

Abstract
Macrophage death in advanced atherosclerosis promotes necrosis and plaque destabilization. A likely cause of macrophage death is accumulation of free cholesterol (FC) in the ER, leading to activation of the unfolded protein response (UPR) and C/EBP homologous protein (CHOP)–induced apoptosis. Here we show that p38 MAPK signaling is necessary for CHOP induction and apoptosis. Additionally, two other signaling pathways must cooperate with p38-CHOP to effect apoptosis. One involves the type A scavenger receptor (SRA). As evidence, FC loading by non-SRA mechanisms activates p38 and CHOP, but not apoptosis unless the SRA is engaged. The other pathway involves c-Jun NH2-terminal kinase (JNK)2, which is activated by cholesterol trafficking to the ER, but is independent of CHOP. Thus, FC-induced apoptosis requires cholesterol trafficking to the ER, which triggers p38-CHOP and JNK2, and engagement of the SRA. These findings have important implications for understanding how the UPR, MAPKs, and the SRA might conspire to cause macrophage death, lesional necrosis, and plaque destabilization in advanced atherosclerotic lesions.
Introduction
Macrophage death in advanced atherosclerotic lesions promotes lesional necrosis, which, in turn, is associated with plaque instability and acute atherothrombotic vasculature occlusion (Ball et al., 1995; Ross, 1995; Libby et al., 1996; Mitchinson et al., 1996; Kolodgie et al., 2000; Feng et al., 2003b). One of the postulated causes of macrophage death in advanced lesions is intracellular accumulation of unesterified, or “free,” cholesterol (FC) (Lupu et al., 1987; Tabas, 2002; Feng et al., 2003b). Whereas macrophages in early lesions store most lipoprotein-derived cholesterol in the esterified form because of the action of acyl-coenzyme A-cholesterol acyltransferase (ACAT), macrophages in late lesions accumulate increasing amounts of FC, probably as the result of combined deficiencies of ACAT activity and cholesterol efflux (Tabas, 2002). In various cell culture models, which use macrophages incubated with acetylated low-density lipoprotein (ac-LDL) under conditions of deficient ACAT activity, FC accumulates and induces apoptosis (Kellner-Weibel et al., 1998; Yao and Tabas, 2000, 2001). Ac-LDL is a model lipoprotein that, like various types of atherogenic lipoproteins in atherosclerotic lesions, enters macrophages by way of the type A scavenger receptor (SRA) (Henriksen et al., 1981; Brown and Goldstein, 1985). In this model, mitochondrial- and Fas-dependent pathways of apoptosis are induced as measured by caspase activation, loss of mitochondrial membrane potential, and increased TUNEL staining (Yao and Tabas, 2000, 2001).
We recently showed that FC trafficking to the ER triggers the unfolded protein response (UPR) (Feng et al., 2003a). FC enrichment of the normally cholesterol-poor and fluid ER membrane causes an increase in order parameter, or packing, of the membrane phospholipids. Stiffening of the ER membrane by this process is associated with the dysfunction of a particular integral membrane protein, sarcoendoplasmic reticulum ATPase, and it is likely that other ER membrane proteins are adversely affected (Li et al., 2004). These events almost certainly contribute to the induction of the UPR. Most importantly, FC trafficking to the ER and activation of the C/EBP homologous protein (CHOP) branch of the UPR are required for a significant portion of FC-induced apoptosis (Feng et al., 2003a). Moreover, in vivo studies support the relevance of this model to macrophage apoptosis and lesional necrosis in advanced atherosclerotic lesions (Feng et al., 2003b).
In ongoing studies in the laboratory, we found that FC loading of macrophages activates the p38 MAPK and c-Jun NH2-terminal kinase (JNK) pathways (Li et al., 2005). In view of this finding and previous literature suggesting possible links between MAPK activation and the UPR and apoptosis (Wang et al., 1996; Maytin et al., 2001; Matsuzawa et al., 2002; Yamamoto et al., 2003; Li and Holbrook, 2004), we sought to determine whether p38 and/or JNK activation played a role in these processes in FC-loaded macrophages. We found that the MAP kinase kinase 3–p38 (MKK3-p38) pathway, but not the JNK pathway, is necessary for CHOP induction. However, p38 and JNK are necessary for apoptosis in our model of FC-induced macrophage death. Most importantly, we discovered that MAPK and UPR activation are not sufficient to trigger apoptosis without another “hit,” namely engagement SRA. We found that SRA-mediated triggering of apoptosis is not unique to FC loading, and can serve as a general trigger for apoptosis in ER-stressed cells. These data provide evidence for a “multiple hit” paradigm that is related to how ER stress can lead to cellular apoptosis, and suggest a novel role for the SRA and MAPKs in macrophage death, lesional necrosis, and plaque disruption in advanced atherosclerotic lesions.
Results
MKK3/p38 MAPK activation is necessary for CHOP induction in cholesterol-enriched macrophages
Ongoing work in our laboratory has revealed that p38 MAPK is phosphorylated and activated in the early, preapoptotic stages of FC loading. p38 activation is downstream of MKK3 as indicated by the absence of p38 phosphorylation in FC-loaded macrophages from Mkk3 −/− mice (Fig. 1 A and Li et al., 2005). Because the commonly used p38 pathway inhibitors (e.g., SB203580) are inhibitors of cholesterol trafficking (unpublished data), Mkk3-deficient macrophages are an essential tool to determine the consequences of FC-induced p38 activation.
Figure 1.
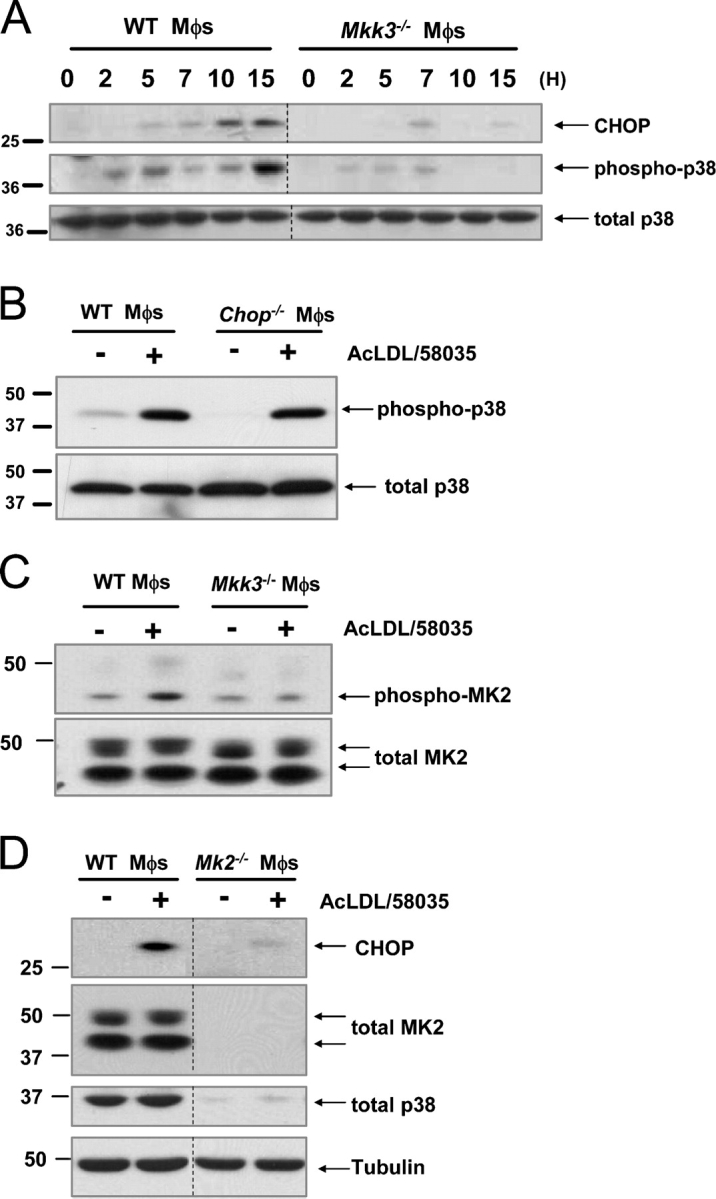
Activation of Mkk3/p38 MAPK is necessary for CHOP induction in FC-loaded macrophages. (A) WT and Mkk3 − / − macrophages (Mφs) were FC-loaded for 0, 2, 5, 7, 10, and 15 h using 100 μg/ml ac-LDL plus the ACAT inhibitor 58035. Whole cell lysates were prepared as described under “Materials and methods” and immunoblotted for CHOP (top panel), activated phospho-Thr180/Tyr182 p38 MAPK (phospho-p38, middle panel), and total p38 (bottom panel). Lines indicate areas of the same gel that were spliced together. (B) WT and Chop − / − macrophages were left untreated (−) or were FC-loaded (+) for 5 h, and lysates were immunoblotted for phospho-p38 and total p38. (C) WT and Mkk3 − / − macrophages were left untreated (−) or were FC-loaded for 7 h (+). In some experiments, WT cells were treated with ac-LDL alone, which showed only minimal MK2 phosphorylation (not depicted). Whole cell lysates were immunoblotted for activated phospho-Thr334 MK2 (phospho-MK2, top panel) and total MK2 (bottom panel). (D) WT and Mk2 − / − macrophages were left untreated (−) or were FC-loaded for 7 h (+). Whole cell lysates were immunoblotted for CHOP (top panel), total MK2 (second panel), total p38 (third panel), or tubulin as a loading control (bottom panel). Lines indicate areas of the same gel that were spliced together.
In this context, we tested whether the MKK3-p38 pathway was necessary for induction of the UPR effector, CHOP, in macrophages that were loaded with FC by the standard protocol (incubation with ac-LDL plus an ACAT inhibitor [58035]). As shown in Fig. 1 A (top panel), CHOP was induced markedly after 10 h of FC loading in macrophages from wild-type (WT) mice, but not in macrophages from Mkk3 −/− mice. Fig. 1 A (middle panel) shows that p38 was phosphorylated early in the course of FC loading in WT, but not in Mkk3 − / −-deficient, macrophages. As expected, p38 phosphorylation correlated with p38 activation, as determined by in vitro phosphorylation of the p38 substrate, ATF-2 (unpublished data). Note that p38 phosphorylation showed a biphasic, pattern: moderate expression 2–5 h after FC loading, then a decrease between 5–10 h, followed by an increase by 15 h. In contrast, CHOP expression first became substantial at 10 h after FC loading, and persisted to the 15-h time point. As a control, the blot was reprobed for total p38, which did not change substantially under any of the conditions (Fig. 1 A, bottom panel). These data are consistent with the MKK3-p38 pathway being upstream of CHOP induction. This point is supported further by the data in Fig. 1 B, which show similar p38 phosphorylation in FC-loaded WT and Chop −/− macrophages.
