1× SSC = 0.15 M sodium chloride/0.015 M sodium citrate, pH 7) onto a nitrocellulose membrane by using a PosiBlot 30–30 pressure blotter (Stratagene) and crosslinked to the membrane by UV irradiation. After rinsing in 5× SSC, the membrane was prehybridized overnight at 42°C in prehybridization solution [6× SSC/20 mM NaH2PO4/0.4% SDS/5× Denhardt’s solution (1× = 0.02% polyvinylpyrrolidone/0.02% Ficoll/0.02% BSA), and 100 μg/ml salmon sperm DNA], and then probed for 72 hr at 42°C with hybridization solution (4× SSC/25 mM NaH2PO4/10% dextran sulfate/4× Denhardt’s solution/100 μg/ml salmon sperm DNA) containing probes corresponding to the 92-bp region deleted in the human cDNA (PR, based on the chimpanzee sequence), the sequence immediately 5′ of this region in the cDNA (P5), and the sequence immediately 3′ of this region (P3). After 3 washes with 2× SSC/0.1%SDS for 1 hr at 42°C, the membrane was exposed onto a PhosphoImager screen (Molecular Dynamics) for 72 hr and then analyzed. Probes were generated by primer extension with α-32P-labeled dNTPs from NEN. Probe PR was generated with primers PR/5 (GCA CAA CTG CAA ATT AGA TGT GAG CAC CAT GAA GTA TAT CAA CC) and PR/3 (CAG AAG CTT TCC GGA GGG TTG ATA TAC TTC ATG GTG); P5 was generated with primers P5/5 (AAG CAC CAA GGA GGC CTG TTC ATA AAA GAT) and P5/3 (CCT CCG GCT AAA TCC TCG ATA TCT TTT ATG); and P3 was generated with primers P3/5 (TTG TTG AAA TGG ATG AAA ACA ACG GAC TTT), P3/3 (GAT TCA GTT CTA AAA GCA AAA GTC CGT TGT).
Human/Rodent Somatic-Cell Hybrid Mapping.
Using 200 ng of DNA from each sample from the NIGMS cell repository human/rodent somatic-cell hybrid mapping panel 2 (Coriell Cell Repositories), PCR amplification and analysis were carried out as above. A radiation-hybrid panel derived from chromosome 6p was screened by using PCR. Primers were designed from the 3′ UTR of the human hydroxylase gene (CHR6.1-F CGA ATC CAA TCA CAG AGG AAA CAG G; CHR6.1-R-GCA TTA TTG TTA TGC ATG TGA GCG G; CHR6.2-F CAA TGG TGG AAT TTG TCT CC; CHR6.2-R CTC ACC ATA GAC ATT CTG ACC TAC; CHR6.3-F CTG AGA TCC TGT TGT GCC TAT CAC C; and CHR6.3-R CCT TGG TGC TTG CAC ATA TTC TTG CAT GCC). Primer pairs were used as follows: CHR6.1-F/CHR6.1-R; CHR6.2-F/CHR6.2-R; CHR6.3-F/CHR6.3-R; and CHR6.1-F/CHR6.2-R. Primer pair CHR6.3-F/CHR6.3-R produced a PCR product in hamster the same size as the human product. All other primer combinations were informative. PCR was carried out in 25 μl reactions using 100 ng of DNA as template. Reactions were performed in 1× Perkin–Elmer PCR buffer/0.1 mM dNTPs (1μM each primer)/5% dimethyl sulfoxide with 1.25 units of Taq polymerase. Amplifications were 30 sec each at 94°C, 57°C, and 72°C, for 30 cycles.
Metaphase Chromosome Preparation.
A transformed normal male lymphoblast cell line was cultured in RPMI medium 1640 supplemented with 10% fetal bovine serum. Cell cultures were treated with Colcemid and harvested by using standard protocols. Fixed-cell suspensions were dropped onto glass slides and were then aged for at least 48 hr at −20°C before use.
Fluorescence in Situ Hybridization (FISH) Analysis.
One microgram of BAC DNA was labeled by nick-translation with digoxigenin 11-dUTP in the presence of reduced dTTP (Boehringer Mannheim), ethanol-precipitated, and resuspended in 10 μl of a 65% (vol/vol) formamide hybridization solution. For FISH, 250 ng of labeled DNA was combined with 1 μg of human COT-1 DNA (GIBCO/BRL) to block repetitive sequences. The probe mixture was denatured at 70°C for 10 min and renatured at 37°C for approximately 30 min. FISH was performed according to standard procedures. Slides were dehydrated in ethanol and denatured in 70% formamide/2× SSC at 70°C for exactly 2 min. Slides were dehydrated again and hybridized with the BAC 443N17 probe overnight at 37°C in a humidified chamber. The slides were washed in 50% formamide/2× SSC and in 2× SSC at 43°C, and BAC hybridization signals were detected with anti-digoxigenin-rhodamine (Boehringer Mannheim) at 37°C. Chromosomes were counterstained with 4′,6-diamidino-2′-phenylindole dihydrochloride (DAPI, Boehringer Mannheim) in Vectashield mounting medium (Vector Laboratories), and viewed under a Zeiss Axioskop fluorescence microscope equipped with a triple band pass filter.
