

Abstract
Amodel that explains both the origin and sporadic nature of cancer argues that cancer cells are a chance result of events that cause genomic and epigenetic variability. The prevailing view is that these events are mutations that affect chromosome segregation or stability. However, genomic and epigenetic variability is also triggered by cell fusion, which is often caused by viruses. Yet, cells fused by viruses are considered harmless because they die. We provide evidence that a primate virus uses both viral and exosomal proteins involved in cell fusion to produce transformed proliferating human cells. Although normal cells indeed fail to proliferate after fusion, expression of an oncogene or a mutated tumor suppressor p53 in just one of the fusion partners is sufficient to produce heterogeneous progeny. We also show that this virus can produce viable oncogenically transformed cells by fusing cells that are otherwise destined to die. Therefore, we argue that viruses can contribute to carcinogenesis by fusing cells.
Introduction
Two models have been provided to explain the sporadic nature of cancer. One posits that cancer is a result of accumulated random mutations and the other that it is a consequence of events that cause genomic instability. The second model was formulated a century ago (Hansemann, 1890; for review see Boveri, 1929) by postulating that cancer results from aberrant mitoses that produce aneuploid cells, in which chromosomes are abnormal in their number or structure. Although most of these abnormalities are lethal, this model argues that some of their combinations produce a cancerous cell. This model has regained interest because of the accumulating evidence that chromosomes of tumor cells are nearly invariantly abnormal in their number, structure, or both, although whether aneuploidy is a cause or consequence of oncogenic transformation is still being debated (for review see Duesberg et al., 2004; Rajagopalan and Lengauer, 2004).
Mutations that affect cell cycle progression, chromosomes, or the mitotic machinery are often considered to be the main, if not exclusive, causes of aneuploidy (Rajagopalan and Lengauer, 2004). However, another well-documented cause is cell fusion, which produces a wide range of chromosomal aberrations, including chromosomal loss, chromosome disjunctions, and translocations (Ringertz and Savage, 1976; for review see Duelli and Lazebnik, 2003). Although mutations and cell fusion can both lead to aneuploidy, progeny of cell fusion may be more viable and diverse. Fusion combines centrosomes of the fusion partners, which increases the likelihood of multiple mitoses that often produce aneuploid cells. Fusion also immediately doubles the number of chromosomes, thereby decreasing the chances that the loss of a chromosome will kill the cell. Furthermore, fusion of cells that are in different phases of the cell cycle often results in premature chromosome condensation, which leads to massive chromosome fragmentation, with the fragments distributed among the daughter cells (Ringertz and Savage, 1976). Finally, fusion between phenotypically distinct cells produces hybrids that not only are genetically diverse but also have unique sets of properties resulting from an apparently random rearrangement of epigenetic and other regulatory networks of the fusion partners (Ringertz and Savage, 1976).
Given that cell fusion causes aneuploidy and that aneuploidy may cause cancer (Rajagopalan and Lengauer, 2004), it follows that cell fusion has the potential to produce cancerous cells. This hypothesis was also proposed a century ago (Klebs, 1887; Aichel, 1911), based on the observation that both cell hybrids and cancer cells are aneuploid and on a conjecture, which proved to be correct, that both fusion and oncogenic transformation cause an unusual reassortment of epigenetic regulatory mechanisms. More recent studies have provided a substantial body of evidence in support of this model, including the observation that the metastatic potential of tumor cells can be increased by fusing them with macrophages or dendritic cells (for review see Duelli and Lazebnik, 2003).
In the body, fused cells originate from physiological and illicit cell fusion. Physiological fusion is required to produce the zygote and several types of multinuclear somatic cells, such as myotubes and osteoclasts (for review see Chen and Olson, 2005). Mechanisms of physiological cell fusion are only beginning to emerge but already prove to be complex (for review see Chen and Olson, 2005). Importantly, with the exception of the zygote and, perhaps, hybrids produced from stem cells (O'Malley and Scott, 2004), cells produced by physiological cell fusion are terminally differentiated and do not proliferate.
