

Abstract
The scattering of Madin-Darby canine kidney cells in vitro mimics key aspects of epithelial–mesenchymal transitions during development, carcinoma cell invasion, and metastasis. Scattering is induced by hepatocyte growth factor (HGF) and is thought to involve disruption of cadherin-dependent cell–cell junctions. Scattering is enhanced on collagen and fibronectin, as compared with laminin1, suggesting possible cross talk between integrins and cell–cell junctions. We show that HGF does not trigger any detectable decrease in E-cadherin function, but increases integrin-mediated adhesion. Time-lapse imaging suggests that tension on cell–cell junctions may disrupt cell–cell adhesion. Varying the density and type of extracellular matrix proteins shows that scattering correlates with stronger integrin adhesion and increased phosphorylation of the myosin regulatory light chain. To directly test the role of integrin-dependent traction forces, substrate compliance was varied. Rigid substrates that produce high traction forces promoted scattering, in comparison to more compliant substrates. We conclude that integrin-dependent actomyosin traction force mediates the disruption of cell–cell adhesion during epithelial cell scattering.
Introduction
Epithelial–mesenchymal transition (EMT) is a critically important process in embryonic development, as well as in tumor invasion and metastasis. EMT is characterized by the disruption of epithelial cell–cell adhesions and the induction of a migratory phenotype that allows cells to escape the surrounding epithelium and invade other tissues (Thiery, 2002). A well-known in vitro model system for EMT uses MDCK epithelial cells, which can be induced to scatter using hepatocyte growth factor (HGF). HGF is the ligand for the transmembrane receptor tyrosine kinase c-Met that, in its oncogenic form (v-Met), strongly contributes to the invasive potential of tumor cells (Birchmeier et al., 2003). Scattering recapitulates many of the events that occur during cancer invasion and metastasis, as tightly clustered epithelial cells break their cell–cell junctions and become single, migrating, invasive cells.
The stages of cell migration are reasonably well defined for many different cell types, including epithelial cells that undergo EMT (Ridley et al., 2003). Migration starts with the formation of a protrusion at the cell's leading edge, which is driven by actin polymerization. Beneath the protrusion, integrin-mediated adhesions to the ECM are initiated, which are then reinforced in response to tension applied by the actomyosin cytoskeleton (Sheetz et al., 1998). The cytoskeleton connects to integrins through complexes of structural and signaling proteins that are called focal complexes or focal adhesions, depending on their life span and constituents (Zamir and Geiger, 2001). Myosin-based contraction of the actin cytoskeleton is transmitted to the adhesion complexes to establish the local traction force that relocates the cell body and contributes to the disassembly of integrin–ECM adhesions in the back of the cell.
How cell–cell adhesion is disrupted during EMT is less well understood. The key event is the loss of E-cadherin–mediated cell–cell adhesion, and several underlying mechanisms may exist. Although mutations in the E-cadherin gene are not often found in human cancer, invasive and metastatic tumors commonly down-regulate E-cadherin transcription, stability, or surface levels (Gumbiner, 2000). Many other tumors, however, retain E-cadherin surface levels, suggesting that there are other ways to regulate E-cadherin adhesive function. Conformational control of E-cadherin adhesiveness during HGF-induced MDCK scattering, analogous to affinity modulation of integrins (Kinbara et al., 2003), has been hypothesized, but no evidence has been presented to support this idea. Cadherin clustering has been shown to promote its adhesive function, and the actin cytoskeleton is a potential regulator of this phenomenon (Yap et al., 1998). E-cadherin is linked to actin through α- and β-catenin, and disruption of this complex results in the loss of E-cadherin function (Gumbiner, 2000). Also, disruption of the actin cytoskeleton by cytochalasin completely abolishes E-cadherin function (Angres et al., 1996). In certain types of cancer, mutation of the α-catenin gene is associated with high metastatic potential (Kobielak and Fuchs, 2004). However, in cells that undergo a rapid loss of cell–cell adhesion, like scattering MDCK cells, biochemical changes in the cadherin–catenin complex are usually not observed (Gumbiner, 2000).
Both cell migration and cell–cell adhesion during EMT have been investigated extensively; however, they have mainly been addressed in separate studies. Nevertheless, there is clear evidence of cross talk between integrin- and cadherin-based adhesive structures. For instance, the ECM on which cells are adhered plays a significant role in modulating the scattering of MDCK cells; collagen (Cn) has been found to promote migration and scattering, whereas other ECM proteins such as fibronectin (Fn), laminin (Ln), and vitronectin (Vn) favor the epithelial phenotype (Clark, 1994; Sander et al., 1998). These effects were hypothesized to be due to a Cn-specific signal, mediated most likely by integrin α2β1. In addition, during embryonic development, increased adhesion of integrins to Fn drives the deregulation of E-cadherin adhesions during branching morphogenesis of salivary glands (Sakai et al., 2003), whereas integrin β1–dependent Fn adhesion regulates convergent extension in the developing Xenopus laevis embryo via modulation of C-cadherin adhesion (Marsden and Desimone, 2003). Finally, in colon cancer cells, the disruption of cell–cell junctions by oncogenic Src critically depends on the presence of integrin-mediated ECM adhesion (Avizienyte et al., 2002). Thus, ECM modulation of cadherin function during cell scattering and morphogenic rearrangements is important for multiple physiological and pathological processes.
Therefore, we set out to investigate the mechanism of cross talk between integrin- and E-cadherin–mediated adhesion in HGF-induced MDCK cell scattering. Our results suggest that different ECM conditions influence scattering by modulation of actomyosin cytoskeletal organization and contractility. Thus, cellular mechanical forces, rather than changes in cadherin function, play a major role in the regulation of scattering by ECM.
Results
HGF does not down-regulate E-cadherin function
It has generally been supposed that HGF disrupts E-cadherin function to induce the loss of cell–cell adhesion and increase cell migration. To investigate the effect of HGF on cell–cell adhesion in the absence of cell migration, cells were plated either densely enough to form a continuous monolayer or sparsely enough to allow the formation of isolated islands of cells. In both cases, coverslips were coated with 3 μg/ml Cn, which strongly promotes scattering, and cells were plated with HGF 24 h before stimulation. 20 h after HGF stimulation, cells were fixed and stained for E-cadherin and F-actin. In densely plated cells, E-cadherin clearly remained in cell–cell junctions at an intensity similar to that in unstimulated cells (Fig. 1 A). When sparsely plated, most cells, as expected, broke contacts with their neighboring cells and moved out of the epithelial islands so that the area of cell–cell contact decreased. However, the remaining cell–cell junctions clearly retained E-cadherin also at an intensity similar to that in unstimulated cells. These results suggest that the disruption of E-cadherin–based cell–cell junctions by HGF is dependent on the ability of cells to migrate and move apart from one another and does not involve direct reduction in E-cadherin function.
Figure 1.
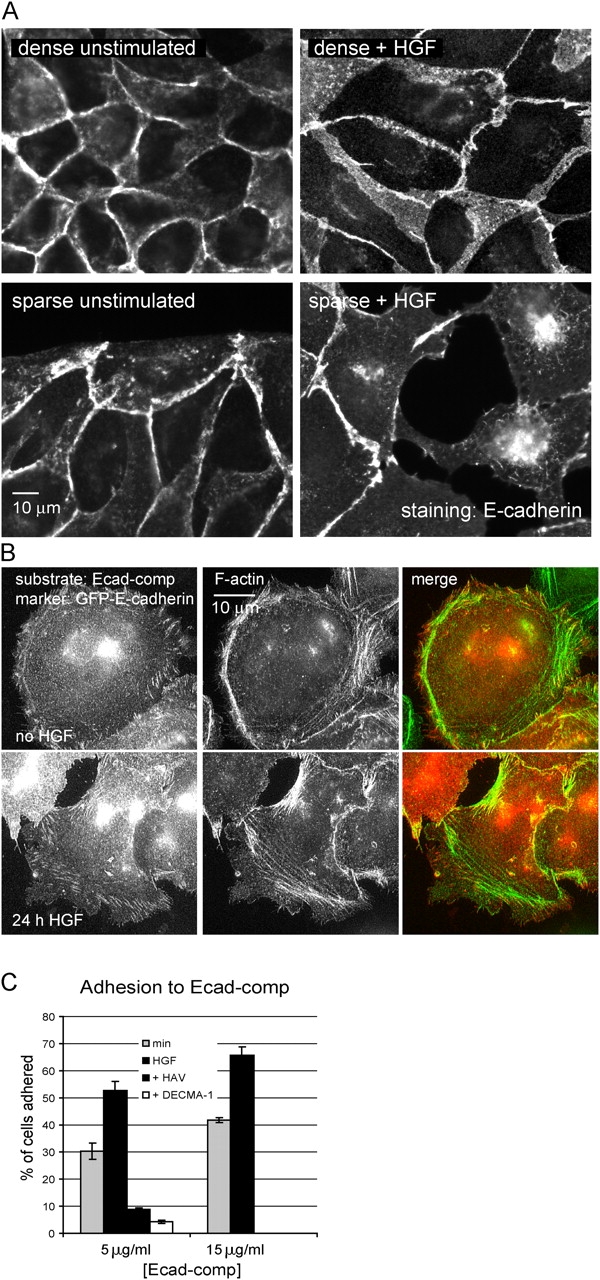
HGF does not inhibit the ability of E-cadherin to form homotypic interactions. (A) Lack of effect of HGF on E-cadherin in cell–cell junctions. MDCK cells plated at high or low density on Cn-coated coverslips were treated with HGF for 24 h. Cells were fixed and E-cadherin was immunolocalized. (B) Lack of effect of HGF on GFP-E-cadherin distribution on E-cadherin–coated coverslips. GFP-E-cadherin–expressing MDCK cells were plated on Ecad-comp in the absence of HGF for 3 h or in the presence of HGF for 24 h, fixed, and stained for F-actin. (C) HGF increases adhesion to Ecad-comp. MDCK cells were plated on the indicated amounts of Ecad-comp in the absence or presence of HGF or inhibitors (DECMA-1 or HAV-peptide) and treated and quantified as described in Materials and methods. Data are means ± SD; n = 3.