RESULTS
Human EST Databases Contain Sequences Homologous to the 3′ Region of the Mouse CMP-Neu5Ac Hydroxylase.
Current human EST databases were found to contain several sequences with 75–85% homology to the previously reported mouse CMP-Neu5Ac hydroxylase cDNA. One of these ESTs (clone number 257329) encoded an ORF with as much as 90% identity (with no gaps) to the carboxy-terminal half of the mouse hydroxylase cDNA (see Fig. 1). The only major difference noted is a carboxyl-terminal extension of the ORF that is not present in the mouse cDNA. Two other ESTs (clones 244303 and 701123) contained generally similar sequences, but included some insertions and deletions that prematurely terminate the ORF (data not shown). We reasoned that all of these cDNAs represent alternatively spliced messages derived from the single gene (50) encoding the corresponding human hydroxylase.
Figure 1.

Comparison of nucleotide and derived amino acid sequences of CMP-Neu5Ac hydroxylase cDNAs from mouse, chimpanzee, and human. (A) Comparison of nucleotide sequences. Sequencing of multiple full-length cDNA clones was carried out as described. The ClustalW program (http://www2.ebi.ac.uk/clustalw/) was used to align the sequences. The 92-bp gap in the human cDNA and differences at the 3′ end are indicated with dotted lines. The start and stop codons of the primary ORF are underlined. (B) Comparison of the ORFs of the cDNAs. The putative Reiske iron-sulfur binding region of the mouse and chimpanzee hydroxylase are underlined.
Chimpanzee and Human CMP-Neu5Ac Hydroxylase cDNA Homologues Expressed in Lymphoblastoid Cells Are Different in Both the 5′ Region and the 3′ Tail.
We had earlier found that although human B lymphoblastoid cells do not express Neu5Gc, cells from both the bonobo and the chimpanzee do express substantial amounts of this sialic acid. We therefore used the human EST database sequences to obtain complete cDNAs for the chimpanzee and human hydroxylase. We first carried out 5′ RACE using mRNA from lymphoblastoid cells and the sequence information from the 5′ region of the EST clones. The major 5′ RACE product in the human was ≈500 bp in size, whereas the corresponding fragment from the chimpanzee was slightly larger (data not shown). Sequencing indicated that the difference is the result of a 92-bp deletion within the 5′ region of the human cDNA (Fig. 1A). The 5′ and 3′ sequences were then used to obtain the complete human and chimpanzee hydroxylase cDNAs by RT-PCR, using a high-fidelity polymerase (Pwo). As shown in Fig. 1, the complete chimpanzee cDNA sequence is ≈85% identical to that of the mouse. It includes an ORF that is ≈90% identical to the mouse, with the only major difference being at the 3′ end, where the chimpanzee has a 13-aa extension. In contrast to the mouse and chimpanzee cDNAs, a 92-bp deletion in the human cDNA results in a frameshift, which would cause a premature truncation of the coding sequence (Fig. 1B). The deletion includes sequences coding for the putative Reiske iron-sulfur binding region of the murine hydroxylase protein (46), and the resulting truncated molecule is therefore highly unlikely to have any hydroxylase activity. This deletion in the cDNA is identical to the one recently reported by Irie et al. (50). However, these authors did not isolate the complete 5′ region of the human cDNA that includes the primary initiator methionine; therefore, they came to a different conclusion regarding the nature of the truncated protein produced (see Discussion).
The cDNA Change in Humans Is Caused by a Genomic Alteration That Is Not Found in Any of the African Great Apes.
Genomic PCR analysis was carried out using primers corresponding to the 5′ and 3′ sequences of the 92-bp region of the chimpanzee cDNA. As shown in Fig. 2, we obtained a corresponding 92-bp PCR product from total genomic DNA of all of the African great apes, but not from the human. This suggests that this region of the human genome is either deleted or markedly altered in its sequence. Sequences from the 3′ UTR of human ESTs were therefore used to screen a human genomic BAC array library. PCR screening of BAC pools resulted in the identification of two BAC clones, 443N17 and 541O17. Upon colony purification, DNA of these BAC clones consistently produced an expected 359-bp PCR product. PCR and Southern blot analysis of clone 443N17 with primers and probes representing the 5′ and 3′ ends of the human cDNA indicate that this BAC clone includes the complete coding region for the hydroxylase (data not shown). Further Southern blot analysis with probes corresponding to the sequences immediately 5′ and 3′ of the 92-bp human cDNA deletion indicates that they are present in the BAC clone (Fig. 3). However, the BAC failed to hybridize with a chimpanzee cDNA probe corresponding to the 92-bp human cDNA deletion (Fig. 3). PCR analysis similar to that shown in Fig. 2 was performed by using the BAC clone as template. Again, no 92-bp product was found, and the higher molecular weight bands seen in the human lane in Fig. 2 were either absent or much less prominent, suggesting that they are nonspecific (data not shown). Further PCR studies (not shown) indicated that the intronic regions on either side of the putative 92 bp-deletion are very large (>10kb) and will require extensive subcloning and sequencing to define precisely.