Illicit cell fusion is caused by a variety of agents, including viruses and chemicals, or by abnormal expression of cellular proteins. Cell fusion caused by such agents as polyethylene glycol (PEG) or inactivated Sendai virus has been used routinely as an experimental tool. Despite the evidence that experimental cell fusion can produce abnormal cells, such as hybridomas or radiation hybrids, the potential pathological consequences of illicit cell fusion are poorly understood.
Viruses are of particular interest as pathological fusogens because they are ubiquitous, diverse, and amplifiable; can persist in cells inconspicuously; and have been implicated in carcinogenesis. Cell fusion is a side effect of viral proteins, such as retroviral Env, that mediate fusion between the virus and the plasma membrane.
If intact viruses can produce aneuploid hybrids in vivo, as inactivated Sendai virus does in vitro, then viruses should be considered causative agents of aneuploidy, even if they are otherwise harmless. However, a current view is that cells fused by intact viruses either die or do not proliferate (Zhivotovsky and Kroemer, 2004). This view has been established sufficiently to use virus-induced cell fusion to kill cancer cells as a therapeutic approach (Peng et al., 2002). The cytotoxic and cytostatic effects of virus-induced fusion are attributable to a response either to fusion by itself or to the resulting aneuploidy, as well as the response to the virus (Zhivotovsky and Kroemer, 2004).
We report that a primate virus that shares properties with exosomes can produce transformed proliferating hybrids from human cells. Although normal cells indeed failed to proliferate after fusion with this virus, they did produce heterogeneous proliferating progeny if one of the fusion partners expressed an oncogene or a mutant of the tumor suppressor p53. We also demonstrate that this virus produced viable oncogenically transformed cells by fusing cells destined to die. Overall, we argue that cell fusion can be one of the processes that links viruses and carcinogenesis.
Results
This study resulted from using cell fusion as a tool to investigate how expression of the oncogenes Myc or adenoviral E1A sensitizes cells to chemotherapy-induced apoptosis (Duelli and Lazebnik, 2000). In one of the experiments, we needed to monitor cells produced by fusion of normal and E1A-expressing cells. We used PEG to fuse normal human fibroblasts that expressed a puromycin-resistance gene (I0P cells) with fibroblasts that expressed both a hygromycin-resistance gene and E1A (IEH cells). As expected, I0P cells were killed by hygromycin and IEH cells by puromycin (Fig. 1 A). Therefore, we expected only hybrids produced by PEG to proliferate in the presence of both drugs. To our surprise, we found that the number of proliferating cells did not depend on the PEG treatment (Fig. 1 B). One possible explanation was that cells fused spontaneously. We were intrigued by this possibility because of the link between fusion and carcinogenesis, the numerous reports that cancer cell lines are inexplicably fusogenic (for review see Duelli and Lazebnik, 2003), our finding that transformed cells acquire resistance to anticancer agents through fusion to normal cells (Duelli and Lazebnik, 2000), and the observation that cells recovered from individual colonies vary widely in their properties (unpublished data).
Figure 1.
