

Abstract
During cell migration, the physical link between the extracellular substrate and the actin cytoskeleton mediated by receptors of the integrin family is constantly modified. We analyzed the mechanisms that regulate the clustering and incorporation of activated αvβ3 integrins into focal adhesions. Manganese (Mn2+) or mutational activation of integrins induced the formation of de novo F-actin–independent integrin clusters. These clusters recruited talin, but not other focal adhesion adapters, and overexpression of the integrin-binding head domain of talin increased clustering. Integrin clustering required immobilized ligand and was prevented by the sequestration of phosphoinositole-4,5-bisphosphate (PI(4,5)P2). Fluorescence recovery after photobleaching analysis of Mn2+-induced integrin clusters revealed increased integrin turnover compared with mature focal contacts, whereas stabilization of the open conformation of the integrin ectodomain by mutagenesis reduced integrin turnover in focal contacts. Thus, integrin clustering requires the formation of the ternary complex consisting of activated integrins, immobilized ligands, talin, and PI(4,5)P2. The dynamic remodeling of this ternary complex controls cell motility.
Introduction
Integrins are transmembrane receptors linking the actin cytoskeleton to the ECM. This connection is dynamically reorganized in response to mechanical, chemokine, and growth factor signals. The lateral assembly of integrins into small stationary focal complexes within the leading edge of the cell and their continuous incorporation into sliding focal contacts at the rear of migrating cells exemplify the dynamics of reversible integrin clustering.
Integrins are noncovalently linked heterodimeric receptors that exist in low- and high-affinity states (Hynes, 2002). In the low-affinity state, the ectodomains of the α and β subunits are in a folded configuration with laterally associated transmembrane and cytoplasmic domains. Upon integrin activation, the distal parts of both ectodomains swing open in a switchblade motion, followed by the separation of the transmembrane and cytoplasmic domains (Xiao et al., 2004). This allosteric switch, or “integrin activation,” can be induced by effector binding to the cytoplasmic tail of the α and β subunit (Vinogradova et al., 2002; Katagiri et al., 2003; Tadokoro et al., 2003) or by ligand binding to the ectodomain (Takagi et al., 2002). Once the integrin heterodimer is in its open configuration, the cytoplasmic domain of the β integrin chain is linked via cytoplasmic adaptor proteins, such as talin, to the actin cytoskeleton (Liddington and Ginsberg, 2002; Critchley, 2004).
The molecular mechanisms that control the lateral assembly of integrins (“clustering”) to form focal adhesions are controversial. It has been proposed that the process of integrin clustering is intimately linked to the switch in its affinity state. In vitro studies have suggested that the activation-induced separation of the integrin transmembrane domains can induce integrin clustering by the respective homooligomerization of the α and β transmembrane domains (Li et al., 2003). However, this model has been contested based on disulfide bond scanning of the exofacial portions of the transmembrane domains of activated integrins expressed in living cells and extensive mutagenesis of the transmembrane domains (Luo et al., 2004; Partridge et al., 2005). Therefore, it remains an open question whether (and how) integrin activation is coupled to integrin clustering and focal contact remodeling in living cells. We analyzed this issue by expressing wild-type (WT) and constitutively activated EGFP-tagged αvβ3 integrins within living cells and investigating the requirement of extracellular ligand binding, recruitment of cytoplasmic adaptors, and membrane lipid composition for the clustering of activated integrins. In addition, we determined by FRAP how constitutive activation of integrins alters their dynamic remodeling within focal contacts.
Results
Integrin activation is a prerequisite for clustering
Lateral diffusion and clustering of integrins is required for the formation of focal adhesion sites. However, it is not known if integrins need to be activated to induce lateral association. Therefore, we analyzed lateral clustering of integrins after manipulation of the affinity state of αvβ3 integrins. High-affinity integrins were obtained by the addition of manganese (Mn2+), a known powerful activator of integrins (Mould et al., 2002; Chen et al., 2003), or by mutation of the β3 integrin chain. Intracellular and extracellular activation of integrins was induced by mutation of a cytoplasmic salt bridge (D723A) and by introduction of a “glycan wedge” in the β3 ectodomain (N305T), respectively (Hughes et al., 1996; Pampori et al., 1999; Vinogradova et al., 2002; Luo et al., 2003). The mutation of D723A perturbs the lateral association of the transmembrane and cytoplasmic domains resulting in their unclasping and the subsequent activation of the integrin (Hughes et al., 1996; Partridge et al., 2005). The introduction of threonine at residue 305 of the mature integrin sequence induces an N-linked glycosylation at the conserved asparagine 303. This asparagine is located at the interface of the I-like and hybrid domain in β integrins, and N-linked glycosylation at this residue forces these domains to assume an extended conformation that results in the activation of β3, β1, and β7 integrins and increased adhesion in cells expressing these mutant integrins (Luo et al., 2003; Chen et al., 2004; Xiao et al., 2004). An inactive form of αvβ3 integrin, which is no longer able to bind ligand, was obtained by mutation of the β3 integrin ectodomain (D119Y), previously identified in a patient suffering from Glanzman thrombasthenia (Loftus et al., 1990). To visualize integrin clustering in living cells, all of these integrins were expressed as EGFP fusion proteins (Ballestrem et al., 2001).
In adherent cells, WT β3-EGFP integrins formed brightly fluorescent clusters at the cell periphery, representing focal contacts (Ballestrem et al., 2001; Fig. 1 A). In addition, nonclustered integrins were found evenly dispersed within the plasma membrane (Fig. 1 A). Because the population of cell surface integrins is in equilibrium between the activated and nonactivated forms, it is not clear to what extent activated integrins are present in the clustered or nonclustered state. To shift the equilibrium toward activated states of integrins, we treated cells with Mn2+. Only minutes after Mn2+ activation, we observed the formation of numerous irregularly shaped integrin clusters underneath the main cell body, in addition to the preexisting clusters of integrins at the periphery of the cell (Fig. 1 B). Similar to Mn2+-activated WT β3 integrin, the activated β3 integrin mutants D723A and N305T formed clusters underneath the entire cell body and in the periphery of the cell (Fig. 1, C and D). In contrast, the inactive β3 integrin mutant (D119Y), lacking the ability to bind ligand, was found evenly dispersed at the cell surface, exhibiting no apparent integrin clustering (Fig. 1 E). These results demonstrate that integrin activation correlates with integrin clustering and suggest a role for ligand binding in this process (Miyamoto et al., 1995).
Figure 1.
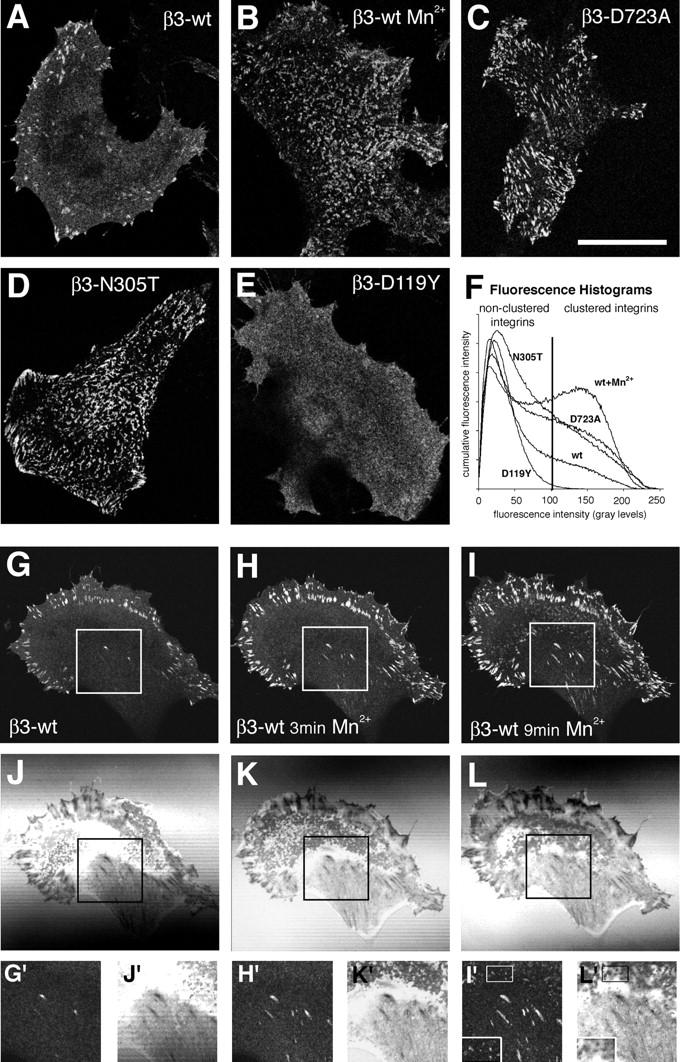
Increased clustering of high-affinity integrins. Confocal images at the level of the ventral cell surface of mouse B16F1 melanoma cells stably transfected with WT (A and B), D723A (C), N305T (D), or D119Y (E) mutant β3-EGFP integrin. Cells were grown in control medium (A and C–E) or stimulated for 20 min in 0.5 mM of Mn2+-containing medium (B). Note the increased number of integrin clusters at the cell surfaces underlying the main cell body in B–D. (F) Quantification of the relative amount of integrin clustering by histogram analysis. Averaged (n > 20) cumulative histograms from cells as shown in A–E (Fig. S1). The vertical line represents the fluorescence threshold corresponding to 99% of the histogram area of the inactive integrin D119Y mutant. Confocal (G–I) and corresponding interference reflection images (J–L) of Mn2+-induced integrin clustering in a B16F1 cell cultured overnight in serum-containing medium. (G and J) The cell before Mn2+ addition; (H and K) the cell after 3 min of Mn2+ addition; (I and L) the cell after 9 min of Mn2+ addition. Magnified views of the area underneath the main cell body representing de novo–formed dot- and streaklike integrin clusters are represented in insets G′–L′. Bars: (A–E) 25 μm; (G–L) 37.5 μm.
