

Abstract
Macroautophagy, which is a lysosomal pathway for the turnover of organelles and long-lived proteins, is a key determinant of cell survival and longevity. In this study, we show that neuronal macroautophagy is induced early in Alzheimer's disease (AD) and before β-amyloid (Aβ) deposits extracellularly in the presenilin (PS) 1/Aβ precursor protein (APP) mouse model of β-amyloidosis. Subsequently, autophagosomes and late autophagic vacuoles (AVs) accumulate markedly in dystrophic dendrites, implying an impaired maturation of AVs to lysosomes. Immunolabeling identifies AVs in the brain as a major reservoir of intracellular Aβ. Purified AVs contain APP and β-cleaved APP and are highly enriched in PS1, nicastrin, and PS-dependent γ-secretase activity. Inducing or inhibiting macroautophagy in neuronal and nonneuronal cells by modulating mammalian target of rapamycin kinase elicits parallel changes in AV proliferation and Aβ production. Our results, therefore, link β-amyloidogenic and cell survival pathways through macroautophagy, which is activated and is abnormal in AD.
Introduction
Macroautophagy is a constitutive mechanism for the turnover of cytoplasmic constituents that is activated under conditions of trophic stress or nutritional deprivation (Mortimore and Schworer, 1977). Its activation reduces the size of cells, thereby decreasing their metabolic burden while generating new substrates for energy and cellular remodeling (Mortimore and Schworer, 1977; Seglen et al., 1986). The better understood form of autophagy, macroautophagy, is initiated when an “isolation” membrane is created under the direction of multiple proteins, including microtubule-associated light chain 3-II (LC3-II), that, together, orchestrate membrane elongation and sequestration of a region of cytoplasm and organelles into a double membrane–limited autophagic vacuole (AV) or autophagosome (Asanuma et al., 2003). This sequestration process is controlled by the mammalian target of rapamycin (mTOR) kinase pathway, which is regulated by insulin via the PI3 kinase–Akt (protein kinase B) pathway and by specific amino acids (e.g., Leu or His) via AMP kinase (Petiot et al., 2000). Autophagosomes mature to single membrane autophagolysosomes (Dunn, 1990b) by fusing with lysosomes or other mature AVs, whereupon they acidify and acquire proteolytic enzymes (Meijer and Codogno, 2004). In addition, endocytosed constituents can also enter the autophagic pathway when late endosomes fuse with autophagosomes to generate an amphisome and may be retained in the cell without complete digestion of the compartment's contents (Gordon and Seglen, 1988).
As a system that is induced by nutritional stress or cell injury, macroautophagy may be activated in pathologic states (Rubinsztein et al., 2005). Macroautophagy may protect cells from apoptosis (Boya et al., 2005), probably by eliminating damaged mitochondria (Brunk and Terman, 2002), and is required for supranormal longevity in Caenorhabditis elegans (Melendez et al., 2003). Moreover, macroautophagy is involved in degrading mutated and aggregated proteins that are implicated in neurodegenerative diseases, including Parkinson's (Cuervo et al., 2004) and Huntington's disease (Ravikumar et al., 2004). Defective removal of these proteins has been linked to disease progression in these disorders, and stimulating macroautophagy in a model of Huntington's disease reduced abnormal protein aggregation and improved neurological function (Ravikumar et al., 2004). Although macroautophagy appears to be neuroprotective in these settings, sustained overactivity during embryonic development (Clarke, 1990), response to ER stress, exposure to toxins (Kanzawa et al., 2004), or dysfunction of the autophagic pathway in pathological states mediate a caspase-independent form of cell death that shares certain features with apoptosis (Bursch, 2001; Gozuacik and Kimchi, 2004). Despite the importance of the macroautophagy pathway in cell survival and its connections to other protein-trafficking pathways that are implicated in Alzheimer's disease (AD; Nixon et al., 2000), the involvement of macroautophagy in AD has received limited attention. Recent immuno-EM (IEM) analyses, however, have shown that autophagosomes and late AVs appear in neurons in the AD brain and accumulate markedly within dystrophic neurites, suggesting a progressive dysfunction of macroautophagy-mediated protein turnover (Nixon et al., 2005).
In AD, extensive neuronal atrophy and loss is preceded by the intraneuronal formation of neurofibrillary tangles, which is composed mainly of aggregated tau protein, and by the extracellular deposition of β-amyloid (Aβ) peptide. Aβ is generated predominantly as a 40–42–amino acid peptide from β- and γ-secretase cleavage of the Aβ precursor protein (APP; Cupers et al., 2001). Both β-site APP cleaving enzyme and γ-secretase, which is a protein complex containing presenilin (PS), nicastrin, PEN2, and Aph-1 (Edbauer et al., 2003), reside in one or more compartments of the central vacuolar apparatus, which includes the ER–Golgi, plasma membrane, endosomes, and lysosomes (Cupers et al., 2001; Pasternak et al., 2003). We recently identified APP, the β-cleaved APP product (β–carboxy-terminal fragment [βCTF]), PS1, and nicastrin in purified AVs from livers of wild-type APP yeast artificial chromosome mice (Yu et al., 2004), raising the possibility that AVs may also be able to generate Aβ.
In this study, we demonstrate that macroautophagy is induced in the brain at early stages of sporadic AD and in an animal model of AD pathology, the PS1/APP transgenic mouse (Holcomb et al., 1998). In the PS1/APP model, macroautophagy induction is evident in vulnerable neuronal populations before the extracellular deposition of Aβ, and, as in AD, AVs of varying types accumulate abnormally within dystrophic neurites, indicating a progressive impairment of AV maturation to lysosomes. We also show that AVs are highly enriched in both the components and enzyme activity of the γ-secretase complex, contain APP and βCTF, and are a major source of intracellular Aβ in the AD brain. Finally, we demonstrate in both neuronal and nonneuronal cell lines that macroautophagy is a previously unrecognized pathway for Aβ generation under conditions in which AVs accumulate. The early and persistent induction of macroautophagy and the later pathological accumulation of AVs in AD and related mouse models, therefore, potentially link β-amyloidogenic and neurodegenerative mechanisms in AD.
Results
Macroautophagic induction and pathological AV accumulation in AD and PS1/APP mouse brain
AVs are rare in neurons of the normal adult brain (Nixon et al., 2005). In AD, however, AVs appear in neocortical and hippocampal pyramidal neurons and accumulate markedly within the dendritic arbors of these affected cells (Nixon et al., 2005). We observed similar pathological AV accumulation in PS1/APP animals, which is a mouse model of AD that expresses human mutant PS1 and the Swedish variant of Aβ (Duff et al., 1996). PS1/APP mice begin to deposit Aβ after 10 wk of age and progressively develop many neuritic plaques that mainly consist of Aβ and grossly swollen dendrites and axons (Holcomb et al., 1998). AVs were rarely found in the neuropil of nontransgenic (NTg) mice (Fig. 1 A). In contrast, the numbers of AVs were at least 23-fold higher in the neurons of 9-mo-old PS1/APP mice (13.17 ± 1.91 SEM) compared with age-matched NTg controls (0.57 ± 0.08 SEM) based on ultrastructural morphometric analyses of AV numbers in a series of 100 EM images. AV pathology in 9-mo-old mice (Fig. 1, B–D), like that in AD brains (Fig. 1 B, inset), ranged from small numbers of AVs in relatively normal-appearing dendrites (Fig. 1 D) to striking pathologic accumulations of AVs within dystrophic neurites, where AVs were usually the predominant organelles (Fig. 1, B and C). A significant proportion of AVs met the morphologic criteria for autophagosomes, including a size >0.5 μm in diameter, a double limiting membrane, and the presence within a single vacuole of multiple membranous organelle-derived structures (Fig. 1 B; Dunn, 1990a). Other AVs included translucent and dense multivesicular and multilamellar bodies with single or double outer membranes (Fig. 1 C), reflecting later stages of macroautophagy. In AD brain, we identified these AVs as autophagolysosomes (“late” AVs; Dunn, 1990b), which contain acid hydrolases but are distinct from lysosomal dense bodies that are small (<0.3 μm) and uniformly dense (Nixon et al., 2005). Therefore, we documented a robust accumulation of both early and late AVs in AD and PS1/APP brains, reflecting marked macroautophagic induction, failed maturation of AVs to lysosomes, or both (Nixon et al., 2005).
Figure 1.
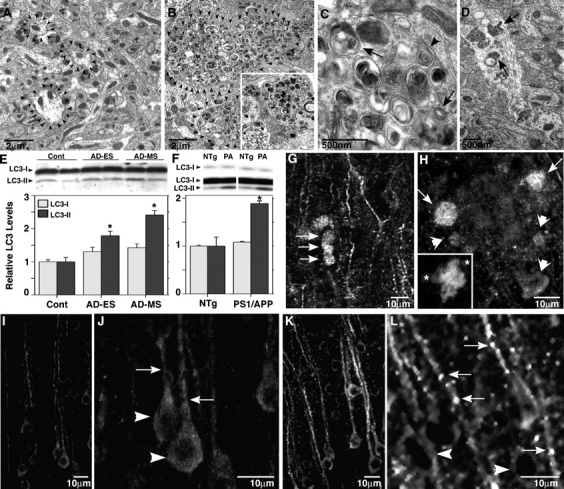
Increased macroautophagy in PS1/APP mice and human brains. (A–D) EM images of cortical neuropil show an absence of AVs and normal neurite profile in 9-mo-old NTg mouse brains (A, arrowheads outline normal neurites) and a marked accumulation of AVs within enlarged or dystrophic neurites in PS1/APP mice (B, arrowheads outline dystrophic neurite profiles in C) and biopsied brain material from an AD patient (B, inset). At higher magnification (C), AVs include autophagosomes (arrows) and multilamellar bodies (arrowhead). In normal dendrites of PS1/APP mice, multiple AVs are frequently seen (D, arrows). (E and F) LC3 quantification analyzed from immunoblots of LC3-I and LC3-II (top) in prefrontal cortical homogenates from cases of nonaffected (Cont), early stage (preclinical) AD (AD-ES), and moderate AD (AD-MS; E), and from brains of 18–22-mo-old PS1/APP (PA) mice (n = 3; F) compared with nontransgenic (NTg) controls (n = 3; *, P < 0.01). Error bars represent SEM. (G–L) LC3 immunofluorescence in 9-mo-old PS1/APP mice can be seen mainly as puncta in dystrophic dendrites of the cortex (G, arrows) and along adjacent dendrites. LC3 (H, arrows) is strong in dystrophic neurites in the periphery (asterisks) of a thioflavin S–labeled plaque core (H, inset) but is less so in neurites closest (H, arrowheads) to the Aβ deposit. LC3 is diffuse and uniform in neurons of NTg mice (I and J) but is predominantly vesicular and distributed more to the dendrites (arrows) than the cell soma (arrowheads) in 9-mo-old PS1/APP cortex (K and L).