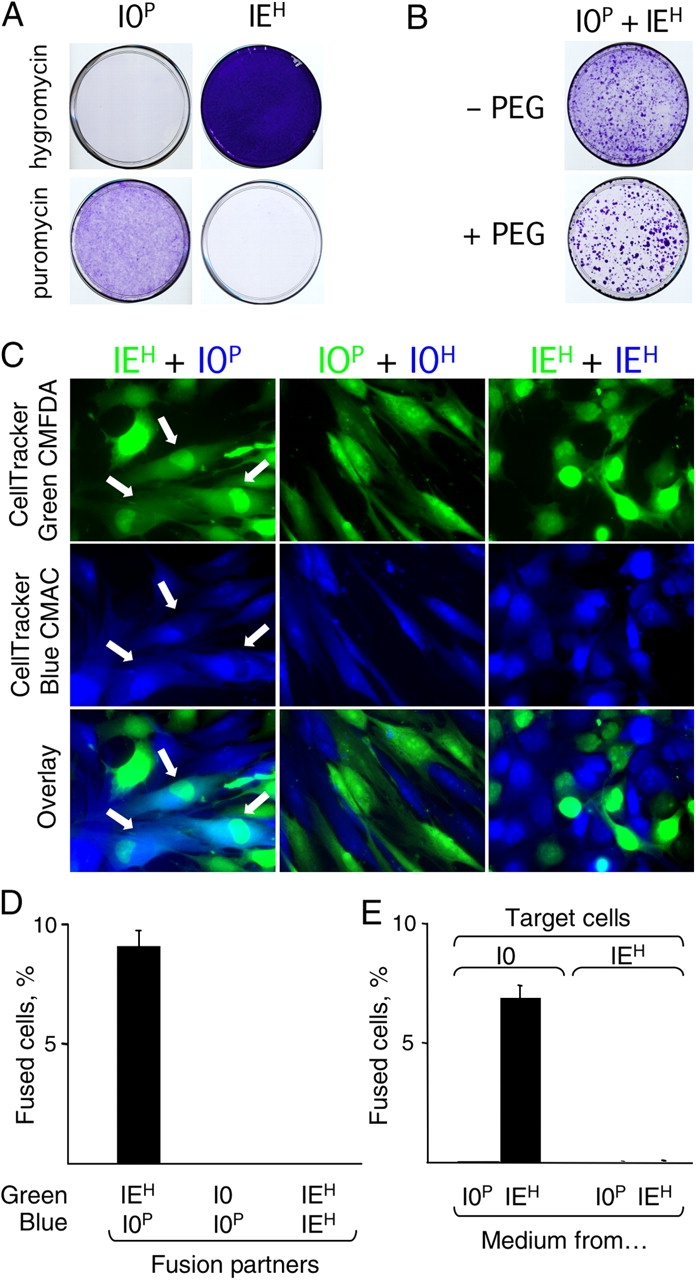
Spontaneous fusion produces proliferating hybrids. (A) IMR90 cells (I0) resistant to puromycin (I0P) or I0 cells transformed with E1A and resistant to hygromycin (IEH) were cultured as indicated and stained with crystal violet to visualize the cells. (B) Co-culture of I0P and IEH cells results in cells resistant to both drugs. Cells were plated together, treated with PEG or left untreated, cultured for 20 d, and visualized as in A. (C and D) IEH cells fuse to each other but not to themselves. I0P and IEH cells were dyed and cultured as indicated for 16 h and visualized by fluorescence microscopy. Heterokaryons (indicated by arrows in C and scored in D) contained both green and blue dyes and at least one green and one blue nucleus. (E) Tissue culture medium from IEH cells induces cell fusion. Tissue culture medium conditioned by either I0P or IEH cells for 16 h was tested in the fusion assay (see Materials and methods). All experiments were performed at least three times. The data in D and E are from three independent experiments, and the error bars indicate SD.
Cells fuse spontaneously
To test whether cells fused, we used an assay in which fusion partners were stained with vital fluorescent dyes of different colors, and the fused cells were identified as heterokaryons that contained both dyes in the cytoplasm and at least two differently colored nuclei (Duelli and Lazebnik, 2000; see also Materials and methods). We stained IEH and I0P cells, cultured them together for 16 h, and identified heterokaryons by fluorescence microscopy (Fig. 1 C, left). IEH and I0P cells indeed fused with each other (Fig. 1, C and D). We noticed that fused cells were mostly dikaryons, which are more likely to produce proliferating progeny than cells containing more nuclei (Ringertz and Savage, 1976); only rarely contained several nuclei; and were never found as syncytia, which are indicative of viral infections. Remarkably, neither IEH nor I0P cells fused to their own kind (Fig. 1 D), which suggested either that these cells collaborated to fuse, as happens in myogenesis, or that one of the lines made a fusogen that was unable to fuse the cells that produced it.
Cells release a fusogen associated with exosomes
Consistent with the second possibility, tissue culture medium conditioned by IEH cells fused I0P cells and several other cell lines that we tested, whereas the medium conditioned by I0P cells did not (Fig. 1 E and not depicted). Enriching the fusogenic activity by filtration and sequential centrifugation yielded a preparation we called P70 (Fig. 2 A) and indicated that the fusogen was 100–200 nm in size. Because trypsin treatment of the P70 abolished the fusogenicity (Fig. 2 B), we identified the digested peptides by mass spectrometry. About half of the reliably identified proteins were reported as components of exosomes (Table S1, available at http://www.jcb.org/cgi/content/full/jcb.200507069/DC1).
