

Abstract
Cdc25 phosphatases are essential for the activation of mitotic cyclin–Cdks, but the precise roles of the three mammalian isoforms (A, B, and C) are unclear. Using RNA interference to reduce the expression of each Cdc25 isoform in HeLa and HEK293 cells, we observed that Cdc25A and -B are both needed for mitotic entry, whereas Cdc25C alone cannot induce mitosis. We found that the G2 delay caused by small interfering RNA to Cdc25A or -B was accompanied by reduced activities of both cyclin B1–Cdk1 and cyclin A–Cdk2 complexes and a delayed accumulation of cyclin B1 protein. Further, three-dimensional time-lapse microscopy and quantification of Cdk1 phosphorylation versus cyclin B1 levels in individual cells revealed that Cdc25A and -B exert specific functions in the initiation of mitosis: Cdc25A may play a role in chromatin condensation, whereas Cdc25B specifically activates cyclin B1–Cdk1 on centrosomes.
Introduction
The master regulator of mitosis in mammalian cells is the protein complex consisting of cyclin B1 and Cdk1 (Cdc2). Cyclin B1–Cdk1 accumulates in the cytoplasm and on centrosomes in G2 phase and is believed to initiate centrosome separation and mitotic spindle formation. Jackman et al. (2003) made the important observation that the centrosome is the site for the initial activation of cyclin B1–Cdk1. Later in prophase, cyclin B1–Cdk1 translocates to the nucleus (Pines and Hunter, 1991), where a larger pool of activated complexes is detected (De Souza et al., 2000; Jackman et al., 2003).
Cyclin A also plays a role in mitosis. Cyclin A antibody microinjections or small interfering RNA (siRNA) against cyclin A delay cells in G2 phase, whereas injection of active cyclin A–Cdk2 shortens G2 phase, but the exact action of cyclin A in mitotic entry is not clear (Pagano et al., 1992; Furuno et al., 1999; Mitra and Enders, 2004). Cyclin A–Cdk2 has been suggested not only to promote progression into prophase independently of cyclin B1–Cdk1 (Furuno et al., 1999) but also to increase cyclin B1–Cdk1 activity by activating Cdc25B (Mitra and Enders, 2004) or by stabilizing cyclin B (Lukas et al., 1999).
As the cyclin B1–Cdk1 complex accumulates in the S and G2 phases of the cell cycle, Myt1 and Wee1 kinases keep Cdk1 inactive by phosphorylating two residues, Thr-14 and Tyr-15, in Cdk1. A key event in the activation of cyclin B1–Cdk1 is the removal of these two inhibitory phosphates. In Schizosaccharomyces pombe the dual-specificity phosphatase Cdc25 is responsible for the removal of the inhibitory phosphate on Tyr-15, thus promoting cell cycle progression. In mammalian cells, there are three Cdc25 isoforms—Cdc25A, -B, and -C—all of which have been implicated in the regulation of mitosis (Obaya and Sedivy, 2002; Donzelli and Draetta, 2003). A fundamental question, therefore, is which of the Cdc25 isoforms is responsible for the activation of cyclin B1–Cdk1 at the centrosome.
The mammalian Cdc25s share a conserved COOH-terminal catalytic domain (identity 60%), whereas the NH2-terminal parts of the proteins are less similar (identity 20–25%). The noncatalytic part of the phosphatase mediates regulation of activity and intracellular trafficking through the presence of phosphorylation motifs, 14-3-3 binding sites, and nuclear export and import signals. This region is also subject to alternative splicing, leading to different splice versions of all Cdc25 isoforms. For Cdc25A and -B, an additional level of regulation exists through their rapid degradation, keeping interphase protein levels low. Cdc25A is present from late G1 phase and throughout mitosis and Cdc25B appears during S phase and disappears in mitosis, whereas Cdc25C is present throughout the cell cycle (Donzelli and Draetta, 2003; Kristjansdottir and Rudolph, 2004).
Cdc25C was the first mammalian Cdc25 isoform to be studied (Sadhu et al., 1990; Millar et al., 1991; Girard et al., 1992). Microinjection of Cdc25C antibodies or overexpression of dominant-negative Cdc25C cause a block at G2/M, suggesting a role for Cdc25C in the initiation of mitosis (Millar et al., 1991; Lammer et al., 1998). Mammalian Cdc25C is inactive during most of the cell cycle but is activated in mitosis by hyperphosphorylation mediated by cyclin B1–Cdk1, suggesting a positive feedback loop (Hoffmann et al., 1993; Strausfeld et al., 1994).
Cdc25B activity peaks before Cdc25C activity in mitosis, which has led to the suggestion that the autoamplification loop is initiated by Cdc25B (Lammer et al., 1998). As shown for Cdc25C, Cdc25B antibodies or a dominant-negative version of Cdc25B can block cells in G2 (Gabrielli et al., 1996; Lammer et al., 1998). Cdc25B's role in the initiation of mitosis is also suggested by the findings that Cdc25B is more efficient than Cdc25C in inducing premature mitosis in HeLa cells when overexpressed (Karlsson et al., 1999) and that this phenotype is associated with abnormal minispindles (Gabrielli et al., 1996; Karlsson et al., 1999).
Cdc25A was originally thought to be active only at the G1/S transition (Hoffmann et al., 1994; Jinno et al., 1994) but has recently been implicated as a mitotic regulator as well. Cdc25A levels increase through G2 and mitosis (Bernardi et al., 2000; Donzelli et al., 2002; Mailand et al., 2002), and Cdc25A overexpression induces mitotic events (Molinari et al., 2000). Further, the Cdc25A protein is stabilized by phosphorylation in mitosis that is mediated by cyclin B1–Cdk1, and it contributes to the activation of cyclin B1–Cdk1 (Donzelli et al., 2002; Mailand et al., 2002). Down-regulation of Cdc25A by siRNA was found to cause delays at both G1/S and G2/M transitions (Mailand et al., 2002).
The subcellular localizations of Cdc25 isoforms can provide some information on which cyclin–Cdk complex they activate. We and others have shown that both Cdc25A and -B are mainly nuclear, although a fraction of Cdc25B accumulates in the cytoplasm in G2 phase (Hoffmann et al., 1994; Gabrielli et al., 1997b; Lindqvist et al., 2004; Källström et al., 2005). Nuclear export of Cdc25B is needed for it to efficiently induce premature mitosis, suggesting an important role of the cytoplasmic pool of Cdc25B (Lindqvist et al., 2004). A recent study reports the activation of Cdc25B by Aurora A kinase and a centrosomal localization of activated Cdc25B (Dutertre et al., 2004). Cdc25C was initially reported to be nuclear (Millar et al., 1991; Girard et al., 1992), but later studies suggest that Cdc25C is cytoplasmic in interphase and enters the nucleus concomitantly with cyclin B1 in late prophase (Heald et al., 1993; Dalal et al., 1999). However, all three Cdc25 phosphatases could have access to both nuclear cyclin A–Cdk2 as well as centrosomal cyclin B1–Cdk1 because they all shuttle across the nuclear membrane (Karlsson et al., 1999; Graves et al., 2001; Källström et al., 2005).
In this paper we address the question of which Cdc25 is needed for the initiation of mitosis in human cells by systematically targeting Cdc25s alone or in combination using RNA interference (RNAi). We demonstrate that targeting Cdc25A or -B leads to a delay in G2 phase. Cells lacking both Cdc25A and -B are unable to enter mitosis, suggesting that Cdc25C alone cannot catalyze entry into mitosis. Based on analyses of single cells, conducted by both time-lapse microscopy and quantification of immunofluorescence, we conclude that Cdc25A and -B both mediate entry into mitosis but play partly different roles in this process. In particular, we found that Cdc25B is specifically needed for the activation of cyclin B1–Cdk1 at the centrosomes.
Results
siRNA-mediated targeting of Cdc25A or -B delays entry into mitosis
All three human Cdc25 phosphatases are suggested to play a role in mitosis. To evaluate the role of the individual Cdc25 isoforms in mitotic entry, we used RNAi to reduce the levels of Cdc25A, -B, and -C in HeLa and HEK293 cells. The siRNA oligo sequences were designed to target all splice versions of each Cdc25 homologue. Western blots on whole cell lysates of G2 HeLa cells, 40 h after transfection with siRNA, showed that the protein level of each Cdc25 could be specifically and efficiently reduced by the siRNA treatment (Fig. 1 A).