Several downstream targets of p38 have been identified, such as mitogen-activated protein kinase–activated protein kinase (MK)-2 (Engel et al., 1998; Rane et al., 2001), MK3 (McLaughlin et al., 1996), MK5 (Seternes et al., 2002), and mitogen and stress-activated protein kinases 1 and 2 (Deak et al., 1998). p38 was shown to be directly responsible for phosphorylation and regulation of MK2 activity and localization during conditions of cell stress, leading to up-regulation of cytokines, such as TNFα and interleukin-6 and -1 (Engel et al., 1998; Ueda et al., 2004). Therefore, we sought to determine if MK2 was activated in response to FC loading. As shown in Fig. 1 C (top panel), unloaded WT macrophages demonstrated a low basal level of MK2 phosphorylation, which was increased after 5 h of FC loading. In contrast, the basal level of MK2 phosphorylation in Mkk3 −/− macrophages was not increased with FC loading. Note in the bottom blot that both isoforms of total MK2 remained relatively constant under all conditions. MK2 phosphorylation was much less in cells that were incubated with ac-LDL alone (unpublished data). In an attempt to determine the functional importance of MK2 in FC-induced CHOP expression, we conducted a series of experiments with macrophages from Mk2 −/− mice. CHOP expression was diminished almost completely in Mk2 −/−-deficient macrophages (Fig. 1 D, top panel). However, MK2 deficiency also led to loss of p38 protein (Fig. 1 D, third panel), which probably is related to its ability to stabilize p38 in a positive feedback manner (Kotlyarov et al., 2002). Therefore, although these data cannot be used to demonstrate a direct role of MK2 in CHOP induction, they provide further evidence for a role of p38.
To determine directly whether p38 was required for CHOP induction, we repeated these experiments in p38α-deficient macrophages. The macrophages were obtained from p38αflox mice that had been crossed with LysMCre mice, which express Cre recombinase in macrophages (Clausen et al., 1999). As shown in Fig. S1 (middle panel; available at http://www.jcb.org/cgi/content/full/jcb.200502078/DC1), p38 was undetectable in these macrophages. Consistent with the data in Fig. 1, CHOP induction was diminished markedly in p38α-deficient macrophages by FC loading (Fig. S1, top panel). These data further establish the importance of p38 in CHOP induction of FC-loaded macrophages.
To determine whether an activator of the p38 pathway, other than FC loading would induce CHOP expression, we compared macrophages that were incubated under FC-loading conditions with those that were incubated with the known p38 activator, staurosporine (Yamaki et al., 2002). Treatment with 50 or 100 μM staurosporine for 6 h resulted in strong p38 activation, which was similar to that observed with 6 h of FC loading (Fig. 2, first panel). In addition, treatment with 100 μM staurosporine resulted in MK2 activation that was similar to that seen with FC loading (Fig. 2, second panel). However, neither concentration of staurosporine led to CHOP induction (Fig. 2, third panel). These data indicate that activation of the p38/MK2 pathway is not sufficient for CHOP induction.
Figure 2.

Staurosporine activates p38 MAPK and MK2, but does not induce CHOP in macrophages. Macrophages were left untreated (−), FC loaded for 6 h (+) using ac-LDL plus 58035, or treated for 6 h with 50 or 100 μM staurosporine. Immunoblot analysis was performed for phospho-p38 or phospho-MK2 (first and second panels), CHOP (third panel), and actin (bottom panel).
Other inducers of the UPR, such as the protein glycosylation inhibitor, tunicamycin, and the sarcoendoplasmic reticulum calcium ATPase inhibitor, thapsigargin, are known to activate p38 (Hung et al., 2004; Li and Holbrook, 2004). Therefore, we questioned whether p38 activation was necessary for CHOP induction by these two UPR inducers. As shown in Fig. 3 (top panel) p38 was phosphorylated after 5 h of treatment under FC-loading conditions or with tunicamycin or thapsigargin in WT macrophages. Phosphorylation of p38 under all three conditions was blocked completely in Mkk3 −/− macrophages. Most importantly, only CHOP induction by FC loading was blocked by MKK3 deficiency (Fig. 3, third panel). Thus, the requirement for p38 activation in FC-induced CHOP expression is a unique feature of this particular inducer of the UPR.
Figure 3.

MKK3-dependent CHOP induction in macrophages is unique to FC loading. WT and Mkk3 − / − macrophages were left untreated, FC loaded using ac-LDL plus 58035, or treated with 2.5 μg/ml tunicamycin or 2 μM thapsigargin for 7 h. Whole cell lysates were immunoblotted for phospho-p38 (top panel), total p38 (second panel), CHOP (third panel), and actin (bottom panel).
Activation of the p38 MAPK/MK2 pathway is necessary for FC-induced apoptosis
CHOP is required for apoptosis in macrophages that are loaded with FC by ac-LDL plus an ACAT inhibitor; MKK3 is required for the induction of CHOP under these conditions. Therefore, we hypothesized that the MKK3/p38 pathway is required for FC-induced apoptosis. As shown in Fig. 4 A, incubation of WT macrophages with ac-LDL plus 58035 caused a substantial increase in apoptosis, but apoptosis was diminished markedly in FC-loaded Mkk3 −/− macrophages. These results cannot be explained by a decrease in ac-LDL uptake or trafficking of ac-LDL–derived cholesterol to the ER in Mkk3 −/− macrophages, because ac-LDL–induced cholesterol esterification was very similar between the WT and Mkk3 −/− macrophages (Fig. 4 B). Similar results were shown using p38α-deficient (Fig. 4 C) and Mk2 −/− macrophages (Fig. S2; available at http://www.jcb.org/cgi/content/full/jcb.200502078/DC1). Therefore, activation of the MKK3/p38 pathway by FC loading of macrophages is required for CHOP induction and subsequent apoptosis.
Figure 4.
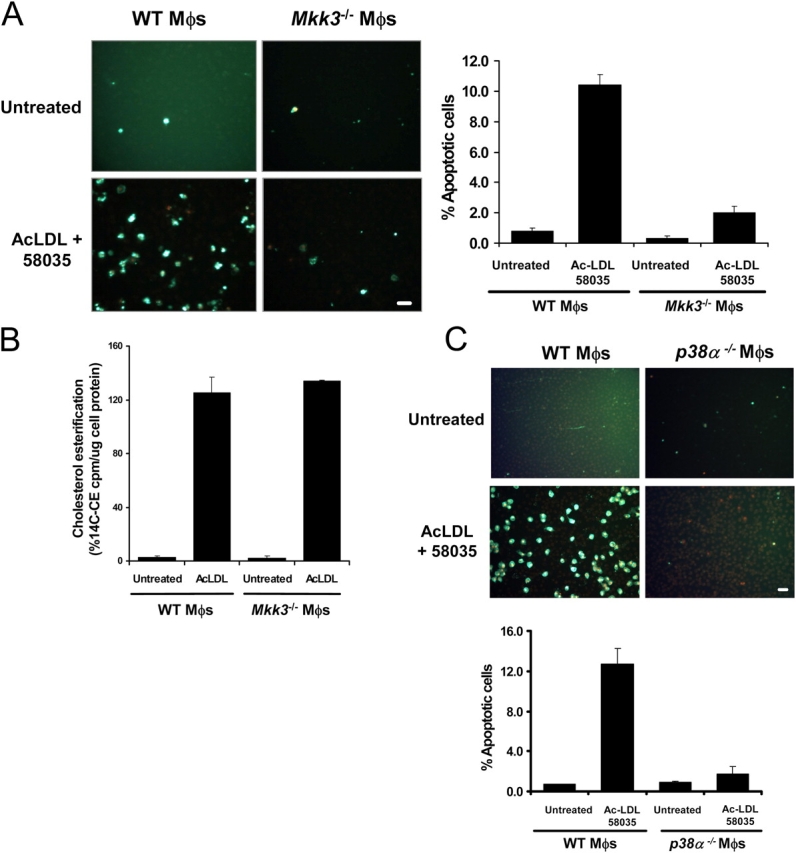
The MKK3/p38 MAPK pathway is necessary for FC-induced macrophage apoptosis. WT, Mkk3 − / − (A), and p38α-deficient (C) macrophages were left untreated or were FC loaded (ac-LDL plus 58035) for 16–18 h. The cells were stained with Alexa 488 Annexin V (green) and propidium iodide (red). Representative fluorescent images and quantitative apoptosis data from four fields of cells for each condition are shown. The data are expressed as the percent of total cells that stained with Annexin V and propidium iodide. Data are expressed as mean ± SEM (n = 4). Bar, 25 μm. (B) Esterification of [14C] oleate in WT or Mkk3 − / − macrophages incubated 5 h with medium containing [14C] oleate with or without 100 μg/ml ac-LDL. Shown is the mean ± SEM (n = 3) of cholesteryl [14C] oleate cpm per μg of total protein.
FC loading of macrophages by CD-cholesterol induces p38 phosphorylation and activates the UPR but does not induce apoptosis
Previous data from our laboratory showed that loading macrophages with cholesterol by a nonlipoprotein route—cholesterol-saturated methyl-β-cyclodextrin (CD-cholesterol) plus an ACAT inhibitor—resulted in a similar level of FC mass accumulation as that observed in macrophages that were loaded by ac-LDL plus an ACAT inhibitor (Feng et al., 2003a). FC loading by this method did not induce apoptosis, which we originally believed was due to poor trafficking of CD-derived cholesterol to the ER (Feng et al., 2003a). However, subsequent experiments revealed that cholesterol derived from CD was esterified by ACAT to a similar degree as lipoprotein-cholesterol, which indicated that the lack of apoptosis with CD-cholesterol loading required another explanation. We reasoned that elucidation of this apparent paradox could provide important insight into the mechanism of apoptosis observed in macrophages loaded with ac-LDL–derived FC.