Figure 2.

Fusion activity is associated with exosomes. (A) Fusogenic activity is enriched by sequential centrifugation. Conditioned medium from IEH cells (S0) was sequentially centrifuged at 500, 16,000, 70,000, and 110,000 g. A pellet from each centrifugation was reconstituted and used in the fusion assay with I0 cells. (B) P70 obtained as in A was treated with trypsin or buffer (see Materials and methods), and the fusogenicity of the resulting preparations was determined as in A. (C) Electron micrograph of P70 from IEH cells. (D and E) P70 was fractionated by floating in a sucrose gradient, and the fractions were analyzed for CD81 (D) and in the fusion assay with I0 cells (E). (F) Exosomes from I0 cells are not fusogenic. Indicated concentrations of exosomes from either I0 or IEH cells were tested in the fusion assay with I0 cells. The experiments in A, B, and F were performed at least three times. The data in D and E are from one of three experiments, and the data in A and B are from the same experiment. The error bars in A, B, and F indicate SDs.
Exosomes are vesicles of characteristic shape, protein composition, and size that are released by many cells, in particular tumor cell lines (Couzin, 2005). These vesicles have been used in clinical trials to induce T cell–mediated tumor rejection (for review see Chaput et al., 2004), although the function of exosomes in vivo, if any, is unclear. Exosomes may serve as shuttles by delivering proteins and lipids to cells and carry proteins implicated in cell fusion, such as CD9 and -81 (Hemler, 2003; Fevrier and Raposo, 2004). To verify that the fusogenic activity is associated with exosomes, we analyzed other parameters that define these vesicles.
The electron microscopy of P70 from IEH cells revealed uniform particles (Fig. 2 C) whose cuplike shape and size was consistent with that described for exosomes (Thery et al., 2001). Fractionation of P70 from both I0 and IEH cells by flotation in a sucrose gradient provided particles with a density (1.18 g/ml), appearance, and CD81 content (Fig. 2 D) consistent with that of exosomes (Thery et al., 2001). The fusogenic activity from IEH cells co-migrated with exosomes in the gradients (Fig. 2 E). However, exosome preparations from I0 cells fused none of the cell lines that we tested at any concentration used (Fig. 2 F and not depicted), indicating that either fusogenic activity is not associated with exosomes or the exosomes from I0P and IEH cells are different.
Fusogenic exosomes contain viral proteins
A possible difference was suggested by the Trojan exosome hypothesis, which states that retroviruses, many of which are fusogenic, can be released in exosomes. Using this mechanism, the virus may expand its host range by using cellular proteins incorporated in its membrane to facilitate fusion and to avoid immune surveillance (Gould et al., 2003). Therefore, we repeated the mass spectrometry analysis, this time using total lysates of purified exosomes (Fig. 3 A). Major polypeptides recovered from the fractions with fusogenic activity (fraction 7; Fig. 3, A and C) were encoded by the Mason-Pfizer monkey virus (MPMV; Table S2, available at http://www.jcb.org/cgi/content/full/jcb.200507069/DC1; for review see Fine and Schochetman, 1978), suggesting that exosomes carried this virus.
Figure 3.