Figure 1.
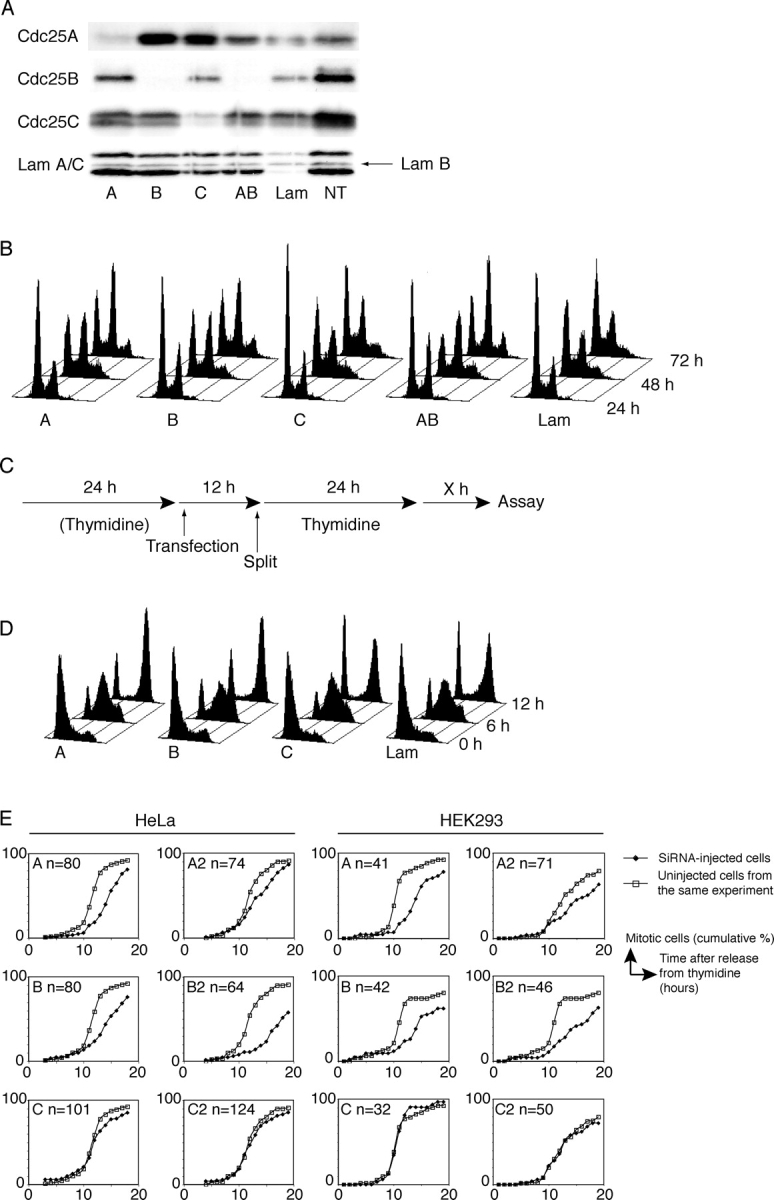
Cells with reduced Cdc25A or -B levels are delayed in G2/M progression. (A) Specific siRNA targeting of Cdc25 isoforms. HeLa cells were transfected with siRNA to Cdc25A (A), -B (B), -C (C), -A and -B (AB), or Lamin A/C (Lam) in the first release of a double thymidine block. 9 h after the second release (40 h after transfection), cells were harvested for Western blot. The antibodies used for immunoblotting are indicated to the left of and transfected siRNAs below the figure. The levels of Lamin B (arrow) indicate equal protein loading (not affected by Lamin A/C silencing). NT, not transfected. (B) FACS profiles of unsynchronized cells 24, 48, and 72 h after transfection of siRNA to Cdc25A (A), -B (B), -C (C), -A and -B (AB), or Lamin A/C (Lam). The x axis is logarithmic. (C) Outline of experimental setup used when transfecting siRNA for collection of synchronized cells at different time points. Cells were subjected to either a single or a double thymidine block. (D) FACS profiles of synchronized cells. HeLa cells were transfected with siRNA to Cdc25A (A), -B (B), -C (C), or Lamin A/C (Lam). Samples were taken for FACS analysis at 0, 6, and 12 h after release from a thymidine block. The x axis is linear. (E) Time-lapse microscopy of synchronized cells. SiRNA-injected HeLa or HEK293 cells were followed with time-lapse microscopy after release from a thymidine block. The timing of mitosis of microinjected cells was compared with the timing of mitosis of uninjected cells in the same dish. The siRNA used for injection and the number of monitored cells is indicated in the graphs. Two different sets of siRNA oligos were used (A, B, and C; A2, B2, and C2). For an example of images from time-lapse microscopy, see Fig. 3 B.
To monitor the effect of Cdc25 siRNA targeting on the cell cycle, we analyzed the DNA content of unsynchronized, siRNA-transfected HeLa cells by FACS at 24, 48, and 72 h after transfection. After 24 h, the DNA profiles looked similar, but after 48 and 72 h, an increased fraction of cells treated with siRNA to Cdc25A or -B showed a 4N content, corresponding to an accumulation in G2 or M phase (Fig. 1 B). Lamin A/C siRNA was used as a control because Lamin A/C silencing does not influence cell cycle progression (Harborth et al., 2001). We did not observe any difference between cells transfected with siRNA to Lamin A/C and Cdc25C. We also analyzed cells that were synchronized in early S phase using thymidine after the siRNA transfection and then harvested for FACS after release from the thymidine block (Fig. 1 C). We found that cells transfected with siRNA to either Cdc25A or -B again accumulated with a 4N DNA content (Fig. 1 D). Synchronized cells transfected with siRNA to Cdc25C behaved as cells transfected with the control Lamin A/C siRNA (Fig. 1 D) or untransfected cells (not depicted). No delay in progression through S phase was observed with any siRNA.
To monitor the timing of mitosis, time-lapse experiments were performed. HeLa and HEK293 cells were microinjected with siRNA, synchronized by a thymidine block, and followed by time-lapse differential interference contrast microscopy. As shown in Fig. 1 E, in both HeLa and HEK293 cells, the accumulation of cells in mitosis was delayed after microinjection of siRNA to Cdc25A or -B, whereas cells microinjected with siRNA to Cdc25C behaved as uninjected cells. In this experiment, two different sets of oligos were used to rule out unspecific off-target effects. As demonstrated in Fig. 1 E, the results from the second set (A2, B2, and C2) were very similar to the results from the first set (A, B, and C). Thus, the time-lapse experiments confirm that RNAi-mediated reduction of Cdc25A or -B levels leads to a delay in the G2/M transition.
Cdc25A and -B cooperate to induce mitosis
SiRNA targeting of Cdc25A or -B caused a delay, but not a block of the cell cycle, which may indicate some redundancy between the isoforms. Therefore, we were interested in knowing the effect of suppressing the expression of more than one Cdc25 in the cell. In an experiment similar to the ones depicted in Fig. 1, we targeted the three Cdc25 homologues in all possible combinations. Combining siRNA to Cdc25C with siRNA to either Cdc25A or -B did not further increase the time of the delay seen when targeting Cdc25A or -B separately (unpublished data). Simultaneous targeting of Cdc25A and -B, however, caused a more prominent G2/M accumulation than targeting Cdc25A and -B individually (unsynchronized cells, Fig. 1 B; synchronized cells, Fig. 2). In fact, very few of the cells cotransfected with siRNA to Cdc25A and -B were able to go through mitosis and enter the next cell cycle during the time course of the experiment (Fig. 2). This suggests that Cdc25A and -B cooperate, and at least partly can compensate for each other, in catalyzing entry into mitosis.
Figure 2.

Combining Cdc25A and -B siRNA leads to a cell cycle block. HeLa cells were transfected with siRNA to Cdc25A (A), -B (B), -C (C), -A and -B (AB), or Lamin A/C (Lam). Samples were harvested for FACS analysis at the indicated time points after release from a thymidine block. The left graph shows the combined fractions of cells in G1 and S phases, and the right graph shows cells in G2 or M phases.