To begin, we determined whether CD-cholesterol loading led to p38 phosphorylation and UPR induction. As shown in Fig. 5 A, p38 and MK2 were phosphorylated in CD-cholesterol–loaded macrophages. Early activation of p38 by CD-cholesterol (i.e., at 5–8 h) was observed independently of ACAT inhibition. This finding may be related to a direct effect of CD-derived cholesterol on the plasma membrane; this is in contrast to the situation with ac-LDL plus the ACAT inhibitor, which involves trafficking of lipoprotein-derived cholesterol to the ER (Li et al., 2005). However, activation of p38 at 24 h was dependent on ACAT inhibition, which suggested that this later phase of activation may be dependent on intracellular cholesterol trafficking. Most importantly, CD-cholesterol led to induction of CHOP and phospho-PERK (ds-RNA–activated protein kinase-like ER kinase) by 24 h, indicating activation of the UPR (Fig. 5 B). Given these data, we considered the possibility that the inability of CD-cholesterol plus the ACAT inhibitor to induce apoptosis, even at 48 h (unpublished data), was due to the later activation of the UPR. For example, it is possible that the gradual activation of the UPR by CD-cholesterol could “precondition” the cells, and thus, render them resistant to UPR-induced apoptosis, as has been observed in other systems (Hung et al., 2003), In this regard, we incubated ACAT-inhibited macrophages with β–very low density lipoprotein (VLDL), a cholesterol-rich lipoprotein that is endocytosed rapidly by macrophages. As shown in Fig. 5 C, FC loading using β-VLDL caused relatively rapid induction of CHOP and activation of p38, similar to the kinetics using ac-LDL; however, apoptosis was not induced (Fig. 5 D). CHOP induction by β-VLDL plus 58035 was diminished in p38α-deficient macrophages (Fig. S3; available at http://www.jcb.org/cgi/content/full/jcb.200502078/DC1), which demonstrated its dependence on p38 signaling. Thus, even rapid activation of CHOP and p38 is unable to induce apoptosis when a lipoprotein other than ac-LDL is used. Therefore, we considered an alternative hypothesis—that FC-induced p38/UPR activation is necessary, but not sufficient, for apoptosis, and apoptosis of UPR-activated macrophages requires another “hit” effected by ac-LDL, but not by CD-cholesterol or β-VLDL.
Figure 5.
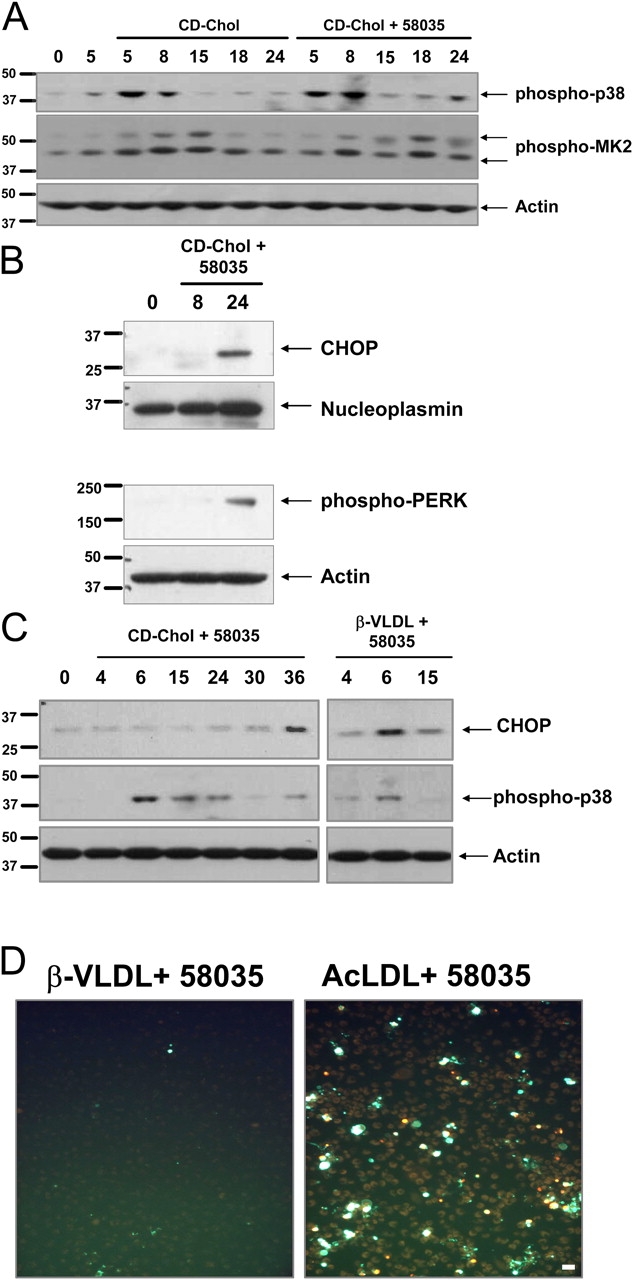
FC loading using CD-cholesterol or β-VLDL induces p38 MAPK phosphorylation and UPR induction. (A) Macrophages were left untreated for 5 h or were incubated with CD-cholesterol (CD-Chol) ± 58035 for the indicated number of hours. Whole cell lysate was immunoblotted for phospho-p38, phospho-MK2, and actin. (B) Macrophages were left untreated or incubated with CD-cholesterol plus 58035 for 8 and 24 h. Nuclear extracts were isolated as described in “Materials and methods” and were subjected to immunoblot analysis for CHOP and nucleoplasmin (as a loading control). Cytosolic extracts were immunoblotted for phospho-Thr980 PERK and actin. (C) Macrophages were incubated with CD-cholesterol or 50 μg/ml β-VLDL plus 58035 for the indicated number of hours. Whole cell lysates were subjected to immunoblot analysis for CHOP, phospho-p38, and actin. (D) Macrophages were incubated with 50 μg/ml β-VLDL or 100 μg/ml ac-LDL plus 58035 for 24 h and then stained with Alexa 488 Annexin V (green) and propidium iodide (red). Bar, 25 μm.
SRA engagement induces apoptosis in FC-enriched and UPR-activated macrophages
One of the differences between ac-LDL and CD-cholesterol or β-VLDL is that only ac-LDL is a ligand for the SRA (Goldstein et al., 1979). Thus, ac-LDL might trigger apoptosis because it delivers cholesterol and engages the SRA, whereas CD-cholesterol and β-VLDL only affect the cholesterol loading step. To test this idea, we determined whether apoptosis could be “reconstituted” by incubating macrophages with a CD-cholesterol (or β-VLDL) plus a noncholesterol-containing ligand for the SRA. As shown in Fig. 6 A, neither fucoidan, a ligand for the SRA, nor CD-cholesterol, induced a substantial degree of apoptosis when the reagents were added individually. However, the combination of reagents caused a marked apoptotic response. The data in Fig. 6 B show similar results with another SRA ligand, carboxymethyllysine-BSA (CML-BSA), that has possible relevance to diabetic macrovascular disease. CML-BSA is an advance glycation end-product (AGE) that may be formed during the oxidation of lipoproteins in the vascular wall (Fu et al., 1996; Miyazaki et al., 2002). In addition, the same results were obtained when the source of non-SRA cholesterol was β-VLDL, instead of CD-cholesterol (Fig. 6 C). To assess the relationship of CD-cholesterol/fucoidan-induced apoptosis with ac-LDL–induced apoptosis further, we tested dependency on the MKK3/p38 MAPK pathway. As shown in Fig. 6 D, CD-cholesterol/fucoidan-induced apoptosis, like ac-LDL–induced apoptosis, was decreased markedly in Mkk3 − / − macrophages; similar results were found in Mk2 − / − macrophages (not depicted).
Figure 6.
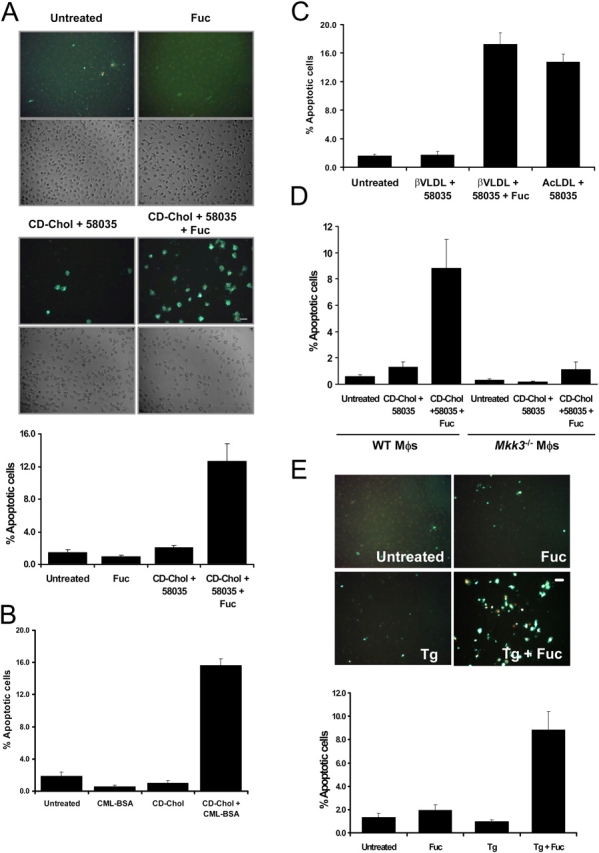
SRA ligands induce apoptosis in macrophages that are FC loaded by non-SRA sources of cholesterol, or treated with low-dose thapsigargin. (A) Macrophages were left untreated or incubated with 25 μg/ml fucoidan alone (Fuc), CD-cholesterol plus 58035 alone (CD-Chol), or CD-cholesterol/58035 plus fucoidan for 24 h. The cells were stained with Alexa 488 Annexin V (green) and propidium iodide (red). Representative fluorescent and bright field images are shown. Quantitative apoptosis data for each condition are shown as described in the legend for Fig. 4. Data are expressed as mean ± SEM (n = 8). Bar, 25 μm. (B) The experiment was conducted as in (A) except the macrophages were incubated for 24 h with 100 μg/ml of CML-BSA, CD-cholesterol/58035, or CD-cholesterol/58035 plus CML-BSA. Bar, 25 μm. As a control, FC-loaded macrophages also were treated with nonmodified BSA, which did not induce apoptosis (not depicted). (C) Quantitative apoptosis data for macrophages left untreated or incubated for 24 h with 50 μg/ml β-VLDL plus 58035, β-VLDL plus 58035 plus fucoidan (25 μg/ml), or 100 μg/ml ac-LDL plus 58035. Data are expressed as mean ± SEM (n = 8). (D) Quantitative apoptosis data for WT or Mkk3 − / − macrophages left untreated or incubated for 24 h with CD-cholesterol/58035 or CD-cholesterol/58035 plus fucoidan. Cells were stained with Alexa 488 Annexin V and propidium iodide. Representative quantitative apoptosis data are from four fields of cells under each condition. Data are expressed as mean ± SEM (n = 4). (E) Quantitative apoptosis data for macrophages left untreated or incubated for 24 h with fucoidan (25 μg/ml), 0.5 μM thapsigargin (Tg), or fucoidan plus thapsigargin. Data are expressed as mean ± SEM (n = 8). Representative fluorescent and bright field images are shown. Bar, 25 μm.