To determine whether the degree of integrin clustering may represent a readout of the activation state of integrins in a given cell, we quantified the relative amount of clustered versus nonclustered integrins under these different experimental conditions. We used a nonbiased histogram analysis of the fluorescence intensity of confocal images of WT or mutant integrin expressing cells, where low fluorescence intensity represents nonclustered integrins and high fluorescence intensity represents clustered integrins (Fig. 1 F and Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200503017/DC1). Using an arbitrary intensity threshold corresponding to 99% of the histogram area of the inactive D119Y β3 integrin mutant, we determined that 21% of WT integrins were above this threshold, and hence in a clustered state. Constitutive integrin activation increased this value to 33, 38, and 48% for the mutant N305T, mutant D723A, and Mn2+-treated WT cells, respectively. Interestingly, we noted that Mn2+ treatment of the D723A mutant further increased the degree of clustering to Mn2+ levels, suggesting that this mutation does not fully activate integrins (unpublished data). In contrast, the relatively low degree of clustering of the activated N305T mutation is biased because a large number of the activated cell surface integrins were shed from the cell surface onto the substrate before analysis (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200503017/DC1). These data confirm a direct correlation between integrin activation and the formation of integrin clusters.
Time course of integrin clustering
The presence of clusters of activated integrins under the main cell body is unusual. Because focal contacts and the proximity of the cell membrane to the substrate can be revealed by interference reflection microscopy (IRM), we analyzed the time course of Mn2+-induced integrin clustering in respect to the IRM image. Before Mn2+ stimulation, β3 integrins were clustered within peripherally located focal contacts, appearing dark in corresponding IRM images (Fig. 1, G and J; Verschueren, 1985). Streaklike IRM-dark structures located toward the center of the cell did not exhibit β3 integrin clustering (Fig. 1, G and J, insets). Similarly, dotlike close contacts located underneath the main cell body were not positive for β3 integrins. Shortly after Mn2+ activation, the fluorescence intensity of preexisting peripheral focal contacts increased, and previously β3-negative, streaklike dark contacts appeared β3 positive (Fig. 1, H and K). Subsequently, dotlike close contacts located underneath the main cell body appeared β3 integrin positive, although with lower fluorescence intensity compared with peripheral focal contacts (Fig. 1, I and L). Hence, de novo–activated integrins gradually appear in streak- and dotlike dark and close contacts underneath the main cell body.
Specific talin recruitment to F-actin–independent integrin clusters
Clusters of Mn2+ or mutational activated integrins were found underneath the main cell body, a cellular localization rarely populated by focal contacts. Because integrins clustered within focal adhesions are mechanically linked to the actin cytoskeleton, we analyzed whether actin and/or focal adhesion adaptor proteins are recruited to de novo–formed integrin clusters. We analyzed whether phalloidin reactive F-actin was localized to Mn2+-induced integrin clusters. Mn2+ activation resulted in the formation of integrin clusters in cellular regions devoid of F-actin (Fig. 2, A and A', inset). De novo formation of integrin clustering independent of F-actin was confirmed by treatment of spread cells with cytochalasin D (cD) before Mn2+ activation. Despite the destruction of the actin cytoskeleton by cD, Mn2+-activated β3 integrins formed clusters (Fig. 2, B and B'). Because actin fibers were dispensable for integrin clustering, we asked whether adaptor proteins were recruited to clusters of activated integrins. Immunofluorescence staining with antitalin antibodies revealed an overlap with all clustered EGFP integrins in Mn2+-treated cells (Fig. 2, C and C'). This confirms the data that talin–integrin association is important for the formation of focal adhesions (Priddle et al., 1998). In contrast to talin, the focal adhesion adaptor proteins vinculin, paxillin, and FAK, as well as antiphosphotyrosine antibodies, did not associate with de novo–formed clusters of activated integrins (Fig. 2, D–G). An identical result was obtained in β3 integrin negative CS-1 hamster melanoma cells that had been transfected with non-EGFP–tagged β3 integrins (Fig. 2, H and I). This suggests that the observed clustering of activated integrins and selective talin recruitment is not influenced by the EGFP tag.
Figure 2.
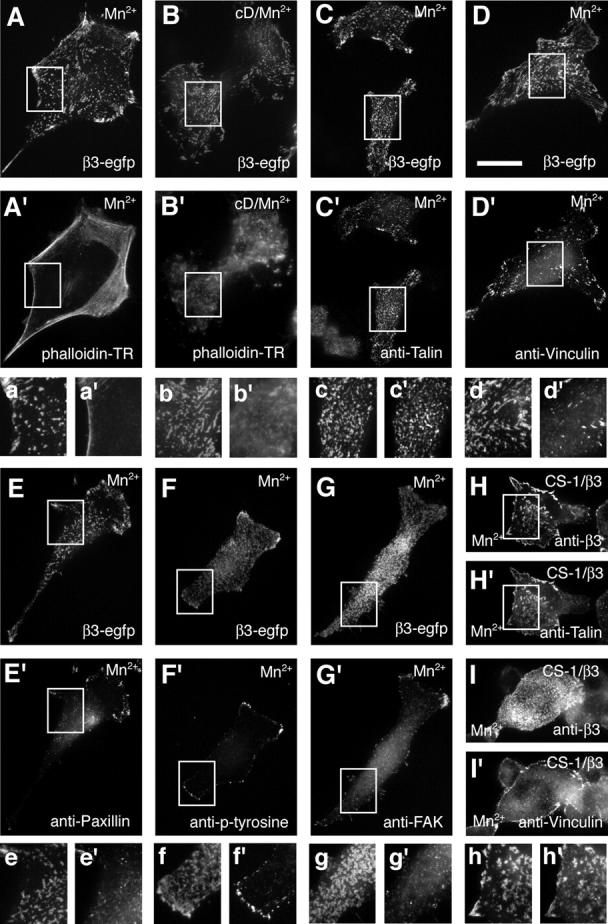
F-actin–independent integrin clustering and talin recruitment. Epifluorescence of fixed B16F1 and CS-1 cells exhibiting de novo formed β3 integrin clusters. Integrin fluorescence (A and B) and phalloidin stained F-actin (A' and B') in 0.5 mM of Mn2+-treated cells (20 min; A and A') or 10 μg/ml of cD/Mn2+-treated B16F1 cells (25 min of cD, followed by 20 min of cD/Mn2+; B and B'). Immunohistochemical analysis of focal adhesion adaptor proteins recruited to Mn2+-induced clusters of β3-EGFP integrins in B16F1 cells (C–G) and nontagged β3 integrins in CS-1 cells (H and I). Pairs of images show the distribution of the EGFP integrin fluorescence (C–G) or anti-β3 staining (H and I) and the respective immunohistochemical localization of talin (C' and H'), vinculin (D' and I'), paxillin (E'), phosphotyrosine (F'), and FAK (G'). Corresponding magnified views of the boxed areas in A–H and A'–H' are shown below each image pair (a–h and a'–h'). Bar, 25 μm.