Figure 2.

PCR analysis of genomic DNA from humans and African great apes. Genomic DNA from human (Hu), chimpanzee (Ch), bonobo (Bo), or gorilla (Go) was subjected to PCR analysis with primers corresponding to the 5′ and 3′ ends of the 92-bp sequence missing from the human cDNA. S, molecular weight standards; +, positive control, using chimpanzee cDNA as template; −, negative control, without template. Some higher molecular weight background bands seen with great ape DNA were more prominently amplified in the human sample.
Figure 3.
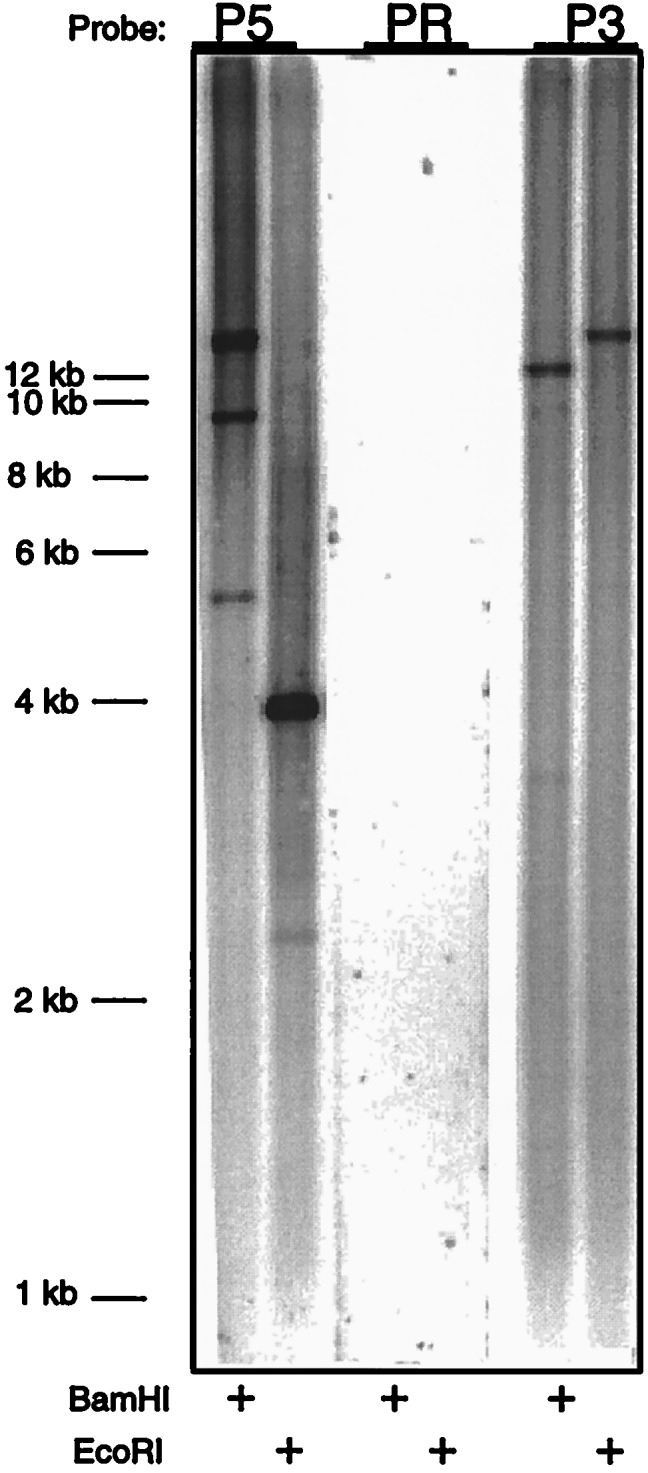
Southern blot analysis of a BAC clone encompassing the human hydroxylase gene. BAC 443N17 DNA (10 μg) was digested with BamHI (HI) or EcoRI (RI) and subjected to Southern blot analysis using probes corresponding to the missing region in the human cDNA (PR, based on chimpanzee sequence), and the sequences immediately 5′ (P5) and 3′ (P3) of this region. All three probes were equally sensitive when used to probe Southern blots of the chimpanzee hydroxylase cDNA (data not shown).
The 92-bp Deletion Is Found in Diverse Human Populations.
Our previous biochemical evidence for Neu5Gc deficiency in humans included a survey of more than 70 individuals from many backgrounds (37). To ensure that the hydroxylase mutation is not simply a human polymorphism, we carried out a similar PCR analyses on DNA samples from 18 “Caucasians,” 4 African Americans, 4 !Kung bushmen, 4 Khwe pygmies, and 6 Japanese. All of these samples failed to give the 92-bp PCR product, whereas control samples from chimpanzees and bonobos showed the product (data not shown).