To specifically assay the effects of HGF on E-cadherin adhesion without interference from other adhesion receptors, we coated glass coverslips with a fusion of the extracellular domain of E-cadherin and cartilage oligomeric protein (Ecad-comp), which forms pentamers and thus mimics E-cadherin clusters at the cell membrane (Pertz et al., 1999). MDCK cells stably expressing GFP-E-cadherin were plated for 3-h. Fluorescence imaging revealed the formation of elongated E-cadherin–containing plaques located at the termini of F-actin bundles, much like the N-cadherin–based adhesion described by Gavard et al. (2004). These adhesions also contained vinculin and α-, β-, and p120-catenin, but did not contain the integrin-binding protein paxillin (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200506152/DC1). Subsequent treatment of these cells with HGF on Ecad-comp for 24 h did not result in any reduction of the amount of adhesions or intensity of GFP-E-cadherin in these adhesions (Fig. 1 B), again suggesting that HGF does not directly alter E-cadherin function. To quantitatively investigate the effects of HGF on E-cadherin adhesion, short-term adhesion to Ecad-comp was measured. Cells were plated for 1.5 h on nontissue culture–treated plastic that was coated with Ecad-comp at two different concentrations and blocked with 1% heat-denatured BSA to prevent nonspecific adhesion. Unbound cells were washed, and bound cells were quantified by acid phosphatase activity. Surprisingly, the presence of HGF increased rather than decreased MDCK adhesion to this substrate. Both the E-cadherin–blocking antibody DECMA-1 and the HAV peptide, which interferes in E-cadherin homotypic interaction, abolished adhesion, demonstrating that adhesion was E-cadherin specific. Together, these experiments show that the ability of E-cadherin to engage in homotypic adhesion is not inhibited, and may even be activated, by HGF.
Cell–cell junctions are pulled apart during scattering
To examine the dynamic behavior of junctional E-cadherin during scattering, MDCK cells stably expressing GFP-E-cadherin were examined by time-lapse fluorescence imaging. GFP-E-cadherin showed localization similar to endogenous E-cadherin at all times during scattering, and expression of GFP-E-cadherin did not inhibit scattering or cell migration (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200506152/DC1), thus confirming the observations by Iino et al. (2001) that GFP-E-cadherin behaves essentially like the endogenous protein. We observed that E-cadherin junctions were dynamic, undergoing continual rearrangement even in the absence of HGF (Fig. 2 A and Video 2). Soon after the addition of HGF, before junctions were disrupted, E-cadherin rearranged into linear structures perpendicular to cell edges. These structures broke down and fluorescence intensity abruptly dropped only as cells pulled apart, with no apparent drop in fluorescence intensity before that point. Time-lapse imaging of a GFP fusion of zona-occludens-1 (ZO-1), to label tight junctions, revealed similar behavior in which linear streaks of fluorescence aligned perpendicular to the cell edge and remained present until the cells pulled apart (Fig. 2 B and Video 3). The linear structures and the mechanism of junction breakdown suggest that increased centripetal tension, perpendicular to the junctions, is pulling the cells apart. Furthermore, as migrating cells contacted one another, new junctions formed at the sites of contact, with GFP-E-cadherin fluorescence immediately accumulating at these sites, as was described for newly forming contacts in the absence of HGF (Adams et al., 1998). This result again indicates that the ability of E-cadherin to form homotypic interactions is not impaired by HGF.
Figure 2.
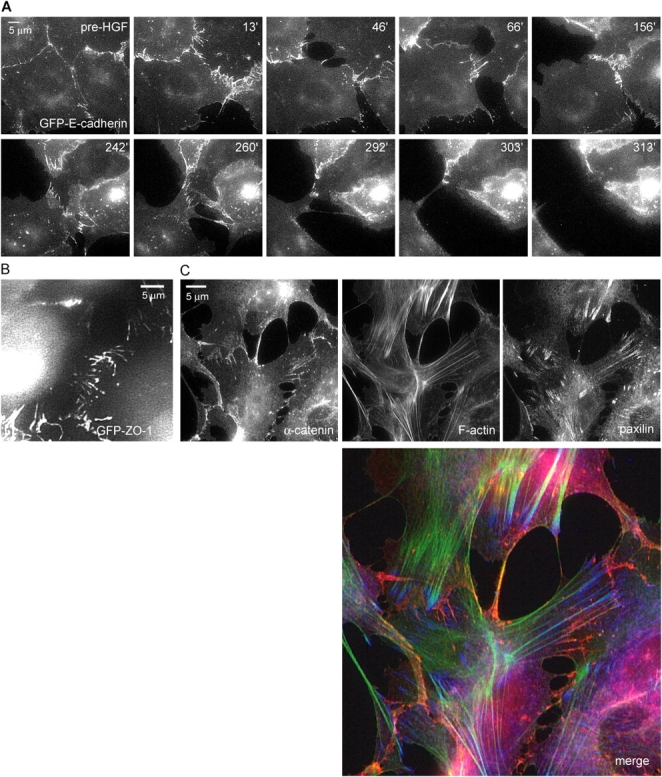
Cell–cell adhesions are pulled apart during scattering. (A) Visualizing GFP-cadherin during scattering. MDCK cells stably expressing GFP-E-cadherin on 3 μg/ml Cn were imaged by time-lapse epifluorescence microscopy. HGF was added after 2 h and imaging continued. Images are from time-lapse Video 2. Images show dynamic adherens junctions that reorganized into radial streaks before breakage. (B) Scattering MDCK cells expressing GFP–ZO-1 were observed in the same manner as in A (Video 3). Cells just before the moment of cell–cell disruption are shown, demonstrating similar radial streaks as seen in GFP-E-cadherin. (C) Cells treated with HGF for 5 h were fixed and stained for α-catenin (red), paxillin (blue), and F-actin (green), to simultaneously visualize adherens junction, focal adhesions, and the actin cytoskeleton. In cells with disrupting adherens junctions, F-actin bundles terminate in focal adhesions just adjacent to cell–cell junctions.
To determine if the cytoskeleton was organized in a way that could promote centripetal tension on junctions, cells at an early stage of scattering were fixed and triple-labeled to localize F-actin, α-catenin (as a marker for adherens junctions), and the focal adhesion protein paxillin. Bundles of actin were clearly arranged perpendicular to the cell–cell junctions (Fig. 2 C), similar to the streaks of GFP junctional markers described in Fig. 2 (A and B). Surprisingly, the thick F-actin bundles that appeared to reach areas of cell–cell adhesion all terminated in the paxillin-rich focal adhesions near the adherens junctions. α-Catenin staining revealed that only thin actin bundles reached the remaining areas of cell–cell adhesion. Together, these results suggest that during scattering cells pull apart from one another through a joint action of the actin cytoskeleton and focal adhesions. Exactly how tension is transmitted physically to the cell–cell junctions remains unclear.