Because Mn2+ is able to activate different types of integrins, we asked whether other integrins (e.g., β1 integrins) are recruited into αvβ3 integrin clusters. Because ligand binding is crucial for integrin clustering, exemplified by the inability of αvβ3 integrins to form Mn2+-induced clusters on a Laminin-1 substrate (unpublished data), we created a GFP-integrin chimera consisting of the β3 extracellular domain and β1 cytoplasmic and transmembrane domains. This integrin formed clusters when activated by Mn2+ treatment (unpublished data). When expressed in combination with a monomeric RFP-tagged form of β3 integrins, both integrins perfectly overlapped in Mn2+-induced clusters (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200503017/DC1). From these results we suggest that talin is the cytoskeletal adaptor protein first recruited to high-affinity β3 and β1 integrins and may play a critical role in their clustering. More importantly, the absence of other cytoskeletal adaptor proteins and the lack of F-actin in these integrin clusters suggest that these de novo–formed clusters are precursors of focal adhesions.
Integrin clustering requires PI(4,5)P2
It has been demonstrated that peptides, representing the cytoplasmic tail of integrins, bind to talin. In addition, phosphoinositol-4,5-bisphosphate (PI[4,5]P2) binding to talin induces a conformational change that facilitates the interaction of the talin head domain with the cytoplasmic tail of β3 integrins (Martel et al., 2001). Therefore, we asked whether PI(4,5)P2 is a critical cofactor in the clustering of high-affinity integrins. We treated β3 integrin–expressing cells with increasing doses of neomycin sulfate, a drug known to selectively bind and sequester PI(4,5)P2 (Arbuzova et al., 2000; Laux et al., 2000). The sequestration of PI(4,5)P2 by 10 mM neomycin dramatically reduced the formation of β3 integrin clusters in the periphery of control cells (Fig. 3 A), as well as Mn2+-induced, de novo integrin clusters underneath the main cell body (Fig. 3 B). Despite the reduction of integrin clustering under the main cell body, some remaining integrin clusters were found in the cell periphery often associated with filopodia. These thin, fingerlike integrin clusters were in contact with the substrate as suggested by the corresponding IRM image (Fig. 3, A and B, insets). Similar to Mn2+-stimulated WT cells, 10 mM neomycin suppressed peripheral focal contacts and integrin clusters under the main cell body in D723A and N305T mutant β3 integrin–transfected cells (unpublished data). To better characterize the effect of neomycin on integrin clustering, we performed dose response analysis with WT β3 integrin–expressing cells. Mn2+-induced integrin clustering was efficiently prevented at 1 mM neomycin sulfate (Fig. 3 C). To exclude the possibility that the neomycin sulfate–dependent inhibition of integrin clustering was indirectly caused by the drug's effect on the actin cytoskeleton (Laux et al., 2000; Kwik et al., 2003), we treated cells with cD before Mn2+ stimulation. Irrespective of the state of the actin cytoskeleton, neomycin sulfate prevented the formation of Mn2+-induced integrin clusters (unpublished data). To test whether PI(4,5)P2 was also important for the maintenance of integrin clusters, we tested whether its sequestration would affect previously clustered integrins. The addition of 10 mM neomycin sulfate to Mn2+-stimulated cells dispersed integrin clusters within 1 h (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200503017/DC1). Nevertheless, the dispersed but activated integrins were still able to link the cells to the substrate as revealed by IRM (unpublished data). These results demonstrate that PI(4,5)P2 is involved in the induction and stabilization of the lateral association of integrins, either by increasing the affinity of talin for integrins (Martel et al., 2001) or by oligomerization of the integrin–talin complex through interaction with lipid domains containing PI(4,5)P2.
Figure 3.
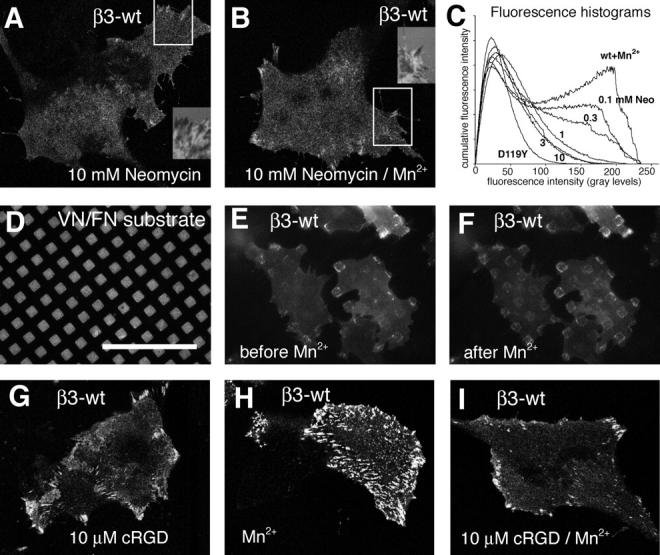
PI(4,5)P 2 and immobilized ligand are required for integrin clustering. (A and B) Stable β3-EGFP integrin–expressing B16F1 cells were cultured in neomycin sulfate–containing medium for 36 h. Control (A) or Mn2+-stimulated (B) cells were fixed and analyzed by confocal microscopy. The corresponding inset represents the IRM image of the boxed region of the cell in A and B, respectively. (C) Averaged histograms (n > 20) of Mn2+-induced integrin clustering in respect to increasing concentrations of neomycin sulfate. The histogram analysis of the inactive integrin mutant D119Y (Fig. 1 F) is shown as a negative reference. (D–F) Integrin clustering on a micropatterned substrate of vitronectin/fibronectin. Epifluorescence images of Alexa 568–labeled fibronectin (D) and corresponding β3-EGFP integrin fluorescence of B16F1 cells before Mn2+ addition (E). The same cells were imaged 10 min after treatment with Mn2+ (F). Note the exclusive accumulation of de novo integrin clusters on ECM-coated, but not noncoated, surfaces. (G–I) Inhibition of de novo integrin clustering in the presence of cRGD. Stable β3-EGFP integrin–expressing B16F1 cells were cultured overnight in complete medium and fixed 20 min after the addition of 10 μM cRGD (G), 0.5 mM Mn2+ (H), or both reagents (I). Confocal images of the integrin fluorescence at the level of the glass coverslip indicate the inhibition of Mn2+-induced integrin clustering by cRGD. Bars: (A,B, and G–I) 35 μm; (D–F) 50 μm.
Integrin clustering requires interaction with immobilized ligands
The specific PI(4,5)P2-dependent recruitment of talin to activated integrins demonstrated that the cytoplasmic tail of β3 integrin is critical for clustering. We now asked whether ligand binding to the extracellular domain is equally important to integrin clustering, as suggested by the phenotype of the D119Y mutation. To visualize ligand-specific clustering, we plated cells on glass coverslips coated with a micropattern of fibronectin/vitronectin. With this setup, we can compare integrin clustering on ligand-containing or control surfaces within one cell. In control medium, integrin clusters formed in the periphery of the cell but were spatially restricted to ligand-coated areas (Fig. 3, D and E). After Mn2+ stimulation, de novo integrin clusters formed underneath the main cell body, but exclusively in regions coated with extracellular ligand (Fig. 3 F). These data demonstrate that activated, high-affinity integrins require extracellular ligands to form integrin clusters.
To determine whether integrin clustering requires “immobilized” in contrast to “soluble” extracellular ligands, we performed Mn2+-induced integrin stimulation in the presence of increasing doses of cyclic RGD (cRGD) peptides. Importantly, we chose a concentration of cRGD that did not completely inhibit the recruitment of αvβ3 integrins into peripheral focal adhesions (Fig. 3 G). At concentrations of 10 μM cRGD, which inhibited binding of soluble αvβ3 integrin to immobilized ligand in an ELISA-type assay (Legler et al., 2001), the formation of Mn2+-induced de novo integrin clusters underneath the main cell body was completely blocked (Fig. 3 I). These data demonstrate that both the cytoplasmic and the extracellular ligand-binding domains are essential for the clustering of αvβ3 integrins. Moreover, immobilized extracellular ligands are required to stabilize nascent clusters of activated integrins at the cell surface.