Chromosomal Localization of the Human Hydroxylase Gene and Comparison with the Great Ape Genes.
A variety of techniques were used to localize the human hydroxylase gene. PCR amplification of human genomic DNA using primers 244MF and 244MR resulted in a PCR product with the expected 359-bp fragment size from the 3′ UTR of EST244303. Amplification of a human/rodent somatic-cell hybrid mapping panel showed the expected product amplified from human genomic DNA and no signal with either mouse or hamster DNA (Fig. 4A). Only sample 6 also gave a product of 359 bp, indicating that the locus maps to human chromosome 6.
Figure 4.
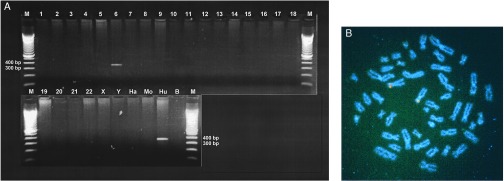
Chromosomal localization of the human CMP-sialic acid hydroxylase gene. (A) Human/rodent somatic-cell hybrid mapping. Ethidium bromide-stained agarose gel analyzing the products of PCR amplification of samples of the NIGMS human/rodent somatic-cell hybrid mapping panel 2. Lanes labeled 1–22, X, and Y reflect DNA from a chromosome 6 hybrid clone containing the designated human chromosomes. Hamster (Ha), mouse (Mo), and human (Hu) are total genomic DNA from those species used to create the hybrid panel. A negative control (B) is shown as well as 100-bp marker lanes with the 300- and 400-bp bands indicated. (B) FISH mapping. Metaphase FISH localization of genomic BAC clone 443N17 containing the CMP-sialic acid hydroxylase gene to the distal short arm of human chromosome 6. Detection of digoxigenin-labeled probes was achieved with anti-digoxigenin antibodies conjugated to rhodamine (giving the red signal), followed by 4′,6-diamidino-2′-phenylindole dihydrochloride (DAPI) counterstaining of the chromosomes and viewing under an epifluorescence microscope with a triple band pass filter.
FISH experiments against metaphase chromosomes from a normal human male showed that BAC clone 443N17 maps to the distal short arm of chromosome 6 (Fig. 4B). PCR primers designed to complement the 3′ UTR of the gene were then used to screen a radiation hybrid panel specific to human 6p (52). This allowed the map position of hydroxylase to be further narrowed to 6p22-p23, to an interval including the marker D6S89 and SCA1 (data not shown). FISH studies using chimpanzee, bonobo, and gorilla metaphase chromosomes showed that the map position is conserved with the human (data not shown). Thus, the human hydroxylase gene is not located at the site of any of the gross chromosomal rearrangements known to have occurred in hominoid evolution (53).
DISCUSSION
Our data indicate that the human CMP-Neu5Ac hydroxylase gene underwent a major mutation since the time of the last common ancestor with the bonobo and chimpanzee. The loss of a 92-bp segment in the coding region results in a frameshift mutation that can account for the lack of hydroxylase activity and deficiency of Neu5Gc in humans. We have also isolated the complete human hydroxylase gene, and have shown by Southern blot analysis that it does not contain the 92-bp region. We have not fully characterized the gene because of its very large size. In parallel studies, Irie et al. (50) have reported an analysis of the same region of the human gene, and conclude that a section corresponding to exon 6 of the mouse hydroxylase gene is deleted in humans.
Sialic acids are common structural components of the glycoconjugates of all animals of the deuterostome lineage (23–26) and are known to mediate cell–cell interactions by specific sialic acid-binding lectins (27–31) such as the selectins and the siglecs. A variety of structural modifications of Neu5Ac such as Neu5Gc (23–25) can modulate recognition in endogenous interactions as well as in exogenous microbial recognition (23–26). The expression of Neu5Gc is widespread in animals from sea urchins to mammals, and also shows tissue-specific and developmentally regulated expression (23, 24, 54–56). A change from Neu5Ac to Neu5Gc is likely to affect several of the known endogenous and exogenous receptors for sialic acids (27–29). For example, the presence or absence of Neu5Gc would affect the interactions of microbial pathogens such as influenza A and B viruses (57–59) and Escherichia coli K99 (60–63). There are many other microbes that utilize sialic acids as specific binding sites on mammalian cells, including major pathogens such as Helicobacter pylori and Plasmodium falciparum (22, 30, 64, 65). In most such instances, the consequences of having Neu5Gc rather than Neu5Ac have not yet been pursued. Because sialic acids are also ligands for a variety of endogenous vertebrate lectins (27–33), the loss of hydroxylation could have complex effects on the growth, development, and function of multiple systems. For example, CD22 on B lymphocytes, sialoadhesin on macrophages, and myelin-associated glycoprotein on neuronal axon myelin sheaths can show preferences for or lack of binding to Neu5Gc (27). Thus, the genetic defect in humans causing loss of or reduction in Neu5Gc expression could potentially influence a wide variety of biological and pathological processes.