Scattering depends on the type and concentration of ECM
Previous studies have suggested that scattering is dependent on the type of ECM provided or the specific integrins engaged (Clark, 1994; Sander et al., 1998; Gimond et al., 1999). To further investigate these effects, we first systematically assayed the influence of ECM on scattering in our model system. For that purpose, we developed a multiwell plate live-cell microscopy assay on an automated digital microscope system with a robotic stage. MDCK cells were plated in 48-well plates in which the wells were coated with a range of concentrations of different ECM proteins (type 1 Cn, Fn, and Ln1). HGF-induced scattering was then followed by time-lapse phase-contrast imaging for all conditions simultaneously (Fig. 3 A and Video 4, available at http://www.jcb.org/cgi/content/full/jcb.200506152/DC1). For quantification, scattering initiation was scored as the percentage of cell islands (groups of 5–16 cells that form when epithelial cells are cultured subconfluently) in which three or more cells simultaneously had disrupted contacts with neighboring cells. Sample time courses for the evolution of scattering on different ECM proteins are shown in Fig. 3 B (bottom). To compare the effects of the three different matrix proteins over a range of concentrations, we determined the time at which 50% of the islands had initiated scattering (t 1/2 of scattering). Increasing concentration of any type of ECM caused faster scattering (lower t 1/2) to a saturation point (Fig. 3 B, top). The minimal t 1/2 of scattering depended on the type of ECM, with cells scattering fastest on Cn, followed closely by Fn, and scattering slowest on Ln1 (Fig. 3 B, top). In addition to the delay in achieving half-scattering, the maximal extent of scattering was also decreased on Ln1 (Fig. 3 A and Video 4). When the number of junctions with neighboring cells was scored after 14 h in the presence of HGF (the time of maximal scattering), cells on Ln1 had significantly more cell contacts (i.e., less scattering) than cells on either Fn or Cn (Fig. 3 C). Thus, as previously observed (Clark, 1994; Sander et al., 1998), scattering is induced by different ECM proteins to different extents. Importantly, we find that in all cases scattering is promoted by increasing concentrations of ECM.
Figure 3.

Scattering is promoted by increasing ECM concentration and is more efficient on Cn and Fn than on Ln 1. MDCK cells plated on the indicated ECM proteins were imaged by time-lapse phase-contrast microscopy. HGF was added after 2 h and imaging continued for 16 h. (A) Representative images from the time-lapse series (Video 4) showing the differences between scattering on the three different ECM proteins at saturating concentrations (3 μg/ml type I Cn, 10 μg/ml Fn, 10 μg/ml Ln1). By 6 h, cells have begun to scatter on Cn and Fn, but have not initiated on Ln1. (B) For all matrices, t 1/2 of scattering is reached faster on increasing matrix concentration and is saturable. Quantification of scattering from three time-lapses per condition from one representative experiment is shown. The top graph shows the time at which 50% of the islands initiated scattering (as measured by the disruption of at least three cell–cell junctions per island). The bottom graph depicts the progression of scattering at saturating ECM concentrations. Scattering progresses with similar kinetics on Fn and Cn, and much more slowly on Ln1. (C) Cells on Ln 1 do not scatter as completely as cells on other matrices. The extent of scattering, quantified as the average number of cell–cell contacts per cell at 14 h of HGF on the indicated matrix (when scattering was complete) or in the absence of HGF on 3 μg/ml Cn. Data are means ± SEM, and at least 250 cells were counted per condition. (D) Scattering is not specific to β1 integrins. Cells were induced to scatter as in A on 3 μg/ml Cn or 3 μg/ml Vn in the absence or presence of the β1 integrin–blocking antibody AIIB2 (10 μg/ml) and followed by time-lapse imaging. Three time-lapse series with identical results were obtained (Video 5) and representative images at 12 h after HGF are shown (right). As a control, the effect of AIIB2 on the adhesion to Cn and Vn was measured using an adhesion assay (left), showing that β1 integrins are not involved in the adhesion to Vn. Data are means ± SD; n = 3.
Previous studies showed that overexpression of β1 integrins in certain cell types is sufficient for scattering (Gimond et al., 1999). To test whether scattering is strictly dependent on β1 adhesion in MDCK cells or can also be mediated by other integrins, we used a blocking antibody to β1 integrins (10 μg/ml AIIB2) and studied its effect on adhesion and scattering on Cn (3 μg/ml), a known β1 ligand, and on Vn (3 μg/ml), which mainly binds to β3 integrins. As shown (Fig. 3 D and Video 5, available at http://www.jcb.org/cgi/content/full/jcb.200506152/DC1), adhesion and scattering is blocked by AIIB2 on Cn, but not on Vn. This shows that scattering is not specific to β1 integrins.
Scattering correlates with strength of adhesion, not migration velocity
One explanation for the ECM dependence of cell scattering is that stronger substrate adhesion may promote disruption of cell–cell junctions. To investigate the relationship between ECM concentration and cell adhesion, we measured MDCK cell adhesion to the same 48-well plates used in the live-cell scattering assays coated with a range of ECM protein concentrations. 1 h after plating, unbound cells were washed from the wells and bound cells were quantified, as before. As expected, adhesion was dose dependent and saturable for each ECM protein, with Cn inducing the most efficient cell adhesion, followed by Fn and Ln1 (Fig. 4 A). Comparing these adhesion curves with Fig. 3 (A–C) shows that increasing adhesion, as measured in this assay, correlates well with the efficiency of scattering. To determine whether the different scattering efficiencies observed for cells plated on different substrates could be due to integrin-specific effects of HGF on adhesive activity, we compared adhesion in the presence or absence of HGF. Adhesion was increased by HGF to a similar extent on all substrates (Fig. 4 B), showing that, like cadherin-mediated adhesion (Fig. 1 C), HGF promotes adhesion to ECM irrespective of the specific integrins engaged.
Figure 4.

Scattering on increasing ECM correlates with adhesion strength, not migration velocity. (A and B) MDCK cells plated on different amounts of ECM, as in Fig. 3, were allowed to adhere in the presence or absence of HGF for 1 h. Unbound cells were removed and bound cells were quantified. Data are means ± SD; n = 3. Results show that adhesion on all matrices is saturable, is in the order Cn > Fn > Ln1, and is increased by HGF. (C) The average velocity of single cells on saturating concentrations of each ECM was determined from the same data used for Fig. 3 B. For all matrices, HGF induces an increase in cell migration, similar in timing and magnitude. (D) Cell velocity exhibits a classic biphasic response to increasing concentrations of all matrices. The average single cell velocity between 12–16 h after HGF was calculated for increasing ECM concentrations. At this interval, velocity had reached its maximum in all cases. Data are means ± SEM; at least 30 single cells per condition were included in this measurement.
The ECM dependence of scattering efficiency could also be due to the ECM dependence of cell migration speed. To test this hypothesis, we measured migration speed under a variety of ECM conditions. We used custom-written, automated cell-tracking software that segments phase-contrast images, detects cells, determines whether cells are single or in groups, and tracks their trajectories in consecutive time-lapse images (see Materials and methods). Only noncontacted, single cells were considered in the velocity measurements to negate effects of cell–cell adhesion on cell speed. At ECM concentrations corresponding to the fastest scattering half-times, HGF increased the migration speed of single cells by the same extent and with similar timing on Cn, Fn, and Ln1 (Fig. 4 C). To examine the concentration dependence, single cell migration speed was measured from 12 to 16 h after HGF exposure, at which time maximal velocity was reached for all ECM conditions. Unlike the t 1/2 of scattering (Fig. 3 A) and cell adhesion (Fig. 4 A), which both reached plateaus at high concentrations of ECM, migration velocity was biphasic, showing highest velocity at intermediate ECM concentration (Fig. 4 D), similar to published results (Palecek et al., 1997). Surprisingly, Ln1 showed the highest velocity despite being the weakest inducer of scattering. Together, these results show that scattering correlates with cell adhesion and not with cell migration velocity.
Focal adhesions and F-actin organization differ on different ECM proteins
To directly investigate cytoskeletal organization under different ECM conditions, cells were fixed and stained for F-actin and the focal adhesion protein paxillin. On Fn or Cn, which promote efficient adhesion and rapid scattering, large, intensely stained focal adhesions extended to within a few micrometers of the cell edge (Fig. 5). In contrast, on Ln1, where adhesion was less efficient and scattering was slower, the cell periphery had only weak paxillin staining of small, thin, elongated adhesions, whereas more intensely stained, elongated structures were concentrated in the central region of the cells. Accordingly, in cells on Fn or Cn, F-actin bundles connecting to focal adhesions were thick and straight, whereas F-actin bundles in the periphery of cells on Ln1 were very thin, and F-actin structures connecting to the focal adhesions were often not discernible. Focal adhesion size is proportional to tension in the connecting actin bundles, whereas reduced cytoskeletal tension results in small focal adhesions (Schoenwaelder and Burridge, 1999; Ballestrem et al., 2001). Thus, these results indicate that efficient scattering correlates with a distinct cytoskeletal phenotype that suggests high transmission of tension to the cell periphery.
Figure 5.