The head domain of talin induces integrin clustering
As demonstrated in Fig. 2, the specific recruitment of talin to Mn2+-induced integrin clusters suggests that talin is critically involved in the formation of integrin clusters. Moreover, because talin exists as an anti-parallel dimer with the integrin-binding head domains positioned at both ends (Isenberg and Goldmann, 1998), talin represents a bona fide intracellular integrin cross-linker. To test whether the dimeric form of talin is required for integrin clustering, we overexpressed the monomeric integrin binding head domain of talin (residues 1–435; Yan et al., 2001) as a CFP-tagged chimera in stable β3-GFP integrin–expressing cells, with the intention to dominantly suppress integrin clustering. Instead, the overexpression of the isolated head domain of talin induced an increase in integrin clustering (Fig. 4, A–C). Similarly, Mn2+ stimulation of cells that overexpressed the head domain of talin induced a greater increase in integrin clustering, covering more than half of the cell surface (Fig. 4, D and E). In most cells, the cytoplasmic expression of talin was very high, making it impossible to determine whether the talin head domain only activated integrins or was also involved in their clustering. However, after Mn2+ stimulation, high levels of the talin head domain were no longer required for integrin activation, which nevertheless resulted in efficient clustering of integrin at low talin expression levels (Fig. 4, G and H). In these clusters (Fig. 4 G), we detected a colocalization with the head domain of talin (Fig. 4 H). These data suggest that the monomeric talin head domain possesses the ability to induce integrin activation and clustering. Because talin-dependent integrin clustering is sensitive to neomycin treatment (unpublished data), integrin clustering may be induced by the binding of the talin head domain to multivalent PI(4,5)P2 lipid domains.
Figure 4.

The head domain of talin induces β3 integrin clustering. Cells stably expressing β3-EGFP integrin were transiently transfected with enhanced cyan fluorescent protein (ECFP)-tagged human NH2-terminal talin head domain and cultured in complete medium. 48 h after transfection, cells were fixed and observed by confocal microscopy. Talin head domain transfected control (A and B) or Mn2+-treated cells (20 min; 0.5 mM final concentration; D and E) revealed extensive integrin clustering (A and D). (C) Histogram analysis of integrin clustering in talin head transfected cells with or without the addition of Mn2+. (F) Analysis of the relative cell surface occupied by β3-EGFP integrin clusters in cells transfected with ECFP-talin head domain or empty ECFP vector and treated with or without Mn2+. Error bars represent the standard deviation of at least 20 double-transfected cells. (G and H) Confocal images of β3-EGFP integrin and ECFP-talin head domain in a Mn2+-stimulated, weakly talin-expressing cell. Note the similar staining pattern between integrins and the talin head domain (G' and H'). Data for graphs in C and F are from one out of three similar experiments. Bar, 27 μm.
Dynamics of integrin clustering
In migrating cells, stationary focal complexes at the front of the cell exhibit slow integrin exchange rates, whereas sliding and stationary focal contacts at the rear of the cell demonstrate rapid integrin exchange rates (Ballestrem et al., 2001). To determine a possible link between integrin exchange rates and the integrin affinity switch, we tested whether blocking αvβ3 integrin in high-affinity states modifies the dynamics of integrin exchange in stress fiber–linked peripheral focal contacts exhibiting high integrin fluorescence intensities (Ballestrem et al., 2001). We performed FRAP experiments with WT-, D723A-, N305T-, or Mn2+-activated β3-EGFP integrin–transfected cells (White and Stelzer, 1999; Ballestrem et al., 2001). Integrin activation by the intracellular mutation D723A or treatment with Mn2+ did not affect integrin exchange rates in focal contacts (Fig. 5, A–C, H, and I; and Videos 1–3, available at http://www.jcb.org/cgi/content/full/jcb.200503017/DC1). In contrast, integrin activation by the ectodomain mutation N305T resulted in a fivefold slower integrin exchange rate (Fig. 5, F and H; and Video 5). These data suggest that the rate-limiting step in the integrin exchange in peripheral focal contacts is determined by the flexibility of the integrin ectodomain. It further suggests that the facilitated interactions of the cytoplasmic tail of integrins, with its intracellular adaptors, do not influence the rate of integrin exchange in focal contacts.
Figure 5.
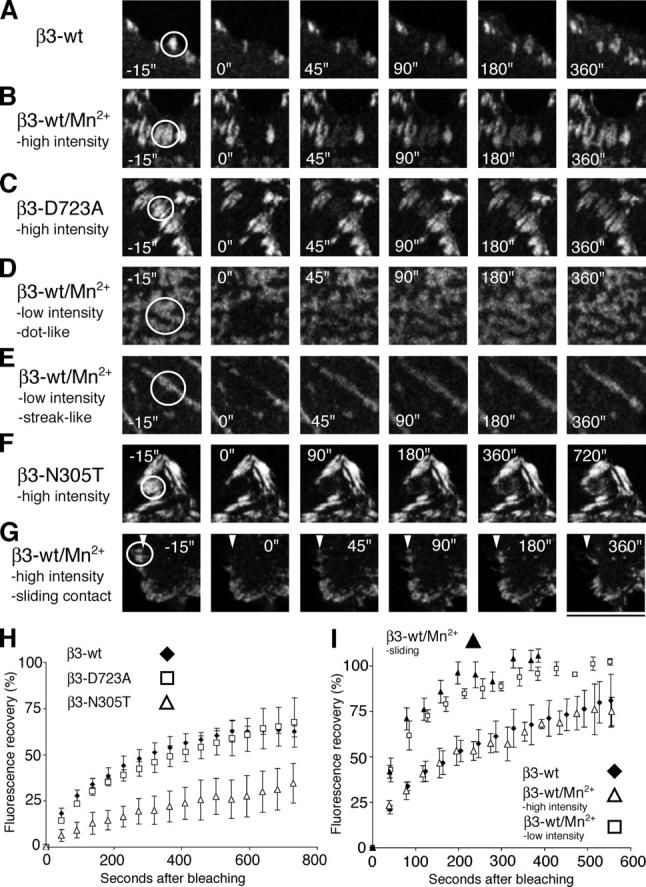
FRAP of activated integrins. FRAP analysis of β3 integrin dynamics within stationary or sliding peripheral high-intensity focal contacts and de novo formed dot- or streaklike integrin clusters in B16F1 cells. The analysis was performed in stably transfected EGFP-tagged WT (β3-wt), D723A (β3-D723A), or N305T (β3-N305T) cells. FRAP sequences of peripheral focal contacts in control cells (β3-wt; A), Mn2+-treated cells (β3-wt/Mn2+; B), and cells expressing D723A-activated β3 integrin (β3-D723A; C; Videos 1–3). FRAP sequences of de novo formed dot- (D) and streaklike (E) low-intensity integrin clusters in Mn2+-treated cells (Video 4). FRAP sequences of stationary peripheral focal contacts in cells expressing N305T-activated β3 integrin (β3-N305T; F; Video 5). Note the twofold expanded time range in F. FRAP sequence of WT β3 integrins in sliding focal contacts of Mn2+-stimulated cells (β3-wt/Mn2+; G; Video 6). Arrowheads in G indicate the position of the inner edge of the sliding contact before bleaching. (H) Comparison of the FRAP curves of peripheral focal contacts in WT (β3-wt, diamonds) or by mutationally activated β3-EGFP integrins (β3-D723A, squares; β3-N305T, open triangles), respectively. (I) Comparison of FRAP curves of control (diamonds) or Mn2+-treated (open triangles) peripheral high intensity focal contacts, as well as Mn2+-induced de novo, dotlike, low-intensity integrin clusters (squares). Superimposed on this graph is the increase in integrin fluorescence at the inner edge of bleached, inward-sliding focal contacts (filled triangles). Error bars correspond to the standard deviation of three independent experiments with each comprising at least three cells. Bars: (A–C) 7.4 μm; (D and E) 5.4 μm; (F) 8 μm; (G) 10.7 μm.
Because the forced exposure of the cytoplasmic tail of β3 integrins (D723A mutation), involving the facilitated access to actin cytoskeleton-linked adapters, did not influence integrin exchange dynamics, we asked whether F-actin–independent integrin clusters exhibited the same integrin dynamics as found in focal contacts. Compared with integrins localized in peripheral focal contacts, both dot- and streaklike de novo integrin clusters exhibiting low integrin fluorescence demonstrated a fivefold faster integrin exchange rate (t 1/2 = 45 s) when measured by FRAP (Fig. 5 D, E, and I; and Video 4, available at http://www.jcb.org/cgi/content/full/jcb.200503017/DC1). Based on the few molecular components that build up F-actin–independent integrin clusters, we propose that this fast exchange rate reflects the association–dissociation equilibrium between the cytoplasmic tail of integrin with the PI(4,5)P2–talin complex and between the extracellular ligand and the integrin ectodomain. We further predict that the recruitment and lateral association of activated integrins into preexisting focal adhesions would follow this binding kinetics. To test this hypothesis, we analyzed the rate of integrin incorporation into focal contacts undergoing sliding. We had previously suggested that focal adhesion sliding is a macroscopic reflection of integrin assembly at the inner perimeter and integrin disassembly at the outer perimeter of an inward-sliding focal contact (Wehrle-Haller and Imhof, 2002). When sliding contacts are bleached, the majority of the new fluorescent integrin molecules accumulate at the inner edge of preexisting focal contacts (Wehrle-Haller and Imhof, 2002). Therefore, we measured the increase in integrin fluorescence intensity at inner edges of bleached focal contacts undergoing sliding. Surprisingly, the kinetics of integrin fluorescence accumulation paralleled the FRAP curve of Mn2+-induced actin-independent integrin clusters (Fig. 5, G and I; and Video 6). This data suggests that (a) the rate limiting step for focal contact sliding is the lateral association of high-affinity integrins and (b) the recruitment rate of high-affinity integrins in sliding contacts is defined by the association–dissociation reaction of the ternary complex of ligand–integrin–PI(4,5)P2–talin.