Despite the present finding of a major mutation in the human hydroxylase gene, human fetal tissues (47) and certain human tumors (47–49) have been reported to contain Neu5Gc in small amounts. Some humans with malignancies or with certain inflammatory or infectious diseases are also known to spontaneously develop antibodies against Neu5Gc (66–68). We have also noted small traces of an HPLC peak corresponding to Neu5Gc in some human tissues (37). Taken together, these data indicate that the capacity to synthesize Neu5Gc may not be lost completely in humans. Further studies will be needed to ascertain whether this represents the up-regulation of another minor pathway for the synthesis of Neu5Gc, such as the suggested use of the donor glycolyl-CoA (69, 70), or the incorporation of Neu5Gc from dietary sources (71).
In this study, we have isolated a human hydroxylase cDNA that includes the codons corresponding to the initiator methionine of the mouse and chimpanzee hydroxylase. In contrast, the recent study of Irie et al. reported an incomplete cDNA that does not include this 5′ region (50). This 5′-truncated cDNA gave them a protein product in an in vitro translation reaction, leading to the assumption that a methionine codon downstream of the 92-bp deletion is the primary initiator for translation of a truncated protein representing >80% of the carboxyl end of the hydroxylase. They went on to show that such a truncated protein had no residual hydroxylase activity when expressed in COS cells. However, because we find an intact primary initiator methionine codon corresponding to those of the mouse and chimpanzee proteins, we predict a much shorter truncated polypeptide involving the amino terminus of the protein. Thus, it remains to be seen whether the protein product studied in vitro by Irie et al. (50) actually exists in vivo.
We should also note that there is a difference between the 3′ ends of the major cDNAs reported by the two groups. However, we also isolated an alternative human transcript with identity to the 3′ region reported in their paper, as well as other clones with various deletions and insertions. Although we did not conduct an extensive search in a chimpanzee library, we encountered only one such anomaly in the chimpanzee clones that we isolated. We speculate that the many abnormalities and variations of the 3′ region of the human hydroxylase are reflective of a gene that became nonfunctional in recent evolutionary times, and is undergoing a random degeneration. Regardless, if it were possible to determine when the loss of Neu5Gc expression occurred in relation to hominid evolution, this may shed light on the controversial question of the origin of modern humans (72). Unfortunately, autosomal DNA from fossils >30,000 years B.P. is extremely difficult to recover (73).
Finally, given the DNA melting curves and gene sequence data indicating an almost 99% identity of the chimpanzee genome to that of the human (9–16), it has been suggested that the genetic changes that result in morphological and functional differences between these species are subtle, e.g., minor changes in the promoter regions of critical genes and/or specific changes in the amino acid sequence of some gene products (4). However, the present data emphasize that the genetic differences between humans and the great apes can include major gene alterations. It remains to be seen whether the mutation in CMP-sialic acid hydroxylase can explain some of the dramatic morphological and functional changes that occurred during the evolution of humans. In this regard, perhaps the most intriguing observation is that no matter what the level of Neu5Gc was in other parts of the body, the amounts in the brain were always very low in a variety of animals studied (34–36), including the chimpanzee (37). This correlates with very low levels of hydroxylase message in the mouse brain (45). These data indicate that, for unknown reasons, Neu5Gc expression is not desirable in the vertebrate brain. Of course, in humans, the last traces of Neu5Gc in the brain are eliminated, because of the genomic mutation in the hydroxylase. It remains to be seen whether this results in any evolutionary advantage for human brain development or function.
Acknowledgments
We thank Peter Parham (Stanford University) for providing the lymphoblastoid lines, Alanna McCall and Huda Zoghbi for the chromosome 6 hybrid panel, and A. Craig Chinault and Barbara Boggs for assistance with FISH studies. This work was supported by US Public Health Service Grant RO1-GM32373 and the Mathers Foundation. S.T.W is an Investigator of the Howard Hughes Medical Institute.