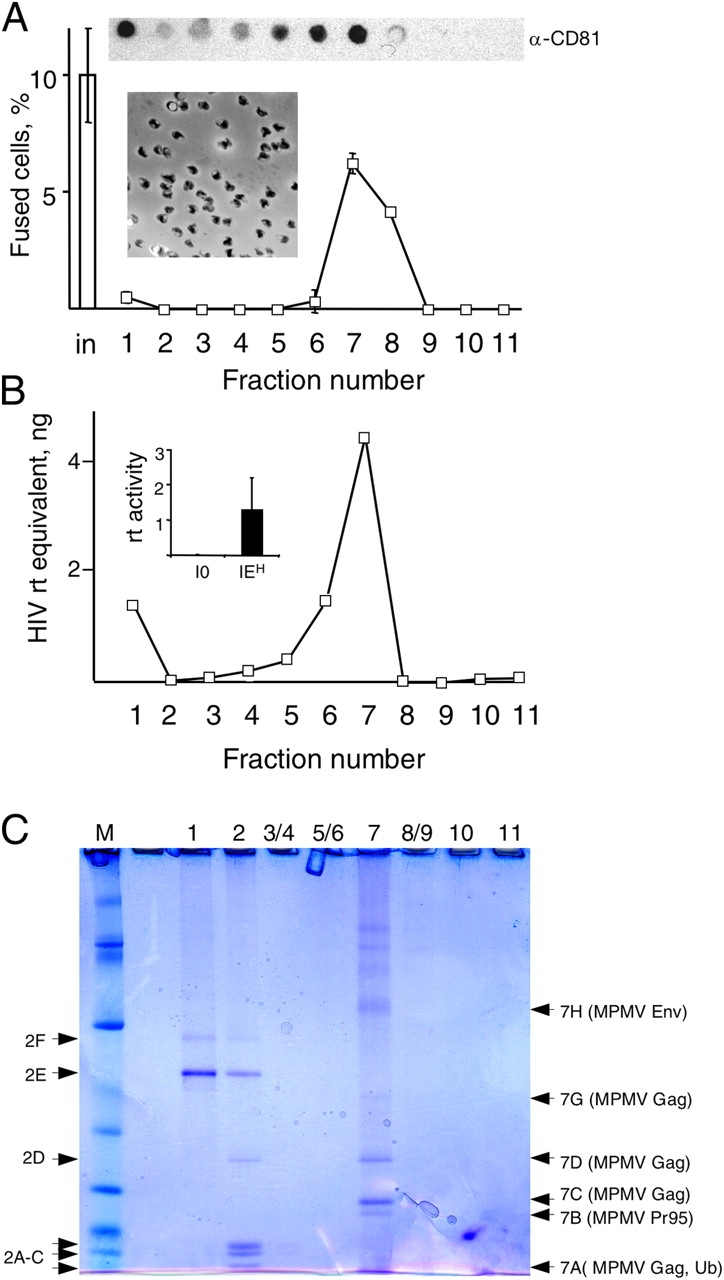
Fusogenicity is associated with an exosome-like virus. (A and B) Exosomes were fractionated by sedimentation in a velocity gradient (see Materials and methods), and aliquots of the fractions were analyzed in the fusion assay with I0 cells (A) or reverse transcriptase activity (B). The insets in A show an electron micrograph of fraction 7 and the content of CD81 in the fractions analyzed by dot blotting. The inset in B compares the reverse transcriptase activity in P70 from I0 and IEH cells. (C) The fractions obtained in A were analyzed by electrophoresis and the indicated polypeptides by mass spectrometry (Table S2). The data in A and in the inset in B are from three independent experiments, and the data in B are the mean of two independent experiments. The error bars indicate SD.
MPMV is the prototype D type retrovirus with a host range largely restricted to primates. The virus was originally isolated from a rhesus monkey breast carcinoma and then detected in humans (Bohannon et al., 1991; Ford et al., 1992) and in many human cell lines (Robert-Guroff et al., 1996). MPMV can cause simian acquired immunodeficiency syndrome but has no identified pathogenic effect in humans or in cultured cells. Ectopic expression of the MPMV Env is sufficient to induce cell fusion (Song and Hunter, 2003), which indicates that the fusogenicity of IEH cells could be associated with this virus. Because our observation suggested that proliferating hybrids could be produced by a virus, we verified that the fusion that we observed was indeed caused by MPMV.