Discussion
High-affinity state is essential, but not sufficient, for integrin clustering
Recent crystallographic, nuclear magnetic resonance, and electronmicroscopic studies have revealed an allosteric switch in αvβ3 integrin that is induced upon activation (Xiong et al., 2001, 2002; Hynes, 2002; Takagi et al., 2002; Vinogradova et al., 2002; Xiao et al., 2004). Despite this detailed model of integrin activation, the mechanisms that result in its lateral clustering (e.g., during the formation of focal complexes) remain controversial. Our data demonstrate that the high-affinity state of integrins is an important prerequisite but is not sufficient for lateral clustering. Clustered integrins persist in a dynamic equilibrium that is influenced by the local concentration of clustering-competent, high-affinity integrins, the presence of PI(4,5)P2 within the plasma membrane, and the recruitment of talin. In addition, the formation of stable integrin clusters requires immobilized ligand. Our data demonstrate that integrin clustering is the result of at least four different parameters (immobilized ligand, activated integrin, presence of PI(4,5)P2, and availability of talin) that have to be fulfilled before the actin cytoskeleton is able to “use” clustered integrins as anchoring points for adhesion and migration.
The driving force of lateral integrin association
The nature of the driving force that induces the lateral association of activated integrins is a matter of debate. One possibility put forward by Li et al. (2003) is that homophilic interactions between the transmembrane domains of activated integrins drive lateral clustering. This model implies that integrin clustering can occur independently of other integrin-binding proteins as long as the transmembrane domains of the α and β integrin subunit are physically separated from each other (e.g., separation induced by integrin activation). Other results, however, do not support the view that activated integrins can cluster spontaneously. Electron microscopic images of purified, activated αIIbβ3 integrins that are incorporated into lipid vesicles or planar membranes give no evidence for spontaneous clustering of activated integrins (Erb et al., 1997). In addition, Mn2+-activated αvβ3 integrins do not cluster spontaneously when exposed to a laminin-1 substrate to which they are unable to bind (unpublished data). Moreover, the disulfide bond scanning of the exofacial portions of the integrin αIIb and β3 transmembrane domains did not reveal a specific interaction of these domains after integrin activation in living cells (Luo et al., 2004). We now demonstrate that activated αvβ3 integrins require immobilized substrate, PI(4,5)P2 lipids, and the focal adhesion adaptor protein talin for clustering. Our data suggest that integrin clustering is controlled by a simple association–dissociation reaction that is influenced by the density of activated integrins in the plasma membrane. However, the equilibrium of this reaction can be shifted depending on the availability of immobilized substrate, PI(4,5)P2, and talin (Fig. 6 A).
Figure 6.

Model of αvβ3 integrin activation and clustering. (A) Schematic view of a sequence of integrin activation and clustering. Talin is recruited and activated by membrane exposed PI(4,5)P2 lipids. Binding of the talin–PI(4,5)P2 complex to the cytoplasmic domain of integrin induces its “inside-out” activation (unfolding) allowing it to bind to immobilized extracellular ligands. Alternatively, integrins can interact with its extracellular ligands leading to “outside-in” activation and subsequent recruitment of PI(4,5)P2-bound talin, resulting in the formation of an “integrin preadhesion complex.” Subsequent clustering of this ternary complex is mediated by multivalent PI(4,5)P2-containing lipid domains. (B) Alternative modes of integrin activation, such as by the head domain of talin in response to calpain-mediated proteolytic cleavage or by low concentrations of soluble ligand (Legler et al., 2001), Mn2+, and activating mutations (N305T and D723A). These activation pathways can be modulated by the sequestration (Laux et al., 2000), synthesis, or degradation of PI(4,5)P2 lipids or by high doses of soluble ligands.
Although the formation of the ternary integrin–ligand–PI(4,5)P2–talin complex seems plausible, we can only speculate about which binding reactions eventually result in lateral clustering. In fact, to induce integrin clustering, one or several components within this ternary complex are required to form bi- or multivalent interactions with components of other complexes. It has been demonstrated that PI(4,5)P2 binds to the talin head domain and increases its affinity for the cytoplasmic domain of integrins (Martel et al., 2001). Alternatively, calpain cleavage of talin into COOH-terminal rod and globular head domains increases the affinity of the head domain of talin to the cytoplasmic tail of β3 integrin (Yan et al., 2001). We demonstrate that the PI(4,5)P2-induced association of talin head domains with integrins is sufficient to induce the formation of integrin clusters, which is probably mediated by multivalent PI(4,5)P2-containing lipid domains (Fig. 6 B). Because the head domain of talin is sufficient for integrin clustering, the rod domains and the second head domain of the talin dimer may have additional important functions. Notably, the rod domain of talin contains several binding sites for the actin-binding adaptor protein vinculin and a major F-actin–binding site (Critchley, 2004). In addition, the integrin-binding site in the head domain of talin can also interact with PIPKIγ by recruiting it to immobilized integrins, which would create a local focal adhesion–associated source of PI(4,5)P2 (Di Paolo et al., 2002; Ling et al., 2002; Barsukov et al., 2003). Our model predicts two critical roles of talin in maintaining the functional integrity of integrin clusters and their dynamic interaction with the actin cytoskeleton. First, the presence of “monovalent” talin head domains is sufficient to induce integrin clusters. Second, full-length talin links these integrin clusters to the actin cytoskeleton. In fact, calpain-mediated cleavage of talin would disrupt the interaction of integrins with the actin cytoskeleton without affecting integrin activation and dynamic clustering, maintaining the dynamic state of focal contacts. Accordingly, the treatment of cells with calpain inhibitors reduces the rate of integrin exchange in focal contacts (unpublished data). This is in agreement with the phenotype of slow focal adhesion turnover in cells expressing calpain-resistant talin (Franco et al., 2004).
In addition to talin cleavage, the reversible phosphorylation of PI(4)P may be a way to control focal adhesion assembly or disassembly (Fig. 6 B). Signal-dependent recruitment of PIPKIγ to the talin head domain could create a local focal adhesion–associated source of PI(4,5)P2 that in turn would stimulate talin recruitment and integrin activation (Di Paolo et al., 2002; Ling et al., 2002; Barsukov et al., 2003). Thus, PIPKIγ activity may represent the driving force for integrin remodeling in sliding focal adhesions at the cell rear.
Independent of the “intracellular” regulation of integrin dynamics in focal contacts, our results demonstrate that the rate-limiting step for the assembly and disassembly of integrin clusters is the time during which the activated integrin is bound to immobilized extracellular ligands. This is supported by the slow integrin exchange dynamics of the N305T ectodomain mutant, suggesting that the flexibility, and hence the extracellular ligand affinity, of the ectodomain controls integrin cluster dynamics.
Integrin clusters are precursors of focal adhesions
De novo Mn2+-induced β3 integrin clusters specifically recruit talin but not other adaptor proteins such as vinculin, paxillin, or FAK. This is in contrast to the recruitment of FAK and other signaling components, but not talin, to antibody cross-linked RGD peptide-activated clusters of α5β1 integrin (Miyamoto et al., 1995). It is currently not known which mechanisms are responsible for these different patterns of integrin adaptor recruitment. In the case of αvβ3 integrin however, we propose that the integrin–PI(4,5)P2–talin complex represents an intermediary step during the assembly of focal adhesions, which are rendered visible by the forced activation of large numbers of integrins. Accordingly, when cells are treated for a prolonged time with Mn2+ (e.g., 2 h), an increasing fraction of integrin clusters can be found to associate with vinculin and F-actin (unpublished data). Consequently, the integrin–PI(4,5)P2–talin complex should be found in locations where focal complexes normally assemble, such as in the lamellipodium. In fact, comparable talin- and integrin-containing immature focal complexes have been found at the leading edge of advancing lamellipodia (DePasquale and Izzard, 1991; Lee and Jacobson, 1997). These structures further mature by recruiting vinculin, α-actinin, and FAK (DePasquale and Izzard, 1991; Lee and Jacobson, 1997). This evolution suggests that talin-associated activated integrins are the progenitors of focal complexes within the lamellipodium. This view is supported by a recent report demonstrating the recruitment of talin into β3 integrin– containing focal complexes before the appearance of vinculin or FAK (Zaidel-Bar et al., 2003). As an exception to the rule, paxillin is present in focal complexes of migrating endothelial cells at the same time as β3 integrins (Zaidel-Bar et al., 2003). The difference to our data could be explained by the specific recruitment of paxillin to α4 integrins that may cluster together with αvβ3 integrin at the leading edge of endothelial cells (Goldfinger et al., 2003).