We next tested whether fucoidan could induce apoptosis in macrophages in which UPR activation was effected by means other than cholesterol. For this purpose, macrophages were incubated with thapsigargin in the absence or presence of fucoidan. Thapsigargin is an inhibitor of sarcoendoplasmic reticulum ATPase and is an inducer of the UPR (Wong et al., 1993; Bertolotti et al., 2000). As shown in Fig. 6 E, 0.5 μM thapsigargin alone and fucoidan alone caused very little apoptosis, but the combination of these two reagents led to a marked increase in macrophage apoptosis. Therefore, the ability of fucoidan to trigger apoptosis in UPR-activated cells does not depend upon cholesterol-loading per se. In this context, we also showed that the ability of fucoidan to trigger apoptosis in UPR-activated cells was dependent on CHOP; apoptosis induced by β-VLDL/58035 plus fucoidan or by thapsigargin plus fucoidan was blocked almost completely in Chop − / − macrophages (Fig. S4; available at http://www.jcb.org/cgi/content/full/jcb.200502078/DC1).
Neither fucoidan nor CML-BSA is specific for the SRA (Bucciarelli et al., 2002). Therefore, it was necessary to determine if the apoptosis-triggering effect of these molecules was dependent on the SRA. As shown in Fig. 7, CD-cholesterol/fucoidan–induced apoptosis was diminished markedly in Sra − / − macrophages. Apoptosis that was induced by β-VLDL plus fucoidan, or thapsigargin plus fucoidan, also was blocked in Sra − / − macrophages (unpublished data). Moreover, similar results were found using a blocking anti-SRA antibody with WT macrophages, which also was effective at blocking 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI)–ac-LDL uptake (Fig. S5, A and B; available at http://www.jcb.org/cgi/content/full/jcb.200502078/DC1). Thus, fucoidan and CML-BSA induce apoptosis primarily through interaction with the SRA. We also found that FC loading by ac-LDL plus 58035 was blocked completely in Sra − / − macrophages, which confirmed that ac-LDL is taken up primarily through SRA (Fig. S5 C). Thus, the ability of ac-LDL (plus 58035) to induce apoptosis in macrophages can be explained by the ability of this lipoprotein to both activate the UPR (i.e., by way of FC loading of the cell) and engage the SRA.
Figure 7.

Induction of apoptosis by fucoidan plus CD-cholesterol/58035 in macrophages requires SRA expression. WT and Sra − / − macrophages were left untreated or were incubated with CD-cholesterol (CD-Chol)/58035 plus fucoidan (Fuc) for 18 h. The cells were stained with Alexa 488 Annexin V (green) and propidium iodide (red). Representative fluorescent images are shown. Quantitative apoptosis data for each condition are shown as described in the legend for Fig. 6. Data are expressed as mean ± SEM (n = 4). Bar, 25 μm.
To determine whether SRA deficiency prevented activation of p38 or induction of CHOP by CD-cholesterol, we assayed these parameters in WT versus Sra − / − macrophages. As shown in Fig. S5, D–F, p38 phosphorylation, MK2 phosphorylation, and CHOP induction were similar in WT and Sra − / − macrophages under these conditions. Thus, FC-induced p38 activation and CHOP induction occur independently of the SRA.
In the experiments described thus far, the SRA ligand was added to the macrophages at the same time as the source of cholesterol. To determine whether addition of the SRA ligand after cholesterol loading could trigger apoptosis, macrophages were loaded with CD-cholesterol (plus 58035) for 24 h, and then treated with fucoidan for an additional 8 h. As shown in Fig. 8 A, cells that were treated with CD-cholesterol for 24 h and left untreated for an additional 8 h had very little induction of apoptosis. However, when the cholesterol-loaded cells were treated subsequently with fucoidan for 8 h, there was a marked increase in apoptosis. As expected, cells that were treated with CD-cholesterol plus fucoidan for the entire 32-h period had an even greater induction of apoptosis. Similar results were obtained using β-VLDL instead of CD-cholesterol (Fig. 8 B). Apoptosis also was seen when the macrophages were incubated with fucoidan, and then with β-VLDL plus 58035 (unpublished data). Thus, engagement of the SRA before, or subsequent to, cholesterol loading is able to induce apoptosis in FC-loaded, UPR-activated macrophages.
Figure 8.
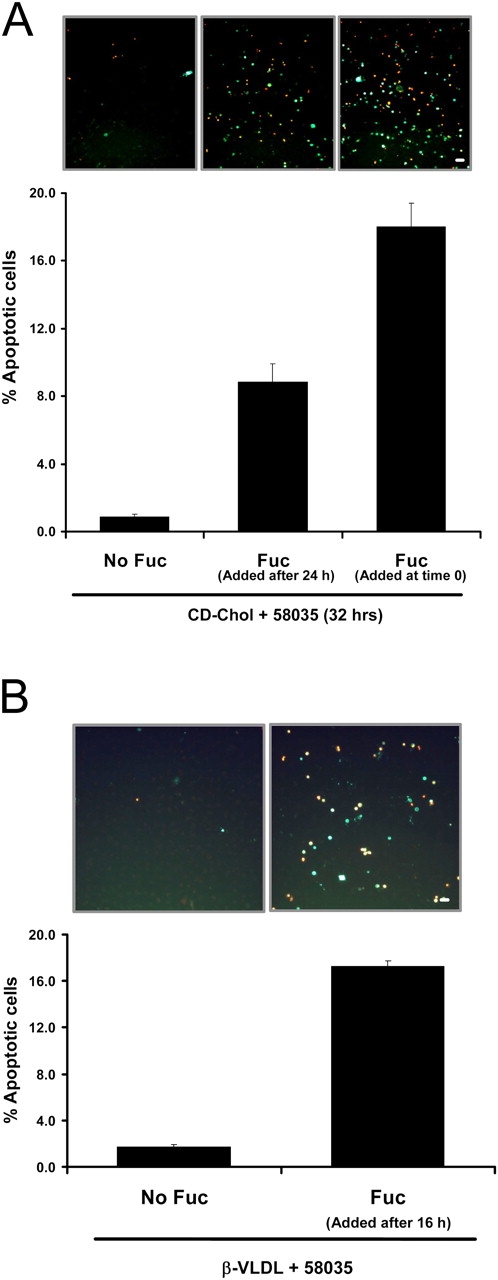
SRA engagement triggers apoptosis in macrophages that have been FC loaded before exposure to SRA ligand. (A) Macrophages were incubated with CD-cholesterol (CD-Chol)/58035 for 32 h. Some of the cells received no fucoidan (Fuc), some received fucoidan (25 μg/ml) at the beginning of the incubation period, and some received fucoidan after 24 h of incubation. (B) Macrophages were incubated with 10 μg/ml β-VLDL plus 58035 for 24 h. Some of the cells received no fucoidan and some received fucoidan after 16 h of incubation. For both sets of experiments, the cells were stained with Alexa 488 Annexin V (green) and propidium iodide (red). Representative fluorescent images are shown. Quantitative apoptosis data for each condition are shown as described in the legend for Fig. 4. Data are expressed as mean ± SEM (n = 4). Bar, 25 μm.
Activation of JNK2 is necessary for UPR-SRA–dependent apoptosis in macrophages
Previous studies suggested roles for JNK in SRA-induced cytokine production and SRA internalization (Hsu et al., 2001; Ricci et al., 2004). Therefore, we considered the possibility that JNK signaling played a role in SRA-dependent apoptosis in UPR-activated macrophages. In this regard, we showed recently that JNK is activated when macrophages are FC-loaded with ac-LDL plus 58035 (Li et al., 2005). To test a possible role for JNK in UPR-SRA–induced apoptosis, we first used the JNK inhibitor SP600125 (Bennett et al., 2001). As shown in Fig. 9, A and B, pretreatment of macrophages with SP600125 blocked apoptosis that was induced by ac-LDL plus 58035 and by thapsigargin plus fucoidan. To assess the specific role of JNK2, we used Jnk2 − / − macrophages instead of the JNK inhibitor, and found a similar inhibition of apoptosis (Fig. 9 C). Thus, JNK activation, in general, and JNK2 activation, in particular, is necessary for UPR-SRA–dependent apoptosis in macrophages.
Figure 9.

JNK2 is necessary for apoptosis, but activation is not dependent on SRA engagement. (A) Macrophages were preincubated for 30 min with 10 μM SP600125 or with vehicle and then incubated for 18 h in medium alone or medium containing ac-LDL plus 58035 ± SP600125. The cells were stained with Alexa 488 Annexin V (green) and propidium iodide (red). Representative fluorescent images and quantitative apoptosis data from four fields of cells for each condition are shown. The data are expressed as the percent of total cells that stained with Annexin V and propidium iodide. Data are expressed as mean ± SEM (n = 4). Bar, 25 μm. (B) Macrophages were preincubated for 30 min with 15 μM SP600125 (SP) or with vehicle and then incubated for 18 h in medium containing 25 μg/ml fucoidan alone (Fuc), 0.5 μM thapsigargin (Tg) alone, or both compounds. Quantitative apoptosis data for each condition are shown as described in (A). Data are expressed as mean ± SEM (n = 4). (C) WT or Jnk2 −/− macrophages were incubated for 18 h in medium alone or medium containing ac-LDL plus 58035, 0.5 μM thapsigargin, or 0.5 μM thapsigargin plus 25 μg/ml fucoidan. Quantitative apoptosis data for each condition are shown as described in (A). Data are expressed as mean ± SEM (n = 4). (D) Macrophages were incubated with 0.5 μM thapsigargin or 0.5 μM thapsigargin plus 25 μg/ml fucoidan for 0, 1, 2, and 3 h. Whole cell lysates were prepared as described in “Materials and methods,” and were immunoblotted for activated phospho-Thr 183/Tyr185 JNK (phospho-JNK, top panel) and total JNK (bottom panel). (E) Macrophages were preincubated for 30 min with medium alone or medium containing the anti-SRA antibody 2f8 or isotype control IgG2b (30 μg/ml). The cells were incubated for 3 h in medium alone or medium containing 0.5 μM thapsigargin plus 25 μg/ml fucoidan. Whole cell lysates were prepared as described in “Materials and methods,” and were immunoblotted for activated phospho-Thr 183/Tyr185 JNK (top panel), and total JNK (bottom panel). (F) WT and Jnk2 − / − macrophages (Mφs) were FC loaded for 0 or 12, and 15 h using 100 μg/ml ac-LDL plus the ACAT inhibitor 58035. Whole cell lysates were prepared as described in “Materials and methods,” and were immunoblotted for CHOP (top panel), total JNK (middle panel), and actin (bottom panel).