Several observations supported this possibility. The genome of IEH but not that of I0 cells contained MPMV sequences (unpublished data). The P70 from IEH but not from I0 cells had reverse transcriptase activity (Fig. 3 B, inset). Also, I0 cells became fusogenic (Fig. S1, A and C, available at http://www.jcb.org/cgi/content/full/jcb.200507069/DC1) and acquired expression of MPMV p27CA after incubation with P70 from either IEH or CMMT, which is the prototypical cell line that produces MPMV (for review see Fine and Schochetman, 1978; Fig. S1 B and not depicted). Immunoblotting confirmed that P70 of IEH cells contained the exosomal proteins CD9, CD81, and major histocompatibility complex class I (MHCI; Thery et al., 1999) and the MPMV proteins gp20 Env and the capsid p27CA Gag (Fig. 4 A). Nearly all particles in P70 from IEH cells contained the MPMV capsid protein p27CA (Fig. 4 B) in its mature form (Fig. 4 A), indicating that nearly all observed vesicles contained MPMV.
Figure 4.
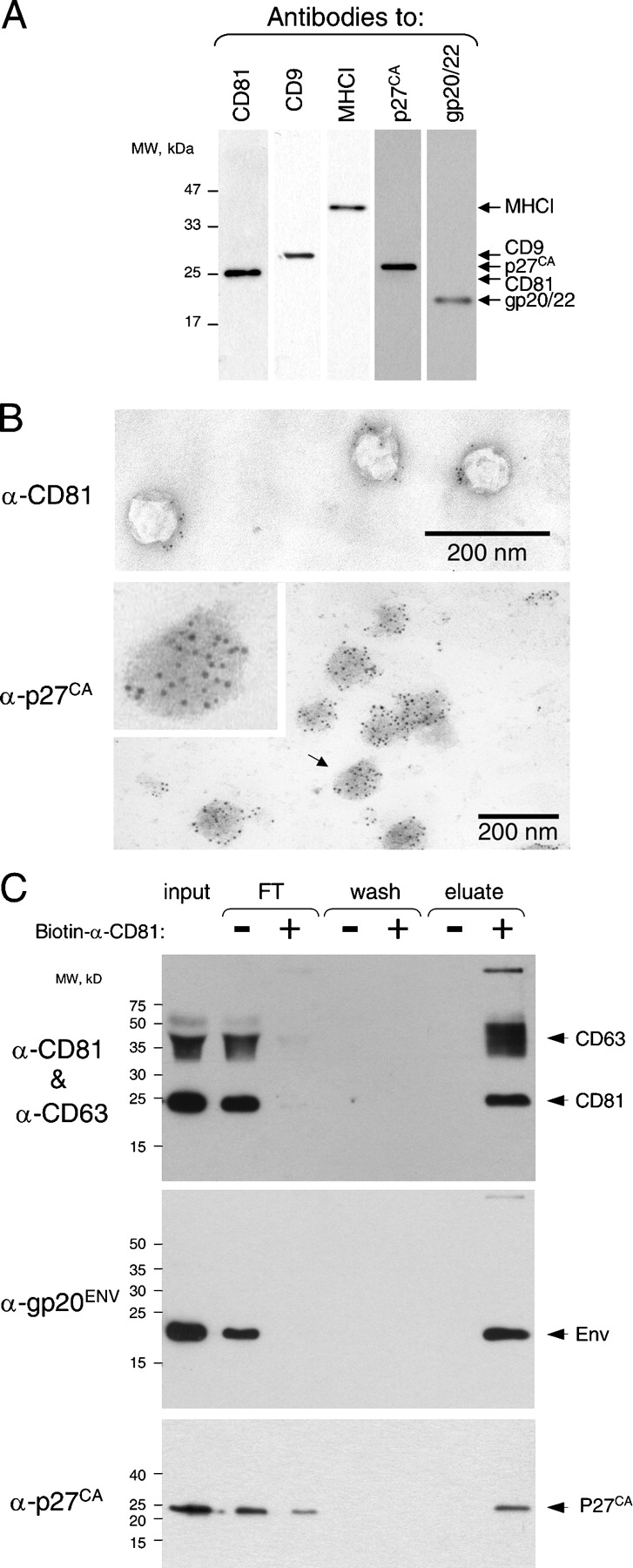
Fusogenic exosomes are associated with MPMV. (A) MPMV Env, Gag, and exosomal CD9, CD81, and MHCI were detected by immunoblotting in P70 (see Materials and methods). (B) CD81 is detected on the surface and p27Ca Gag of MPMV inside of exosomes by immuno-EM (see Materials and methods). The inset is an enlargement of the particle indicated by the arrow. (C) MPMV Env and p27 capsid are associated with exosomal markers CD81 and -63. P70 preparation was incubated with or without biotinylated anti-human CD81 antibody and fractionated in a capture assay using magnetic streptavidin microbeads (see Materials and methods). The unbound fraction (FT), the final wash, and the bound fraction (eluate) were analyzed by immunoblotting. Antibodies to CD63 and -81 were used together for convenience because both identify distinct polypeptides. The experiments in A and B were done multiple times and the experiment in C twice.