We propose, therefore, that the formation of focal complexes within a lamellipodium includes several steps: PI(4,5)P2 is synthesized in the lamellipodium in a Rac1- and ARF6-dependent manner (Honda et al., 1999). PI(4,5)P2 induces a conformational change in talin that associates and activates β3 integrins (Martel et al., 2001; Calderwood et al., 2002), forming “preadhesion complexes.” Simultaneously, PI(4,5)P2 stimulates vinculin and ezrin–radixin–moesin family proteins to bind to talin and to link the actin cytoskeleton to the plasma membrane, respectively (Yin and Janmey, 2003), which is a process facilitated by the transient interaction of vinculin with the actin nucleation factor Arp2/3 (DeMali et al., 2002). Therefore, a focal complex forms in the lamellipodium as a result of the intersection of several intra- and extracellular systems requiring immobilized ligand, integrin activation, actin polymerization, and PI(4,5)P2 synthesis.
Materials and methods
cDNAs and site-directed mutagenesis
cDNA encoding full-length mouse β3-EGFP integrin fusion protein in a pcDNA3 expression vector has been described in Ballestrem et al. (2001). The D119Y, N305T, and D723A substitutions were introduced by PCR amplification using PfuTurbo DNA polymerase followed by DpnI digestion (Stratagene). A monomeric red fluorescent protein variant of mouse β3 integrin was generated by the exchange of the EGFP with the monomeric red fluorescent protein sequence (obtained from R.Y. Tsien, University of California, San Diego, La Jolla, CA). The COOH-terminal fragment, including the transmembrane and cytoplasmic domain of the chicken β1 integrin (obtained from G.G. Gundersen, Columbia University, New York, NY), was amplified with PfuTurbo DNA polymerase and cloned between an EcoRV and PinAI site in the β3-EGFP integrin fusion protein using the primers ATAGATATCATACCCATTGTAGCTGG and AATACCGGTGATTTTCCCTCATATTTAGGA. The NH2-terminal fragment of human Talin1 (residues 1–435) was amplified with PfuTurbo DNA polymerase from IMAGE clone 3844238 (available from GenBank/EMBL/DDBJ under accession no. BE732988) and cloned into the XhoI and EcoRI sites of pECFP-C1 (CLONTECH Laboratories, Inc.) using the primers GATCTCGAGCCATGGTTGCACTTTCACTG and TATGAATTCTATTGCTGCTGCAGGACTG. DNA sequence analysis was performed for all constructs to ensure error-free amplification and proper base replacement.
Cell culture, transfection, and inhibitor treatment
Mouse B16F1 melanoma cells and hamster CS-1 melanoma cells were grown in DME containing 10% FCS, glutamine, and antibiotics (Thomas et al., 1993; Ballestrem et al., 2001). Cells were transfected using Fugen 6 (Roche) and selected for stably expressing cells in the presence of 1.2 mg/ml B16F1 or 0.6 mg/ml CS-1 G418 (Becton Dickinson). Stably transfected B16F1 cells were FACS sorted, and clones were selected for their expression of EGFP fluorescence and cell surface–exposed αvβ3 integrins using the Kistrin-CD31 fusion protein SKI-7 and the rat anti-CD31 mAb GC51, as previously described (Ballestrem et al., 2001; Legler et al., 2001) or using hamster anti–mouse β3 integrin in the case of the D119Y mutant (2C9.G2; BD Biosciences). Nontagged β3 integrin–transfected CS-1 cells were selected for their SKI-7 reactivity and their capacity to adhere to tissue culture dishes. Neomycin sulfate, cD (both from Sigma-Aldrich), and cRGD peptides (cRGD; Bachem; Legler et al., 2001) were prepared as stock solutions in PBS. Inhibitor studies were all performed in complete medium.
Measurement of integrin clustering
WT and mutant B16F1 β3-EGFP–expressing cells were cultured overnight in complete medium in glass bottom dishes, fixed for 10 min with 4% PFA, and rinsed with PBS. Mn2+ activation (0.5 μM Mn2+) of B16F1 or CS-1 cells was performed for 20 min in complete or inhibitor-containing complete medium. EGFP fluorescence and respective IRM images were acquired with identical excitation and exposure settings using a Plan Neofluar 63× NA 1.4 oil-immersion objective on an inverted confocal microscope (model LSM510; Carl Zeiss MicroImaging, Inc.), focusing on the glass coverslip by using the maximal reflection of the laser light. Intensity histograms of cells were obtained after smoothing (3 × 3 kernel), background subtraction, and manual selection of the cell surface using MetaMorph software (Molecular Devices) and exported to Excel (Microsoft) for further analysis. Histograms were normalized in respect to the cell surface area and aligned according to the peak of the histograms corresponding to the membrane fluorescence of the cell before averaging (n > 20). Cumulative intensity histograms were obtained by multiplication of the number of pixels (in percent) with the respective gray value (Fig. S1).
The relative area of integrin clusters in respect to the entire cell surface was obtained from total internal reflection fluorescence (TIRF) images of at least 20 cells per condition. TIRF images of β3-EGFP integrin fluorescence were obtained with a Plan Neo-Fluar 100× NA 1.45 objective mounted on an Axiovert 100M (both from Carl Zeiss MicroImaging, Inc.) equipped with a 12-bit digital charge-coupled device camera (model Orca 4742–95; Hamamatsu Photonics) controlled by the Openlab software (Improvision). Intraobjective TIRF was obtained with a 488-nm laser through a TIRF adaptor (TILL Photonics). Image analysis was performed after manually setting of the intensity threshold using MetaMorph software.
Measurement of integrin intensities
Relative integrin fluorescence intensities in control and Mn2+- or neomycin sulfate–treated cells were essentially measured as described in Ballestrem et al. (2001). In brief, 10-bit-wide field fluorescent images were acquired with identical settings on an Axiovert 100TV using Openlab software. For each image, the fluorescence intensity was determined from triplicate measures of the background outside the cell, inside bright peripheral contacts, in the cell membrane, and, when appropriate, in Mn2+-induced integrin clusters. The respective integrin fluorescence was obtained by deduction of the background.
Antibodies, immunofluorescence, and the digital processing of images
Cells grown overnight in complete medium were fixed with 4% PFA in PBS for 10 min, permeabilized, and blocked in 0.1% Triton X-100 and 1% BSA in PBS for 20 min. Mouse mAbs to vinculin, talin recognizing the rod domain of talin (both from Sigma-Aldrich), P-Tyr (4G10; Upstate Biotechnology), FAK (F15020), and paxillin (P13520; both from BD Transduction Laboratories) were applied in 1% BSA-PBS for 1 h. After being washed in blocking solution, Texas–red conjugated goat anti–mouse antibodies (Jackson ImmunoResearch Laboratories) were applied for 1 h and subsequently washed as above. Preparations were stored in PBS and images were collected at RT using a Ph3 Plan-Apochromat 63× NA 1.40 objective on an Axiovert 100TV microscope equipped with a 10-bit digital charge-coupled device camera controlled by Openlab software. Confocal images were obtained as described in Measurement of integrin clustering on a confocal microscope. Due to technical reasons, confocal images of EGFP-integrin and ECFP-talin double-transfected cells were acquired individually after changing the dichroic mirror. For publication, a Gaussian blur (0.4) was applied to all confocal images. In addition, background and contrast were adjusted using the adjust Level command in Photoshop (Adobe).
Substrate patterns
Substrate patterns of 5 × 5 μm2 squares were prepared using a variation of the microcontact printing technique (Csucs et al., 2003). Polydimethylsiloxane stamps carrying the corresponding pattern (Csucs et al., 2003) were incubated for 1 h with a mixture of 20 μg/ml of Alexa Fluor 568–labeled fibronectin and 10 μg/ml of nonlabeled vitronectin in PBS. Inked stamps were placed onto glass coverslips for 30 s to ensure contact between the stamp and the glass surface. After rinsing the coverslip with PBS, noncoated areas of the coverslip were blocked for 20 min in 1 mg/ml of polylysine-g-polyethyleneglycol copolymer in 10 mM Hepes, pH 7.4, which is proven to be efficient in repelling serum-protein absorption to the glass coverslip (Huang et al., 2001; VandeVondele et al., 2003). Vitronectin and fibronectin were obtained from Sigma-Aldrich and fibronectin was labeled with an Alexa Fluor 568 labeling kit as recommended by the manufacturer (Molecular Probes). Cells were plated overnight in BSA containing medium and were shifted to 3% FCS containing medium 1 h before imaging.