ABBREVIATIONS
- Neu5Gc
N-glycolylneuraminic acid
- Neu5Ac
N-acetylneuraminic acid
- BAC
bacterial artificial chromosome
- FISH
fluorescence in situ hybridization
- EST
expressed sequence tag
- TdT
terminal deoxynucleotidyltransferase
- EBV
Epstein–Barr virus
- RACE
rapid amplification of cDNA ends
- UTR
untranslated region
Footnotes
References
- 1. Darwin C. The Descent of Man, and Selection in Relation to Sex. New York: D. Appleton and Co.; 1871. [Google Scholar]
- 2.Sarich V M, Wilson A C. Science. 1967;158:1200–1203. doi: 10.1126/science.158.3805.1200. [DOI] [PubMed] [Google Scholar]
- 3.Doolittle R F, Wooding G L, Lin Y, Riley M. J Mol Evol. 1971;1:74–83. doi: 10.1007/BF01659395. [DOI] [PubMed] [Google Scholar]
- 4.King M C, Wilson A C. Science. 1975;188:107–116. doi: 10.1126/science.1090005. [DOI] [PubMed] [Google Scholar]
- 5.Goodman M, Braunitzer G, Stangl A, Schrank B. Nature (London) 1983;303:546–548. doi: 10.1038/303546a0. [DOI] [PubMed] [Google Scholar]
- 6.Sibley C G, Ahlquist J E. J Mol Evol. 1984;20:2–15. doi: 10.1007/BF02101980. [DOI] [PubMed] [Google Scholar]
- 7.Miyamoto M M, Slightom J L, Goodman M. Science. 1987;238:369–373. doi: 10.1126/science.3116671. [DOI] [PubMed] [Google Scholar]
- 8.Sibley C G, Ahlquist J E. J Mol Evol. 1987;26:99–121. doi: 10.1007/BF02111285. [DOI] [PubMed] [Google Scholar]
- 9.Caccone A, Powell J R. Evolution. 1989;43:925–942. doi: 10.1111/j.1558-5646.1989.tb02540.x. [DOI] [PubMed] [Google Scholar]
- 10.Goodman M, Tagle D A, Fitch D H, Bailey W, Czelusniak J, Koop B F, Benson P, Slightom J L. J Mol Evol. 1990;30:260–266. doi: 10.1007/BF02099995. [DOI] [PubMed] [Google Scholar]
- 11.Sibley C G, Comstock J A, Ahlquist J E. J Mol Evol. 1990;30:202–236. doi: 10.1007/BF02099992. [DOI] [PubMed] [Google Scholar]
- 12.Ruvolo M, Disotell T R, Allard M W, Brown W M, Honeycutt R L. Proc Natl Acad Sci USA. 1991;88:1570–1574. doi: 10.1073/pnas.88.4.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horai S, Satta Y, Hayasaka K, Kondo R, Inoue T, Ishida T, Hayashi S, Takahata N. J Mol Evol. 1992;35:32–43. doi: 10.1007/BF00160258. [DOI] [PubMed] [Google Scholar]
- 14.Goodman M, Bailey W J, Hayasaka K, Stanhope M J, Slightom J, Czelusniak J. Am J Phys Anthropol. 1994;94:3–24. doi: 10.1002/ajpa.1330940103. [DOI] [PubMed] [Google Scholar]
- 15.Shoshani J, Groves C P, Simons E L, Gunnell G F. Mol Phylogenet Evol. 1996;5:102–154. doi: 10.1006/mpev.1996.0009. [DOI] [PubMed] [Google Scholar]
- 16.Ruvolo M. Mol Biol Evol. 1997;14:248–265. doi: 10.1093/oxfordjournals.molbev.a025761. [DOI] [PubMed] [Google Scholar]
- 17.McClure H M. Am J Phys Anthropol. 1973;38:425–429. doi: 10.1002/ajpa.1330380243. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt R E. J Med Primatol. 1975;7:274–318. doi: 10.1159/000459914. [DOI] [PubMed] [Google Scholar]
- 19.Krawczynski K, Prince A M, Nowoslawski A. J Med Primatol. 1979;8:222–232. doi: 10.1159/000460202. [DOI] [PubMed] [Google Scholar]
- 20.Shouval D, Chakraborty P R, Ruiz-Opazo N, Baum S, Spigland I, Muchmore E, Gerber M A, Thung S N, Popper H, Shafritz D A. Proc Natl Acad Sci USA. 1980;77:6147–6151. doi: 10.1073/pnas.77.10.6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Novembre F J, Saucier M, Anderson D C, Klumpp S A, O’Neil S P, Brown C R I, Hart C E, Guenthner P C, Swenson R B, McClure H M. J Virol. 1997;71:4086–4091. doi: 10.1128/jvi.71.5.4086-4091.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ollomo B, Karch S, Bureau P, Elissa N, Georges A J, Millet P. Am J Trop Med Hyg. 1997;56:440–445. doi: 10.4269/ajtmh.1997.56.440. [DOI] [PubMed] [Google Scholar]