Cdc25A and -B are needed before prophase
To better define the cell cycle delay caused by Cdc25A or -B siRNA, we harvested synchronized siRNA-transfected cells for Western blot analysis with an experimental setup as depicted in Fig. 1 C. We chose to monitor cyclin A levels because cyclin A is degraded in prometaphase (den Elzen and Pines, 2001; Geley et al., 2001). In untransfected cells or in cells transfected with siRNA-targeting Cdc25C, a reduction of cyclin A levels was observed after ∼11 h (Fig. 3 A). In cells transfected with siRNA to either Cdc25A or -B, cyclin A levels remained high after 12 h, indicating that these cells had not entered prometaphase. In cells transfected with siRNA targeting both Cdc25A and -B, cyclin A levels did not decrease, demonstrating an accumulation of cells arrested before prometaphase. Cyclin B1 degradation starts in metaphase, and a pattern similar to the one described for cyclin A was observed when using cyclin B1 antibodies (unpublished data).
Figure 3.

The delay caused by siRNA to Cdc25A or -B occurs before prophase. (A) Transfection of siRNA to Cdc25A or -B leads to a delay in cyclin A degradation. HeLa cells were transfected and synchronized, and at the indicated time points, samples were taken for immunoblotting using cyclin A antibodies. The time after release from a double thymidine block is indicated below and siRNAs to the left of the figure. NT, not transfected. (B) Cells microinjected with siRNA to Cdc25A or -B are delayed in G2 with uncondensed DNA. Time-lapse images were acquired as in Fig. 1 E of synchronized, siRNA-injected HeLa cells. Cells on a single glass-bottomed dish were microinjected with or without Cdc25B siRNA together with a pYFP-histone H2B plasmid. The first and last images show YFP-histone H2B fluorescence at the start and at the end of the experiment. The middle images were acquired with differential interference contrast. Bar, 10 μm.
These results are in agreement with observations from the time-lapse experiments on living siRNA-treated cells described in Fig. 1 E. As shown for cells treated with siRNA to Cdc25B, these delayed cells have not entered prophase because the cells remain flat and the DNA is not condensed (Fig. 3 B). Together, these experiments demonstrate that the activities of Cdc25A and -B are required before prophase.
Cyclin accumulation is regulated by Cdc25A and -B
It was previously reported that suppression of Cdc25A protein expression leads to a reduction of cyclin A levels (Turowski et al., 2003), which caused us to more carefully monitor by immunofluorescence the accumulation of cyclin A and B1 in siRNA-treated cells. We microinjected cells with siRNA together with a plasmid to mark microinjected cells. The cells were then synchronized, fixed, and stained with antibodies to cyclin A and B1, as well as with Cdc25B antibodies, to monitor the efficiency of the RNAi treatment. The protocol for acquiring comparable images for quantification is described in Materials and methods. The fluorescence of between 46 and 77 cells was measured for each siRNA that was microinjected. Fig. 4 B shows the average fluorescence of Cdc25B, cyclin A, and cyclin B1 in siRNA-treated cells. We noticed that cyclin B1 levels were reduced in cells treated with either Cdc25A or -B siRNA, but not in cells treated with siRNA to Cdc25C or CD46 (control) or in untreated cells (Fig. 4 B). Also, average cyclin A levels were slightly lower in cells treated with Cdc25A siRNA. As expected, Cdc25B levels were greatly reduced in cells microinjected with siRNA to Cdc25B.
Figure 4.

Cyclin accumulation is delayed in cells injected with siRNA to Cdc25A or -B. HeLa cells were microinjected with pCFP-Golgi as a marker for injected cells, together with siRNA to Cdc25A, -B, or -C, and fixed 8 h after release from a thymidine block. Cells were stained with antibodies to cyclin A, cyclin B1, and Cdc25B and the fluorescence was quantified as described in Materials and methods. (A) Example of images (maximum intensity projections) of cells injected with siRNA to Cdc25B. Microinjected cells (which express CFP-Golgi) are indicated by arrows. Bar, 10 μm. (B) Average fluorescence levels of Cdc25B (left), cyclin A (middle), and cyclin B1 (right) in siRNA-treated cells. The horizontal lines show the average, whereas the vertical lines visualize the quartiles of the quantified fluorescence. (C) Cyclin A and B1 fluorescence in single cells. Nuclear cyclin A levels (x axis) plotted against cytoplasmic cyclin B1 levels (y axis) for individual cells. In each graph the injected siRNA and number of quantified cells are shown. To facilitate the comparison, the area that includes all uninjected cells is marked in red and transferred to all graphs. As shown, a subset of cells injected with siRNA to Cdc25B express very little cyclin B1 but contain high cyclin A levels. NI, not injected.
To compare cyclin A and B1 levels in individual cells, we plotted the cyclin A signal against the cyclin B1 signal for each cell. A clear correlation between cyclin A and B1 levels was observed (Fig. 4 C). At a certain cellular cyclin A level, cyclin B1 staining starts to appear. After this threshold, the cyclin A and B1 levels display an approximate linear correlation. Notably, however, in cells injected with Cdc25B siRNA, a subpopulation of cells displayed high cyclin A levels with very low cyclin B1 levels. The high cyclin A levels in these cells suggest that the lack of cyclin B1 accumulation is not a secondary effect of a G2 delay/arrest. Instead, it points to Cdc25B activity being specifically needed for cyclin B1 accumulation. Microinjecting siRNA to Cdc25A or -C did not affect the correlation between cyclin A and B1 levels (Fig. 4 C). The subcellular localization of cyclin A or B1 was not affected by microinjection of any siRNA (unpublished data). In summary, this experiment suggests that Cdc25A and -B activities are needed for the accumulation of cyclin B1 and that Cdc25A is also required for the accumulation of cyclin A.
siRNA to Cdc25A and -B have different effects on chromosome condensation and centrosome separation
The activation of cyclin B1–Cdk1 is initiated at the centrosomes, when centrosome separation starts (Jackman et al., 2003). In HeLa cells, the first visible sign of chromosome condensation occurs when the centrosomes have separated. Immediately after chromosome condensation becomes visible, cyclin B1–Cdk1 translocates to the nucleus, where it induces breakdown of nuclear lamins and enhances chromosome condensation. Because we showed in Figs. 1 and 3 that Cdc25A and -B both have a role in the beginning of mitosis, we wished to investigate their involvement in early events, such as centrosome separation and chromosome condensation.
We microinjected siRNA together with plasmids coding for YFP-histone H2B and Discosoma red fluorescent protein (dsRED)-γ-tubulin in synchronized HeLa cells to be able to follow DNA condensation and centrosome separation with three-dimensional (3D) time-lapse microscopy. Images were collected every 12 min. In almost all cells microinjected with only the marker plasmids, the centrosomes separated at between one and four time points (corresponding to 0–60 min) before chromatin condensation was visible. The majority of cells microinjected with siRNA to Cdc25A or -B behaved in a manner similar to that of control cells. However, in a subset of cells microinjected with siRNA to Cdc25A, the time between centrosome separation and DNA condensation was prolonged. In these cells, chromosomes had not condensed 1 h or more after the start of centrosome separation (Fig. 5, A and B).
Figure 5.