We also used an antibody against the AV marker protein, LC3, to identify the specific subpopulation of autophagosomes, which is the earliest compartment of macroautophagy. LC3-II, a phosphatidylethanolamine-modified isoform of the microtubule-associated protein LC3-I, is generated and translocated to nascent autophagosomes when macroautophagy is induced, and its presence, therefore, is a putative index of macroautophagy induction (Mizushima et al., 2004). Very low levels of LC3-II were detected by Western blot analysis in cortical gray matter of normal control human brains. LC3-II levels were significantly elevated at early stages of AD and, to a greater extent, at a mild/moderate stage of AD as compared with neuropathologically normal age-matched elderly controls (P < 0.05 and P < 0.005, respectively; Fig. 1 E). The level of LC3-I, which was present in the brain at much higher amounts than LC3-II, rose modestly but not significantly (Fig. 1 E). This biochemical change implied an induction of macroautophagy in early AD.
Similarly, adult (18–22 mo old) PS1/APP mice also displayed nearly twofold higher levels of LC3-II but not LC3-I (Fig. 1 F, top) in the frontal cortex as compared with age-matched NTg mice (n = 3 each; P < 0.01; Fig. 1 F). Moreover, by immunofluorescence labeling, total LC3 was higher in the dendrites relative to the perikarya of neocortical pyramidal neurons in PS1/APP mice (Fig. 1, G, H, K, and L) as compared with NTg mice (Fig. 1, I and J). LC3 in PS1/APP dendrites was frequently vesicular (Fig. 1, K and L), indicating the presence of LC3-II, which is associated with nascent AVs (Fig. 1, G, K, and L). LC3-positive vesicles were numerous in the dystrophic portions of dendrites, particularly those at the periphery of plaques (Fig. 1 H). Dystrophic neurites close to Aβ deposits contained high numbers of AVs (Fig. 1 H), but these were mostly LC3 negative (Fig. 1 H, arrowheads), reflecting a later maturational state of AV populations in these presumably older dystrophic neurites.
Macroautophagy is induced very early in AD and PS1/APP mice
Elevation of LC3-II in AD brains at preclinical stages of AD (Fig. 1 E) suggested that macroautophagic induction is an early response in the disease process. To investigate early induction in PS1/APP mice, we also analyzed LC3-II levels in these mice at an age (8–9 wk) that precedes known neuropathology, including Aβ deposition (“predepositing” mice; Matsuoka et al., 2001). AVs were visible in the cell bodies and neurites of predepositing PS1/APP mice (Fig. 2, A and B) but were significantly less frequent in NTg animals (Fig. 2 C). Based on the EM fields that were surveyed from NTg and PS1/APP mice at 8–9 wk, >95% of images from NTg mice had zero or one AV present, whereas >80% of EM images from PS1/APP mice had at least one AV per field (Fig. 2 C). Quantitative ultrastructural analyses confirmed an increased incidence of AVs in the neocortex of these predepositing PS1/APP mice, where AVs were fivefold more numerous (n = 3 each; P < 0.001) than in age-matched NTg mice (Fig. 2 D). LC3-II levels in predepositing PS1/APP brains, as determined by Western blot analysis, were modestly yet significantly elevated (n = 6; P < 0.05) in the absence of a significant change in LC3-I levels (Fig. 2 E). Furthermore, LC3 immunoreactivity, which was present predominantly in neuronal cell bodies in NTg mice (Fig. 2, F and I), was distributed to both hippocampal cell bodies and dendrites in 9-mo- (Fig. 2, G and H) and 9-wk-old (Fig. 2, J and K) PS1/APP mice. The staining pattern for LC3 in many dendrites was more frequently punctate in PS1/APP than in NTg mice (Fig. 2, I and J), although less so than in older PS1/APP mice (Fig. 2, compare H with K). Collectively, these observations demonstrate that macroautophagy is induced at a prepathological stage of disease in PS1/APP mice and that, in addition, different subtypes of AVs accumulate pathologically as neuritic dystrophy develops in older mice, as in AD.
Figure 2.
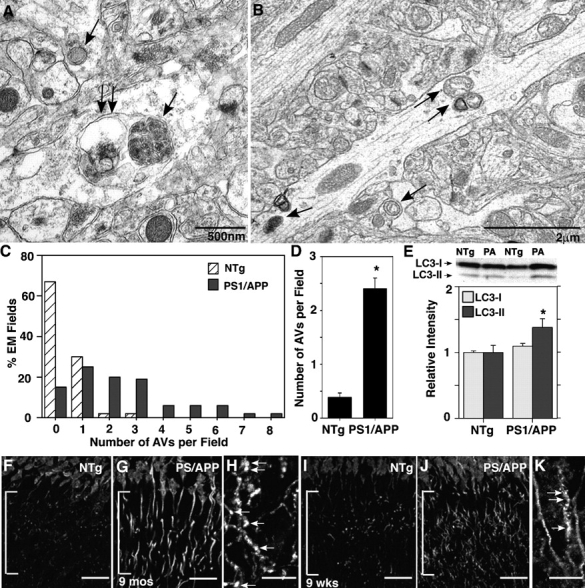
Identification of macroautophagy in the hippocampus of predepositing PS1/APP mice. Ultrastructural inspection of brain tissue from PS1/APP mice (A–D) shows that AVs (A and B, arrows) are five times more frequent in the dendrites of 8-wk-old PS1/APP than in those of age-matched NTg mice. The frequency of AVs per EM field (C) and mean number of AVs per EM field (D) within the hippocampal molecular layer (n = 3) are shown. LC3 immunoblot and analysis (E) and immunofluorescent labeling (F–K) of the hippocampal dendrites (brackets) in 8–9-wk-old PS1/APP and NTg mice show LC3-II elevation (P < 0.05) in 8-wk-old PS1/APP compared with NTg mice (E). (D) *, P < 0.001. (E) *, P < 0.05. Error bars represent SEM. LC3 immunoreactivity in pyramidal cell dendrites is increased in 9-mo-old (F–H) and 9-wk-old (I–K) PS1/APP mice and frequently exhibits a punctate labeling pattern, which is more evident at 9 mo than at 9 wk (H and K, arrows) and is uncommon in NTg mice (F and I). Bars (F, G, I, and J), 20 μm; (H and K), 10 μm.
Modulation of macroautophagy markedly influences Aβ production
Multiple tubulovesicular compartments that were implicated in APP metabolism, such as the ER–Golgi and endosomes, become components of AVs either as substrates or by contributing to the limiting membrane of autophagosomes (Dunn, 1990a). Moreover, APP, β-site APP cleaving enzyme, and PS1 are reported to be abundant within neuritic plaques (Leuba et al., 2005), raising the possibility that these components are localized in AVs. We confirmed that neuritic plaques of PS1/APP mice are intensely labeled with a monoclonal antibody to PS1 (Fig. 3, A and B). Based on IEM, AVs were the principle immunoreactive structures within the dystrophic neurite, accounting for >90% of PS1 immunolabeling (Fig. 3, C–F). In the brains of both PS1/APP mice (Fig. 3, C and D) and AD patients (Fig. 3, E and F), PS1 antibody predominantly decorated the double-limiting membranes of AVs or single or double membranes within AVs (Fig. 3, arrows). Tubulovesicular membranes in the adjacent normal neurites were the only other immunolabeled structures that were detected in the neuropil (Fig. 3 D, arrowheads), which is consistent with the expected contribution of PS1-rich smooth ER to AV formation (Culvenor et al., 1997). Mitochondria, plasma membranes, small vesicles, and cytoplasm were essentially devoid of gold, underscoring the specificity of immunolocalization.
Figure 3.
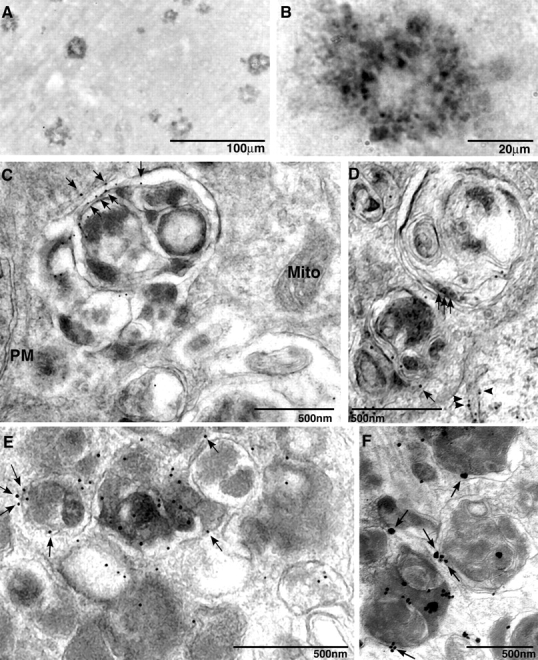
Immunolocalization of PS1 in plaques and AVs within dystrophic neurites in AD and PS1/APP mice. Cingulate cortex from 9-mo-old PS1/APP mice immunolabeled with PS1 antibody and NT1 showed that PS1 localized to plaques (A). At higher magnification, anti-PS1 antibodies strongly labeled neuritic profiles that were distributed within the plaque corona (B). PS1 immunoreactivity is identified by IEM in AVs within dystrophic neurites of PS1/APP animals (C and D) and human brain (E and F) by IEM. Arrowheads identify tubulovesicular membrane labeling. PS1 (C–F, arrows) was localized on the outer limiting membrane of the AV but not in mitochondria (Mito) or on plasma membranes (PM). IEM followed by silver stain enhancement for PS1 was performed on a human brain that was diagnosed for AD (F).