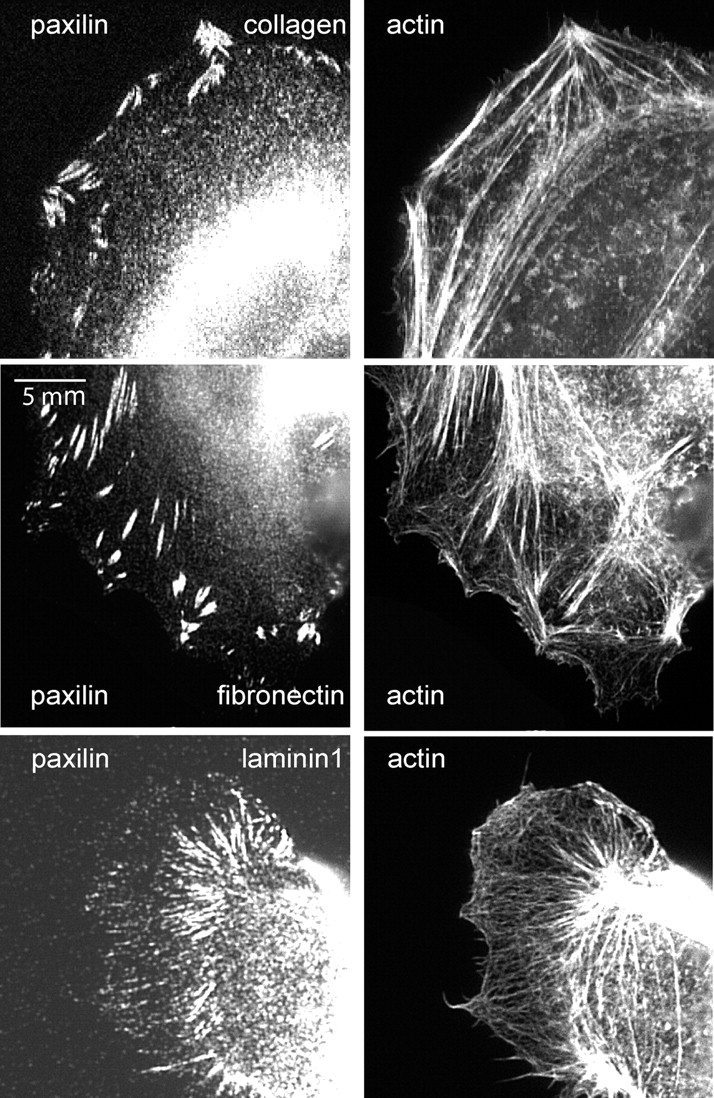
Focal adhesions and F-actin on different types of ECM. Paxillin and F-actin localization in cells grown on 3 μg/ml Cn, 10 μg/ml Fn, or 10 μg/ml Ln1. In cells on Fn and Cn, larger peripheral focal adhesions are associated with dense actin bundles. On Ln1, peripheral focal adhesions and actin bundles are nearly absent, although elongated central adhesions are associated with actin bundles.
Myosin II activation during scattering is influenced by the ECM
To further investigate whether the differential scattering of MDCK cells on ECM correlates with cytoskeletal contraction, we investigated the phosphorylation of the myosin II regulatory light chain (MLC), which is the main regulatory event leading to actomyosin contractility (Somlyo and Somlyo, 1994; Bresnick, 1999). Western blotting for MLC phosphorylated at serine 19 (pMLC) showed that increasing concentrations of Cn increased pMLC (Fig. 6 A), which correlates with better scattering on high ECM coating concentrations (Fig. 3 A). Similar results were obtained with Fn and Ln1 (unpublished data). Moreover, at ECM concentrations corresponding to maximal scattering efficiency, pMLC was higher on Cn than on Ln1 (Fig. 6 B), again correlating with scattering efficiency on the two substrate types (Fig. 3 C). The addition of HGF induced a transient increase in total levels of pMLC on all ECM substrates (Fig. 6 C; Fn not depicted). These results suggest that myosin II–dependent contractility correlates with the differences in scattering observed for different ECM conditions.
Figure 6.
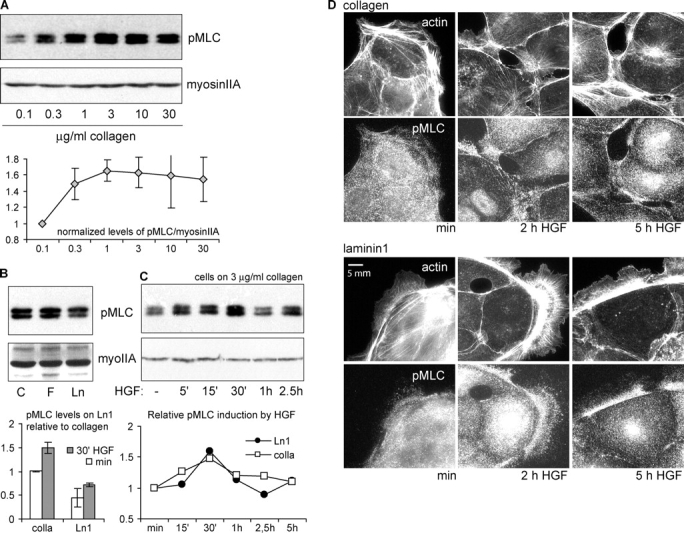
Myosin II regulatory light chain phosphorylation and distribution are regulated by ECM type and concentration. (A) MLC phosphorylation increases with concentration of matrix. Lysates from cells plated on the indicated ECM were analyzed for pMLC by Western blotting. Total myosin IIA heavy chain was analyzed as a loading control. The induction of pMLC by increasing Cn was quantified relative to the lowest concentration used. Data are means ± SD; n = 3. (B) pMLC is greater in cells plated on Cn or Fn than on Ln 1. pMLC content was compared in cells on saturating concentrations of 3 μg/ml Cn, 10 μg/ml Fn, and 10 μg/ml Ln1. Levels of pMLC on Ln1 relative to Cn in the absence and presence of HGF (30 min) were calculated. Data are means ± SD; n = 5. (C) HGF induces a transient increase in pMLC that peaks at 30 min after application. Cells on Cn (3 μg/ml) or Ln1 (10 μg/ml) were stimulated with HGF for the indicated periods of time. Levels of pMLC relative to unstimulated samples on either Cn or Ln1 are shown. (D) HGF induces localization of pMLC to actin bundles near cell–cell junctions on Cn, but not on Ln1. Cells were plated on either 3 μg/ml Cn- or 10 μg/ml Ln1-coated coverslips and stimulated for the indicated times with HGF. Cells were fixed, and actin and pMLC were localized.
Because many of our results pointed to cytoskeletal contraction as an important factor in scattering, it was surprising that HGF induced only a transient increase in pMLC that peaked at 30 min on all ECM substrates in our bulk biochemical assay (Fig. 6 C, bottom; Fn not depicted). After 2–5 h of HGF (the time at which scattering begins), pMLC did not significantly exceed the levels in unstimulated cells. To investigate if myosin II activity was affected by HGF regionally in individual cells, we used the same antibody used in immunoblots for immunolocalization of pMLC in MDCK cells. In cells plated on Cn, pMLC localization dramatically reorganized over time. Before HGF stimulation, pMLC staining was punctate throughout the cytoplasm, with some concentration of puncta along thicker F-actin bundles in the lamella and cell body (Fig. 6 D, top). Starting at 30 min after HGF (unpublished data) and lasting for at least 5 h, pMLC puncta became more heavily concentrated on F-actin bundles that terminated in areas of cell–cell adhesion, consistent with increased tension on cell–cell junctions. In contrast, in cells on Ln1, where scattering efficiency was low, the redistribution of punctate pMLC to areas of cell–cell adhesion was much less pronounced. Thus, the concentration of pMLC in areas of cell–cell adhesion upon HGF stimulation correlates well with the efficiency of scattering on different ECM substrates, suggesting that ECM modulates contractility in the cytoskeletal network locally, near cell–cell junctions.
To directly investigate the effect of modulating contractility on scattering and cell–cell adhesion, we used the pharmacological inhibitor ML-7 to inhibit MLC kinase and blebbistatin to directly inhibit myosin II ATPase. Even in the absence of HGF, however, these inhibitors disrupted cell–cell adhesion (Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200506152/DC1), indicating that myosin activity is necessary for the stability of cell–cell adhesion. Using cytochalasin D to disrupt F-actin resulted in a strong inhibition of cell migration, as well as a loss of cell–cell adhesion (unpublished data), confirming the inhibitory effect on scattering observed in fixed cells (Rosen et al., 1990). These results precluded direct testing of the effects of myosin and F-actin inhibitors on scattering. However, together with the aforementioned results, they suggest that both F-actin and myosin II activity are required for maintaining cell–cell adhesion, whereas proper spatiotemporal regulation of contractile activity is responsible for breaking cell–cell adhesions.