- 23.Schauer R. Sialic Acids: Chemistry, Metabolism and Function, Cell Biology Monographs. New York: Springer; 1982. , Volume 10. [Google Scholar]
- 24.Varki A. Glycobiology. 1992;2:25–40. doi: 10.1093/glycob/2.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kelm S, Schauer R. Int Rev Cytol. 1997;175:137–240. doi: 10.1016/S0074-7696(08)62127-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ye J, Kitajima K, Inoue Y, Inoue S, Troy F A, II. Methods Enzymol. 1994;230:460–484. doi: 10.1016/0076-6879(94)30029-1. [DOI] [PubMed] [Google Scholar]
- 27.Kelm S, Schauer R, Manuguerra J-C, Gross H-J, Crocker P R. Glycoconjugate J. 1994;11:576–585. doi: 10.1007/BF00731309. [DOI] [PubMed] [Google Scholar]
- 28.Powell L D, Varki A. J Biol Chem. 1994;269:10628–10636. [PubMed] [Google Scholar]
- 29.Collins B E, Kiso M, Hasegawa A, Tropak M B, Roder J C, Crocker P R, Schnaar R L. J Biol Chem. 1997;272:16889–16895. doi: 10.1074/jbc.272.27.16889. [DOI] [PubMed] [Google Scholar]
- 30.Varki A. FASEB J. 1997;11:248–255. doi: 10.1096/fasebj.11.4.9068613. [DOI] [PubMed] [Google Scholar]
- 31.Crocker P R, Feizi T. Curr Opin Struct Biol. 1996;6:679–691. doi: 10.1016/s0959-440x(96)80036-4. [DOI] [PubMed] [Google Scholar]
- 32.Varki A. Proc Natl Acad Sci USA. 1994;91:7390–7397. doi: 10.1073/pnas.91.16.7390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Powell L D, Varki A. J Biol Chem. 1995;270:14243–14246. doi: 10.1074/jbc.270.24.14243. [DOI] [PubMed] [Google Scholar]
- 34.Tettamanti G, Bertona L, Berra B, Zambotti V. Nature (London) 1965;206:192. doi: 10.1038/206192a0. [DOI] [PubMed] [Google Scholar]
- 35.Ecsedy J A, Manfredi M G, Yohe H C, Seyfried T N. Cancer Res. 1997;57:1580–1583. [PubMed] [Google Scholar]
- 36.Mikami T, Kashiwagi M, Tsuchihashi K, Daino T, Akino T, Gasa S. J Biochem (Tokyo) 1998;123:487–491. doi: 10.1093/oxfordjournals.jbchem.a021962. [DOI] [PubMed] [Google Scholar]
- 37.Muchmore, E. A., Diaz, S. and Varki, A. (1998) Amer. J. Phys. Anthropol., in press. [DOI] [PubMed]
- 38.Shaw L, Schauer R. Biol Chem Hoppe-Seyler. 1988;369:477–486. doi: 10.1515/bchm3.1988.369.1.477. [DOI] [PubMed] [Google Scholar]
- 39.Muchmore E A, Milewski M, Varki A, Diaz S. J Biol Chem. 1989;264:20216–20223. [PubMed] [Google Scholar]
- 40.Kozutsumi Y, Kawano T, Yamakawa T, Suzuki A. J Biochem (Tokyo) 1990;108:704–706. doi: 10.1093/oxfordjournals.jbchem.a123268. [DOI] [PubMed] [Google Scholar]
- 41.Shaw L, Schneckenburger P, Carlsen J, Christiansen K, Schauer R. Eur J Biochem. 1992;206:269–277. doi: 10.1111/j.1432-1033.1992.tb16925.x. [DOI] [PubMed] [Google Scholar]
- 42.Kawano T, Kozutsumi Y, Takematsu H, Kawasaki T, Suzuki A. Glycoconjugate J. 1993;10:109–115. doi: 10.1007/BF00731194. [DOI] [PubMed] [Google Scholar]
- 43.Shaw L, Schneckenburger P, Schlenzka W, Carlsen J, Christiansen K, Jürgensen D, Schauer R. Eur J Biochem. 1994;219:1001–1011. doi: 10.1111/j.1432-1033.1994.tb18583.x. [DOI] [PubMed] [Google Scholar]
- 44.Takematsu H, Kawano T, Koyama S, Kozutsumi Y, Suzuki A, Kawasaki T. J Biochem (Tokyo) 1994;115:381–386. doi: 10.1093/oxfordjournals.jbchem.a124347. [DOI] [PubMed] [Google Scholar]
- 45.Kawano T, Koyama S, Takematsu H, Kozutsumi Y, Kawasaki H, Kawashima S, Kawasaki T, Suzuki A. J Biol Chem. 1995;270:16458–16463. doi: 10.1074/jbc.270.27.16458. [DOI] [PubMed] [Google Scholar]
- 46.Schlenzka W, Shaw L, Kelm S, Schmidt C L, Bill E, Trautwein A X, Lottspeich F, Schauer R. FEBS Lett. 1996;385:197–200. doi: 10.1016/0014-5793(96)00384-5. [DOI] [PubMed] [Google Scholar]
- 47.Hirabayashi Y, Kasakura H, Matsumoto M, Higashi H, Kato S, Kasai N, Naiki M. Jpn J Cancer Res. 1987;78:251–260. [PubMed] [Google Scholar]