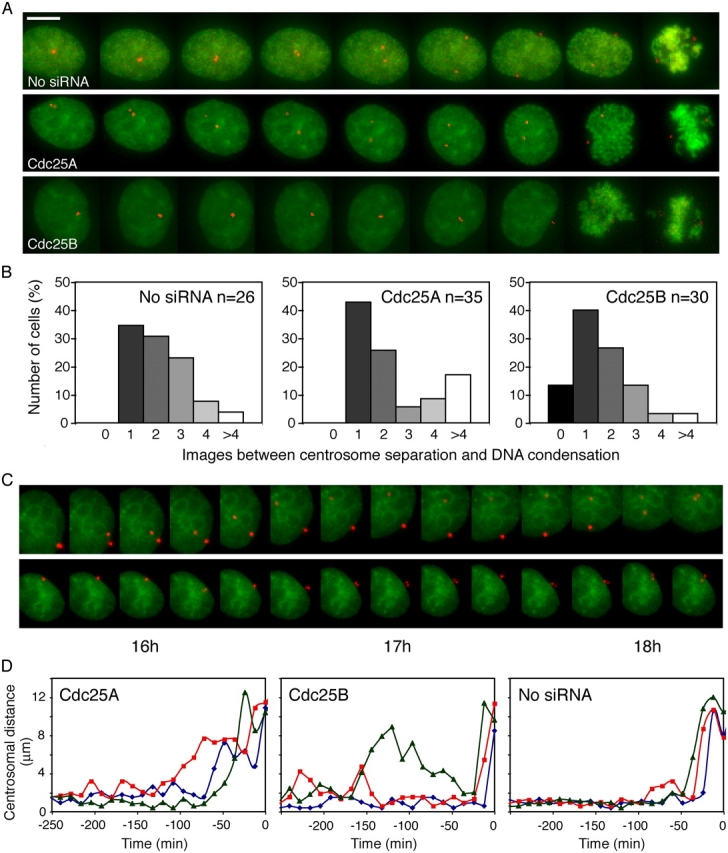
Decreased time between centrosome separation and DNA condensation in a fraction of cells injected with siRNA to Cdc25B. HeLa cells were microinjected with the marker plasmids pYFP-histone H2B and pdsRED-γ-tubulin alone or together with siRNA to Cdc25A or -B. After release from a thymidine block, injected cells were followed by 3D time-lapse microscopy. (A) Examples of the behavior of cells entering mitosis. Images show maximum intensity projections of YFP-histone H2B and dsRED-γ-tubulin fluorescence. The time between images is 12 min. (top) Normal mitotic progression of a cell injected with pYFP-histone H2B and pdsRED-γ-tubulin. (middle) Delay between centrosome separation and chromosome condensation in cell microinjected with siRNA to Cdc25A. (bottom) Less than 12 min between centrosome separation and chromosome condensation in cell injected with siRNA to Cdc25B. Bar, 10 μm. (B) Time between centrosome separation and DNA condensation in a larger number of siRNA-treated cells. The number of images between centrosome separation and DNA condensation is shown below. The distance between images is 12 min. (C) Centrosomes separate but reunite in a subset of cells injected with siRNA to Cdc25B. Example of two cells microinjected with siRNA to Cdc25B that do not enter mitosis in the time frame of the experiment. The time after release from a thymidine block is indicated below the figure. (D) Quantification of centrosome distances before entry into mitosis. Each graph presents the behavior of three single cells. The distance between centrosomes was measured in 3D and plotted against time. Time 0 is defined as first time point when DNA condensation is clearly visible.
The opposite was found for cells microinjected with siRNA to Cdc25B. Here, a fraction of cells separated their centrosomes very late in relation to chromatin condensation. As exemplified in Fig. 5 (A and B), in these cells centrosome separation and chromatin condensation became visible in the same image. This was never observed in cells microinjected with only the plasmids (Fig. 5, A and B) or a control siRNA (not depicted).
Some cells injected with siRNA to Cdc25A or -B did not enter mitosis in the time frame of the experiment (20 h after release from thymidine block). In this case, all cells microinjected with siRNA to Cdc25A were delayed before centrosome separation had started. In contrast, some of the cells microinjected with siRNA to Cdc25B separated the centrosomes and then reunited them (Fig. 5 C). This behavior could also be observed in cells that divided during the experiment. To exclude the possibility that centrosomes on opposite sides of the nucleus were perceived as unseparated, we measured the distance between centrosomes in 3D (Fig. 5 D). The centrosome separation seen here is clearly distinguished from normal centrosome movement both in duration of the separation and in distance between the centrosomes (Fig. 5, C and D).
These experiments suggest that Cdc25A and -B play different roles in mitotic entry. Cdc25A may take a larger part in regulating chromosome condensation, whereas Cdc25B is needed for centrosome separation.
Cyclin A–Cdk2 and cyclin B1–Cdk1 activities are lower in cells treated with siRNA to Cdc25A and -B
Next, we sought to investigate the molecular mechanism behind the G2 delay caused by siRNA to Cdc25A and -B in our experiments. Cyclin B1–Cdk1 and cyclin A–Cdk2 are the main cyclin–Cdk complexes active at the G2/M transition (Pines and Hunter, 1989, 1990), and both can be dephosphorylated by Cdc25A and -B in vitro, whereas Cdc25C can only efficiently dephosphorylate cyclin B1–Cdk1 (Rudolph et al., 2001; Mailand et al., 2002). We analyzed the phosphorylation shift of Cdk1 by Western blot of synchronized siRNA-transfected cells. A slower migrating form of Cdk1, corresponding to the inactive kinase, appeared at the same time as cyclin B1, and followed the increase in cyclin B1 levels in intensity. Approximately 11 h after release from a thymidine block, the slower migrating form of Cdk1 disappeared in untransfected cells and in cells transfected with siRNA to Cdc25C, demonstrating Cdk1 activation. In contrast, inactive Cdk1 persisted in Cdc25A or -B siRNA-transfected cells (Fig. 6 A), suggesting that both Cdc25A and -B take part in the activation of Cdk1 at the G2/M transition. This is in agreement with previous studies (Gabrielli et al., 1997a; Lammer et al., 1998; Donzelli et al., 2002; Mailand et al., 2002).
Figure 6.

Reduced activities of both cyclin A–Cdk2 and cyclin B1–Cdk1 in lysates of cells transfected with siRNA to Cdc25A or -B. (A) Delayed dephosphorylation of Cdk1 in cells treated with siRNA to Cdc25A or -B. SiRNA-transfected synchronized cells were subjected to Cdk1 immunoblotting. Arrows indicate the faster migrating unphosphorylated Cdk1 (bottom band) and the slower migrating phosphorylated Cdk1 (top band). siRNAs are indicated to the left. A quantification of the ratios of inactive versus active Cdk1 is available in Fig. S1 (available at http://www.jcb.org/cgi/content/full/jcb.200503066/DC1). (B) Reduced activation of cyclin A–Cdk2 in cells treated with siRNA to Cdc25A or -B. Cyclin A was immunoprecipitated from siRNA-transfected cells 9 h after release from thymidine block. The ability of the immunoprecipitate to phosphorylate histone H1 as well as the amount of Cdk2 in the immunoprecipitate was assessed. Bars show average from three independent experiments of normalized ratio between cyclin A–Cdk2 activity and amount of Cdk2.
We also investigated the effect of Cdc25 protein reduction on cyclin A–Cdk2 complexes. Because we were unable to reproducibly detect a phosphorylation shift of Cdk2, we performed histone H1 kinase assays. We synchronized siRNA-transfected HeLa cells in late G2 and immunoprecipitated cyclin–Cdk complexes with cyclin A antibodies. The activity of the complexes was assessed by their ability to phosphorylate recombinant histone H1 and was correlated to the amount of immunoprecipitated Cdk2. The activity of cyclin A–Cdk2 was decreased in cells transfected with Cdc25A and -B siRNA as compared with cells transfected with Cdc25C and Lamin A/C siRNA (Fig. 6 B). Thus, cyclin A–Cdk2 seems to be activated by both Cdc25A and -B at G2/M as previously suggested (Lammer et al., 1998). It should be noted, however, that rather small differences in Cdk2 activity are observed after Cdc25A and -B siRNA treatment. This could be attributable to inhibitory phosphorylation being less crucial for the regulation of Cdk2 than for that of Cdk1, as suggested previously (Chow et al., 2003).
Cdc25B activates cyclin B1–Cdk1 on the centrosome
The two experiments depicted in Fig. 6 indicate that neither cyclin B1–Cdk1 nor cyclin A–Cdk2 is efficiently activated in the absence of Cdc25A or -B. However, these experiments cannot provide information on Cdk activation in different subcellular locations or relate the cyclin–Cdk activation to cyclin levels in individual cells. Therefore, we developed a method by which we could follow the Tyr-15 phosphorylation status of Cdk1 in individual cells through the G2/M transition by immunofluorescence. We microinjected different siRNAs together with a marker plasmid on a single coverslip. After synchronization, cells were fixed and stained with cyclin B1 and Y15P Cdk1 (Cdk1-P) antibodies. The fluorescence signal was quantified in subcellular compartments of individual cells.