The original hypothesis was that JNK involvement would be related to SRA signaling. However, additional experiments revealed that JNK plays a role independent of the SRA. First, thapsigargin alone induced JNK phosphorylation, which is consistent with previous data in the literature (Li and Holbrook, 2004), and this effect was not augmented by fucoidan (Fig. 9 D). Second, pretreatment of the cells with a blocking SRA antibody (2f8), which effectively inhibited DiI–ac-LDL uptake in these cells (see Fig. S5 B), did not block JNK phosphorylation (Fig. 9 E). Third, previous work in our laboratory showed that cholesterol trafficking to the ER is necessary for JNK activation (Li et al., 2005), which further suggests that JNK is activated by ER cholesterol overload.
This last point raised an important issue—whether JNK signaling was involved in the UPR–CHOP pathway. Previous work showed that JNK activation was not downstream of CHOP, because JNK was phosphorylated to the same extent in FC-loaded Chop +/+ and Chop −/− macrophages (Li et al., 2005). To determine whether JNK was upstream of CHOP, FC-induced CHOP expression was assayed in Jnk2 −/− macrophages. As shown in Fig. 9 F, CHOP induction was the same in FC-loaded WT and Jnk2 −/− macrophages. Similar results were observed using the JNK inhibitor SB600125 (unpublished data). Thus, JNK2 activation is necessary for UPR-SRA–dependent apoptosis and, in the case of FC-loaded macrophages, is part of an ER-cholesterol pathway that is independent of the CHOP branch of the UPR.
In summary, the results of this study support a model in which at least two other “hits” must conspire with the p38-UPR-CHOP pathway to effect apoptosis in macrophages: engagement of the SRA and activation of JNK2 (Fig. 10). These hits can be activated by separate inducers, as depicted in the figure, or by the same inducer, as is the case with ac-LDL.
Figure 10.

Model of how ER stress, MAPK pathways, and SRA engagement conspire to induce apoptosis in FC-loaded macrophages. Excess FC loading of the ER induces an ER stress response that leads to activation of p38 and JNK. Activation of p38, in turn, induces the UPR effector, CHOP. Three pathways—p38-CHOP, JNK, and engagement of the SRA—combine to induce apoptosis. Ac-LDL is able to trigger all three pathways, because it delivers cholesterol to the cells and is a ligand for the SRA.
Discussion
Studies of advanced atherosclerotic plaques in experimental animals and humans showed a strong correlation between FC enrichment, macrophage death, and lesional necrosis (Ball et al., 1995; Ross, 1995; Libby et al., 1996; Mitchinson et al., 1996; Kolodgie et al., 2000; Feng et al., 2003b). Lesional necrosis, which is caused directly by macrophage death and likely results from postapoptotic necrosis of these cells in the absence of phagocytic clearance (Kockx, 1998), is believed to be a precipitating event in plaque rupture (Mitchinson et al., 1996). Plaque rupture, in turn, leads to acute atherothrombotic vascular occlusion and tissue infarction (Kolodgie et al., 2004). Thus, elucidating the mechanisms of macrophage death in advanced atherosclerotic lesions is an important step in understanding the most critical stage of atherosclerotic vascular disease.
The connection between late lesional macrophage death and FC enrichment has been suggested by several studies (Small et al., 1984; Lundberg, 1985). Our recent work in this area provided evidence for UPR activation and the necessity of the UPR effector, CHOP, in FC-induced macrophage death (Feng et al., 2003a). We have provided in vivo data suggesting the importance of this pathway in advanced lesional necrosis, and other groups also found evidence of UPR activation in advanced atherosclerotic lesions (Feng et al., 2003b; Austin et al., 2004). Nonetheless, as we probed these processes further, it became apparent that FC-induced UPR activation and apoptosis were more complex than originally appreciated. In that context, the work herein has revealed several critical steps in these processes, depicted in Fig. 10. This new model has important cell biologic and pathophysiologic implications.
Several new questions are raised by the findings in this study. For example, future studies will need to address the upstream molecules in the MKK3–p38 and JNK pathways that are stimulated by FC loading, and the downstream molecules that lead to UPR activation and apoptosis. Typically, the MKK3–p38 and JNK pathways are initiated by one of several upstream kinases, including MEKK1–4, MLK2–3, DLK, ASK1, Tpl2, and Tak1, that are activated by various forms of cellular stress (Nishitoh et al., 2002; Roux and Blenis, 2004; Kaneto et al., 2005). Examples of cellular stresses that are known to activate these kinases are oxidative stress, inflammatory cytokines, hypoxia/ischemia, and UV irradiation (Roux and Blenis, 2004). Of relevance to the current study, p38 was shown to be phosphorylated by ER stressors, such as tunicamycin and thapsigargin (Hung et al., 2004; Li and Holbrook, 2004); however, the molecules that link ER stress to upstream activators of p38 have not been identified. JNK also was shown to be activated by ER stress–in this case by an ASK1-dependent mechanism (Nishitoh et al., 2002)—but further details in this pathway remain to be elucidated. In FC-loaded macrophages, sustained p38 phosphorylation and JNK phosphorylation were inhibited by 70 nM U18666A, which selectively blocks cholesterol trafficking to the ER (Feng et al., 2003a; Li et al., 2005). However, p38 and JNK phosphorylation were not blocked in Chop − / − macrophages. These data raise the interesting possibility that a non-CHOP branch of the UPR (e.g., the Ire1–XBP-1 branch) or an ER stress pathway, other than the UPR (e.g., the PKR pathway [Gardner et al., 2005]), helps to initiate or sustain p38 or JNK phosphorylation. The molecular basis of these findings, as well as the downstream pathways linking p38 and JNK to CHOP induction and apoptosis, will be the subject of future studies.
One of the critical new issues raised by this study is the mechanism by which SRA engagement triggers apoptosis in FC-loaded/UPR-activated macrophages. Previous reports suggested possible signaling effects of the SRA. Miki et al. (1996) showed that incubation of THP-1 human macrophages with ac-LDL induced tyrosine phosphorylation of several cellular proteins. In particular, the tyrosine kinase, Lyn, was phosphorylated and activated within 30 s of ac-LDL binding, and Lyn was found to be associated with the SRA by immunoprecipitation experiments. Hsu et al. (1998) found similar results using lipoprotein and nonlipoprotein ligands of the SRA. In addition, these investigators showed that PLCγ1 and PI3-kinase were phosphorylated, and that cellular PKC activity was increased by SRA engagement. In this study, one of the downstream effects of kinase activation was induction of urokinase-type plasminogen activator (Hsu et al., 1998). However, studies by Kim et al. (2003) showed that tyrosine phosphorylation of PI3-kinase was not SRA dependent, but required CD-14. In a follow-up study, SRA engagement in J774 murine macrophages was implicated in signaling cascades involving the tyrosine kinase, Src, which activated Rac1 and p21-activated kinase. These pathways activated JNK and p38 with subsequent induction of interleukin-1 (Hsu et al., 2001). Finally, a recent study by Ricci et al. (2004) presented data that implicated JNK in the phosphorylation and endocytic function of the SRA. Our data suggest that JNK activation is necessary for ER stress–SRA-dependent apoptosis, but the role of JNK is in the ER-stress component, not the SRA component. Future work will be directed at determining whether these previously reported non-JNK SRA signaling events, or others, are involved in the apoptosis-triggering effect of SRA ligands in FC-loaded/UPR-activated macrophages.
On a more fundamental level, we will need to determine whether SRA ligand internalization is needed for the effect, and whether triggering is mediated by SRA alone or by the interaction of SRA with another “signaling receptor.” This latter scenario might occur by SRA-mediated recruitment of another receptor upon engagement, or by multivalent binding of ligands to SRA with one or more other receptors. In this regard, fucoidan and CML-BSA are known to interact with other receptors.
The multiple-hit model of apoptosis described here has potentially important cell biologic implications related to ER stress. Our current view is that ER stress strikes a delicate balance between survival and apoptosis, which, in the absence of other “hits,” favors survival. Cell survival in the initial stages of ER stress is likely necessary to give repair processes a chance to work. For example, one or more effectors downstream of PERK activation are involved in cell survival, because genetic deficiency of PERK markedly accelerates cell death, including that induced by FC loading (Feng et al., 2003a). Conversely, ER-stressed cells need to be prepared to undergo apoptosis if the repair processes do not succeed (Kadowaki et al., 2004). Thus, ER stress induces several anti- and proapoptotic processes that keep the cells alive for a while, but enable rapid onset of apoptosis when needed. In this context, FC/UPR-activated macrophages may be balanced in favor of cell survival; engagement of the SRA might tip the balance toward apoptosis by inhibiting one or more of the survival mechanisms and/or by inducing additional proapoptotic mechanisms.
The major SRA-ligand lipoprotein in atherosclerotic lesions is oxidized low density lipoprotein (LDL), but this lipoprotein has complex effects on macrophage viability. For example, at low concentrations, oxidized LDL promotes macrophage survival by induction of the Akt pathway (Hundal et al., 2001). Although higher concentrations of oxidized LDL do induce apoptosis (Wintergerst et al., 2000), it is questionable whether these high levels exist in lesions. Moreover, antioxidant trials in humans failed to show beneficial effects on coronary artery disease (Meagher and Rader, 2001). Conversely, advanced atherosclerotic lesions have massive amounts of non-SRA–ligand lipoproteins that are very effective at loading macrophages with cholesterol, such as remnant lipoproteins (which is modeled by β-VLDL) and aggregated lipoproteins (Hoff and Morton, 1985; Guyton and Klemp, 1996). Moreover, these lesions also have abundant molecules that can bind to the SRA, such as matrix proteins. Of particular interest is the finding that elevated levels of advanced glycosylation end-products (AGEs) and advanced lipoxidation end-products (Baynes and Thorpe, 2000) are found in diabetic and nondiabetic patients who have coronary artery disease (Kilhovd et al., 1999). Immunohistochemical analysis of human atherosclerotic lesions demonstrated AGE deposition extracellularly and intracellularly in macrophages (Nakamura et al., 1993). The function of AGEs as apoptosis triggers may be particular relevant given the high incidence of vulnerable plaque formation and atherothrombotic vascular disease in diabetes.