In light of these observations, we further investigated the possibilities that macroautophagy is a pathway capable of producing Aβ and that AVs are a site of Aβ generation. We used murine fibroblast-like L cells that were stably transfected with APP695 (murine L cell type + wild-type human alkaline phosphatase [L/APP]), human (SH-SY5Y), and murine (N2a) neuroblastoma cell lines to determine the effect of macroautophagy on APP metabolism. In both nonneuronal (Fig. 4 A, top, L/APP) and neuronal (Fig. 4 A, bottom, SH-SY5Y; N2a, not depicted) cell models, inducing macroautophagy via the mTOR kinase pathway (Seglen et al., 1996; Petiot et al., 2000; Kadowaki and Kanazawa, 2003) by treating cells with rapamycin, which is a specific inhibitor of mTOR phosphorylation and an inducer of macroautophagy (Noda and Ohsumi, 1998), or by depriving them of Leu and/or His (Kanazawa et al., 2003) substantially increased the number of AVs (Fig. 4 A, right), including autophagosomes (Fig. 4 B) relative to that in cells cultured under baseline conditions (10% serum; Fig. 4 A, left). Monodansylcadaverine, a fluorescent compound that preferentially accumulates in multilayer membranous structures such as AVs (Biederbick et al., 1995), was localized in large vacuolar structures after the deprivation of Leu, His, or both (Fig. 4 C, bottom left). Similarly, as expected with macroautophagy induction, LC3 immunofluorescence redistributed from the cytoplasm of untreated cells (Fig. 4 C, top middle) to a population of large vacuoles in Leu/His-deprived L/APP cells (Fig. 4 C, bottom middle) and serum-deprived SH-SY5Y cells (Fig. 4 C, bottom right), confirming our ultrastructural evidence of autophagosome generation (Fig. 4, A and B). Cells that were cultured in complete medium are shown in the top panels (Fig. 4 C). Serum or specific amino acid (Leu or His) deprivation induce autophagy through the AMP kinase pathway (Kadowaki and Kanazawa, 2003). We found a similar induction of autophagy in all of the cell lines (L/APP, N2a, and SH-SY5Y) after Leu/His deprivation or 10 nM rapamycin treatment (Fig. 4 D). Both treatments elevated levels of LC3-II (Fig. 4 D) and reduced levels of phosphorylated mTOR but not total mTOR relative to the values measured in untreated or 3-methyladenine (3MA)–treated cells as expected (Shigemitsu et al., 1999).
Figure 4.
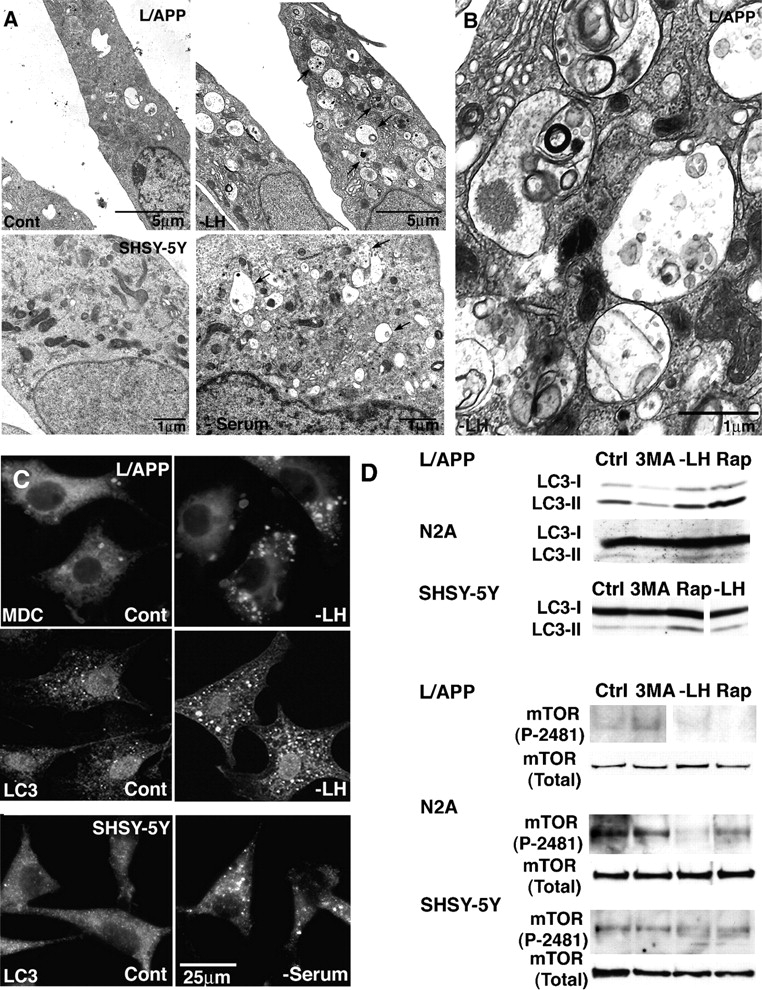
Induction of macroautophagy in L/APP, SH-SY5Y, and N2a cells. (A and B) EM images showing changes in the number of AVs (arrows) in L/APP-overexpressing APP695 (L/APP cells) grown in complete medium (A, top left) or in medium lacking Leu and His (A, top right) for 6 h and in SH-SY5Y cells grown in the presence (A, bottom left) or absence (A, bottom right) of serum. At higher magnification, early and late AVs with typical morphologies are seen in a Leu/His-deprived L/APP cell (B). (C) Fluorescent and immunofluorescent labeling of large vesicles by the AV marker monodansylcadaverine (0.5 μg/ml for 30 min; left) and LC3 antibody (middle, L/APP; right, SH-SY5Y) in macroautophagy-induced cells (bottom), which are much less abundant in cells grown in complete medium (top). (D) Western blots confirm the cytochemical evidence for increased LC3-II levels as well as phospho-mTOR (P-2481) but not total mTOR after macroautophagy induction by Leu and His deprivation or 10 nM rapamycin (Rap) and macroautophagy inhibition by 5 mM 3MA in L/APP, N2a, and SH-SY5Y cells. Immunoblots for LC3 in SH-SY5Y cells and P-2481 mTOR in L/APP cells have been spliced but are derived from the same blot.
A relationship between macroautophagy induction and Aβ generation (Fig. 5, A and B) was established when we observed that suppressing macroautophagy in L/APP cells for 6 h with either 5 mM 3MA or 4:1 mM Leu/His decreased Aβ40 secretion into the medium by 39 (n = 6 each; P < 0.01) and 26–41% (n = 6 each; P < 0.05) relative to untreated cells, respectively, as measured by sandwich ELISA and standardized to total protein. Conversely, inducing macroautophagy by depriving L/APP cells of Leu or His stimulated Aβ40 secretion by 22 and 39%, respectively (n = 6 each; P < 0.05) compared with untreated cells (Fig. 5 A) and 70–90% over the levels in macroautophagy-suppressed cells. An analysis of covariance revealed a highly significant relationship between macroautophagy suppression by amino acid supplementation and reduction in Aβ levels (Aβ40, P < 0.01; Aβ42, P < 0.001) and between macroautophagy induction by Leu/His deprivation and increased Aβ production (Aβ40, P < 0.01; Aβ42, P < 0.0001). These results were confirmed with a second method of macroautophagy induction, rapamycin, which also effectively increased Aβ production nearly twofold in L/APP cells. Effects on Aβ40 levels were similar (Fig. 5 B), although there was a nonsignificant trend toward greater effects on Aβ42 production, as indicated by an increase in the ratio of Aβ42 to Aβ40. Modulating other amino acids, such as Gly or Val, in the medium had no effect on Aβ levels (Fig. 5, A and B); this finding is consistent with the known negligible influence of these amino acids on macroautophagy (Kadowaki and Kanazawa, 2003). The levels of βCTF and full-length APP levels were not significantly altered by these macroautophagy modulations (Fig. 5, C and D). We also examined the effect of macroautophagy modulators on Aβ40 and Aβ42 generation in the neuronal cell lines SH-SY5Y (Fig. 5, E and F) and N2a (Fig. 5, G and H). Inducing macroautophagy with rapamycin or Leu/His deprivation significantly increased Aβ40 in both cell lines (52–132%; P < 0.01; Fig. 5, E and G) and Aβ42 in N2a cells (98%; P < 0.05; Fig. 5 H), whereas suppressing macroautophagy with 3MA or Leu and His supplementation decreased Aβ levels by 12–54% (P < 0.05; Fig. 5 H).
Figure 5.

Aβ generation in cells after autophagic modulation. Levels of Aβ40 (A), Aβ42 (B), βCTF (C), and APP (D) measured by sandwich ELISA after the incubation of L/APP cells (6 h) in conditions that block autophagy (+Leu, +His, +Leu/+His, and 5 mM 3MA), activate macroautophagy (−Leu, −His, −Leu/−His, and rapamycin), or do not affect autophagy (complete media and enrichment of the deprivation of Gly or Val). Values reported as percent difference of control ± SEM; *, P < 0.05 (at least). In similar experiments, Aβ40 and Aβ42 levels from SH-SY5Y (E and F) and N2a cells (G and H) after various treatments. Error bars represent SEM.