To evaluate the Cdk1-P antibody in immunofluorescence, we first stained cells not treated with any siRNA. The Cdk1-P staining appeared in the cytoplasm and on the centrosomes together with cyclin B1 staining in G2 cells but was close to background levels in metaphase cells, showing that the antibody does not recognize unphosphorylated Cdk1 in immunofluorescence (Fig. 7 A and Fig. S2 D, available at http://www.jcb.org/cgi/content/full/jcb.200503066/DC1). We then plotted the quantified Cdk-P fluorescence against the cyclin B1 fluorescence signal. In uninjected cells or in cells treated with a control siRNA, the cytoplasmic Cdk1-P staining increased as cyclin B1 levels increased (Fig. 7 B, left). This indicates that as the cyclin B1–Cdk1 complex accumulates, Cdk1 is inactivated by phosphorylation as expected. Thus, we believe that the Cdk1-P antibody staining correctly represents the pool of inactive cyclin B1–Cdk1 in the cell. When cells microinjected with siRNA to Cdc25A or -B were observed 8 h after release from the thymidine block, the accumulation of Cdk1-P was observed to be similar to that in control cells both in the cytoplasm (Fig. 7 B) and on centrosomes (not depicted). However, the cyclin B1 accumulation was delayed in these cells, confirming our previous finding (Fig. 5 B). Thus, 8 h after release from a thymidine block, the siRNA-injected cells did not differ in Cdk1 Y15 phosphorylation but, rather, in the amount of cyclin B1–Cdk1 complexes. Therefore, we turned our focus to a later time point, when cyclin B1 had accumulated. 12 h after release from a thymidine block, most of the cells injected with siRNA to Cdc25C had entered mitosis, whereas a large portion of cells injected with siRNA to Cdc25A or -B had still not divided (Fig. 7 A and not depicted). We quantified cytoplasmic as well as centrosomal cyclin B1 and Cdk1-P immunofluorescence signals and measured the distance between the centrosomes in the same cell. Centrosomes were considered separated when the distance between them exceeded 2 μm.
Figure 7.

Cdc25B dephosphorylates cyclin B1–Cdk1 on the centrosomes. HeLa cells were microinjected with pCFP-Golgi together with siRNA to Cdc25A, -B, -C, or CD46 after release of a double thymidine block and fixed 8 or 12 h after release. Cells were stained with antibodies to phosphorylated Cdk1 (Y15P Cdk1) and to cyclin B1. The specificity of the Cdk1-P antibody is demonstrated in Fig. S2 (available at http://www.jcb.org/cgi/content/full/jcb.200503066/DC1). (A) Examples of cells microinjected with siRNA to Cdc25B and stained with Y15P Cdk1 12 h after release from thymidine block. The Y15P Cdk1 staining is high on centrosomes in G2 cells but low in metaphase cells. Most surrounding cells have passed through mitosis. Bar, 25 μm. (B) Cytoplasmic cyclin B1 (x axis) and Y15P Cdk1 (y axis) fluorescence 8 h after release from a thymidine block. Each dot corresponds to one cell. The diagonal line indicates the levels of cyclin B1 and Y15P at which the control cells are starting to activate Cdk1 (the Y15P Cdk1 accumulation slows down). The fraction of cells above this line is indicated in percent and in numbers of cells in each graph. Cytoplasmic (C) and centrosomal (D) cyclin B1 (x axis) and Y15P Cdk1 (y axis) fluorescence in G2 cells 12 h after release from a thymidine block. Red dots represent cells with separated centrosomes and blue dots cells where centrosomes are not separated. The diagonal line indicates the levels of cyclin B1 and Y15P above which the majority of control cells contain separated centrosomes. The fraction of cells with unseparated centrosomes above this line is indicated in percent and in numbers of cells in each graph. Note that cells situated above the diagonal line in B have not separated their centrosomes and most probably correspond to cells below the diagonal line in C.
When measuring the cytoplasmic fluorescence, we noticed that cells with unseparated centrosomes injected with siRNA to Cdc25B had slightly higher levels of both cyclin B1 and phosphorylated Cdk1, compared with cells injected with siRNA to Cdc25A or -C (Fig. 7 C). This pattern was even more prominent when we quantified the occurrence of cyclin B1 and phosphorylated Cdk1 on the centrosomes. In cells with separated centrosomes, the centrosomal amount of Cdk1-P versus cyclin B1 is slightly reduced compared with cells with unseparated centrosomes. This most likely reflects the activation of a pool of cyclin B1–Cdk1 in these cells. In uninjected cells and in cells injected with siRNA to Cdc25A or -C, most cells had separated their centrosomes before reaching a threshold level of cyclin B1–Cdk1-P (Fig. 7 D). In cells injected with siRNA to Cdc25B, however, 45% of the cells with unseparated centrosomes had exceeded this threshold level (Fig. 7 D). Thus, these cells accumulate with higher levels of inactive cyclin B1–Cdk1 complex, suggesting that they are impaired in their ability to dephosphorylate cyclin B1–Cdk1 on centrosomes. Our results therefore strongly imply that Cdc25B activates centrosomal cyclin B1–Cdk1.
Discussion
It has been known for almost 20 yr that Cdc25 phosphatases regulate mitotic entry by activation of cyclin-dependent kinases (Russell and Nurse, 1986, 1987). The original discovery of Cdc25 in yeast led to the identification of three mammalian Cdc25 isoforms: A, B, and C. Subsequently, all three isoforms were associated with the regulation of mitosis, but their individual roles have remained obscure. In this paper, we have used RNAi to show that Cdc25A and -B are required for entry into mitosis in two different human cell lines. We also demonstrate that Cdc25B is the Cdc25 isoform responsible for the initial activation of cyclin B1–Cdk1 on centrosomes.
We were somewhat surprised that we did not see a G2 delay after siRNA-mediated targeting of Cdc25C. These results are in contrast to recent ones reporting that interfering with Cdc25C protein expression using antisense oligonucleotides or RNAi caused a G2 delay (Bulavin et al., 2003; van Vugt et al., 2004). The siRNA oligos used here efficiently reduce the levels of Cdc25C expression (Fig. 1 A), and we can also monitor a functional Cdc25C depletion in time-lapse experiments where siRNA to Cdc25C causes a prometaphase delay in HeLa cells (unpublished data). However, with regard to mitotic entry, cells treated with siRNA to Cdc25C behave as control cells as observed in two different human cell lines (HeLa and HEK293). Moreover, transfection of siRNA to Cdc25C together with Cdc25A or -B siRNA did not further increase the G2 delay seen in the FACS analysis after targeting the A or B isoforms alone. Thus, we conclude that Cdc25C is less crucial for entry into mitosis than are Cdc25A and -B.
Experiments on knockout mice have shown that neither Cdc25B nor -C is required for mouse development (Chen et al., 2001; Lincoln et al., 2002). Moreover, the combined disruption of Cdc25B and -C leads to normal mouse development and normal somatic cell cycles in isolated cultures (Ferguson et al., 2005). Thus, in mice, Cdc25A, or possibly other phosphatases, can compensate for the loss of the B and C isoforms. It remains to be understood whether there is a species difference in the requirement for different Cdc25 isoforms, or if there is an adaptation occurring in the developing animal as has been suggested for other knockouts (Sage et al., 2003).
To analyze siRNA-mediated silencing in individual cells, we have set up a novel experimental system, where microinjection of different siRNAs on a single coverslip, in combination with immunostaining, allows us to quantify phosphorylation status and amount of proteins in a comparative manner. We were able to confirm that Cdk1 is phosphorylated and thus inactive on the centrosomes as long as the centrosomes remain unseparated and that centrosome separation correlates with a portion of Cdk1 being dephosphorylated. These findings are consistent with earlier observations (De Souza et al., 2000; Jackman et al., 2003).
We further observe that cells treated with siRNA to Cdc25B accumulate with high levels of inactive cyclin B1–Cdk1 and unseparated centrosomes. This suggests that Cdc25B is involved in activating cyclin B1–Cdk1 on centrosomes. Other data presented here point to the same conclusion: in 3D time-lapse microscopy experiments on living cells, we were able to measure the distance between centrosomes and correlate the process of centrosome separation to that of chromatin condensation. In contrast to the behavior of cells treated with siRNA to Cdc25A or -C, a subset of Cdc25B siRNA-treated cells separated their centrosomes immediately before DNA condensation, suggesting that centrosome separation is delayed in these cells. In some cells microinjected with siRNA to Cdc25B, centrosomes separated and then reunited without any sign of chromosome condensation. Because microinjection of siRNA (as well as transfection) gives a distribution of the efficiency of RNAi, it is possible that these cells had enough Cdc25B activity to initiate centrosomal Cdk1 activation and centrosome separation but not enough to sustain the activity.