In summary, the data herein shed new light on the fundamental issue of how ER stress leads to apoptosis and on how the UPR, MAPKs, and the SRA might conspire to cause macrophage apoptosis, lesional necrosis, and plaque instability in advanced atherosclerosis. These findings will help in the design of future mechanistic and in vivo studies to elucidate further the cell biologic and pathophysiologic implications of this work.
Materials and methods
Materials
Falcon tissue culture plastic and 11-mm coverslips were purchased from Fisher Scientific. Tissue culture media, cell culture reagents, and heat-inactivated FBS (GIBCO BRL) were purchased from Invitrogen. The acyl-coenzyme A-cholesterol acyltransferase (ACAT) inhibitor 58035 (3-[decyldimethylsilyl]-N-[2-(4-methylphenyl)-1-phenylethyl]propanamide (Ross et al., 1984) was from J. Heider, formally of Sandoz (East Hanover, NJ). A 10-mg/ml stock was made in DMSO and used at a concentration of 10 μg/ml in all experiments. All other chemical reagents, including tunicamycin, thapsigargin, methyl-β-cyclodextrin (CD), cholesterol-oleate, concanavalin A, and fucoidan, were purchased from Sigma-Aldrich. Methyl-β-CD was saturated with cholesterol as previously described (Christian et al., 1997). The mouse monoclonal antibodies against GADD 153 (CHOP) and tubulin were purchased from Santa Cruz Biotechnology, Inc. Antibodies against p38 MAPK, phospho-(Thr180/Tyr182) p38 MAPK, phospho-(Thr334) MAPKAPK-2, MAPKAPK-2, phospho-(Thr183/Tyr185) c-Jun NH2-terminal kinase (JNK)1/2, and JNK1/2 were purchased from Cell Signaling Technology. The mouse monoclonal antibody to actin was purchased from CHEMICON International. Antibody 2f8 directed against the type A scavenger receptor (SRA) and isotype control antibodies were purchased from Serotec. The HRP-conjugated donkey anti–mouse and donkey anti–rabbit IgG secondary antibodies were purchased from Jackson ImmunoResearch Laboratories. The compound SP600125 was purchased from Biosource International. The Vybrant Annexin V/Propidium Iodide Apoptosis Assay kit #2 was from Molecular Probes. Carboxymethyllysine-modified (CML)-BSA (24.5% lysine residues modified as CML) and control BSA (<0.5% lysine modified as CML) were provided by S. Thorp (University of South Carolina, Columbia, SC) (Reddy et al., 1995). DiI–acetyl LDL was purchased from Molecular Probes.
Mice
Mkk3 − / − mice on a C57BL/6J background were generated as described previously (Wysk et al., 1999). Chop − / − mice on a C57BL/6J background were provided by D. Ron (New York University, New York, NY) (Zinszner et al., 1998) and R. Burke (Columbia University, New York, NY). Female Balb/c and C57BL6J mice were purchased from The Jackson Laboratory. Female Mapkapk − / − (Mk2 − / −) mice, reported previously by Kotlyarov et al. (1999), were provided by Y. Wang (UCLA, Los Angeles, California). Sra − / − (Msr − / −) mice on the C57BL/6J background were generated as described previously (Sakai et al., 1996). Macrophages from female 8–10-wk-old C57BL/6J mice were used as wild-type (WT) controls in most experiments. Jnk2 − / − mice on the C57BL/6J background were generated as described previously (Yang et al., 1998). Macrophages deficient in p38 were obtained from p38flox/flox mice (Engel et al., 2005) crossed with LysMCre/C57BL6 mice (Clausen et al., 1999).
Preparation of lipoproteins and CD-cholesterol
Low density lipoprotein (d, 1.020–1.063 g/ml) from fresh human plasma was isolated by ultracentrifugation (Havel et al., 1955). Acetylated-LDL (ac-LDL) was prepared from a reaction with acetic anhydride as described previously (Basu et al., 1976), and used at a concentration of 100 μg/ml in all experiments. β-Very low density lipoprotein (VLDL) was isolated from a New Zealand white rabbit that was fed a 2% cholesterol diet for 3 wk and fasted the night before phlebotomy (Tabas et al., 1990). CD-cholesterol was prepared as described (Feng et al., 2003a).
FC loading of mouse peritoneal macrophages
Peritoneal macrophages from adult female C57BL6J mice—and all mutant mice used in this study—were harvested 3 d after i.p. injection of concanavalin A (Feng et al., 2003a), or 4 d after i.p. injection of methyl-BSA (mBSA) in mice that were immunized previously with this antigen (Cook et al., 2003). For the latter method, 2 mg/ml mBSA in 0.9% saline was emulsified in an equal volume of Complete Freund's adjuvant (CFA; DIFCO). Mice were immunized intradermally with 100 μl emulsion. 14 d later, the immunization protocol was repeated, except incomplete Freund's adjuvant was used instead of CFA. 7 d later, the mice were injected i.p. with 0.5 ml PBS containing 100 μg mBSA. Macrophages were harvested 4 d later by peritoneal lavage. All macrophages were grown in full medium containing DME, 10% FBS, and 20% L-cell conditioned medium. The medium was replaced every 24 h until cells reached 90% confluency. On the day of the experiment, the cells were washed three times in warm PBS and incubated as described in each figure legend. WT and mutant macrophages were FC-loaded by incubation with full medium containing 10 μg/ml of the ACAT inhibitor 58035 plus 100 μl/ml of ac-LDL, 5 mM methyl-β-CD/cholesterol (5:1 mass ratio), or 10–50 μg/ml β-VLDL. Cells were labeled with DiI–ac-LDL at 10 μg/ml in DME/0.02% BSA at 37°C for 60 min. Cells were washed with PBS and incubated with 2% PFA in medium for 10 min, washed, and viewed by fluorescent microscopy as described below.
Cell death assays
After FC loading, macrophages were assayed for early- to mid-stage apoptosis by staining with Alexa 488–conjugated Annexin V (green), and for late-stage apoptosis by costaining with propidium iodide (red), as described previously (Yao and Tabas, 2000). Cells were viewed immediately at room temperature using an Olympus IX-70 inverted fluorescent microscope equipped with a mercury 100W lamp (CHIU Tech. Corp.), filter wheels, fluorescent filters (Chroma Technology Corp.), an Olympus LCPlanF1 20× objective, Imaging software (Roper Scientific), and a Cool Snap CCD camera (RS Photometrics). Representative fields (4–6 fields containing ∼1,000 cells) were photographed for each condition. The number of Annexin V/propidium iodide–positive were counted and expressed as a percent of the total number of cells in at least four separate fields from duplicate wells.
Whole-cell cholesterol esterification assay
Macrophages were incubated for 5 h with ac-LDL or 18 h with CD-cholesterol, all in the presence of 0.1 mM 14C-labeled oleate; esterification was assayed as described (Tabas et al., 1987).
Immunoblot analysis
Cells were lysed in a buffer containing 2% SDS, 62.5 mM Tris-HCl (pH 6.8), 10% glycerol, 50 mM DTT, and 0.01% bromophenol blue, and were boiled at 100°C for 5 min. Cytosolic and nuclear extracts were isolated using the Nuclear Extraction Kit (PANOMIC) according to the manufacturer's protocol. Approximately 100 μg lysate protein was separated on a 4–20% gradient SDS-PAGE gel (Invitrogen), and electrotransferred to 0.45-μm nitrocellulose membrane using a Bio-Rad Laboratories mini-transfer tank. Membranes were incubated with primary antibodies overnight, and the protein bands were detected with HRP-conjugated secondary antibodies and SuperSignal West Pico enhanced chemiluminescent solution (Pierce Chemical Co.). Membranes were stripped with Restore Western Blot Stripping Buffer (Pierce Chemical Co.) for 15 min at room temperature, and reprobed with antibodies to actin to control for differences in loading.
Statistics
Values are given as means ± SEM; no error bars in the bar graphs signify that SEM values were smaller than the graphic symbols.
Online supplemental material
Fig. S1 shows that CHOP induction by FC loading is blocked in macrophages deficient in p38 MAPK. Fig. S2 shows that apoptosis is blocked in macrophages deficient in MK2. Fig. S3 shows that p38 MAPK is necessary for CHOP induction by β-VLDL. Fig. S4 shows that CHOP is necessary for induction of apoptosis by fucoidan. Fig. S5 shows that induction of apoptosis by ac-LDL plus 58035, fucoidan, or CML-BSA in cholesterol-enriched macrophages is mediated by the SRA. Online supplemental material available at http://www.jcb.org/cgi/content/full/jcb.200502078/DC1.
Acknowledgments
We thank Drs. P. Gough and E. Raines for helpful discussions related to p38 in the early stages of the project; Drs. S. Thorpe and J. Baynes for generously providing CML-BSA; Drs. T. Kodama and H. Suzuki for creating Sra −/− mice; and Drs. K. Moore and M. Freeman for providing the Sra −/− mice on the C57BL6/J background.
This work was supported by National Institutes of Health grants DK07715 (T. DeVries-Seimon awarded through the Institute of Human Nutrition), HL75662 and HL57560 (I. Tabas), and HL062311 (Y. Wang). Dr. DeVries-Seimon also was supported by American Heart Association (AHA) Post-doctoral training grant AHA-0425805T, and Dr. Y. Li was supported by AHA grant 0435364T.
The authors have no conflicting financial interests.
Abbreviations used in this paper: ACAT, acyl-coenzyme A-cholesterol acyltransferase; ac-LDL, acetylated low density lipoprotein; AGE, advanced glycation end-product; CD, cyclodextrin; CHOP, C/EBP homologous protein; CML-BSA, carboxymethyllysine modified BSA; DiI, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate; FC, free cholesterol; JNK, c-Jun NH2-terminal kinase; LDL, low density lipoprotein; MK2, mitogen-activated protein kinase–activated protein kinase 2; MKK3, MAP kinase kinase 3; PERK, ds-RNA–activated protein kinase-like ER kinase; SRA, scavenger receptor type A; UPR, unfolded protein response; VLDL, very low density lipoprotein.