Ultrastructural localization of Aβ and γ-secretase complex components
Using immunogold labeling and EM in Leu- and His-deprived L/APP cells, we observed that Aβ and γ-secretase components were preferentially localized to AVs in situ and, as previously described (Cupers et al., 2001), were localized to tubulovesicular compartments corresponding morphologically to ER/Golgi/endosomes. Immunolabeling for Aβ40 or Aβ42 using COOH-terminal–specific antibodies (Mathews et al., 2002) preferentially decorated the same compartments (Fig. 6, A and B, respectively). PS1 and nicastrin antibodies directed against endogenously expressed proteins also principally decorated both internal and limiting membrane components of AVs in L/APP cells (Fig. 6, C and D, respectively). Quantitative analysis of gold particles showed high proportions of immunogold staining for Aβ40, Aβ42, PS1, and nicastrin that were associated with AVs (43, 35, 48, and 35%, respectively) and tubulovesicular compartments (31, 37, 33, and 35%, respectively; Fig. 6 F). We found that the AV and tubulovesicular compartments comprise 27.0 ± 11.0 and 19.2 ± 7.2%, respectively, of the total cell area, indicating that PS1 signal in these organelles was five times higher than in other subcellular compartments and was similar to the organellar distribution in the PS1/APP brain (Fig. 3, C and D). Immunolabeling of brains from 9-mo-old PS1/APP mice with Aβ40 antibody also indicated a significant localization of Aβ40 in AVs (Fig. 6, G and H).
Figure 6.
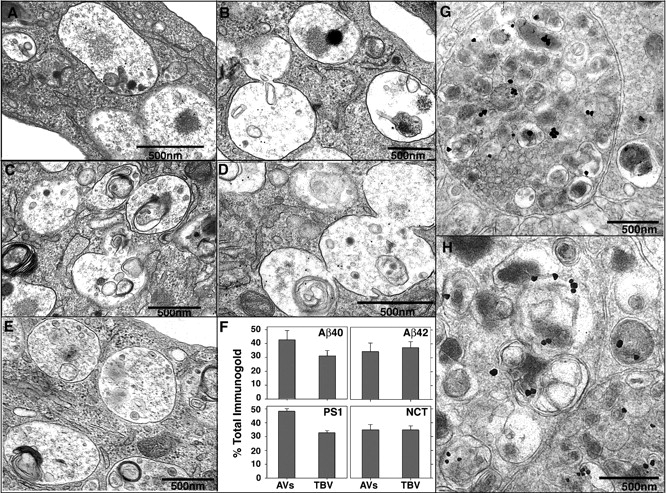
Immunolocalization of Aβ in AVs from L/APP cells and PS1/APP brains and γ-secretase components (PS1 and nicastrin) in L/APP cells. Immunogold localization of Aβ40 (A), Aβ42 (B), PS1 (C), nicastrin (D), and in the absence of primary antibody (E) in L/APP cells grown for 6 h in the absence of Leu and His. (F) Quantification of gold particle frequency in AV or tubulovesicular compartments (TBV), which comprise 27.0 ± 11.0 and 19.2 ± 7.2%, respectively, of the total cell area. Error bars represent SEM. (G and H) IEM followed by silver stain enhancement for Aβ40 was performed in 9-mo-old PS1/APP mice.
Enrichment of γ-secretase activity and protein components in isolated AVs
In further studies, we assayed for the protein components and the γ-secretase activity that are needed for Aβ generation in highly purified fractions of AVs from serum-deprived L/APP cells. The AV and lysosome fractions were isolated, and the identity and purity of AV and lysosome fractions were confirmed by EM (Fig. 7 A) and by immunoblotting using antibodies to LC3-II and rab24, which is a ras-related GTPase that is associated with macroautophagy (Fig. 7 B; Munafo and Colombo, 2002). Consistent with the immunogold studies of L/APP cells in Fig. 6, AV fractions were highly enriched in levels of PS1 and nicastrin relative to the other subcellular fractions. Purified AVs also contained significant levels of APP and βCTF (Fig. 7 B). APP was enriched in the ER/endosome/Golgi fraction as well as the AV isolates in cells that were treated with media lacking Leu and His and cells grown in serum. In uninduced cells, the total amount of AVs recovered was considerably lower. PS1 was most abundant in the AV (A2) fraction after macroautophagic induction by serum deprivation, with lesser amounts in the A1 and lysosome fractions and in the ER/endosome/Golgi.
Figure 7.

Evidence for the enrichment of PS1-dependent γ-secretase activity in AVs. (A and B) Ultrastructure of AVs (A1 and A2) and lysosomes in subcellular fractions from serum-deprived L/APP cells (A) and Western blot analysis (B) for LC3-II, rab24, APP, βCTF, PS1 (PS1 amino-terminal fragment), and nicastrin (NCT) in L/APP subcellular fractions. AVs, A1 and A2; L, lysosomes; E, tubulovesicular compartments (Golgi/ER/endosomes); P, postnuclear pellet; C, cytosol; M, mitochondria. Empty lanes in the original blot have been removed from the figure and noted with a white line. (C) Rates of cleavage of the fluorogenic substrate in subcellular fractions from L/APP cells grown in −serum media (left) or −Leu/−His media (right). PNP, postnuclear pellet. (D and E) Proportions of the total recovered cell γ-secretase activity in different subcellular fractions after serum deprivation (D) or in uninduced (+serum) or induced (−Leu/−His) cells (E). Comparison of +serum versus −Leu and −His conditions shows the redistribution of γ-secretase activity from the tubulovesicular to AV fractions after macroautophagic induction. G, Golgi; A, AV. (F) Liquid chromatography mass spectrometry analysis of the fluorogenic substrate after incubation with purified AVs. Selected ion chromatogram corresponding to different cleavage products is displayed. Numbers in graph refer to the cleavage/amino acid site from the Aβ peptide. (G) γ-Secretase activity in AV and lysosome fractions from mice blastocysts in which the PS1 and PS2 genes were deleted (PS KO; BD8) or in which human PS1 was stably transfected into the PS KO blastocysts (hPS1; BD8/hPS1). Numbers on x axis are in minutes. (H) Aβ40 and Aβ42 levels in medium from these cells as detected by sandwich ELISA. Values are given as means ± SEM (error bars).
By using a fluorogenic synthetic polypeptide substrate to measure γ-secretase activity after inducing macroautophagy in L/APP cells (serum or Leu + His deprivation), we found that in L/APP cells, AV-enriched fractions contained the highest γ-secretase activity of all fractions analyzed on a per protein basis (Fig. 7 C). Lysosome, Golgi, and microsome (E: ER/endosome/Golgi) fractions also contained significant γ-secretase activity relative to the low activities in the mitochondrial, post nuclear pellet/unbroken cells, and cytosol fractions (Fig. 7 C). Based on the γ-secretase activity per total recovered amount of each compartment, AVs accounted for 20–25% of the total γ-secretase activity recovered from L/APP cells that were deprived of serum (Fig. 7 D). Microsomes (ER and endosome) and Golgi fractions showed less (per protein basis) γ-secretase than AVs (19%), but, because these organelles are more abundant in the cell, they accounted for slightly higher proportions (28 and 20%, respectively) of the total recovered γ-secretase activity in these cells. Similar findings were made in L/APP cells that were grown in −Leu/−His media (Fig. 7 E). In contrast to conditions of macroautophagy induction, total AV yields in noninduced L/APP cells were much lower but exhibited similar properties to those seen in induced cells (unpublished data). In noninduced cells, the γ-secretase activity in AVs was ∼10% of the total cell activity, whereas that in the mixed microsome fraction increased to >60% (Fig. 7 E). In both induced and noninduced conditions, tubulovesicular compartments and AV fractions contained 60–70% of the total γ-secretase activity, suggesting that this activity shifts from tubulovesicular compartments to AVs when macroautophagy is induced.
Mass spectroscopic analysis indicated that the predominant cleavage of the model fluorogenic substrate in AVs occurred at the Aβ42 site (Fig. 7 F), reflecting similar findings of preference for Aβ42 cleavage over Aβ40 with this substrate in other compartments. This Aβ-specific cleavage product did not undergo further cleavage in AVs even after incubating the substrate in AV isolates for 24 h, indicating that the product is not a transient proteolytic intermediate. This is in contrast to the Aβ-specific cleavage product in lysosomal fractions, where the γ-cleaved product was further cleaved to smaller products when the incubation time was extended or when the pH was lowered (Table S1, available at http://www.jcb.org/cgi/content/full/jcb.200505082/DC1).
γ Cleavage in AVs is PS dependent
To confirm that fluorogenic substrate cleavage was PS1 specific, we assayed its activity in AVs that were isolated from PS1 + PS2 knockout (PS KO) blastocysts (BD8; Lai et al., 2003) and BD8 blastocysts that were transfected with human PS1 (BD8/hPS1). Cleavage of the γ substrate was 60–65% lower in AVs from the BD8 cells compared with AVs from PS1-rescued BD8/hPS1 cells (Fig. 7 G), which is comparable with studies in human brain showing that 50% of total substrate activity is inhibited by using specific γ-secretase inhibitors. Although there was some residual cleavage of the substrate in the BD8 double PS KO blastocysts, these cells generated no detectable murine Aβ, whereas the BD8/hPS1 cells produced abundant Aβ40 and Aβ42 (Fig. 7 H). Consistent with these results, a 5-μM IC50 concentration of the selective γ-secretase inhibitor L685,458 inhibited 29.6–66.6% of the secretase activity in AV fractions of hPS1-rescued cells (unpublished data). The lower levels of γ-secretase cleavage that were measured in lysosomal fractions were much less affected by PS deletion (25%) than were the AV fractions, indicating that lysosomes (and to a lesser extent AVs) contain PS-independent protease activity that is capable of γ cleavage of the model substrate. This activity, however, does not support Aβ generation in intact cells based on the negligible Aβ production in double PS KO blastocysts.