FRAP
B16F1 cells were cultured in glass bottom dishes for 24 h in complete culture medium. Medium was replaced by F12 medium containing glutamine, antibiotics and 10% FCS before FRAP analysis. FRAP was performed at 37°C on an LSM510 inverted microscope equipped with a heated stage and CO2 control essentially as described in Ballestrem et al. (2001). To reduce loss of fluorescence due to bleaching during the recovery period, we reduced the laser power of the 488-nm line to 0.5% at a maximal laser output of 50%. To improve light collection, the pin hole was partially opened (to 200 μm). To ensure maintenance of the focus during the recovery period, the IRM was recorded simultaneously.
Online supplemental material
Fig. S1 further explains the procedure used to evaluate integrin clustering from confocal images. In addition, the phenotype of integrin “shedding” observed in the N305T β3 integrin mutant transfected cells is illustrated in Fig. S2. Fig. S3 demonstrates the ability of the cytoplasmic tail of β1 integrin to mediate Mn2+-induced integrin clustering. In Fig. S4 the role of PI(4,5)P2 in maintaining Mn2+-induced integrin clusters is shown by their dispersal in response to neomycin sulfate treatment. Videos 1–6 represent the corresponding FRAP sequences of the cells from which the boxed areas are shown in Fig. 5 (A–G). Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200503017/DC1.
Acknowledgments
We thank Roland Püttmann-Delgado, Marie-Claude Jacquier, and Christian Vesin for excellent technical assistance. We are also grateful to Drs. Junichi Takagi, James A. Weston, Caroline Johnson-Léger, Monique Wehrle-Haller, and Michel Aurrands-Lions for suggestions, discussions, and critical reading of the manuscript.
C. Cluzel and F. Saltel were supported by grants for post-doctoral fellowships of the French Foundation for Medical Research and the French Association for Cancer Research. The work was supported by research grants from the Swiss Cancer Ligue (KFS 412-1-1997) and the Swiss Science Foundation to B.A. Imhof and B. Wehrle-Haller (31-059173.99, 31-64000.00, and 3100A0-103805).
C. Cluzel and F. Saltel contributed equally to this paper.
C. Cluzel's present address is Institut de Biologie et Chimie des Proteines, Lyon Cedex 07, France.
F. Saltel, F. Paulhe, and B. Wehrle-Haller's present address is Dept. of Cellular Physiology and Metabolism, Centre Medical Universitaire, 1211 Geneva 4, Switzerland.
Abbreviations used in this paper: cRGD, cyclic RGD; cD, cytochalasin D; IRM, interference reflection microscopy; PI(4,5)P2, phosphoinositol-4,5-bisphosphate; TIRF, total internal reflection fluorescence; WT, wild type.
References
- Arbuzova, A., K. Martushova, G. Hangyas-Mihalyne, A.J. Morris, S. Ozaki, G.D. Prestwich, and S. McLaughlin. 2000. Fluorescently labeled neomycin as a probe of phosphatidylinositol-4,5-bisphosphate in membranes. Biochim. Biophys. Acta. 1464:35–48. [DOI] [PubMed] [Google Scholar]
- Ballestrem, C., B. Hinz, B.A. Imhof, and B. Wehrle-Haller. 2001. Marching at the front and dragging behind: differential αVβ3-integrin turnover regulates focal adhesion behavior. J. Cell Biol. 155:1319–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsukov, I.L., A. Prescot, N. Bate, B. Patel, D.N. Floyd, N. Bhanji, C.R. Bagshaw, K. Letinic, G. Di Paolo, P. De Camilli, et al. 2003. Phosphatidylinositol phosphate kinase type 1gamma and beta1-integrin cytoplasmic domain bind to the same region in the talin FERM domain. J. Biol. Chem. 278:31202–31209. [DOI] [PubMed] [Google Scholar]
- Calderwood, D.A., B. Yan, J.M. de Pereda, B.G. Alvarez, Y. Fujioka, R.C. Liddington, and M.H. Ginsberg. 2002. The phosphotyrosine binding-like domain of talin activates integrins. J. Biol. Chem. 277:21749–21758. [DOI] [PubMed] [Google Scholar]
- Chen, J., A. Salas, and T.A. Springer. 2003. Bistable regulation of integrin adhesiveness by a bipolar metal ion cluster. Nat. Struct. Biol. 10:995–1001. [DOI] [PubMed] [Google Scholar]
- Chen, J., J. Takagi, C. Xie, T. Xiao, B.H. Luo, and T.A. Springer. 2004. The relative influence of metal ion binding sites in the I-like domain and the interface with the hybrid domain on rolling and firm adhesion by integrin alpha4beta7. J. Biol. Chem. 279:55556–55561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critchley, D.R. 2004. Cytoskeletal proteins talin and vinculin in integrin-mediated adhesion. Biochem. Soc. Trans. 32:831–836. [DOI] [PubMed] [Google Scholar]
- Csucs, G., R. Michel, J.W. Lussi, M. Textor, and G. Danuser. 2003. Microcontact printing of novel co-polymers in combination with proteins for cell-biological applications. Biomaterials. 24:1713–1720. [DOI] [PubMed] [Google Scholar]
- DeMali, K.A., C.A. Barlow, and K. Burridge. 2002. Recruitment of the Arp2/3 complex to vinculin: coupling membrane protrusion to matrix adhesion. J. Cell Biol. 159:881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePasquale, J.A., and C.S. Izzard. 1991. Accumulation of talin in nodes at the edge of the lamellipodium and separate incorporation into adhesion plaques at focal contacts in fibroblasts. J. Cell Biol. 113:1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo, G., L. Pellegrini, K. Letinic, G. Cestra, R. Zoncu, S. Voronov, S. Chang, J. Guo, M.R. Wenk, and P. De Camilli. 2002. Recruitment and regulation of phosphatidylinositol phosphate kinase type 1 gamma by the FERM domain of talin. Nature. 420:85–89. [DOI] [PubMed] [Google Scholar]
- Erb, E.M., K. Tangemann, B. Bohrmann, B. Muller, and J. Engel. 1997. Integrin alphaIIb beta3 reconstituted into lipid bilayers is nonclustered in its activated state but clusters after fibrinogen binding. Biochemistry. 36:7395–7402. [DOI] [PubMed] [Google Scholar]
- Franco, S.J., M.A. Rodgers, B.J. Perrin, J. Han, D.A. Bennin, D.R. Critchley, and A. Huttenlocher. 2004. Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nat. Cell Biol. 6:977–983. [DOI] [PubMed] [Google Scholar]
- Goldfinger, L.E., J. Han, W.B. Kiosses, A.K. Howe, and M.H. Ginsberg. 2003. Spatial restriction of α4 integrin phosphorylation regulates lamellipodial stability and α4β1-dependent cell migration. J. Cell Biol. 162:731–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda, A., M. Nogami, T. Yokozeki, M. Yamazaki, H. Nakamura, H. Watanabe, K. Kawamoto, K. Nakayama, A.J. Morris, M.A. Frohman, and Y. Kanaho. 1999. Phosphatidylinositol 4-phosphate 5-kinase alpha is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell. 99:521–532. [DOI] [PubMed] [Google Scholar]
- Huang, N.-P., R. Michel, J. Voros, M. Textor, R. Hofer, A. Rossi, D.L. Elbert, J.A. Hubbell, and N.D. Spencer. 2001. Poly(L-lysine)-g-poly(ethylene glycol) layers on metal oxide surfaces: surface-analytical characterization and resistance to serum and fibrinogen adsorption. Langmuir. 17:489–498. [Google Scholar]
- Hughes, P.E., F. Diaz-Gonzalez, L. Leong, C. Wu, J.A. McDonald, S.J. Shattil, and M.H. Ginsberg. 1996. Breaking the integrin hinge. A defined structural constraint regulates integrin signaling. J. Biol. Chem. 271:6571–6574. [DOI] [PubMed] [Google Scholar]
- Hynes, R.O. 2002. Integrins: bidirectional, allosteric signaling machines. Cell. 110:673–687. [DOI] [PubMed] [Google Scholar]