References
- Austin, R.C., S. Lentz, and G. Werstuck. 2004. Role of hyperhomocysteinemail in endothelial dysfunction and atherothrombotic disease. Cell Death Differ. 11:S56–64. [DOI] [PubMed] [Google Scholar]
- Ball, R.Y., E.C. Stowers, J.H. Burton, N.R.B. Cary, J.N. Skepper, and M.J. Mitchinson. 1995. Evidence that the death of macrophage foam cells contributes to the lipid core of atheroma. Atherosclerosis. 114:45–54. [DOI] [PubMed] [Google Scholar]
- Basu, SK, J.L. Goldstein, G.W. Anderson, and M.S. Brown. 1976. Degradation of cationized low density lipoprotein and regulation of cholesterol metabolism in homozygous familial hypercholesterolemia fibroblasts. Proc. Natl. Acad. Sci. USA. 73:3178–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baynes, J.W., and S.R. Thorpe. 2000. Glycoxidation and lipoxidation in atherogenesis. Free Radic. Biol. Med. 28:1708–1716. [DOI] [PubMed] [Google Scholar]
- Bennett, B.L., D.T. Sasaki, B.W. Murray, E.C. O'Leary, S.T. Sakata, W. Xu, J.C. Leisten, A. Motiwala, S. Pierce, Y. Satoh, et al. 2001. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA. 98:13681–13686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotti, A., Y. Zhang, L.M. Hendershot, H.P. Harding, and D. Ron. 2000. Dynamic interaction of BiP and ER stress transducers in the unfolded protein response. Nat. Cell Biol. 2:326–332. [DOI] [PubMed] [Google Scholar]
- Brown, M., and J.L. Goldstein. 1985. Scavenger cell receptor shared. Nature. 316:680–681. [DOI] [PubMed] [Google Scholar]
- Bucciarelli, L.G., T. Wendt, W. Qu, Y. Lu, E. Lalla, L.L. Rong, M.T. Goova, B. Moser, T. Kislinger, D.C. Lee, et al. 2002. RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation. 106:2827–2835. [DOI] [PubMed] [Google Scholar]
- Christian, A., P. Haynes, M. Phillips, and G.H. Rothblat. 1997. Use of cyclodextrins for manipulating cellular cholesterol content. J. Lipid Res. 38:2264–2272. [PubMed] [Google Scholar]
- Clausen, B.E, D. Burkhardt, W. Reith, R. Renkawitz, and I. Forster. 1999. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 4:265–277. [DOI] [PubMed] [Google Scholar]
- Cook, A.D., E.L. Braine, and J.A. Hamilton. 2003. The phenotype of inflammatory macrophages is stimulus dependent: implications for the nature of the inflammatory response. J. Immunol. 171:4816–4823. [DOI] [PubMed] [Google Scholar]
- Deak, M., A.D. Clifton, J. Lucocq, and D. Alessi. 1998. Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 17:4426–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel, F.B., M. Schebesta, M.T. Duong, G. Lu, S. Ren, J.B. Madwed, H. Jiang, Y. Wang, and M.T. Keating. 2005. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 19:1175–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel, K., A. Kotlyarov, and M. Gaestel. 1998. Leptomycin B-sensitive nuclear export of MAPKAP kinase 2 is regulated by phosphorylation. EMBO J. 17:3363–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, B., P. Yao, Y. Li, C. Devlin, D. Zhang, H.P. Harding, M. Sweeney, J. Rong, G. Kuriakose, E. Fisher, et al. 2003. a. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat. Cell Biol. 5:781–792. [DOI] [PubMed] [Google Scholar]
- Feng, B., D. Zhang, G. Kuriakose, C.M. Devlin, M. Kockx, and I. Tabas. 2003. b. Niemann-Pick C heterozygosity confers resistance to lesional necrosis and macrophage apoptosis in murine atherosclerosis. Proc. Natl. Acad. Sci. USA. 100:10423–10428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, M.X., J.s.R. Requena, A.J. Jenkins, T.J. Lyons, J.W. Baynes, and S.R. Thorpe. 1996. The advanced glycation end product, N-(Carboxymethyl)lysine, is a product of both lipid peroxidation and glycoxidation reactions. J. Biol. Chem. 271:9982–9986. [DOI] [PubMed] [Google Scholar]
- Gardner, O.S., C.W. Shiau, C.S. Chen, and L.M. Graves. 2005. PPARγ-independent activation of p38 MAPK by thiazolidinediones involves calcium/calmodulin-dependent protein kinase II and protein kinase R: correlation with endoplasmic reticulum stress. J. Biol. Chem. 280(11):10109–10118. [DOI] [PubMed] [Google Scholar]
- Goldstein, J., Y. Ho, S. Basu, and M. Brown. 1979. Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc. Natl. Acad. Sci. USA. 76:333–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyton, J.R., and K.F. Klemp. 1996. Development of the lipid-rich core of human atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 16:4–11. [DOI] [PubMed] [Google Scholar]
- Havel, R., H. Eder, and J. Bragdon. 1955. The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J. Clin. Invest. 34:1345–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen, T., E. Mahoney, and D. Steinberg. 1981. Enhanced macrophage degradation of low density lipoprotein previously incubated with cultured endothelial cells: recognition by receptors for acetylated low density lipoproteins. Proc. Natl. Acad. Sci. USA. 78:6499–6503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff, H.F., and R.E. Morton. 1985. Lipoproteins containing apo B extracted from human aortas. Structure and function. Ann. NY Acad. Sci. 454:183–194. [DOI] [PubMed] [Google Scholar]
- Hsu, H.Y., D.P. Hajjar, K.M. Khan, and D.J. Falcone. 1998. Ligand binding to macrophage scavenger receptor-A induces urokinase-type plasminogen activator expression by a protein kinase-dependent signaling pathway. J. Biol. Chem. 273:1240–1246. [DOI] [PubMed] [Google Scholar]
- Hsu, H.-Y., S.-L. Chiu, M.-H. Wen, K.-Y. Chen, and K.-F. Hua. 2001. Ligands of macrophage scavenger receptor induce cytokine expression via differential modulation of protein kinase signaling pathways. J. Biol. Chem. 276:28719–28730. [DOI] [PubMed] [Google Scholar]
- Hundal, R.S., B.S. Salh, J.W. Schrader, A. Gomez-Munoz, V. Duronio, and U.P. Steinbrecher. 2001. Oxidized low density lipoprotein inhibits macrophage apoptosis through activation of the PI 3-kinase/PKB pathway. J. Lipid Res. 42:1483–1491. [PubMed] [Google Scholar]
- Hung, C.C., T. Ichimura, J.L. Stevens, and J.V. Bonventre. 2003. Protection of renal epithelial cells against oxidative injury by endoplasmic reticulum stress preconditioning is mediated by ERK1/2 activation. J. Biol. Chem. 278:29317–29326. [DOI] [PubMed] [Google Scholar]
- Hung, J.H., I.J. Su, H.Y. Lei, H.C. Wang, W.C. Lin, W.T. Chang, W. Huang, W.C. Chang, Y.S. Chang, C.C. Chen, and M.D. Lai. 2004. Endoplasmic reticulum stress stimulates the expression of cyclooxygenase-2 through activation of NF-κB and p38 mitogen-activated protein kinase. J. Biol. Chem. 279:46384–46392. [DOI] [PubMed] [Google Scholar]
- Kadowaki, H., H. Nishitoh, and H. Ichijo. 2004. Survival and apoptosis signals in ER stress: the role of protein kinases. J. Chem. Neuroanat. 28:93–100. [DOI] [PubMed] [Google Scholar]
- Kaneto, H., T.A. Matsuoka, Y. Nakatani, D. Kawamori, T. Miyatsuka, M. Matsuhisa, and Y. Yamasaki. 2005. Oxidative stress, ER stress, and the JNK pathway in type 2 diabetes. J. Mol. Med. 83:429–439. [DOI] [PubMed] [Google Scholar]
- Kellner-Weibel, G., W.G. Jerome, D.M. Small, G.J. Warner, J.K. Stoltenborg, M.A. Kearney, M.H. Corjay, M.C. Phillips, and G.H. Rothblat. 1998. Effects of intracellular free cholesterol accumulation on macrophage viability: a model for foam cell death. Arterioscler. Thromb. Vasc. Biol. 18:423–431. [DOI] [PubMed] [Google Scholar]
- Kilhovd, B., T. Berg, K. Birkeland, P. Thorsby, and K. Hanssen. 1999. Serum levels of advanced glycation end products are increased in patients with type-2 diabetes and coronary heart disease. Diabetes Care. 22:1543–1548. [DOI] [PubMed] [Google Scholar]
- Kim, W.S., C.M. Ordija, and M.W. Freeman. 2003. Activation of signaling pathways by putative scavenger receptor class A (SR-A) ligands requires CD14 but not SR-A. Biochem. Biophys. Res. Commun. 310:542–549. [DOI] [PubMed] [Google Scholar]
- Kockx, M.M. 1998. Apoptosis in the atherosclerotic plaque: quantitative and qualitative aspects. Arterioscler. Thromb. Vasc. Biol. 18:1519–1522. [DOI] [PubMed] [Google Scholar]
- Kolodgie, F.D., J. Narula, A.P. Burke, N. Haider, A. Farb, Y. Hui-Liang, J. Smialek, and R. Virmani. 2000. Localization of apoptotic macrophages at the site of plaque rupture in sudden coronary death. Am. J. Pathol. 157:1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodgie, F.D., R. Virmani, A.P. Burke, A. Farb, D.K. Weber, R. Kutys, A.V. Finn, and H.K. Gold. 2004. Pathologic assessment of the vulnerable human coronary plaque. Heart. 90:1385–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotlyarov, A., A. Neininger, C. Schubert, R. Eckert, C. Birchmeier, H.D. Volk, and M. Gaestel. 1999. MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nat. Cell Biol. 1:94–97. [DOI] [PubMed] [Google Scholar]
- Kotlyarov, A., Y. Yannoni, S. Fritz, K. Laass, J.B. Telliez, D. Pitman, L.L. Lin, and M. Gaestel. 2002. Distinct cellular functions of MK2. Mol. Cell. Biol. 22:4827–4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J., and N.J. Holbrook. 2004. Elevated gadd153/chop expression and enhanced c-Jun N-terminal protein kinase activation sensitizes aged cells to ER stress. Exp. Gerontol. 39:735–744. [DOI] [PubMed] [Google Scholar]