Discussion
Our data provide strong evidence that Aβ is generated in AVs during macroautophagy. Moreover, macroautophagy is both induced and impaired in AD brain and PS1/APP mice, leading to the pathological accumulation of Aβ-containing AVs within affected neurons. Macroautophagy induction was evidenced by an appearance of autophagosomes, which are only rarely detectable in normal brain (Nixon et al., 2005), and by elevated levels and cytosol-to-vesicle translocation of LC3-II, which is a specific marker of autophagosome formation. These changes are evident at the earliest stage of AD and in 8–9-wk-old PS1/APP mice before Aβ deposition, which suggests that macroautophagy induction is an early response in disease development, although it is not necessarily independent of Aβ influences. The specific pathologic events that induce macroautophagy in predepositing PS1/APP mice are not known, but sources of oxidative stress, including intracellular forms of Aβ (Billings et al., 2005), are possible factors contributing to macroautophagy induction or to later dysfunction of the pathway. The induction of macroautophagy at early stages of AD is consistent with the expected need for increased protein/organelle turnover in injured and regenerating neurites as well as for protection against apoptotic stimuli, such as damaged mitochondria, that are turned over by macroautophagy (Brunk and Terman, 2002).
The most striking feature of the macroautophagy-related pathology in PS1/APP mouse brains was a grossly abnormal accumulation of autophagosomes and other AV subtypes in dystrophic neurites of the cortex and hippocampus, as also seen in AD brain (Nixon et al., 2005). Lysosome-related multilamellar and dense bodies have been described previously in neurological disease states (Suzuki and Terry, 1967; Masliah et al., 1993) and, in part, represent late stages of macroautophagic activity and lysosomal digestion (Nixon et al., 2005). In contrast, we found that dystrophic neurites contain very high proportions of autophagosomes and other immature AVs, implying impairment in the normal maturation of these nascent AVs to lysosomes. During normal neurite outgrowth or regeneration, immature AVs move retrogradely and fuse with lysosomes in the neurite or are more likely near or within the cell body (Hollenbeck, 1993). After this fusion event, the contents of the AV are rapidly degraded, and the AV becomes a lysosome (Overly and Hollenbeck, 1996). This process is normally highly efficient, with little evidence of AV buildup. In dystrophic neurites, however, the striking buildup of autophagosomes and late AVs implies a defect in AV transport, maturation to lysosomes, or both, which is likely to impede lysosomal degradation through this pathway (Nixon et al., 2005).
Our data provide strong evidence that Aβ is generated in one or more subtypes of AVs that build up abnormally in affected neurons. AVs not only contain immunoreactive Aβ and βCTF but are also enriched in PS-dependent γ-secretase activity. Senile plaques in AD and PS1/APP mouse brains are also abundantly immunoreactive for these components. Our data confirm findings that PS1 and nicastrin are enriched in lysosomes (Pasternak et al., 2003) and indicate that AVs, which were not distinguished from lysosomes in these earlier studies, are a more concentrated source of these γ-secretase components. Our findings also explain and are supported by observations that inclusion body myositis, which is the only known condition in which Aβ deposits occur outside the nervous system, involves the accumulation of macroautophagy-related “rimmed” vacuoles containing elevated APP, Aβ, and PS1 (Askanas et al., 1998). Previous studies have identified intracellular Aβ accumulation in endosomes (Cataldo et al., 2004a) and multivesicular bodies (Takahashi et al., 2002) in AD and Down syndrome (Gyure et al., 2001). Macroautophagy could substantially increase pools of intracellular Aβ and contribute to the formation of Aβ oligomers and protofibrils, which is a process that is promoted in the acidic environment of lysosome-related organelles (Vassar and Citron, 2000).
We have also demonstrated, for the first time, that Aβ is generated during macroautophagy. Aβ production rises when macroautophagy is acutely stimulated, and AVs proliferate and fall when macroautophagy is inhibited. AVs are depleted by blocking either of the two independent signaling pathways for macroautophagy that converge on mTOR kinase activity: amino acid–mediated signaling and the PI3-kinase–dependent pathway. Conditions that either stimulate AV production and delay or impair maturation of AVs to lysosomes might be expected to increase the number of AVs and raise intracellular Aβ levels (Fig. 8). Aβ is believed to be generated at several sites within neurons, including endosomes, Golgi, and ER (Cataldo et al., 2004a), and this multiplicity of APP processing routes would account for our observations that considerable Aβ is still secreted from L/APP, SH-SY5Y, and N2a cells when macroautophagy is inhibited. Endocytic and autophagic pathways communicate extensively, and both Golgi and ER are turned over by macroautophagy (Dunn, 1990a), raising the possibility that each of these organelles could contribute to Aβ generation, in part, via macroautophagy. This communication between the macroautophagy and endosomal systems also provides one possible avenue for Aβ that is generated during macroautophagy to be released from cells, because late endosomes also communicate with endocytic recycling compartments (for review see Luzio et al., 2005). Extracellular release of some Aβ from AVs is also possible from exosomes, which is a mechanism proposed for prion release (Fevrier et al., 2005), or from the direct fusion of AVs with the plasma membrane (Jackson et al., 2005). It is worth noting, however, that the inefficient extracellular elimination of autophagy-generated Aβ may imply greater pathogenicity of this pool than the Aβ that is normally secreted because intracellular Aβ appears to be more cytotoxic than extracellular Aβ.
Figure 8.

Proposed models of AV accumulation leading to elevated Aβ levels. The schematic of macroautophagy depicts (A) the usual progression from autophagosomes (AP) to autophagolysosomes (APL) to lysosomes (L). Conditions that result in AV buildup (B and C) are expected to promote Aβ generation and accumulation, including impaired or delayed maturation of autophagosomes to lysosomes (B) or acute maximum induction of macroautophagy (C). Within neurons, AVs normally progress to lysosomes efficiently and are rarely seen in neurons (D). In AD, the disrupted retrograde transport of AVs in dendrites represents one of several possible mechanisms that impede the maturation of AVs to lysosomes, leading to Aβ generation in AVs and its delayed degradation in lysosomes (E).
Based on the low number of AVs that were detected in normal brain, macroautophagy may play a relatively minor role in constitutive Aβ generation (Fig. 8). At low levels of macroautophagy induction, Aβ that was generated in AVs would be subsequently degraded within lysosomes, which contain the necessary proteases (Bendiske and Bahr, 2003). In damaged or regenerating neurites, however, where more APP-rich substrates are diverted into the macroautophagy pathway and AV–lysosome fusion may be delayed, intracellular Aβ is generated within specific subtypes of AVs that accumulate (Fig. 8 B). AV accumulation that is associated with any significant neuritic injury could stimulate local Aβ production, as seen, for example, in traumatic brain injury (Smith et al., 2003). Moreover, in AD, in which large numbers of AVs accumulate and persist without maturing within dystrophic neurites, macroautophagy could contribute substantially to β-amyloidogenesis and especially to intracellular Aβ accumulation. In addition, several risk factors for AD, including aging (Cuervo and Dice, 2000) and PS mutations (unpublished data), impair AV maturation to lysosomes. This accounts, in part, for the aging-related accentuation of autophagic–lysosomal system pathology and β-amyloidogenesis in familial forms of AD that are caused by PS mutations and in PS/APP mice relative to mice overexpressing mutant APP alone (Cataldo et al., 2004b).
Collectively, these studies identify AVs as Aβ-generating compartments that accumulate pathologically in AD brain (Nixon et al., 2005), accounting for a significant source of intracellular Aβ in AD. Macroautophagy, as a pathway for Aβ generation and a mediator of both cell survival and degenerative phenomena (Nixon et al., 2001), represents a new direction for investigations into the pathogenesis and possible therapy of AD.
Materials and methods
Tissues
We examined biopsy specimens from the temporal or frontal cortices of 10 patients, which were collected to confirm the suspected diagnosis of encephalopathy. Seven of these cases (aged 71–86 yr) were later found to meet the neuropathological criteria of AD (Braak and Braak, 1991; Mirra et al., 1991, 1993; Newell et al., 1999), and the remaining three specimens (aged 67–72 yr) were found to be neuropathologically normal (Wegiel et al., 2000). Clinicopathologic characteristics of these cases have been described previously (Nixon et al., 2005). Tissue was fixed in 3% PFA and 1% glutaraldehyde/0.1 M phosphate buffer, pH 7.4, and were postfixed in 1% osmium tetroxide in Sorensen's phosphate buffer. After dehydration in ethyl alcohol, the tissue was embedded in Epon resin. After routine histological inspection, the tissue blocks containing senile plaques were cut serially into ultrathin (0.06 μm) sections, and semithin (0.5 μm) sections were cut every 10th section. Ultrathin sections were placed on formvar-coated grids and were poststained with uranyl acetate and lead citrate.
In addition, we used postmortem fixed and frozen brain tissue (Brodmann areas 8 and 10) that was obtained from 43 elderly individuals of both sexes ranging in age from 41 to 100 yr. Postmortem intervals for all cases that were used were 17 h or less. Using the Consortium to Establish a Registry for AD and Reagan guidelines (Mirra et al., 1991; Newell et al., 1999), Braak staging (Braak and Braak, 1991), and the criteria proposed by Mirra et al. (1993), 14 nondemented individuals from this group were evaluated and diagnosed with early stage AD neuropathology (the neocortex was devoid of plaques, and transentorhinal, entorhinal/hippocampal cortices contained sparse plaques; Braak stages I–II). A second group consisting of 16 age-matched cases met the criteria for mild/moderate stage AD (moderate plaque numbers in the transentorhinal, entorhinal/hippocampal cortices and slightly fewer in the neocortex; Braak stages III–V). A third group consisting of 13 age-matched cases that were determined to be neuropathologically normal (isocortex and entorhinal cortex/hippocampus were devoid of plaques and neurofibrillary tangles) were used as controls. The tissues were obtained from the Bronx Veteran's Administration Medical Center and the Harvard Brain Tissue Resource Center at McLean Hospital. The magnitude of neuropathology was confirmed by histopathological inspection using Nissl stain, hematoxylin and eosin staining, Bielschowsky silver stain, and thioflavin S histofluorescence. Transgenic mice expressing the Swedish mutation of human APP (APPK670M/N671L) and mutant human PS1 (PS1M146L; Duff et al., 1996) were studied at 8–9 wk, 9 mo, and 18–22 mo of age together with age-matched controls.