Nearly all vesicles also had CD81 on their surface (Fig. 4 B), consistent with the possibility that the virus was carried by exosomes. We tested this possibility by capturing exosomes with an antibody to CD81 and assessing whether the viral proteins segregated with this exosomal marker. All Env and most p27CA indeed segregated with CD81, as did another tetraspanin, CD63 (Fig. 4 C; Pelchen-Matthews et al., 2003). Considering our observations, we concluded that MPMV was released as an exosome-like particle and will refer to this form of the virus as MPMVE.
Antibodies to exosomal proteins interfere with fusion
MPMVE contained two sets of proteins implicated in fusion, the MPMV Env and the cellular tetraspanins CD81 and -9, which modulate fusion by a poorly understood mechanism. To test whether tetraspanins are involved in cell fusion induced by MPMVE, we used the observation that antibodies to CD9 and -81 prevent fusion of mouse gametes and myoblasts (Hemler, 2003). We incubated aliquots of MPMVE with antibodies to CD9, CD81, the capsid p27CA, or the MPMV Env gp20/22. To exclude unspecific effects of antibodies, we used total mouse IgG and an antibody to MHCI, an abundant exosome surface protein (Clayton et al., 2001) that has not been linked to fusion (Fig. 4 C and Fig. 5 A). Antibodies to Env, CD9, and CD81, but not to the capsid protein p27CA, did bind MPMVE (Fig. 5 A), confirming that Env and the tetraspanins were on the surface of MPMVE and that the envelope of MPMVE was intact. After washing away the unbound antibodies, we tested whether the treated MPMVE still fused I0 cells.
Figure 5.

Antibodies to tetraspanins CD9 and -81 inhibit fusion by MPMV E . (A) Aliquots of MPMVE were incubated with indicated antibodies, washed, and analyzed by immunoblotting (see Materials and methods). (B) Exosomes were treated with indicated antibodies and tested in the fusion assay with I0 or HepG2 cells, which express no CD81 (C). The experiments were done three times with I0 cells, except that total mouse IgG was used as a control only in two of these experiments, and the experiments with HepG2 cells were done twice. ND, not done. The error bars indicate SD. (C) CD81 is transferred to HepG2 cells by MPMVE. HepG2 cells cultured for 22 h without (left) or with (right) MPMVE from IEH cells were probed with an antibody to CD81 (red). The nuclei were visualized with Hoechst 33342 (blue).
Antibodies to CD9 or -81 blocked fusion, whereas the control antibodies had no effect (Fig. 5 B). Treating MPMVE with the antibody to CD81 also prevented fusion of HepG2 cells (Fig. 5 B), which express no detectable CD81 (Fig. 5 C; Charrin et al., 2001), indicating that the effect of this antibody is associated with its binding to CD81 located on MPMVE rather than on cells. Considering our observations and the reported function of tetraspanins, we concluded that exosomal proteins participate, directly or indirectly, in cell fusion induced by MPMVE. How these proteins affect fusion remains to be determined. The role of MPMV Env also remains unclear, as the ability of the antibody that we used to block membrane fusion or infection in any experimental system is unknown.