Reducing ECM-generated tension inhibits scattering
To test the hypothesis that the modulation of scattering by ECM type is due to ECM-specific effects on cytoskeletal contraction, we sought to alter mechanotransduction between the cytoskeleton and the substrate without changing the type or concentration of ECM. We therefore used polyacrylamide substrates cross-linked with different amounts of bisacrylamide to vary their stiffness. We developed a modified gel that binds ECM protein due to the incorporation of a positively charged acrylamide monomer, and thus does not require covalent attachment of the ECM protein. Gels of various degrees of stiffness were coated with the same amount of Fn (see Materials and methods). It is well established that decreasing substrate rigidity leads to a decrease in tractions applied to the substrate by the cells (Lo et al., 2000). Cells on Fn-coated flexible substrates were examined by time-lapse phase-contrast imaging during HGF-induced scattering. Quantification revealed that both the time course and extent of scattering were inhibited by decreasing substrate rigidity (Fig. 7, A and B; and Video 6, available at http://www.jcb.org/cgi/content/full/jcb.200506152/DC1). Quantifying single cell migration velocity showed a peak at intermediate rigidity (0.15% bisacrylamide) and a decline at higher rigidity (Fig. 7 C), which is similar to the observations of Peyton and Putnam (2005). Therefore, effects on migration speed cannot account for the differences in scattering. Finally, fluorescent staining of paxillin and F-actin showed that increasing substrate rigidity correlated with increases in the size and peripheral distribution of focal adhesions. Indeed, cells on very compliant Fn-coated substrates displayed a cytoskeletal phenotype similar to that observed for cells plated on Ln1-coated glass (compare Fig. 7 D with Fig. 5). Thus, the ability of cells to develop strong tractions promotes HGF-induced cell scattering. Together with the effects of ECM on cytoskeletal organization and scattering, we conclude that ECM modulates disruption of cadherin-dependent cell–cell adhesions and cell scattering through its effects on cytoskeletal contractility.
Figure 7.
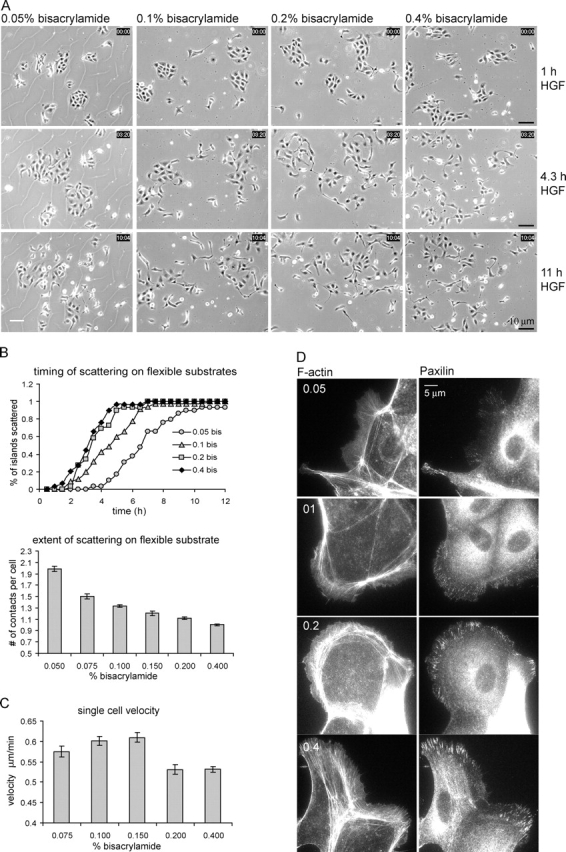
Substrate rigidity regulates scattering. (A–C) Cells were plated for 20 h on Fn-coated acrylamide substrates of increasing rigidity (determined by the percentage of bisacrylamide), stimulated with HGF, and observed by phase-contrast timelapse imaging. (A) Representative pictures of key time points after HGF stimulation show increased scattering on more rigid substrates (B; Video 6). (top) Scattering time course, quantified as in Fig. 3 A. (bottom) Extent of scattering at 10 h HGF, quantified as in Fig. 3 C. Data are means ± SEM. (C) Single cell migration exhibits a biphasic response to substrate stiffness. Velocity was determined at 8–10 h after HGF stimulation, when maximal velocity was reached. Data are means ± SEM. The software was unable to track cells grown at the 0.05% bisacrylamide-containing substrate because of cracks in the substrate (as in A, left) interfering in the segmentation algorithm. (D) Substrate stiffness promotes the formation of larger peripheral adhesions associated with thick actin bundles. Cells on flexible substrates for 16 h were fixed and stained as indicated.
Discussion
The characteristic loss of cell–cell adhesion and the induction of motility upon HGF stimulation have made MDCK cells a well-studied in vitro model for the EMT (Stoker and Perryman, 1985). We report here that HGF does not decrease the ability of E-cadherin to form homotypic adhesions, but instead up-regulates integrin-mediated adhesion, which results in increased myosin-dependent tension at or near cell–cell junctions to induce scattering. These results implicate the actomyosin cytoskeleton as a mediator of cross talk between integrins and cadherins in epithelial cells.
HGF and the down-regulation of cell–cell adhesion
The regulation of E-cadherin–mediated adhesion by HGF has been the subject of much investigation. HGF induces little or no decrease in E-cadherin total protein levels (Weidner et al., 1990), cell surface expression, or the association of E-cadherin with catenin family proteins. In fact, HGF has been found to increase associations between E-cadherin, p120-catenin, and β-catenin (Balkovetz et al., 1997; Balkovetz and Sambandam, 1999). In this study, cell adhesion to Ecad-comp–coated substrates clearly showed no decrease after short- or long-term HGF treatment. This result was corroborated by time-lapse analyses of GFP-E-cadherin and GFP–ZO1 during scattering because their intensity in junctions remained constant and new junctions were able to form when HGF-treated, migrating cells contacted one another. Instead of down-regulation of E-cadherin levels in cell–cell junctions, we found that cell–cell junctions appeared to be physically pulled apart by centripetally oriented actin bundles that terminated in substrate adhesions adjacent to the cell–cell junctions. Although we cannot exclude that cell–cell junctions are weakened by mechanisms beyond our detection in these assays, our data strongly suggest an important role for the tensile forces generated by cytoskeletal contraction against the ECM in the breakdown of cell–cell adhesion.
The role of ECM in the response of MDCK cells to HGF
Our results suggest that the differences in HGF-induced scattering efficiency of MDCK cells on diverse ECM types and concentrations are due primarily to differences in adhesion strength. Although our results cannot exclude contributions from differential integrin signaling, the simplest model that fits the data is that adhesion strength, and hence the distribution of tensile force between the cell–cell and cell–substrate adhesions, is the main mediator of ECM-dependent effects on scattering. Our data exclude that scattering is controlled merely by migratory properties of cells under the different ECM conditions because we found that cell migration velocity is highest on Ln1, which is the poorest substrate for scattering. Furthermore, cell migration exhibits a biphasic response to increasing ECM concentration (Palecek et al., 1997), with fastest migration occurring at intermediate concentrations, whereas scattering is maximal at high ECM concentrations.
Whereas HGF induces scattering on ECM in two-dimensional (2D) cultures, in three-dimensional Cn gels HGF induces tubule formation and branching morphogenesis that is thought to resemble kidney development (Santos et al., 1993). Although cells move extensively, relative to one another, during such tubulogenesis in three-dimensional ECM, they remain adhered to other cells, suggesting that the disruption of cell–cell adhesion by HGF depends on immobilization of the ECM on a 2D substrate. However, our results using 2D ECM substrates of varying rigidities further suggest that the stiffness of the ECM substrate, and not just its dimensionality, is critical in determining whether cells remain adhered to one another.
The ECM controls cytoskeletal forces, which determine tension on cell–cell junctions, to regulate scattering
Our results suggest that organization of contractile forces in the cytoskeleton is critical to scattering. This conclusion is suggested by morphological comparison of cells on substrates that promote HGF-induced scattering to different degrees. Cells on Ln1 (which poorly promotes scattering) had thin actin bundles, suggesting weak contractility, whereas cells on Cn or Fn (which promote efficient scattering) displayed more robust actin bundles, suggesting strong contraction. Similar effects on cytoskeletal phenotype, again correlating with scattering efficiency, were seen on compliant substrates where strong traction cannot develop, versus stiff substrates where traction is high. In addition, MLC phosphorylation, an indicator of myosin II activity (Somlyo and Somlyo, 1994; Bresnick, 1999), was lower in MDCK cells on Ln1, compared with cells on Cn and Fn, and pMLC was augmented by increasing concentrations of any type of ECM. Thus, pMLC correlates well with scattering. Although HGF treatment induced only a transient increase in total pMLC levels, pMLC showed a sustained change in localization to areas near cell–cell adhesions, which was much more apparent in cells on Cn compared with Ln1. Thus, the formation of centripetally oriented actin bundles, as well as MLC phosphorylation and localization to cell–cell junctions, appeared to promote efficient scattering, which is consistent with the conclusion that forces on cell–cell adhesions mediate their breakdown. These results suggest that the spatiotemporal regulation of actomyosin activity, not just the total level, is critical to the regulation of scattering.