Antibodies and reagents
The antibodies used for Aβ ELISAs have been previously described (Mathews et al., 2002) and were a gift from Johnson and Johnson Pharmaceutical Research and Development/Janssen Pharmaceutica (Beerse, Belgium). NT1 was used to identify PS1 in PS1/APP mice. Ab14, which is a pAb recognizing the amino terminus of murine PS1, was used for IEM localization of PS1 in L/APP cells and was a gift from S. Gandy (Thomas Jefferson University, Philadelphia, PA; Petanceska et al., 2000). An antinicastrin pAb was obtained from P. Fraser and P. St George-Hyslop (University of Toronto, Toronto, Canada). mAbs, 4G8 recognizing residues 17–24 of the Aβ peptide, 6E10 recognizing residues 1–16 of the Aβ peptide, βCTF, and APP were purchased from Signet. mTOR antibodies (total and P-2481) were purchased from Cell Signaling Technology, and rab24 was obtained from BD Biosciences. Organelle immunomarkers included anti-rab5 affinity-purified rabbit pAb (Santa Cruz Biotechnology, Inc.) and a polyclonal EEA1 (gift from S. Corvero, University of Massachusetts, Worcester, MA) for endosomes, a mAb clone p58K-9 for Golgi (Sigma-Aldrich), a pAb calnexin (StressGen Biotechnologies) for ER, and an anticathepsin D polyclonal for lysosomes (Cataldo et al., 1996). A pAb to LC3 was made that recognized native (LC3-I) and postautophagic-induced protein (LC3-II; Kabeya et al., 2000, 2004; Mizushima et al., 2004). 500 μg/ml monodansylcadaverine was added to living cells for 30 min to accumulate in AVs (Biederbick et al., 1995).
Modulation of macroautophagy
Cell maintenance and human wild-type APP695 expression in L/APP cells have been previously described (Mathews et al., 2002). Upon the induction of APP expression, L/APP cells were treated with various agents to modulate macroautophagy for 6 h. SH-SY5Y and N2a cells were maintained in DME + 10% FBS. PS-null and human PS1–transfected PS-null blastocysts (a gift from A. Bernstein, Mount Sinai Hospital, Toronto, Canada) were cultured in DME + 15% FBS with 10 nM β-mercaptoethanol. Media were formulated as listed by Invitrogen for DME and was supplemented with sodium pyruvate, vitamins, and 10% FBS dialyzed at 10 kD (all were obtained from Invitrogen). In some experiments, the media either lacked one amino acid of interest or contained fivefold excess of this amino acid from Sigma-Aldrich. 10 nM rapamycin was used to induce macroautophagy, whereas 5 mM 3MA (Sigma-Aldrich) was used to inhibit AV formation (Seglen and Gordon, 1984).
After treatment, cells that were used for immunofluorescence analysis were fixed in 2% PFA (Electron Microscopy Sciences) in PBS, pH 7.4, and were washed with PBS. Cells intended for EM were fixed in 4% PFA/1% glutaraldehyde in 0.1 M sodium cacodylate buffer.
Immunochemical analyses
Immunofluorescence microscopy, sandwich ELISA for APP and its metabolites, βCTF, Aβ40, Aβ42, and Western blot analyses on cells were performed as previously described (Mathews et al., 2002; Grbovic et al., 2003). Western blot analysis was also performed on L/APP, N2a, and SH-SY5Y cell lysates probing for LC3-I/-II, phospho-mTOR (P-2481), and total mTOR. Analysis was also performed on AV isolates from L/APP cells, and cells were probed for LC3, rab24, APP, βCTF, PS1 amino-terminal fragment, and nicastrin. Immunofluorescence was visualized on either a microscope (Axiovert 200; Carl Zeiss MicroImaging, Inc.) with a 100× objective equipped with a digital camera (AxioCam HRC; Carl Zeiss MicroImaging, Inc.) and Axiovision 4.2 software (Carl Zeiss MicroImaging, Inc.) or on a laser scanning confocal microscope (model TCSNT; Leica) using a 100× objective and was analyzed using Leica Confocal Software. Western blots were imaged on film, and the film was scanned with a scanner (Duoscan T1200; Agfa). Denstitometry analysis was performed using a National Institutes of Health image (version 1.59) with samples that were within the linear range of the software. In some cases, lanes from the same Western blot were rearranged or, if not relevant, were eliminated to enable alignments of multiple gels in a composite figure. Splicing within a given blot is indicated by a white line between spliced portions.
Mouse brains were harvested after intracardiac perfusion with 1% PFA and were sucrose embedded overnight, frozen, and cryostat sectioned at 40 μm of thickness before immunolabeling with LC3. Fluorescent labeling was identified using the laser scanning confocal microscope (Leica) equipped with a 100× objective.
IEM
Ultrathin sections from Epon-embedded blocks were placed on copper grids for structural analysis or on nickel grids for immunogold labeling and were air-dried and etched briefly with 1% sodium metaperiodate in PBS followed by washing in filtered double distilled water and were incubated in 1% BSA in PBS for 2 h. Sections were incubated overnight in primary antibody (calnexin or protein disulfide isomerase; both were obtained from Stressgen) in a humidified chamber at 4°C, spin washed several times in PBS, and incubated in 5–20 nm gold-conjugated secondary antibody (anti–mouse or anti–rabbit IgG; GE Healthcare) for 2 h at RT. Silver enhancement (GE Healthcare) of PS1 in human brain and Aβ42 in mouse brain was performed after IEM. In negative control experiments, primary antibody was substituted with normal rabbit or mouse serum (1:20) depending on the primary antibody used (polyclonal or monoclonal) and was incubated in a humidified chamber at 4°C. The grids were extensively washed with PBS and incubated for 2 h at RT with 10 nm gold-labeled secondary antibody (GE Healthcare). Grids were washed again and briefly stained with uranyl acetate and lead citrate before being examined with an electron microscope (model CM 10; Philips). Images were captured on a camera (model C4742-95; Hamamatsu) and on Advantage CCD Camera System software (Advanced Microscopy Techniques Corporation). Some EM images (Fig. 4, A and B; L/APP cells) were captured on film, printed on Kodak glossy paper, and scanned with a scanner (StudioScan II; Agfa) at 600 dpi.
Quantitative analysis of AVs from EM images
For 9-wk-old PS1/APP and NTg mice images (n = 3 each), 30 randomly selected EM images per animal were captured at a final magnification of 10,500×, and the number of AVs in each captured field was counted by visual inspection using criteria for identification that were previously established (Dunn, 1990b; Nixon et al., 2005). For 9-mo-old mice, the same procedure was applied, but 50 fields/mouse with two mice per group were used.
Preparation of AVs
For each cell line (L/APP, PS KO–BD8, or hPS1–BD8/hPS1), 500 million cells were serum deprived overnight to induce autophagic activity (Fuertes et al., 2003). In additional experiments, AVs were isolated from L/APP cells with normal serum and media lacking Leu and His. By using a protocol that was modified from Marzella et al. (1982), the cells were harvested, disrupted by nitrogen cavitation, homogenized, and separated by differential centrifugation to first produce a (low speed) pellet containing the nuclear fraction and (up to 30%) unbroken cells (postnuclear pellet) and a second (supernatant in higher speed) pellet that was enriched in AVs, lysosomes, and mitochondria. From the AV/lysosome/mitochondrial fraction, lysosomes were separated from the two AV fractions by using a discontinuous metrizamide gradient (A1, 10% metrizamide; A2, 20% metrizamide). A cytosol fraction was obtained by centrifuging the supernatant from the AV/lysosome/mitochondrial fraction at 100,000× g for 1 h at 4°C. The pellet from this centrifugation yielded an enriched microsome fraction that predominantly contained ER but also contained small proportions of endosomes. Fractions were pelleted and either immersed in a cacodylate fixation buffer for EM or analyzed directly by Western blot or enzyme assay as described below. To isolate liver AVs, three C57BL/6 mice were starved for 12 h and injected intraperitoneally with 5 mg/100 g of body weight of vinblastine in normal saline (0.9% NaCl) 3 h before killing (Marzella et al., 1982). Livers were harvested, minced, and homogenized using a polytron Teflon homogenizer before differential and density gradient centrifugation as described above for the isolation of AVs from individual cell lines.
γ-Secretase assay and mass spectrometry
A 0.5-μg aliquot of protein from each subcellular fraction was plated onto an opaque 96-well microplate. Samples were diluted to a final concentration of 50 mM Tris, pH 6.5, 2 mM EDTA, and 0.25% CHAPS (Sigma-Aldrich). In tandem, another set of samples was pretreated with 5 μM of the selective γ-secretase inhibitor L685,458 (Bachem) for 15 min. A fluorogenic γ-secretase substrate (Nma-GGVVIATVK[DNP]-rrr-NH2; Farmery et al., 2003) was added at a 10-μM final concentration to the plate, and the samples were read in a fluorescent plate reader (Wallac Victor2; PerkinElmer) using a 355-nm excitation filter and a 440-nm emission filter over a period of 4 h. γ-Secretase activity was measured as increased fluorescence, which is achieved only when the fluorogen is cleaved (Farmery et al., 2003). Using liquid chromatography mass spectrometry, samples from the secretase assay containing solubilized membranes and peptide probe were injected onto a liquid chromatography packings reverse phase column (model 75 ID C18; Dionex). Samples were eluted over 30 min with distilled water/0.2% formic acid, and 0.2% formic acid/acetonitrile (10–40% gradient) was used as the mobile phase. The column was coupled online to an electrospray ion trap mass spectrometer (model 1100 SL; Agilent Technologies). As a control, samples were also prepared in the absence of incubation.
Online supplemental material
Table S1 details the percentage of peptide product for original peptide, non-Aβ, and Aβ-specific cleavage as determined by mass spectroscopy after incubation of the fluorogenic peptide in autophagic and lysosomal isolates at pH 6.8 and 4.5 for 1 or 24 h or in the presence of the γ-secretase inhibitor L-685,458. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200505082/DC1.