Circumstantial evidence previously suggested that Cdc25B is the Cdc25 isoform responsible for centrosomal cyclin B1–Cdk1 activation. Cdc25B overexpression was correlated with prophase spindle formation (Gabrielli et al., 1996), and we recently demonstrated that interfering with Cdc25B nuclear export leads to less efficient induction of mitosis (Lindqvist et al., 2004). Furthermore, we observe a centrosomal localization of a GFP-Cdc25B fusion protein (unpublished data), whereas Dutertre et al. (2004) observed a centrosomal localization of Cdc25B using a phosphoantibody specific to Cdc25B phosphorylated at Ser-353 by Aurora A kinase. Another recent study suggests that the Chk1 kinase contributes to the timing of mitosis by controlling the activity of centrosomal Cdc25B in an as yet undefined manner (Krämer et al., 2004).
In contrast to cells treated with Cdc25B siRNA, a subset of cells microinjected with Cdc25A siRNA displayed a prolonged time between centrosome separation and chromatin condensation. This suggests that Cdc25A is involved in the initiation of chromatin condensation in the nucleus, although it is unclear which cyclin–Cdk is activated by Cdc25A. By immunofluorescence we did not see any change in inactive Cdk1 levels in the nuclei after treatment with siRNA to Cdc25A (unpublished data). It would be interesting to also analyze the phosphorylation status of Cdk2 by immunofluorescence to determine the effect of Cdc25A silencing on the cyclin A–Cdk2 complex.
It should be noted that a partial overlap of Cdc25A and -B functions is likely. We have previously shown that, like cyclin A and B1, both Cdc25A and -B shuttle between the nucleus and the cytoplasm (Karlsson et al., 1999; Jackman et al., 2002; Lindqvist et al., 2004; Källström et al., 2005). Moreover, many Cdc25A or -B siRNA-injected cells, although delayed, entered mitosis without an abnormal duration between centrosome separation and chromatin condensation.
When measuring cyclin levels in individual siRNA-treated cells, we made the unexpected finding that cyclin B1 levels are lower in cells treated with siRNA to Cdc25A or -B. Thus, the G2 delay caused by reduced Cdc25A or -B levels could also be accounted for by a delay in cyclin B1 accumulation. The accumulation of mitotic cyclins is mainly regulated by transcription (Brandeis and Hunt, 1996; Eward et al., 2004). Transcription factors known to stimulate the activity of the cyclin A and B promoters such as nuclear transcription factor Y (NF-Y) and FoxM1 possess Cdk phosphorylation sites (Yun et al., 2003; Major et al., 2004). In S and G2 phases, Cdk2 phosphorylates the NF-Y subunit YA, which mediates DNA binding of NF-Y and enhances cyclin A and B transcription (Yun et al., 2003; Chae et al., 2004). FoxM1 was shown to require phosphorylation of the Cdk site to recruit the cAMP-response element-binding protein and to induce cyclin B1 transcription (Major et al., 2004). Also, Cdc25B overexpression was found to increase FoxM1-dependent transcription (Major et al., 2004). It is therefore very likely that the observed delay in cyclin accumulation observed here is the result of a reduction of transcription-factor phosphorylation and, thus, reduced transcription.
When cells were transfected with siRNA to both Cdc25A and -B, the G2 delay was longer than when siRNA was transfected to each of these isoforms separately. We suggest that Cdc25A and -B cooperate to induce mitosis by having specific functional capacities. In late G2, Cdc25A activates a nuclear cyclin–Cdk, leading to chromatin condensation, whereas Cdc25B activates cyclin B1–Cdk1 on centrosomes.
Materials and methods
Cell culture and siRNA delivery
HeLa and HEK293 cells were grown in DME containing glutamax (GIBCO BRL), supplemented with 10% FBS and antibiotics. Cells were synchronized by a 24-h thymidine (2.5 mM) block or by two successive 24-h thymidine blocks separated by 12 h. For transfection, cells were grown without antibiotics in 6-cm tissue culture plates. For all time series experiments, cells transfected with a given siRNA were pooled and reseeded to reduce errors caused by variation in transfection efficiency. Transfection was performed with Oligofectamine or Lipofectamine 2000 (Invitrogen), resulting in a final siRNA concentration of 10 pmol/cm2. Microinjection of siRNA was performed as described previously (Lindqvist et al., 2004).
siRNA and antibodies
siRNA was purchased from Dharmacon Research, Inc. The oligos denoted A, B, and C were designed to target a 19-mer fragment in the coding sequence of Cdc25A (CCTGACCGTCACTATGGAC), -B (Lindqvist et al., 2004), and -C (CCTGCTCCTGGAGAGAGAC). The targeting sequences for the Lamin A/C oligos and the second set of Cdc25 oligos (A2, B2, and C2) have been published previously (Harborth et al., 2001; van Vugt et al., 2004). CD46 siRNA was provided by A.-B. Jonsson (Uppsala University, Uppsala, Sweden). Mouse anti–cyclin B1 (GNS1), rabbit anti–cyclin A (sc751), rabbit anti-Cdc25C (sc327), and mouse anti-Cdc25A (sc7389) were obtained from Santa Cruz Biotechnology, Inc. Guinea pig anti-Cdc25B (B1:5) is described in Lindqvist et al. (2004). Mouse anti-Cdk1 (C12720) was purchased from Transduction Labs, mouse anti-Cdk2 (AN4.3) from Upstate Biotechnology, and rabbit anti–Y15-phosphorylated Cdk1 (9112) from Cell Signaling Technology. For immunofluorescence, Alexa488 goat anti–rabbit (Invitrogen), Cy3 donkey anti–mouse (Jackson ImmunoResearch Laboratories), and Cy5 donkey anti–guinea pig (Jackson ImmunoResearch Laboratories) were used as secondary antibodies before mounting in vectashield mounting medium (Vector Laboratories).
FACS analysis
Cells were harvested with trypsin, washed twice in ice-cold PBS, and fixed with −20°C 70% ethanol. After incubation with 4 mg/ml of propidium Iodide and 10 mg/ml RNase A, cells were analyzed for DNA content by flow cytometry on a FACSCalibur (Becton Dickinson).
Cyclin–Cdk activity assay
Cyclin A–Cdk2 complexes were immunoprecipitated from synchronized siRNA-transfected cells, using anti–cyclin A antibodies and protein G–conjugated magnetic beads (Dynal). The immunoprecipitate was incubated with histone H1 (Cdk1 kinase assay kit; Upstate Biotechnology) in the presence of p33-labeled γ-ATP for 30 min at 30°C. The Cdk2 protein levels were assessed by Western blot of the entire reaction mix, after which the membrane was transferred to a phosphoimager (Fuji) for quantification of p33 incorporation in histone H1.
Time-lapse microscopy
Cells growing on a glass-bottomed dish (MatTek) were microinjected with 10 nM siRNA and 0.008 μg/μl YFP-histone H2B (provided by J. Pines, Wellcome Trust/Cancer Research UK, Cambridge, UK) and, for the experiment depicted in Fig. 5, with 0.0016 μg/μl dsRED-γ-tubulin (provided by J. Ellenberg, European Molecular Biology Laboratory, Heidelberg, Germany). To reduce experimental variation, different siRNAs were microinjected into different areas of cells growing in the same dish. After a 24-h thymidine block, cells were moved to an imaging system (Deltavision Spectris; Applied Precision) equipped with a heater and CO2 chamber (Solent Scientific). For Figs. 1 and 3, images were acquired on multiple locations every 15 min using a 20× objective (NA 0.7; Olympus). For Fig. 5, 15 z levels with 1-μm spacing were acquired on multiple locations in the dish every 12 min using a 60× objective (NA 1.4; Olympus). The distance between centrosomes was measured using Softworx (Applied Precision). For figures, a maximum intensity projection was made of the z stack.