Our results suggest a common theme in cell adhesive interactions, in which cytoskeletal organization and its spatiotemporally regulated transmission of tension to transmembrane proteins may regulate the formation and strength of cadherin–catenin complexes much like it is thought to regulate the formation and turnover of integrin-mediated focal adhesions (Chrzanowska-Wodnicka and Burridge, 1996; Ballestrem et al., 2001; Webb et al., 2004). Our finding that myosin inhibition by either ML7 or blebbistatin destabilize cell–cell adhesions suggests that, like integrin-mediated focal adhesions, cadherin-mediated cell–cell adhesions exist within a window of the cellular tension regime. As such, we speculate that a certain amount of tension is required for their formation and maintenance, but too much will pull them from their ligand. This regulation is likely to be complex and involve both mechanically and biochemically mediated feedbacks on ligand affinity. Our results using ECM-coated substrates of differing rigidities further suggest that a “tug-of war” exists between cell–cell and cell–substrate adhesions and that the structure that is able to develop the most tension on its ligand “wins.” Thus, reciprocal modulation of tension applied to cell–cell and cell–ECM adhesions may contribute to the previously described cross talk between these structures (Clark, 1994; Sander et al., 1998; Gimond et al., 1999; Avizienyte et al., 2002; Yano et al., 2004).
Implications
Our results implicate cytoskeletal tension as a regulator of cell–cell adhesions during HGF-induced scattering. It is not known whether cytoskeletal forces also contribute to the mesenchymal phenotype of carcinoma cells in vivo, but indications for such a model clearly exist. Ras-transformed breast epithelial cells display increased pMLC, and pharmacological inhibition of myosin activity restores their E-cadherin–mediated adhesion (Zhong et al., 1997). Ras-transformed MDCK cells have also elevated the activity of Rho, a well-known regulator of contractility (Zondag et al., 2000). Finally, the loss of E-cadherin adhesion in Src-transformed colon carcinoma cells depends on myosin activity as well as integrin-mediated adhesion (Avizienyte et al., 2002, 2004). Both Ras and Src can regulate contractility through the activation of MLC kinase by ERK2 (Klemke et al., 1997; Avizienyte et al., 2004). Additional mechanisms downstream of Ras down-regulate E-cadherin adhesion, including up-regulation of Snail (Peinado et al., 2003) and inhibition of Rho through Rnd3 (Hansen et al., 2000). Thus, several mechanisms may operate in parallel.
In endothelial cells, transient down-regulation of VE-cadherin–mediated cell–cell adhesion is an important event in induction of vascular permeability, transmigration of leukocytes, extravasation of tumor cells, and initiation of neoangiogenesis. Increased contractility of the actin cytoskeleton has been clearly shown to play a crucial role in this process (van Buul and Hordijk, 2004; Stockton et al., 2004; Wittchen et al., 2005), highlighting a striking similarity in the regulation of epithelial cell scattering and endothelial cell–cell adhesion.
Finally, our results implicate the actin cytoskeleton as a mediator of cross talk between integrins and cadherins during epithelial cell scattering. Whether this mechanism of cross talk will apply to other EMT-like cell behaviors remains to be elucidated.
Materials and methods
Cell lines and cell culture
MDCK-II cells were cultured in DME supplemented with 10% FCS, glutamine, and antibiotics. When used for experiments, cells were trypsinized, washed once in DME with 10% FCS, and resuspended at the appropriate concentration in DME containing 0.5% FCS, glutamine, antibiotics, and 10 mM Hepes, pH 7.4. They were plated on glass or plastic that was coated as indicated. For stimulation, recombinant human HGF (Sigma-Aldrich) was used at 5 ng/ml in all experiments. To generate cell lines stably expressing GFP-E-cadherin (A. Kusumi, Nagoya University, Nagoya, Japan) or GFP–ZO-1 (A. Fanning, University of North Carolina, Chapel Hill, NC), transfected cells were selected with Geneticin for 3 wk, after which cells that expressed moderate levels of GFP were isolated by FACS. Western blotting with anti-GFP antibodies (CLONTECH Laboratories, Inc.) revealed expression of full-length GFP constructs without any detectable truncated fusion proteins in the polyclonal cell lines. During experiments the GFP lines were treated exactly the same as the parental MDCK lines and no differences in scattering behavior were observed.
ECM and coating conditions
Cn type 1 from calf skin (Sigma-Aldrich) was stored at 1 mg/ml in 0.1 N HAc and diluted to the appropriate concentration for coating in 0.2 N HAc. Human Ln1 (Sigma-Aldrich) and human Fn (GIBCO BRL) were kept at 1 mg/ml in PBS at −80°C and diluted to the appropriate concentrations for coating in PBS. In all cases, coating was done by incubation for 2 h at 37°C. Unless otherwise indicated, coated surfaces were washed three times with PBS and blocked with 1% heat denatured BSA in PBS for 1 h at 37°C. Ecad-comp was purified as previously described (Pertz et al., 1999). For coating, Ecad-comp was diluted to 15 μg/ml in PBS and incubated for 20 h at 4°C, washed three times in PBS, and blocked for 1 h with 1% heat denatured BSA in PBS at 37°C.
Adhesion assays
To assay cell adhesion, the appropriate ECM or Ecad-comp substrates were prepared as described in 48-well polystyrene plates. MDCK cells were trypsinized, washed once in DMEM containing 10% FCS, and allowed to recover surface proteins for 1 h in suspension in DMEM containing 0.5% FCS, glutamine, antibiotics, and 10 mM Hepes, pH 7.4, at 37°C with constant, gentle shaking. 100,000 cells were plated per well, and adhesion was allowed to proceed for the indicated time at 37°C. Unbound cells were discarded by washing three times with PBS (containing 1 mM calcium in the case of Ecad-comp) preheated to 37°C. Detection of total cellular protein per well was performed by acid phosphatase activity as previously described (Schwartz and Denninghoff, 1994). In brief, cells were lysed in the wells by adding 200 μl of assay buffer containing 0.4% Triton X-100, 50 mM sodium citrate, and 10 mg/ml phosphatase substrate (Sigma-Aldrich). The reaction was incubated for 20 h at 37°C and terminated by addition of 100 μl of 1 N NaOH. Absorbance was measured at 405 nm. Every condition was measured at least in triplicate. DECMA-1 (Sigma-Aldrich) for the inhibition of E-cadherin adhesion was used at 15 μg/ml. HAV peptide (American Peptide Company, Inc.) was used at 10 μg/ml.
Live cell microscopy
Live cell microscopy was performed using an inverted microscope (model Eclipse TE200 and TE300; Nikon) heated with an airstream incubator (Nevtek) to 37°C, as measured inside the imaging chambers. The microscopes were equipped with a robotic stage with linear position feedback encoders on the x, y, and z axes (model MS-2000; Applied Scientific Instruments) to allow image series to be collected at different stage positions over time. Illumination was controlled with an electronic filterwheel/shutter (Sutter Instrument Co.) and a multi-bandpass dichromatic mirror and emission filter (Chroma Technology Corp.). All electronic microscope functions were controlled using MetaMorph software (Universal Imaging Corp.). For imaging of GFP, cells were grown in 35-mm glass-bottom dishes (MatTek Corp.) completely filled with Opti-MEM (GIBCO BRL), containing 0.5% FCS, and were tightly closed using silicon grease and the inverted lid of the dish to prevent evaporation. Fluorescent images were acquired every 2 min using a 60× 1.4 NA Plan APO objective lens and a cooled charge-coupled device (CCD) camera (model ORCA II; Hamamatsu Corporation) operated in the 14-bit mode.
For phase-contrast imaging, cells were grown in nontissue culture–treated polystyrene well plates, completely filled with DMEM containing 0.5% FCS and 10 mM Hepes, pH 7.4, and sealed using silicon grease and a glass plate. Images were acquired every 5 min using a 10× 0.5 NA Plan objective lens and a 0.5 NA ELWD condenser with a 12-bit chilled CCD camera (model Orca 285; Hamamatsu Corporation). Unless otherwise stated, cells were grown for 20 h in the indicated imaging media before mounting on the microscope stage. 5 ng/ml HGF was added to cells on the microscope stage to prevent the loss of the cells of interest. At least three time-lapse series were acquired for each condition in each separate experiment.
Fixation and immunolocalization
Cells were grown and treated as indicated, followed by fixation in 4% PFA for 10 min at RT. Next, cells were permeabilized and blocked in TBS containing 2% BSA and 0.1% Triton X-100 for 30 min at RT. Antibody incubation was done for 1 h in the same buffer at RT, followed by incubation with the appropriate secondary antibody for 30 min at RT. Cells were stained with phalloidin-Alexa488 (Molecular Probes) for 30 min at RT to visualize F-actin. E-cadherin (clone 36), paxillin, and α-catenin antibodies were obtained from BD Biosciences. Phospho–Serine 19 MLC-specific antibody (Sakurada et al., 1998) was a gift from Y. Sasaki (Kitasito University, Tokyo, Japan). Fluorescent images of fixed samples were acquired on an inverted microscope, using a 60× 1.4 Plan APO objective lens and a cooled CCD camera (Hamamatsu Corporation). All electronic microscope functions were controlled and image analysis was performed using MetaMorph software.