Acknowledgments
We would like to thank J.N. Peterson, N.B. Terio, and O. Grbovic for technical assistance and L. Goldberg, H. McAuliff, H. Braunstein, and G. Lardi for manuscript preparation.
This research was supported by the Alzheimer Association (grant TLL-99-1877), National Institutes of Health (grants AG17617-05 and AG021904 to A.M. Cuervo), the Howard Hughes Medical Institutes (grant to A.M. Cuervo), and the Canadian Institute of Health Research (grant to W.H. Yu).
Abbreviations used in this paper: 3MA, 3-methyladenine; Aβ, β-amyloid; AD, Alzheimer's disease; APP, Aβ precursor protein; AV, autophagic vacuole; βCTF, β–carboxy-terminal fragment; IEM, immuno-EM; KO, knockout; L/APP, murine L cell type + wild-type human alkaline phosphatase; LC3, light chain 3; mTOR, mammalian target of rapamycin; NTg, nontransgenic; PS, presenilin.
References
- Asanuma, K., I. Tanida, I. Shirato, T. Ueno, H. Takahara, T. Nishitani, E. Kominami, and Y. Tomino. 2003. MAP-LC3, a promising autophagosomal marker, is processed during the differentiation and recovery of podocytes from PAN nephrosis. FASEB J. 17:1165–1167. [DOI] [PubMed] [Google Scholar]
- Askanas, V., W.K. Engel, C.C. Yang, R.B. Alvarez, V.M. Lee, and T. Wisniewski. 1998. Light and electron microscopic immunolocalization of presenilin 1 in abnormal muscle fibers of patients with sporadic inclusion-body myositis and autosomal-recessive inclusion-body myopathy. Am. J. Pathol. 152:889–895. [PMC free article] [PubMed] [Google Scholar]
- Bendiske, J., and B.A. Bahr. 2003. Lysosomal activation is a compensatory response against protein accumulation and associated synaptopathogenesis-an approach for slowing Alzheimer disease? J. Neuropathol. Exp. Neurol. 62:451–463. [DOI] [PubMed] [Google Scholar]
- Biederbick, A., H.F. Kern, and H.P. Elsasser. 1995. Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur. J. Cell Biol. 66:3–14. [PubMed] [Google Scholar]
- Billings, L.M., S. Oddo, K.N. Green, J.L. McGaugh, and F.M. Laferla. 2005. Intraneuronal Aβ causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 45:675–688. [DOI] [PubMed] [Google Scholar]
- Boya, P., R.A. Gonzalez-Polo, N. Casares, J.L. Perfettini, P. Dessen, N. Larochette, D. Metivier, D. Meley, S. Souquere, T. Yoshimori, et al. 2005. Inhibition of macroautophagy triggers apoptosis. Mol. Cell. Biol. 25:1025–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak, H., and E. Braak. 1991. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. (Berl.). 82:239–259. [DOI] [PubMed] [Google Scholar]
- Brunk, U.T., and A. Terman. 2002. The mitochondrial-lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur. J. Biochem. 269:1996–2002. [DOI] [PubMed] [Google Scholar]
- Bursch, W. 2001. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ. 8:569–581. [DOI] [PubMed] [Google Scholar]
- Cataldo, A.M., J.L. Barnett, D.M. Mann, and R.A. Nixon. 1996. Colocalization of lysosomal hydrolase and beta-amyloid in diffuse plaques of the cerebellum and striatum in Alzheimer's disease and Down's syndrome. J. Neuropathol. Exp. Neurol. 55:704–715. [DOI] [PubMed] [Google Scholar]
- Cataldo, A.M., S. Petanceska, N.B. Terio, C.M. Peterhoff, J.C. Troncoso, R. Durham, M. Mercken, P.D. Mehta, J.D. Buxbaum, V. Haroutunian, and R.A. Nixon. 2004. a. Aβ localization to abnormal endosomes coincides with early increases in soluble Aβ in Alzheimer's disease brain. Neurobiol. Aging. 25:1263–1272. [DOI] [PubMed] [Google Scholar]
- Cataldo, A.M., C.M. Peterhoff, S.D. Schmidt, N.B. Terio, K. Duff, M. Beard, P.M. Mathews, and R.A. Nixon. 2004. b. Presenilin mutations in familial Alzheimer disease and transgenic mouse models accelerate neuronal lysosomal pathology. J. Neuropathol. Exp. Neurol. 63:821–830. [DOI] [PubMed] [Google Scholar]
- Clarke, P.G. 1990. Developmental cell death: morphological diversity and multiple mechanisms. Anat. Embryol. (Berl.). 181:195–213. [DOI] [PubMed] [Google Scholar]
- Cuervo, A.M., and J.F. Dice. 2000. Age-related decline in chaperone-mediated autophagy. J. Biol. Chem. 275:31505–31513. [DOI] [PubMed] [Google Scholar]
- Cuervo, A.M., L. Stefanis, R. Fredenburg, P.T. Lansbury, and D. Sulzer. 2004. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science. 305:1292–1295. [DOI] [PubMed] [Google Scholar]
- Culvenor, J.G., F. Maher, G. Evin, F. Malchiodi-Albedi, R. Cappai, J.R. Underwood, J.B. Davis, E.H. Karran, G.W. Roberts, K. Beyreuther, and C.L. Masters. 1997. Alzheimer's disease-associated presenilin 1 in neuronal cells: evidence for localization to the endoplasmic reticulum-Golgi intermediate compartment. J. Neurosci. Res. 49:719–731. [DOI] [PubMed] [Google Scholar]
- Cupers, P., M. Bentahir, K. Craessaerts, I. Orlans, H. Vanderstichele, P. Saftig, B. De Strooper, and W. Annaert. 2001. The discrepancy between presenilin subcellular localization and γ-secretase processing of amyloid precursor protein. J. Cell Biol. 154:731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff, K., C. Eckman, C. Zehr, X. Yu, C.M. Prada, J. Perez-tur, M. Hutton, L. Buee, Y. Harigaya, D. Yager, et al. 1996. Increased amyloid-β42(43) in brains of mice expressing mutant presenilin 1. Nature. 383:710–713. [DOI] [PubMed] [Google Scholar]
- Dunn, W.A., Jr. 1990. a. Studies on the mechanisms of autophagy: formation of the autophagic vacuole. J. Cell Biol. 110:1923–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn, W.A., Jr. 1990. b. Studies on the mechanisms of autophagy: maturation of the autophagic vacuole. J. Cell Biol. 110:1935–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edbauer, D., E. Winkler, J.T. Regula, B. Pesold, H. Steiner, and C. Haass. 2003. Reconstitution of gamma-secretase activity. Nat. Cell Biol. 5:486–488. [DOI] [PubMed] [Google Scholar]
- Farmery, M.R., L.O. Tjernberg, S.E. Pursglove, A. Bergman, B. Winblad, and J. Naslund. 2003. Partial purification and characterization of gamma-secretase from post-mortem human brain. J. Biol. Chem. 278:24277–24284. [DOI] [PubMed] [Google Scholar]
- Fevrier, B., D. Vilette, H. Laude, and G. Raposo. 2005. Exosomes: a bubble ride for prions? Traffic. 6:10–17. [DOI] [PubMed] [Google Scholar]
- Fuertes, G., J.J. Martin De Llano, A. Villarroya, A.J. Rivett, and E. Knecht. 2003. Changes in the proteolytic activities of proteasomes and lysosomes in human fibroblasts produced by serum withdrawal, amino-acid deprivation and confluent conditions. Biochem. J. 375:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, P.B., and P.O. Seglen. 1988. Prelysosomal convergence of autophagic and endocytic pathways. Biochem. Biophys. Res. Commun. 151:40–47. [DOI] [PubMed] [Google Scholar]
- Gozuacik, D., and A. Kimchi. 2004. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 23:2891–2906. [DOI] [PubMed] [Google Scholar]
- Grbovic, O.M., P.M. Mathews, Y. Jiang, S.D. Schmidt, R. Dinakar, N.B. Summers-Terio, B.P. Ceresa, R.A. Nixon, and A.M. Cataldo. 2003. Rab5-stimulated up-regulation of the endocytic pathway increases intracellular beta-cleaved amyloid precursor protein carboxyl-terminal fragment levels and Aβ production. J. Biol. Chem. 278:31261–31268. [DOI] [PubMed] [Google Scholar]
- Gyure, K.A., R. Durham, W.F. Stewart, J.E. Smialek, and J.C. Troncoso. 2001. Intraneuronal abeta-amyloid precedes development of amyloid plaques in Down syndrome. Arch. Pathol. Lab. Med. 125:489–492. [DOI] [PubMed] [Google Scholar]
- Holcomb, L., M.N. Gordon, E. McGowan, X. Yu, S. Benkovic, P. Jantzen, K. Wright, I. Saad, R. Mueller, D. Morgan, et al. 1998. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med. 4:97–100. [DOI] [PubMed] [Google Scholar]
- Hollenbeck, P.J. 1993. Products of endocytosis and autophagy are retrieved from axons by regulated retrograde organelle transport. J. Cell Biol. 121:305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, W.T., T.H. Giddings Jr., M.P. Taylor, S. Mulinyawe, M. Rabinovitch, R.R. Kopito, and K. Kirkegaard. 2005. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 10.1371/journal. pbio.0030156. [DOI] [PMC free article] [PubMed]
- Kabeya, Y., N. Mizushima, T. Ueno, A. Yamamoto, T. Kirisako, T. Noda, E. Kominami, Y. Ohsumi, and T. Yoshimori. 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19:5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya, Y., N. Mizushima, A. Yamamoto, S. Oshitani-Okamoto, Y. Ohsumi, and T. Yoshimori. 2004. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 117:2805–2812. [DOI] [PubMed] [Google Scholar]