Quantification of fluorescence
Cells growing on a glass-bottomed dish (MatTek) or a coverslip were microinjected with 10 nM siRNA and 0.008 μg/μl plasmid CFP (pCFP)-Golgi as a marker in the first release of a double thymidine block. As for time-lapse microscopy, different siRNAs were introduced in different areas of the same dish or coverslip. After release from the second thymidine block, cells were fixed in 3% PFA and 2% sucrose in PBS and permeabilized by adding −20°C methanol. After antibody labeling, a 30-image z stack with 0.5-μm spacing was acquired of injected cells using an imaging system (Deltavision Spectris; Applied Precision) equipped with a 60× objective, NA 1.4 (Fig. 4), or a 15-image z stack with 1-μm spacing using a 20× objective, NA 0.7 (Fig. 7). To avoid bleaching of antibody fluorescence, cells were focused using the Hoechst labeling. Quantifications were performed on nondeconvolved images. The images were corrected for variation in illumination as determined by a photosensor (Applied Precision) and corrected for flat-field and lightpath alignment invariance. For Fig. 4, the average intensity of a rectangle with a 15-pixel side was measured in the nucleus (cyclin A and Cdc25B) or in the cytoplasm (cyclin B1). For Fig. 7, the peak intensity of the centrosomes and the average intensity of a circle with a 10-pixel diam in the cytoplasm was measured using Softworx. To exclude the possibility that bleaching affected quantification, we quantified cells neighboring the injected cells and compared the intensities from different areas.
Online supplemental material
Fig. S1 shows quantification of the Cdk1 phosphorylation shift in Fig. 6 A, and Fig. S2 shows characterization of the Cdk1-Y15P antibody. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200503066/DC1.
Acknowledgments
We thank Christer Höög, Mattias Karlén, and Elisabeth Schreuder for valuable comments on the manuscript. We are grateful to Ann-Beth Jonsson, Jonathon Pines, and Jan Ellenberg for providing reagents.
This work was supported by the Swedish Cancer Society, the Swedish Research Council, and the Knut and Alice Wallenberg Foundation.
Abbreviations used in this paper: 3D, three-dimensional; dsRED, Discosoma red fluorescent protein; NF-Y, nuclear transcription factor Y; pCFP, plasmid CFP; RNAi, RNA interference; siRNA, small interfering RNA.
References
- Bernardi, R., D.A. Liebermann, and B. Hoffman. 2000. Cdc25A stability is controlled by the ubiquitin-proteasome pathway during cell cycle progression and terminal differentiation. Oncogene. 19:2447–2454. [DOI] [PubMed] [Google Scholar]
- Brandeis, M., and T. Hunt. 1996. The proteolysis of mitotic cyclins in mammalian cells persists from the end of mitosis until the onset of S phase. EMBO J. 15:5280–5289. [PMC free article] [PubMed] [Google Scholar]
- Bulavin, D.V., Y. Higashimoto, Z.N. Demidenko, S. Meek, P. Graves, C. Phillips, H. Zhao, S.A. Moody, E. Appella, H. Piwnica-Worms, and A.J. Fornace. 2003. Dual phosphorylation controls Cdc25 phosphatases and mitotic entry. Nat. Cell Biol. 5:545–551. [DOI] [PubMed] [Google Scholar]
- Chae, H.D., J. Yun, Y.J. Bang, and D.Y. Shin. 2004. Cdk2-dependent phosphorylation of the NF-Y transcription factor is essential for the expression of the cell cycle-regulatory genes and cell cycle G1/S and G2/M transitions. Oncogene. 23:4084–4088. [DOI] [PubMed] [Google Scholar]
- Chen, M.-S., J. Hurov, L.S. White, T. Woodford-Thomas, and H. Piwnica-Worms. 2001. Absence of apparent phenotype in mice lacking Cdc25C protein phosphatase. Mol. Cell. Biol. 21:3853–3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow, J.P.H., W. Yi Siu, H.T.B. Ho, K.H.T. Ma, C.C. Ho, and R.Y.C. Poon. 2003. Differential contribution of inhibitory phosphorylation of CDC2 and CDK2 for unperturbed cell cycle control and DNA integrity checkpoints. J. Biol. Chem. 278:40815–40828. [DOI] [PubMed] [Google Scholar]
- Dalal, S.N., C.M. Schweitzer, J.M. Gan, and J.A. DeCaprio. 1999. Cytoplasmic localization of human Cdc25C during interphase requires an intact 14-3-3 binding site. Mol. Cell. Biol. 19:4465–4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Souza, C.P.C., K.A.O. Ellem, and B.G. Gabrielli. 2000. Centrosomal and cytoplasmic Cdc2/cyclin B1 activation precedes nuclear mitotic events. Exp. Cell Res. 257:11–21. [DOI] [PubMed] [Google Scholar]
- den Elzen, N., and J. Pines. 2001. Cyclin A is destroyed in prometaphase and can delay chromosome alignment and anaphase. J. Cell Biol. 153:121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donzelli, M., and G.F. Draetta. 2003. Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep. 4:671–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donzelli, M., M. Squatrito, D. Ganoth, A. Hershko, M. Pagano, and G.F. Draetta. 2002. Dual mode of degradation of Cdc25 A phosphatase. EMBO J. 21:4875–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutertre, S., M. Cazales, M. Quaranta, C. Froment, V. Trabut, C. Dozier, G. Mirey, J.P. Bouche, N. Theis-Febvre, E. Schmitt, et al. 2004. Phosphorylation of CDC25B by Aurora-A at the centrosome contributes to the G2-M transition. J. Cell Sci. 117:2523–2531. [DOI] [PubMed] [Google Scholar]
- Eward, K.L., M.N. Van Ert, M. Thornton, and C.E. Helmstetter. 2004. Cyclin mRNA stability does not vary during the cell cycle. Cell Cycle. 3:1057–1061. [PubMed] [Google Scholar]
- Ferguson, A.M., L.S. White, P.J. Donovan, and H. Piwnica-Worms. 2005. Normal cell cycle and checkpoint responses in mice and cells lacking Cdc25B and Cdc25C protein phosphatases. Mol. Cell. Biol. 25:2853–2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuno, N., N. den Elzen, and J. Pines. 1999. Human cyclin A is required for mitosis until mid prophase. J. Cell Biol. 147:295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrielli, B.G., C.P.C. DeSouza, I.D. Tonks, J.M. Clark, N.K. Hayward, and K.A.O. Ellem. 1996. Cytoplasmic accumulation of Cdc25B phosphatase in mitosis triggers centrosomal microtubule nucleation in HeLa cells. J. Cell Sci. 109:1081–1093. [DOI] [PubMed] [Google Scholar]
- Gabrielli, B.G., J.M. Clark, A.K. McCormack, and K.A.O. Ellem. 1997. a. Hyperphosphorylation of the N-terminal domain of Cdc25 regulates activity toward cyclin B1/Cdc2 but not cyclin A/Cdk2. J. Biol. Chem. 272:28607–28614. [DOI] [PubMed] [Google Scholar]
- Gabrielli, B.G., J.M. Clark, A.K. McCormack, and K.A.O. Ellem. 1997. b. Ultraviolet light-induced G2 phase cell cycle checkpoint blocks Cdc25-dependent progression into mitosis. Oncogene. 15:749–758. [DOI] [PubMed] [Google Scholar]
- Geley, S., E. Kramer, C. Gieffers, J. Gannon, J.M. Peters, and T. Hunt. 2001. Anaphase-promoting complex/cyclosome-dependent proteolysis of human cyclin A starts at the beginning of mitosis and is not subject to the spindle assembly checkpoint. J. Cell Biol. 153:137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard, F., U. Strausfeld, J.C. Cavadore, P. Russell, A. Fernandez, and N.J.C. Lamb. 1992. Cdc25 is a nuclear protein expressed constitutively throughout the cell cycle in nontransformed mammalian cells. J. Cell Biol. 118:785–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves, P.R., C.M. Lovly, G.L. Uy, and H. Piwnica-Worms. 2001. Localization of human Cdc25C is regulated both by nuclear export and 14-3-3 protein binding. Oncogene. 20:1839–1851. [DOI] [PubMed] [Google Scholar]
- Harborth, J., S.M. Elbashir, K. Bechert, T. Tuschl, and K. Weber. 2001. Identification of essential genes in cultured mammalian cells using small interfering RNAs. J. Cell Sci. 114:4557–4565. [DOI] [PubMed] [Google Scholar]