Analysis of cell scattering in time-lapse movies
The progression of scattering was quantified as the percentage of cell islands in which three or more cells had disrupted cell–cell contacts at the same time. Only islands that contained 6–15 cells were included. The experiments used for quantification were performed three times independently with comparable results, always testing all different ECM conditions simultaneously. Variations in the absolute onset of scattering between separate experiments due to factors such as temperature fluctuations preclude the meaningful calculation of standard deviations between experiments.
The extent of scattering (Figs. 3 C and 7 B) was quantified by counting all cells from three time-lapse image series taken at similar cell density (between 85–115 cells per image) and counting the number of cells that each of these cells maintained contact with at the time of maximal scattering. The results are depicted as the average number of cell–cell junctions per cell.
To describe cell behavior during scattering, software was developed by A. Kerstens and G. Danuser (The Scripps Research Institute, La Jolla, CA) that automatically segments phase-contrast images and detects cells based on their specific features of phase density. It labels each detected cell and records its coordinates and its status as a single cell or cell that is in contact with neighbors. Using nearest neighbor and gap-closing algorithms, it tracks the nuclei throughout time-lapse image series to determine cell velocity. Here, we used this software to detect and track single cells in the time-lapse images of scattering cells and to determine their velocity throughout the process of scattering. Only cells that were faithfully tracked for at least six consecutive frames and stayed “single” during that period of time were considered. The velocity was calculated as the displacement (μm) over three consecutive frames, divided by the elapsed time (10 min). Taking the displacement over three frames, instead of two, minimized measurement of nuclear displacement due to extending and retracting cellular protrusions that did not result in actual movement of the cell.
Western blotting
For biochemical experiments, cells were lysed in buffer containing 1% SDS and 20 mM Tris, pH 7.4. Protein concentration in the lysates was measured using BCA reagent and 10 μg of protein was loaded for each sample. Loading was controlled by blotting with myosin IIA heavy chain antibody (Biomedical Technologies). pMLC was detected using the phospho-Serine 19 MLC-specific antibody. Blots were developed using HRP-coupled secondary antibodies (The Jackson Laboratory) and ECL (GE Healthcare). For quantification, nonsaturated x-ray films exposed to the blots were scanned and the signal ratio of pMLC to myosin IIA heavy chain was determined and normalized to the signal in the indicated reference lysates.
Flexible substrates
Flexible substrates were prepared as described (Pelham and Wang, 1997; Dembo and Wang, 1999), with a slight modification for ECM binding. In brief, 22-mm-round coverslips were coated with amino-silane, washed extensively with water, and activated by 30-min incubation with 0.5% glutaraldehyde at RT. In the mean time, acrylamide solutions were prepared that contained 10% wt/vol of a mixture containing 70% acrylamide and 30% triethylammonium-acrylamide (which introduced a positive charge to allow the direct binding of proteins; Sigma-Aldrich). The concentration of bisacrylamide was varied to control rigidity. Polymerization was induced with 0.01% ammoniumpersulfate and 0.003% N,N,N′,N′-Tetramethylethylenediamine. After brief washing with water, 12 μl of the acrylamide solution was added to the activated surface and covered with a 15-mm-round coverslip to form a flat round gel with a thickness of ∼70 μm in the middle of the 22-mm coverslip. After polymerization for 30 min, the 15-mm coverslip was removed and the gels were washed in PBS and incubated for 30 min in 50 mM Hepes, pH 7.4. Next, the gels were coated with 15 μg/ml Fn in PBS for 2 h at 37°C and washed in PBS. The gel-coated coverslips were adhered to the bottom of wells in 6-well plates, using silicone grease, and cells were plated 20 h before filming in the same well plates to allow simultaneous examination of scattering on different gels. For staining, the samples were fixed and stained as described in Fixation and immunolocalization.
Online supplemental material
Fig. S1 shows a characterization of the E-cadherin–based adhesions formed on Ecad-comp, revealing the presence of the adherens junction components vinculin and p120-catenin, as well as the absence of the focal adhesion protein paxillin. Fig. S2 compares the localization of endogenous and GFP-labeled E-cadherin after 1 h of HGF and indicates how endogenous E-cadherin becomes localized to the same pulling structures as GFP-E-cadherin during scattering. Furthermore, scattering and migration are shown to be unaffected by the stable expression of GFP-E-cadherin in MDCK cells. Video 1 shows the effect of contraction-inhibiting drugs ML7 (3 μm), Blebbistatin (30 μm), and Y27632 (10 μm) on cell–cell adhesion. Video 2 shows the behavior of GFP-E-cadherin during scattering. Video 3 shows the behavior of EGFP–ZO-1 during scattering. Video 4 compares the scattering of MDCK cells on Cn, Fn, and Ln1. Video 5 compares scattering on Vn and Cn in the presence of a β1 integrin–blocking antibody. Video 6 shows the effect of substrate flexibility on scattering. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200506152/DC1.
Acknowledgments
We would like to thank Olivier Pertz for providing ready-for-use Ecad-comp in large amounts, as well as expert advice; Alan Fanning for the generous sharing of unpublished constructs; Torsten Wittmann for extensive help with image acquisition and processing; Michael Adams for technical assistance with respect to microscopes; and the Waterman laboratory members for helpful discussion. Furthermore, we thank Arnoud Sonnenberg and the Netherlands Cancer Institute for providing the excellent support necessary to successfully finish this study.
This work was supported by grants from the National Institutes of Health to C.M. Waterman-Storer, G. Danuser (GM67230), and M. Schwartz (GM47214). J. de Rooij was supported by a fellowship from the Dutch Cancer Society (KWF Kankerbestrijding).
M.A. Schwartz and C.M. Waterman-Storer contributed equally to this paper.
J. de Rooij's present address is Dept. of Cell Biology, Netherlands Cancer Institute, 1066 CX Amsterdam, Netherlands.
A. Kersten's present address is University of Texas at El Paso, College of Engineering, El Paso, TX 79968.
Abbreviations used in this paper: 2D, two-dimensional; CCD, charge-coupled device; Cn, collagen; EMT, epithelial–mesenchymal transition; Fn, fibronectin; HGF, hepatocyte growth factor; Ln, laminin; MLC, myosin II regulatory light chain; Vn, vitronectin; ZO-1, zona-occludens-1.
References
- Adams, C.L, Y.T. Chen, S.J. Smith, and W.J. Nelson. 1998. Mechanisms of epithelial cell-cell adhesion and cell compaction revealed by high-resolution tracking of E-cadherin-green fluorescent protein. J. Cell Biol. 142:1105–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angres, B., A. Barth, and W.J. Nelson. 1996. Mechanism for transition from initial to stable cell–cell adhesion: kinetic analysis of E-cadherin–mediated adhesion using a quantitative adhesion assay. J. Cell Biol. 134:549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avizienyte, E., A.W. Wyke, R.J. Jones, G.W. McLean, M.A. Westhoff, V.G. Brunton, and M.C. Frame. 2002. Src-induced de-regulation of E-cadherin in colon cancer cells requires integrin signalling. Nat. Cell Biol. 4:632–638. [DOI] [PubMed] [Google Scholar]
- Avizienyte, E., V.J. Fincham, V.G. Brunton, and M.C. Frame. 2004. Src SH3/2 domain-mediated peripheral accumulation of Src and phospho-myosin is linked to deregulation of E-cadherin and the epithelial-mesenchymal transition. Mol. Biol. Cell. 15:2794–2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkovetz, D.F., and V. Sambandam. 1999. Dynamics of E-cadherin and gamma-catenin complexes during dedifferentiation of polarized MDCK cells. Kidney Int. 56:910–921. [DOI] [PubMed] [Google Scholar]
- Balkovetz, D.F., A.L. Pollack, and K.E. Mostov. 1997. Hepatocyte growth factor alters the polarity of Madin-Darby canine kidney cell monolayers. J. Biol. Chem. 272:3471–3477. [DOI] [PubMed] [Google Scholar]
- Ballestrem, C., B. Hinz, B.A. Imhof, and B. Wehrle-Haller. 2001. Marching at the front and dragging behind: differential αVβ3-integrin turnover regulates focal adhesion behavior. J. Cell Biol. 155:1319–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchmeier, C., W. Birchmeier, E. Gherardi, and G.F. Vande Woude. 2003. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 4:915–925. [DOI] [PubMed] [Google Scholar]