- Kadowaki, M., and T. Kanazawa. 2003. Amino acids as regulators of proteolysis. J. Nutr. 133:2052S–2056S. [DOI] [PubMed] [Google Scholar]
- Kanazawa, T., I. Taneike, R. Akaishi, F. Yoshizawa, N. Furuya, S. Fujimura, and M. Kadowaki. 2003. Amino acids and insulin control autophagic proteolysis through different signaling pathways in relation to mTOR in isolated rat hepatocytes. J Biol Chem. 279:8452–8459. [DOI] [PubMed] [Google Scholar]
- Kanzawa, T., I.M. Germano, T. Komata, H. Ito, Y. Kondo, and S. Kondo. 2004. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 11:448–457. [DOI] [PubMed] [Google Scholar]
- Lai, M.T., E. Chen, M.C. Crouthamel, J. DiMuzio-Mower, M. Xu, Q. Huang, E. Price, R.B. Register, X.P. Shi, D.B. Donoviel, et al. 2003. Presenilin-1 and presenilin-2 exhibit distinct yet overlapping γ-secretase activities. J. Biol. Chem. 278:22475–22481. [DOI] [PubMed] [Google Scholar]
- Leuba, G., G. Wernli, A. Vernay, R. Kraftsik, M.H. Mohajeri, and K.D. Saini. 2005. Neuronal and nonneuronal quantitative BACE immunocytochemical expression in the entorhinohippocampal and frontal regions in Alzheimer's Disease. Dement. Geriatr. Cogn. Disord. 19:171–183. [DOI] [PubMed] [Google Scholar]
- Luzio, J.P., P.R. Pryor, S.R. Gray, M.J. Gratian, R.C. Piper, and N.A. Bright. 2005. Membrane traffic to and from lysosomes. Biochem. Soc. Symp. 2005:77–86. [DOI] [PubMed]
- Marzella, L., J. Ahlberg, and H. Glaumann. 1982. Isolation of autophagic vacuoles from rat liver: morphological and biochemical characterization. J. Cell Biol. 93:144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah, E., M. Mallory, T. Deerinck, R. DeTeresa, S. Lamont, A. Miller, R.D. Terry, B. Carragher, and M. Ellisman. 1993. Re-evaluation of the structural organization of the neuritic plaques in Alzheimer's disease. J. Neuropathol. Exp. Neurol. 52:619–632. [DOI] [PubMed] [Google Scholar]
- Mathews, P.M., Y. Jiang, S.D. Schmidt, O.M. Grbovic, M. Mercken, and R.A. Nixon. 2002. Calpain activity regulates the cell surface distribution of amyloid precursor protein. J. Biol. Chem. 277:36415–36424. [DOI] [PubMed] [Google Scholar]
- Matsuoka, Y., M. Picciano, J. La Francois, and K. Duff. 2001. Fibrillar β-amyloid evokes oxidative damage in a transgenic mouse model of Alzheimer's disease. Neuroscience. 104:609–613. [DOI] [PubMed] [Google Scholar]
- Meijer, A.J., and P. Codogno. 2004. Regulation and role of autophagy in mammalian cells. Int. J. Biochem. Cell Biol. 36:2445–2462. [DOI] [PubMed] [Google Scholar]
- Melendez, A., Z. Talloczy, M. Seaman, E.L. Eskelinen, D.H. Hall, and B. Levine. 2003. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science. 301:1387–1391. [DOI] [PubMed] [Google Scholar]
- Mirra, S.S., A. Heyman, D. McKeel, S.M. Sumi, B.J. Crain, L.M. Brownlee, F.S. Vogel, J.P. Hughes, G. van Belle, and L. Berg. 1991. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 41:479–486. [DOI] [PubMed] [Google Scholar]
- Mirra, S.S., M.N. Hart, and R.D. Terry. 1993. Making the diagnosis of Alzheimer's disease. A primer for practicing pathologists. Arch. Pathol. Lab. Med. 117:132–144. [PubMed] [Google Scholar]
- Mizushima, N., A. Yamamoto, M. Matsui, T. Yoshimori, and Y. Ohsumi. 2004. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell. 15:1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimore, G.E., and C.M. Schworer. 1977. Induction of autophagy by amino-acid deprivation in perfused rat liver. Nature. 270:174–176. [DOI] [PubMed] [Google Scholar]
- Munafo, D.B., and M.I. Colombo. 2002. Induction of autophagy causes dramatic changes in the subcellular distribution of GFP-Rab24. Traffic. 3:472–482. [DOI] [PubMed] [Google Scholar]
- Newell, K.L., B.T. Hyman, J.H. Growdon, and E.T. Hedley-Whyte. 1999. Application of the National Institute on Aging (NIA)-Reagan Institute criteria for the neuropathological diagnosis of Alzheimer disease. J. Neuropathol. Exp. Neurol. 58:1147–1155. [DOI] [PubMed] [Google Scholar]
- Nixon, R.A., A.M. Cataldo, and P.M. Mathews. 2000. The endosomal-lysosomal system of neurons in Alzheimer's disease pathogenesis: a review. Neurochem. Res. 25:1161–1172. [DOI] [PubMed] [Google Scholar]
- Nixon, R.A., P.M. Mathews, and A.M. Cataldo. 2001. The neuronal endosomal-lysosomal system in Alzheimer's disease. J. Alzheimers Dis. 3:97–107. [DOI] [PubMed] [Google Scholar]
- Nixon, R.A., J. Wegiel, A. Kumar, W.H. Yu, C. Peterhoff, A. Cataldo, and A.M. Cuervo. 2005. Extensive involvement of autophagy in Alzheimer disease: an immunoelectron microscopy study. J. Neuropathol. Exp. Neurol. 64:113–122. [DOI] [PubMed] [Google Scholar]
- Noda, T., and Y. Ohsumi. 1998. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 273:3963–3966. [DOI] [PubMed] [Google Scholar]
- Overly, C.C., and P.J. Hollenbeck. 1996. Dynamic organization of endocytic pathways in axons of cultured sympathetic neurons. J. Neurosci. 16:6056–6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak, S.H., R.D. Bagshaw, M. Guiral, S. Zhang, C.A. Ackerley, B.J. Pak, J.W. Callahan, and D.J. Mahuran. 2003. Presenilin-1, nicastrin, amyloid precursor protein, and γ-secretase activity are co-localized in the lysosomal membrane. J. Biol. Chem. 278:26687–26694. [DOI] [PubMed] [Google Scholar]
- Petanceska, S.S., M. Seeger, F. Checler, and S. Gandy. 2000. Mutant presenilin 1 increases the levels of Alzheimer amyloid beta-peptide Aβ42 in late compartments of the constitutive secretory pathway. J. Neurochem. 74:1878–1884. [DOI] [PubMed] [Google Scholar]
- Petiot, A., E. Ogier-Denis, E.F. Blommaart, A.J. Meijer, and P. Codogno. 2000. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. 275:992–998. [DOI] [PubMed] [Google Scholar]
- Ravikumar, B., C. Vacher, Z. Berger, J.E. Davies, S. Luo, L.G. Oroz, F. Scaravilli, D.F. Easton, R. Duden, C.J. O'Kane, and D.C. Rubinsztein. 2004. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36:585–595. [DOI] [PubMed] [Google Scholar]
- Rubinsztein, D.C., M. DiFiglia, N. Heinta, R.A. Nixon, Z.-H. Qin, B. Ravikumar, L. Stefanis, and A. Tolkovsky. 2005. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy. 1:11–22. [DOI] [PubMed] [Google Scholar]
- Seglen, P.O., and P.B. Gordon. 1984. Amino acid control of autophagic sequestration and protein degradation in isolated rat hepatocytes. J. Cell Biol. 99:435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seglen, P.O., P.B. Gordon, H. Tolleshaug, and H. Hoyvik. 1986. Use of [3H]raffinose as a specific probe of autophagic sequestration. Exp. Cell Res. 162:273–277. [DOI] [PubMed] [Google Scholar]
- Seglen, P.O., T.O. Berg, H. Blankson, M. Fengsrud, I. Holen, and P.E. Stromhaug. 1996. Structural aspects of autophagy. Adv. Exp. Med. Biol. 389:103–111. [DOI] [PubMed] [Google Scholar]
- Shigemitsu, K., Y. Tsujishita, K. Hara, M. Nanahoshi, J. Avruch, and K. Yonezawa. 1999. Regulation of translational effectors by amino acid and mammalian target of rapamycin signaling pathways. Possible involvement of autophagy in cultured hepatoma cells. J. Biol. Chem. 274:1058–1065. [DOI] [PubMed] [Google Scholar]
- Smith, D.H., K. Uryu, K.E. Saatman, J.Q. Trojanowski, and T.K. McIntosh. 2003. Protein accumulation in traumatic brain injury. Neuromolecular Med. 4:59–72. [DOI] [PubMed] [Google Scholar]
- Suzuki, K., and R.D. Terry. 1967. Fine structural localization of acid phosphatase in senile plaques in Alzheimer's presenile dementia. Acta Neuropathol. (Berl.). 8:276–284. [DOI] [PubMed] [Google Scholar]
- Takahashi, R.H., T.A. Milner, F. Li, E.E. Nam, M.A. Edgar, H. Yamaguchi, M.F. Beal, H. Xu, P. Greengard, and G.K. Gouras. 2002. Intraneuronal Alzheimer Aβ42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am. J. Pathol. 161:1869–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar, R., and M. Citron. 2000. Aβ-generating enzymes: recent advances in β- and γ-secretase research. Neuron. 27:419–422. [DOI] [PubMed] [Google Scholar]
- Wegiel, J., K.C. Wang, M. Tarnawski, and B. Lach. 2000. Microglia cells are the driving force in fibrillar plaque formation, whereas astrocytes are a leading factor in plague degradation. Acta Neuropathol. (Berl.). 100:356–364. [DOI] [PubMed] [Google Scholar]
- Yu, W.H., A. Kumar, C. Peterhoff, L. Shapiro Kulnane, Y. Uchiyama, B.T. Lamb, A.M. Cuervo, and R.A. Nixon. 2004. Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: implications for β-amyloid peptide over-production and localization in Alzheimer's disease. Int. J. Biochem. Cell Biol. 36:2531–2540. [DOI] [PubMed] [Google Scholar]