- Isenberg, G., and W.H. Goldmann. 1998. Peptide-specific antibodies localize the major lipid binding sites of talin dimers to oppositely arranged N-terminal 47 kDa subdomains. FEBS Lett. 426:165–170. [DOI] [PubMed] [Google Scholar]
- Katagiri, K., A. Maeda, M. Shimonaka, and T. Kinashi. 2003. RAPL, a Rap1-binding molecule that mediates Rap1-induced adhesion through spatial regulation of LFA-1. Nat. Immunol. 4:741–748. [DOI] [PubMed] [Google Scholar]
- Kwik, J., S. Boyle, D. Fooksman, L. Margolis, M.P. Sheetz, and M. Edidin. 2003. Membrane cholesterol, lateral mobility, and the phosphatidylinositol 4,5-bisphosphate-dependent organization of cell actin. Proc. Natl. Acad. Sci. USA. 100:13964–13969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laux, T., K. Fukami, M. Thelen, T. Golub, D. Frey, and P. Caroni. 2000. GAP43, MARCKS, and CAP23 modulate PI(4,5)P2 at plasmalemmal rafts, and regulate cell cortex actin dynamics through a common mechanism. J. Cell Biol. 149:1455–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J., and K. Jacobson. 1997. The composition and dynamics of cell-substratum adhesions in locomoting fish keratocytes. J. Cell Sci. 110:2833–2844. [DOI] [PubMed] [Google Scholar]
- Legler, D.F., G. Wiedle, F.P. Ross, and B.A. Imhof. 2001. Superactivation of integrin alphavbeta3 by low antagonist concentrations. J. Cell Sci. 114:1545–1553. [DOI] [PubMed] [Google Scholar]
- Li, R., N. Mitra, H. Gratkowski, G. Vilaire, R. Litvinov, C. Nagasami, J.W. Weisel, J.D. Lear, W.F. DeGrado, and J.S. Bennett. 2003. Activation of integrin alphaIIbbeta3 by modulation of transmembrane helix associations. Science. 300:795–798. [DOI] [PubMed] [Google Scholar]
- Liddington, R.C., and M.H. Ginsberg. 2002. Integrin activation takes shape. J. Cell Biol. 158:833–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling, K., R.L. Doughman, A.J. Firestone, M.W. Bunce, and R.A. Anderson. 2002. Type I gamma phosphatidylinositol phosphate kinase targets and regulates focal adhesions. Nature. 420:89–93. [DOI] [PubMed] [Google Scholar]
- Loftus, J.C., T.E. O'Toole, E.F. Plow, A. Glass, A.L. Frelinger III, and M.H. Ginsberg. 1990. A beta 3 integrin mutation abolishes ligand binding and alters divalent cation-dependent conformation. Science. 249:915–918. [DOI] [PubMed] [Google Scholar]
- Luo, B.H., T.A. Springer, and J. Takagi. 2003. Stabilizing the open conformation of the integrin headpiece with a glycan wedge increases affinity for ligand. Proc. Natl. Acad. Sci. USA. 100:2403–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, B.H., T.A. Springer, and J. Takagi. 2004. A specific interface between integrin transmembrane helices and affinity for ligand. PLoS Biol. 2:e153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martel, V., C. Racaud-Sultan, S. Dupe, C. Marie, F. Paulhe, A. Galmiche, M.R. Block, and C. Albiges-Rizo. 2001. Conformation, localization, and integrin binding of talin depend on its interaction with phosphoinositides. J. Biol. Chem. 276:21217–21227. [DOI] [PubMed] [Google Scholar]
- Miyamoto, S., S.K. Akiyama, and K.M. Yamada. 1995. Synergistic roles for receptor occupancy and aggregation in integrin transmembrane function. Science. 267:883–885. [DOI] [PubMed] [Google Scholar]
- Mould, A.P., J.A. Askari, S. Barton, A.D. Kline, P.A. McEwan, S.E. Craig, and M.J. Humphries. 2002. Integrin activation involves a conformational change in the alpha 1 helix of the beta subunit A-domain. J. Biol. Chem. 277:19800–19805. [DOI] [PubMed] [Google Scholar]
- Pampori, N., T. Hato, D.G. Stupack, S. Aidoudi, D.A. Cheresh, G.R. Nemerow, and S.J. Shattil. 1999. Mechanisms and consequences of affinity modulation of integrin alpha(V)beta(3) detected with a novel patch-engineered monovalent ligand. J. Biol. Chem. 274:21609–21616. [DOI] [PubMed] [Google Scholar]
- Partridge, A.W., S. Liu, S. Kim, J.U. Bowie, and M.H. Ginsberg. 2005. Transmembrane domain helix packing stabilizes integrin alphaIIbbeta3 in the low affinity state. J. Biol. Chem. 280:7294–7300. [DOI] [PubMed] [Google Scholar]
- Priddle, H., L. Hemmings, S. Monkley, A. Woods, B. Patel, D. Sutton, G.A. Dunn, D. Zicha, and D.R. Critchley. 1998. Disruption of the talin gene compromises focal adhesion assembly in undifferentiated but not differentiated embryonic stem cells. J. Cell Biol. 142:1121–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadokoro, S., S.J. Shattil, K. Eto, V. Tai, R.C. Liddington, J.M. de Pereda, M.H. Ginsberg, and D.A. Calderwood. 2003. Talin binding to integrin beta tails: a final common step in integrin activation. Science. 302:103–106. [DOI] [PubMed] [Google Scholar]
- Takagi, J., B.M. Petre, T. Walz, and T.A. Springer. 2002. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 110:599–611. [DOI] [PubMed] [Google Scholar]
- Thomas, L., P.W. Chan, S. Chang, and C. Damsky. 1993. 5-Bromo-2-deoxyuridine regulates invasiveness and expression of integrins and matrix-degrading proteinases in a differentiated hamster melanoma cell. J. Cell Sci. 105:191–201. [DOI] [PubMed] [Google Scholar]
- VandeVondele, S., J. Voros, and J.A. Hubbell. 2003. RGD-grafted poly-L-lysine-graft-(polyethylene glycol) copolymers block non-specific protein adsorption while promoting cell adhesion. Biotechnol. Bioeng. 82:784–790. [DOI] [PubMed] [Google Scholar]
- Verschueren, H. 1985. Interference reflection microscopy in cell biology: methodology and applications. J. Cell Sci. 75:279–301. [DOI] [PubMed] [Google Scholar]
- Vinogradova, O., A. Velyvis, A. Velyviene, B. Hu, T. Haas, E. Plow, and J. Qin. 2002. A structural mechanism of integrin alpha(IIb)beta(3) “inside-out” activation as regulated by its cytoplasmic face. Cell. 110:587–597. [DOI] [PubMed] [Google Scholar]
- Wehrle-Haller, B., and B.A. Imhof. 2002. The inner lives of focal adhesions. Trends Cell Biol. 12:382–389. [DOI] [PubMed] [Google Scholar]
- White, J., and E. Stelzer. 1999. Photobleaching GFP reveals protein dynamics inside live cells. Trends Cell Biol. 9:61–65. [DOI] [PubMed] [Google Scholar]
- Xiao, T., J. Takagi, B.S. Coller, J.H. Wang, and T.A. Springer. 2004. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature. 432:59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong, J.P., T. Stehle, B. Diefenbach, R. Zhang, R. Dunker, D.L. Scott, A. Joachimiak, S.L. Goodman, and M.A. Arnaout. 2001. Crystal structure of the extracellular segment of integrin alpha Vbeta3. Science. 294:339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong, J.P., T. Stehle, R. Zhang, A. Joachimiak, M. Frech, S.L. Goodman, and M.A. Arnaout. 2002. Crystal structure of the extracellular segment of integrin alpha Vbeta3 in complex with an Arg-Gly-Asp ligand. Science. 296:151–155. [DOI] [PubMed] [Google Scholar]
- Yan, B., D.A. Calderwood, B. Yaspan, and M.H. Ginsberg. 2001. Calpain cleavage promotes talin binding to the beta 3 integrin cytoplasmic domain. J. Biol. Chem. 276:28164–28170. [DOI] [PubMed] [Google Scholar]
- Yin, H.L., and P.A. Janmey. 2003. Phosphoinositide regulation of the actin cytoskeleton. Annu. Rev. Physiol. 65:761–789. [DOI] [PubMed] [Google Scholar]
- Zaidel-Bar, R., C. Ballestrem, Z. Kam, and B. Geiger. 2003. Early molecular events in the assembly of matrix adhesions at the leading edge of migrating cells. J. Cell Sci. 116:4605–4613. [DOI] [PubMed] [Google Scholar]