- Bresnick, A.R. 1999. Molecular mechanisms of nonmuscle myosin-II regulation. Curr. Opin. Cell Biol. 11:26–33. [DOI] [PubMed] [Google Scholar]
- Chrzanowska-Wodnicka, M., and K. Burridge. 1996. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J. Cell Biol. 133:1403–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, P. 1994. Modulation of scatter factor/hepatocyte growth factor activity by cell-substratum adhesion. J. Cell Sci. 107:1265–1275. [DOI] [PubMed] [Google Scholar]
- Dembo, M., and Y.L. Wang. 1999. Stresses at the cell-to-substrate interface during locomotion of fibroblasts. Biophys. J. 76:2307–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavard, J., M. Lambert, I. Grosheva, V. Marthiens, T. Irinopoulou, J.F. Riou, A. Bershadsky, and R.M. Mege. 2004. Lamellipodium extension and cadherin adhesion: two cell responses to cadherin activation relying on distinct signalling pathways. J. Cell Sci. 117:257–270. [DOI] [PubMed] [Google Scholar]
- Gimond, C., A. Der Flier, S. van Delft, C. Brakebusch, I. Kuikman, J.G. Collard, R. Fassler, and A. Sonnenberg. 1999. Induction of cell scattering by expression of β1 integrins in β1-deficient epithelial cells requires activation of members of the rho family of GTPases and downregulation of cadherin and catenin function. J. Cell Biol. 147:1325–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner, B.M. 2000. Regulation of cadherin adhesive activity. J. Cell Biol. 148:399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, S.H., M.M. Zegers, M. Woodrow, P. Rodriguez-Viciana, P. Chardin, K.E. Mostov, and M. McMahon. 2000. Induced expression of Rnd3 is associated with transformation of polarized epithelial cells by the Raf-MEK-extracellular signal-regulated kinase pathway. Mol. Cell. Biol. 20:9364–9375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iino, R., I. Koyama, and A. Kusumi. 2001. Single molecule imaging of green fluorescent proteins in living cells: E-cadherin forms oligomers on the free cell surface. Biophys. J. 80:2667–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinbara, K., L.E. Goldfinger, M. Hansen, F.L. Chou, and M.H. Ginsberg. 2003. Ras GTPases: integrins' friends or foes? Nat. Rev. Mol. Cell Biol. 4:767–776. [DOI] [PubMed] [Google Scholar]
- Klemke, R.L., S. Cai, A.L. Giannini, P.J. Gallagher, P. de Lanerolle, and D.A. Cheresh. 1997. Regulation of cell motility by mitogen-activated protein kinase. J. Cell Biol. 137:481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobielak, A., and E. Fuchs. 2004. Alpha-catenin: at the junction of intercellular adhesion and actin dynamics. Nat. Rev. Mol. Cell Biol. 5:614–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo, C.M., H.B. Wang, M. Dembo, and Y.L. Wang. 2000. Cell movement is guided by the rigidity of the substrate. Biophys. J. 79:144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden, M., and D.W. Desimone. 2003. Integrin-ECM interactions regulate cadherin-dependent cell adhesion and are required for convergent extension in Xenopus. Curr. Biol. 13:1182–1191. [DOI] [PubMed] [Google Scholar]
- Palecek, S.P., J.C. Loftus, M.H. Ginsberg, D.A. Lauffenburger, and A.F. Horwitz. 1997. Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature. 385:537–540. [DOI] [PubMed] [Google Scholar]
- Peinado, H., M. Quintanilla, and A. Cano. 2003. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. J. Biol. Chem. 278:21113–21123. [DOI] [PubMed] [Google Scholar]
- Pelham, R.J., Jr., and Y. Wang. 1997. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc. Natl. Acad. Sci. USA. 94:13661–13665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertz, O., D. Bozic, A.W. Koch, C. Fauser, A. Brancaccio, and J. Engel. 1999. A new crystal structure, Ca2+ dependence and mutational analysis reveal molecular details of E-cadherin homoassociation. EMBO J. 18:1738–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyton, S.R., and A.J. Putnam. 2005. Extracellular matrix rigidity governs smooth muscle cell motility in a biphasic fashion. J. Cell. Physiol. 204:198–209. [DOI] [PubMed] [Google Scholar]
- Ridley, A.J., M.A. Schwartz, K. Burridge, R.A. Firtel, M.H. Ginsberg, G. Borisy, J.T. Parsons, and A.R. Horwitz. 2003. Cell migration: integrating signals from front to back. Science. 302:1704–1709. [DOI] [PubMed] [Google Scholar]
- Rosen, E.M., L. Meromsky, I. Goldberg, M. Bhargava, and E. Setter. 1990. Studies on the mechanism of scatter factor. Effects of agents that modulate intracellular signal transduction, macromolecule synthesis and cytoskeleton assembly. J. Cell Sci. 96:639–649. [DOI] [PubMed] [Google Scholar]
- Sakai, T., M. Larsen, and K.M. Yamada. 2003. Fibronectin requirement in branching morphogenesis. Nature. 423:876–881. [DOI] [PubMed] [Google Scholar]
- Sakurada, K., M. Seto, and Y. Sasaki. 1998. Dynamics of myosin light chain phosphorylation at Ser19 and Thr18/Ser19 in smooth muscle cells in culture. Am. J. Physiol. 274:C1563–C1572. [DOI] [PubMed] [Google Scholar]
- Sander, E.E., S. van Delft, J.P. ten Klooster, T. Reid, R.A. van der Kammen, F. Michiels, and J.G. Collard. 1998. Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell–cell adhesion or cell migration and is regulated by phosphatidylinositol 3-kinase. J. Cell Biol. 143:1385–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos, O.F., L.A. Moura, E.M. Rosen, and S.K. Nigam. 1993. Modulation of HGF-induced tubulogenesis and branching by multiple phosphorylation mechanisms. Dev. Biol. 159:535–548. [DOI] [PubMed] [Google Scholar]
- Schoenwaelder, S.M., and K. Burridge. 1999. Bidirectional signaling between the cytoskeleton and integrins. Curr. Opin. Cell Biol. 11:274–286. [DOI] [PubMed] [Google Scholar]
- Schwartz, M.A., and K. Denninghoff. 1994. Alpha v integrins mediate the rise in intracellular calcium in endothelial cells on fibronectin even though they play a minor role in adhesion. J. Biol. Chem. 269:11133–11137. [PubMed] [Google Scholar]
- Sheetz, M.P., D.P. Felsenfeld, and C.G. Galbraith. 1998. Cell migration: regulation of force on extracellular-matrix-integrin complexes. Trends Cell Biol. 8:51–54. [DOI] [PubMed] [Google Scholar]
- Somlyo, A.P., and A.V. Somlyo. 1994. Signal transduction and regulation in smooth muscle. Nature. 372:231–236. [DOI] [PubMed] [Google Scholar]
- Stockton, R.A., E. Schaefer, and M.A. Schwartz. 2004. p21-activated kinase regulates endothelial permeability through modulation of contractility. J. Biol. Chem. 279:46621–46630. [DOI] [PubMed] [Google Scholar]
- Stoker, M., and M. Perryman. 1985. An epithelial scatter factor released by embryo fibroblasts. J. Cell Sci. 77:209–223. [DOI] [PubMed] [Google Scholar]
- Thiery, J.P. 2002. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer. 2:442–454. [DOI] [PubMed] [Google Scholar]
- van Buul, J.D., and P.L. Hordijk. 2004. Signaling in leukocyte transendothelial migration. Arterioscler. Thromb. Vasc. Biol. 24:824–833. [DOI] [PubMed] [Google Scholar]
- Webb, D.J., H. Zhang, and A.F. Horwitz. 2004. Cell migration: an overview. Methods Mol. Biol. 294:3–12. [PubMed] [Google Scholar]
- Weidner, K.M., J. Behrens, J. Vandekerckhove, and W. Birchmeier. 1990. Scatter factor: molecular characteristics and effect on the invasiveness of epithelial cells. J. Cell Biol. 111:2097–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittchen, E.S., J.D. van Buul, K. Burridge, and R.A. Worthylake. 2005. Trading spaces: Rap, Rac, and Rho as architects of transendothelial migration. Curr. Opin. Hematol. 12:14–21. [DOI] [PubMed] [Google Scholar]
- Yano, H., Y. Mazaki, K. Kurokawa, S.K. Hanks, M. Matsuda, and H. Sabe. 2004. Roles played by a subset of integrin signaling molecules in cadherin-based cell–cell adhesion. J. Cell Biol. 166:283–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap, A.S., C.M. Niessen, and B.M. Gumbiner. 1998. The juxtamembrane region of the cadherin cytoplasmic tail supports lateral clustering, adhesive strengthening, and interaction with p120ctn. J. Cell Biol. 141:779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamir, E., and B. Geiger. 2001. Molecular complexity and dynamics of cell-matrix adhesions. J. Cell Sci. 114:3583–3590. [DOI] [PubMed] [Google Scholar]
- Zhong, C., M.S. Kinch, and K. Burridge. 1997. Rho-stimulated contractility contributes to the fibroblastic phenotype of Ras-transformed epithelial cells. Mol. Biol. Cell. 8:2329–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zondag, G.C., E.E. Evers, J.P. ten Klooster, L. Janssen, R.A. van der Kammen, and J.G. Collard. 2000. Oncogenic Ras downregulates Rac activity, which leads to increased Rho activity and epithelial–mesenchymal transition. J. Cell Biol. 149:775–782. [DOI] [PMC free article] [PubMed] [Google Scholar]