- Heald, R., M. McLoughlin, and F. McKeon. 1993. Human Wee-1 maintains mitotic timing by protecting the nucleus from cytoplasmically activated Cdc2 kinase. Cell. 74:463–474. [DOI] [PubMed] [Google Scholar]
- Hoffmann, I., P.R. Clarke, M.J. Marcote, E. Karsenti, and G. Draetta. 1993. Phosphorylation and activation of human Cdc25-C by Cdc2-cyclin B and its involvement in the self-amplification of MPF at mitosis. EMBO J. 12:53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann, I., G. Draetta, and E. Karsenti. 1994. Activation of the phosphatase activity of human Cdc25A by a Cdk2-cyclin E dependent phosphorylation at the G1/S transition. EMBO J. 13:4302–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman, M., Y. Kubota, N. den Elzen, A. Hagting, and J. Pines. 2002. Cyclin A- and cyclin E-Cdk complexes shuttle between the nucleus and the cytoplasm. Mol. Biol. Cell. 13:1030–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman, M., C. Lindon, E.A. Nigg, and J. Pines. 2003. Active cyclin B1-Cdk1 first appears on centrosomes in prophase. Nat. Cell Biol. 5:143–148. [DOI] [PubMed] [Google Scholar]
- Jinno, S., K. Suto, A. Nagata, M. Igarashi, Y. Kanaoka, H. Nojima, and H. Okayama. 1994. Cdc25A is a novel phosphatase functioning early in the cell cycle. EMBO J. 13:1549–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Källström, H., A. Lindqvist, V. Pospisil, A. Lundgren, and C.K. Rosenthal. 2005. Cdc25A localisation and shuttling: characterisation of sequences mediating nuclear export and import. Exp. Cell Res. 303:89–100. [DOI] [PubMed] [Google Scholar]
- Karlsson, C., S. Katich, A. Hagting, I. Hoffmann, and J. Pines. 1999. Cdc25B and Cdc25C differ markedly in their properties as initiators of mitosis. J. Cell Biol. 146:573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer, A., N. Mailand, C. Lukas, R.G. Syljuasen, C.J. Wilkinson, E.A. Nigg, J. Bartek, and J. Lukas. 2004. Centrosome-associated Chk1 prevents premature activation of cyclin-B-Cdk1 kinase. Nat. Cell Biol. 6:884–891. [DOI] [PubMed] [Google Scholar]
- Kristjansdottir, K., and J. Rudolph. 2004. Cdc25 phosphatases and cancer. Chem. Biol. 11:1043–1051. [DOI] [PubMed] [Google Scholar]
- Lammer, C., S. Wagerer, R. Saffrich, D. Mertens, W. Ansorge, and I. Hoffmann. 1998. The Cdc25B phosphatase is essential for the G2/M phase transition in human cells. J. Cell Sci. 111:2445–2453. [DOI] [PubMed] [Google Scholar]
- Lincoln, A.J., D. Wickramasinghe, P. Stein, R.M. Schultz, M.E. Palko, A.P. De Miguel, L. Tessarollo, and P.J. Donovan. 2002. Cdc25b phosphatase is required for resumption of meiosis during oocyte maturation. Nat. Genet. 30:446–449. [DOI] [PubMed] [Google Scholar]
- Lindqvist, A., H. Källström, and C.K. Rosenthal. 2004. Characterisation of Cdc25B localisation and nuclear export during the cell cycle and in response to stress. J. Cell Sci. 117:4979–4990. [DOI] [PubMed] [Google Scholar]
- Lukas, C., C.S. Sorensen, E. Kramer, E. Santoni-Rugiu, C. Lindeneg, J.M. Peters, J. Bartek, and J. Lukas. 1999. Accumulation of cyclin B1 requires E2F and cyclin-A-dependent rearrangement of the anaphase-promoting complex. Nature. 401:815–818. [DOI] [PubMed] [Google Scholar]
- Mailand, N., A.V. Podtelejnikov, A. Groth, M. Mann, J. Bartek, and J. Lukas. 2002. Regulation of G(2)/M events by Cdc25A through phosphorylation-dependent modulation of its stability. EMBO J. 21:5911–5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major, M.L., R. Lepe, and R.H. Costa. 2004. Forkhead box M1B transcriptional activity requires binding of Cdk-cyclin complexes for phosphorylation-dependent recruitment of p300/CBP coactivators. Mol. Cell. Biol. 24:2649–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar, J.B.A., J. Blevitt, L. Gerace, K. Sadhu, C. Featherstone, and P. Russell. 1991. p55cdc25 is a nuclear protein required for the initiation of mitosis in human cells. Proc. Natl. Acad. Sci. USA. 88:10500–10504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra, J., and G.H. Enders. 2004. Cyclin A/Cdk2 complexes regulate activation of Cdk1 and Cdc25 phosphatases in human cells. Oncogene. 23:3361–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinari, M., C. Mercurio, J. Dominguez, F. Goubin, and G.F. Draetta. 2000. Human Cdc25 A inactivation in response to S phase inhibition and its role in preventing premature mitosis. EMBO Rep. 1:71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obaya, A.J., and J.M. Sedivy. 2002. Regulation of cyclin-Cdk activity in mammalian cells. Cell. Mol. Life Sci. 59:126–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano, M., R. Pepperkok, F. Verde, W. Ansorge, and G. Draetta. 1992. Cyclin-A is required at 2 points in the human cell-cycle. EMBO J. 11:961–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines, J., and T. Hunter. 1989. Isolation of a human cyclin cDNA: evidence for cyclin mRNA and protein regulation in the cell cycle and for interaction with p34cdc2. Cell. 58:833–846. [DOI] [PubMed] [Google Scholar]
- Pines, J., and T. Hunter. 1990. Human cyclin A is adenovirus E1A-associated protein p60 and behaves differently from cyclin B. Nature. 346: 760-763. [DOI] [PubMed] [Google Scholar]
- Pines, J., and T. Hunter. 1991. Human cyclin A and B1 are differentially located in the cell and undergo cell cycle–dependent nuclear transport. J. Cell Biol. 115:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph, J., D.M. Epstein, L. Parker, and J. Eckstein. 2001. Specificity of natural and artificial substrates for human Cdc25A. Anal. Biochem. 289:43–51. [DOI] [PubMed] [Google Scholar]
- Russell, P., and P. Nurse. 1986. Cdc25+ functions as an inducer in the mitotic control of fission yeast. Cell. 45:145–153. [DOI] [PubMed] [Google Scholar]
- Russell, P., and P. Nurse. 1987. Negative regulation of mitosis by wee1+, a gene encoding a protein kinase homolog. Cell. 49:559–567. [DOI] [PubMed] [Google Scholar]
- Sadhu, K., S.I. Reed, H. Richardson, and P. Russell. 1990. Human homolog of fission yeast Cdc25 mitotic inducer is predominantly expressed in G2. Proc. Natl. Acad. Sci. USA. 87:5139–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage, J., A.L. Miller, P.A. Perez-Mancera, J.M. Wysocki, and T. Jacks. 2003. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature. 424:223–228. [DOI] [PubMed] [Google Scholar]
- Strausfeld, U., A. Fernandez, J.P. Capony, F. Girard, N. Lautredou, J. Derancourt, J.C. Labbe, and N.J.C. Lamb. 1994. Activation of p34cdc2 protein kinase by microinjection of human Cdc25C into mammalian cells. Requirement for prior phosphorylation of Cdc25C by p34cdc2 on sites phosphorylated at mitosis. J. Biol. Chem. 269:5989–6000. [PubMed] [Google Scholar]
- Turowski, P., C. Franckhauser, M.C. Morris, P. Vaglio, A. Fernandez, and N.J.C. Lamb. 2003. Functional Cdc25C dual-specificity phosphatase is required for S-phase entry in human cells. Mol. Biol. Cell. 14:2984–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vugt, M., A. Bras, and R.H. Medema. 2004. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol. Cell. 15:799–811. [DOI] [PubMed] [Google Scholar]
- Yun, J., H.D. Chae, T.S. Choi, E.H. Kim, Y.J. Bang, J.K. Chung, K.S. Choi, R. Mantovani, and D.Y. Shin. 2003. Cdk2-dependent phosphorylation of the NF-Y transcription factor and its involvement in the p53-p21 signaling pathway. J. Biol. Chem. 278:36966–36972. [DOI] [PubMed] [Google Scholar]