- Li, Y., M. Ge, L. Ciani, G. Kuriakose, E.J. Westover, M. Dura, D.F. Covey, J.H. Freed, F.R. Maxfield, J. Lytton, and I. Tabas. 2004. Enrichment of endoplasmic reticulum with cholesterol inhibits sarcoplasmic-endoplasmic reticulum calcium ATPase-2b activity in parallel with increased order of membrane lipids. J. Biol. Chem. 279:37030–37039. [DOI] [PubMed] [Google Scholar]
- Li, Y., R.F. Schwabe, T. DeVries-Seimon, P.M. Yao, M.C. Gerbod-Giannone, A.R. Tall, R.J. Davis, R. Flavell, D.A. Brenner, and I. Tabas. 2005. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-α and interleukin-6. J. Biol. Chem. 280:21763–21772. [DOI] [PubMed] [Google Scholar]
- Libby, P., Y. Geng, M. Aikawa, U. Schoenbeck, F. Mach, S. Clinton, G. Sukhova, and R. Lee. 1996. Macrophages and atherosclerotic plaque stability. Curr. Opin. Lipidol. 7:330–335. [DOI] [PubMed] [Google Scholar]
- Lundberg, B. 1985. Chemical composition and physical state of lipid deposits in atherosclerosis. Atherosclerosis. 56:93–110. [DOI] [PubMed] [Google Scholar]
- Lupu, F., I. Danaricu, and N. Simionescu. 1987. Development of intracellular lipid deposits in the lipid laden cells of atherosclerotic lesions. A cytochemical and ultrastructural study. Atherosclerosis. 67:127–142. [DOI] [PubMed] [Google Scholar]
- Matsuzawa, A., H. Nishitoh, K. Tobiume, K. Takeda, and H. Ichijo. 2002. Physiological roles of ASK1-mediated signal transduction in oxidative stress- and endoplasmic reticulum stress-induced apoptosis: advanced findings from ASK1 knockout mice. Antioxid. Redox Signal. 4:415–425. [DOI] [PubMed] [Google Scholar]
- Maytin, E.V., M. Ubeda, J.C. Lin, and J.F. Habener. 2001. Stress-inducible transcription factor CHOP/gadd153 induces apoptosis in mammalian cells via p38 kinase-dependent and independent mechanisms. Exp. Cell Res. 267:193–204. [DOI] [PubMed] [Google Scholar]
- McLaughlin, M.M., S. Kumar, P.C. McDonnell, S. Van Horn, J.C. Lee, G.P. Livi, and P.R. Young. 1996. Identification of mitogen-activated protein (MAP) kinase-activated protein kinase-3, a novel substrate of CSBP p38 MAP kinase. J. Biol. Chem. 271:8488–8492. [DOI] [PubMed] [Google Scholar]
- Meagher, E., and D. Rader. 2001. Antioxidant therapy and atherosclerosis: animal and human studies. Trends Cardiovasc. Med. 11:162–165. [DOI] [PubMed] [Google Scholar]
- Miki, S., S. Tsukada, S. Aimoto, H. Hojo, B. Sato, M. Yamamoto, and Y. Miki. 1996. Functional and possible physical association of scavenger receptor with cytoplasmic tyrosine kinase Lyn in monocytic THP-1-derived macrophages. FEBS Lett. 399:241–244. [DOI] [PubMed] [Google Scholar]
- Mitchinson, M.J., S. Hardwick, and M. Bennett. 1996. Cell death in atherosclerotic plaques. Curr. Opin. Lipidol. 7:324–329. [DOI] [PubMed] [Google Scholar]
- Miyazaki, A., H. Nakayama, and S. Horiuchi. 2002. Scavenger receptors that recognize advanced glycation end products. Trends Cardiovasc. Med. 12:258–262. [DOI] [PubMed] [Google Scholar]
- Nakamura, Y., Y. Horii, and T. Nishino. 1993. Immunohistochemical localization of advanced glycation end products in coronary atheroma and cardiac tissue in diabetes mellitus. Am. J. Pathol. 143:1649–1656. [PMC free article] [PubMed] [Google Scholar]
- Nishitoh, H., A. Matsuzawa, K. Tobiume, K. Saegusa, K. Takeda, K. Inoue, S. Hori, A. Kakizuka, and H. Ichijo. 2002. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 16:1345–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rane, M.J., P.Y. Coxon, D.W. Powell, R. Webster, J.B. Klein, W. Pierce, P. Ping, and K.R. McLeish. 2001. p38 Kinase-dependent MAPKAPK-2 activation functions as 3-phosphoinositide-dependent kinase-2 for Akt in human neutrophils. J. Biol. Chem. 276:3517–3523. [DOI] [PubMed] [Google Scholar]
- Reddy, H., J. Bichler, K.J. Wells-Knecht, S.R. Thorpe, and J.W. Baynes. 1995. N epsilon-(carboxymethyl)lysine is a dominant advanced glycation end product (AGE) antigen in tissue proteins. Biochemistry. 34:10872–10878. [DOI] [PubMed] [Google Scholar]
- Ricci, R., G. Sumara, I. Sumara, I. Rozenberg, M. Kurrer, A. Akhmedov, M. Hersberger, U. Eriksson, F.R. Eberli, B. Becher, et al. 2004. Requirement of JNK2 for scavenger receptor A-mediated foam cell formation in atherogenesis. Science. 306:1558–1561. [DOI] [PubMed] [Google Scholar]
- Ross, A.C., K.J. Go, J.G. Heider, and G.H. Rothblat. 1984. Selective inhibition of acyl coenzyme A: cholesterol acyltransferase by compound 58-035. J. Biol. Chem. 259:815–819. [PubMed] [Google Scholar]
- Ross, R. 1995. Cell biology of atherosclerosis. Ann. Rev. Physiol. 57:791–804. [DOI] [PubMed] [Google Scholar]
- Roux, P.P., and J. Blenis. 2004. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 68:320–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai, M., A. Miyazaki, H. Hakamata, T. Kodama, H. Suzuki, S. Kobori, M. Shichiri, and S. Horiuchi. 1996. The scavenger receptor serves as a route for internalization of lysophosphatidylcholine in oxidized low density lipoprotein-induced macrophage proliferation. J. Biol. Chem. 271:27346–27352. [DOI] [PubMed] [Google Scholar]
- Seternes, O.M., B. Johansen, B. Hegge, M. Johannessen, S.M. Keyse, and U. Moens. 2002. Both binding and activation of p38 mitogen-activated protein kinase (MAPK) play essential roles in regulation of the nucleocytoplasmic distribution of MAPK-activated protein kinase 5 by cellular stress. Mol. Cell. Biol. 22:6931–6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small, D.M., M.G. Bond, D. Waugh, M. Prack, and J.K. Sawyer. 1984. Physicochemical and histological changes in the arterial wall of nonhuman primates during progression and regression of atherosclerosis. J. Clin. Invest. 73:1590–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas, I. 2002. Consequences of cellular cholesterol accumulation: basic concepts and physiological implications. J. Clin. Invest. 110:905–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas, I., G. Boykow, and A. Tall. 1987. Foam cell-forming J774 macrophages have markedly elevated acyl coenzyme A: cholesterol acyl transferase activity compared with mouse peritoneal macrophages in the presence of low density lipoprotein (LDL) despite similar receptor activity. J. Clin. Invest. 79:418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas, I., S. Lim, X.X. Xu, and F.R. Maxfield. 1990. Endocytosed beta-VLDL and LDL are delivered to different intracellular vesicles in mouse peritoneal macrophages. J. Cell Biol. 111:929–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda, K., H. Kosako, Y. Fukui, and S. Hattori. 2004. Proteomic identification of Bcl2-associated athanogene 2 as a novel MAPK-activated protein kinase 2 substrate. J. Biol. Chem. 279:41815–41821. [DOI] [PubMed] [Google Scholar]
- Wang, X., B. Lawson, J. Brewer, H. Zinszner, A. Sanjay, L. Mi, R. Boorstein, G. Kreibich, L. Hendershot, and D. Ron. 1996. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153). Mol. Cell. Biol. 16:4273–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wintergerst, E.S., J. Jelk, C. Rahner, and R. Asmis. 2000. Apoptosis induced by oxidized low density lipoprotein in human monocyte-derived macrophages involves CD36 and activation of caspase-3. Eur. J. Biochem. 267:6050–6059. [DOI] [PubMed] [Google Scholar]
- Wong, W.L., M.A. Brostrom, G. Kuznetsov, D. Gmitter-Yellen, and C.O. Brostrom. 1993. Inhibition of protein synthesis and early protein processing by thapsigargin in cultured cells. Biochem. J. 289:71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysk, M., D.D. Yang, H.-T. Lu, R.A. Flavell, and R.J. Davis. 1999. Requirement of mitogen-activated protein kinase kinase 3 (MKK3) for tumor necrosis factor-induced cytokine expression. Proc. Natl. Acad. Sci. USA. 96:3763–3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaki, K., J. Hong, K. Hiraizumi, J.W. Ahn, O. Zee, and K. Ohuchi. 2002. Participation of various kinases in staurosporine-induced apoptosis of RAW 264.7 cells. J. Pharm. Pharmacol. 54:1535–1544. [DOI] [PubMed] [Google Scholar]
- Yamamoto, K., H. Hamada, H. Shinkai, Y. Kohno, H. Koseki, and T. Aoe. 2003. The KDEL receptor modulates the endoplasmic reticulum stress response through mitogen-activated protein kinase signaling cascades. J. Biol. Chem. 278:34525–34532. [DOI] [PubMed] [Google Scholar]
- Yang, D., D. Conze, A.J. Whitmarsh, T. Barrett, R.J. Davis, M. Rincon, and R. Flavell. 1998. Differentiation of CD4+ T cells to Th1 cells requires MAP kinase JNK2. Immunity. 9:575–585. [DOI] [PubMed] [Google Scholar]
- Yao, P.M., and I. Tabas. 2000. Free cholesterol loading of macrophages induces apoptosis involving the Fas pathway. J. Biol. Chem. 275:23807–23813. [DOI] [PubMed] [Google Scholar]
- Yao, P.M., and I. Tabas. 2001. Free cholesterol loading of macrophages is associated with widespread mitochondrial dysfunction and activation of the mitochondrial apoptosis pathway. J. Biol. Chem. 276:42468–42476. [DOI] [PubMed] [Google Scholar]
- Zinszner, H., M. Kuroda, X. Wang, N. Batchvarova, R.T. Lightfoot, H. Remotti, J.L. Stevens, and D. Ron. 1998. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 12:982–995. [DOI] [PMC free article] [PubMed] [Google Scholar]