- 48.Devine P L, Clark B A, Birrell G W, Layton G T, Ward B G, Alewood P F, McKenzie I F C. Cancer Res. 1991;51:5826–5836. [PubMed] [Google Scholar]
- 49.Marquina G, Waki H, Fernandez L E, Kon K, Carr A, Valiente O, Perez R, Ando S. Cancer Res. 1996;56:5165–5171. [PubMed] [Google Scholar]
- 50.Irie A, Koyama S, Kozutsumi Y, Kawasaki T, Suzuki A. J Biol Chem. 1998;273:15866–15871. doi: 10.1074/jbc.273.25.15866. [DOI] [PubMed] [Google Scholar]
- 51.Ausubel F, Kingston R, Moore D, Seidman J, Smith J, Struhl K, editors. Current Protocols in Molecular Biology. New York: Wiley; 1988. [Google Scholar]
- 52.Banfi S, Chung M-Y, Kwiatkowski T J, Jr, Ranum L P W, McCall A E, Chinault A C, Orr H T, Zoghbi H Y. Genomics. 1993;3:627–635. doi: 10.1016/s0888-7543(05)80365-9. [DOI] [PubMed] [Google Scholar]
- 53.Yunis J J, Prakash O. Science. 1982;215:1525–1530. doi: 10.1126/science.7063861. [DOI] [PubMed] [Google Scholar]
- 54.Bouhours D, Bouhours J F. J Biol Chem. 1983;258:299–304. [PubMed] [Google Scholar]
- 55.Muchmore E, Varki N, Fukuda M, Varki A. FASEB J. 1987;1:229–235. doi: 10.1096/fasebj.1.3.3623000. [DOI] [PubMed] [Google Scholar]
- 56.Muchmore E A. Glycobiology. 1992;2:337–343. doi: 10.1093/glycob/2.4.337. [DOI] [PubMed] [Google Scholar]
- 57.Higa H H, Rogers G N, Paulson J C. Virology. 1985;144:279–282. doi: 10.1016/0042-6822(85)90325-3. [DOI] [PubMed] [Google Scholar]
- 58.Ito T, Suzuki Y, Mitnaul L, Vines A, Kida H, Kawaoka Y. Virology. 1997;227:493–499. doi: 10.1006/viro.1996.8323. [DOI] [PubMed] [Google Scholar]
- 59.Suzuki T, Horiike G, Yamazaki Y, Kawabe K, Masuda H, Miyamoto D, Matsuda M, Nishimura S I, Yamagata T, Ito T, et al. FEBS Lett. 1997;404:192–196. doi: 10.1016/s0014-5793(97)00127-0. [DOI] [PubMed] [Google Scholar]
- 60.Ouadia A, Karamanos Y, Julien R. Glycoconjugate J. 1992;9:21–26. doi: 10.1007/BF00731174. [DOI] [PubMed] [Google Scholar]
- 61.Kyogashima M, Ginsburg V, Krivan H C. Arch Biochem Biophys. 1989;270:391–397. doi: 10.1016/0003-9861(89)90042-8. [DOI] [PubMed] [Google Scholar]
- 62.Willemsen P T J, de Graaf F K. Infect Immun. 1993;61:4518–4522. doi: 10.1128/iai.61.10.4518-4522.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lanne B, Uggla L, Stenhagen G, Karlsson K-A. Biochemistry. 1995;34:1845–1850. doi: 10.1021/bi00006a004. [DOI] [PubMed] [Google Scholar]
- 64.Karlsson K A. Curr Opin Struct Biol. 1995;5:622–635. doi: 10.1016/0959-440x(95)80054-9. [DOI] [PubMed] [Google Scholar]
- 65.Escalante A A, Barrio E, Ayala F J. Mol Biol Evol. 1995;12:616–626. doi: 10.1093/oxfordjournals.molbev.a040241. [DOI] [PubMed] [Google Scholar]
- 66.Merrick J M, Zadarlik K, Milgrom F. Int Arch Allergy Appl Immunol. 1978;57:477–480. doi: 10.1159/000232140. [DOI] [PubMed] [Google Scholar]
- 67.Nishimaki T, Kano K, Milgrom F. J Immunol. 1979;122:2314–2318. [PubMed] [Google Scholar]
- 68.Morito T, Kano K, Milgrom F. J Immunol. 1982;129:2524–2528. [PubMed] [Google Scholar]
- 69.Vamecq J, Poupaert J H. Biochem Cell Biol. 1990;68:846–851. doi: 10.1139/o90-125. [DOI] [PubMed] [Google Scholar]
- 70.Vamecq J, Mestdagh N, Henichart J-P, Poupaert J. J Biochem (Tokyo) 1992;111:579–583. doi: 10.1093/oxfordjournals.jbchem.a123800. [DOI] [PubMed] [Google Scholar]
- 71.Nohle U, Schauer R. Hoppe-Seylers Z Physiol Chem. 1981;362:1495–1506. doi: 10.1515/bchm2.1981.362.2.1495. [DOI] [PubMed] [Google Scholar]
- 72.Cavalli-Sforza L L, Menozzi P, Piazza A. The History and Geography of Human Genes. Princeton: Princeton Univ. Press; 1994. [Google Scholar]
- 73.Krings M, Stone A, Schmitz R W, Krainitzki H, Stoneking M, Pääbo S. Cell. 1997;90:19–30. doi: 10.1016/s0092-8674(00)80310-4. [DOI] [PubMed] [Google Scholar]