MPMVE infection is not required for fusion
Cells can be fused by proteins of the viral particles, which does not require infection, or by viral proteins that are expressed in the infected cells. Several observations indicated that cell fusion induced by MPMVE did not require infection. MPMVE had to be continuously present in the medium to induce cell fusion (Fig. 6 A), which implies that fusion was directly caused by the added viral particles. Cell fusion was detectable within hours (Fig. 6 A), as opposed to the days required to express MPMV proteins (Fine et al., 1979), which is consistent with the finding that fused cells have no detectable MPMV proteins (Fig. S2 A, available at http://www.jcb.org/cgi/content/full/jcb.200507069/DC1). Azidothymidine, a reverse transcriptase inhibitor, prevented infection but not fusion (Fig. S2 B). Finally, the efficiencies of cell fusion and productive infection were unrelated (Fig. 6 B), confirming that fusion of the virus to the cells and fusion of cells induced by viruses or viruslike particles can be different processes (Schmid et al., 2000) and suggesting that hybrids produced by viruses may have no trace of the virus that created them, meaning that their viral etiology would be impossible to determine.
Figure 6.

Cells are fused by MPMV E particles. (A) MPMVE must be continuously present in the medium to induce fusion. MPMVE was added to I0 cells, and the fused cells were scored at the indicated times (top, curve indicated with filled circles). Alternatively, MPMVE was added to one of the fusion partners for the indicated time, after which the cells were washed three times to remove unbound MPMVE, and the second fusion partner was added and the fused cells scored 16 h later (bottom, curve indicated with filled triangles). (B) The infection and fusion efficiencies of MPMVE do not correlate. Various amounts of MPMVE were added to the fusion assay with I0 cells, which were then incubated for 16 h to score fused cells or for 4 d to score for reverse transcriptase (RT) activity in tissue culture medium. The data are from three independent experiments, and the error bars indicate SD.
Oncogenes abolish a proliferation block caused by fusion
Because our observations contrasted with the view that cells fused by active viruses do not proliferate, we investigated what is required to make hybrids proliferate. None of the normal primary cell lines that we tested, which included human fibroblasts I0, HSF43, SF68, BJ, WI38, and Detroit 551; human umbilical vein endothelial cells; and renal proximal epithelial tubule cells, produced colonies or mononuclear cells after fusion by MPMVE (Fig. 7 A, left, and not depicted). Therefore, we concluded that these cells not only failed to proliferate but did not undergo even a single cell division after fusion. The cells were dikaryons, with occasional polykaryons, and remained attached for at least 20 d and appeared healthy (Fig. S2 C). Therefore, cell fusion was cytostatic but not cytotoxic to normal cells. Hence, we concluded that fusion of normal differentiated cells by MPMVE is unlikely to result in proliferating progeny.
Figure 7.

Expression of E1A or suppression of p53 is sufficient for proliferation of hybrids induced by MPMV E . (A) Detroit 551 cells resistant to puromycin (D0P), hygromycin (D0H), transduced with E1A and resistant to hygromycin (DEH), or transduced with p53 R175H and resistant to hygromycin (Dp53R175HH) were cultured for 16 h in combinations as indicated with or without MPMVE and then for an additional 20 d in fresh medium containing hygromycin and puromycin. The cells were then visualized by staining with crystal violet. (B) Cell colonies from various fields of the bottom right plate in A. Note the diversity in cell morphology among the fields. The data are from one out of three experiments, all of which produced similar results. Bars, 200 μm.
Our initial observation (Fig. 1) suggested that expression of E1A may allow proliferation of fused cells. Indeed, we found that fusing normal and E1A-expressing fibroblasts yielded proliferating hybrids (Fig. 7 A, middle). However, cells transduced with E1A had to be passaged eight or more times to produce viable hybrids, perhaps to select cells that are less prone to apoptosis. Ectopic expression of c-Myc also allowed proliferation, although the efficiency was lower (unpublished data). These observations raised the possibility that fusion can produce malignant cells from cells that have deregulated cell cycles.
Inactivating tumor suppressor p53 abolishes a proliferation block caused by fusion
Cell cycle arrest induced by fusion was reminiscent of arrest caused by incomplete cytokinesis, which can be inactivated by dominant-negative mutants of p53 (Margolis et al., 2003), one of which, p53-R172H (p53-R175H in humans), causes metastatic aneuploid tumors in mice (Hingorani et al., 2005). Therefore, we tested whether expression of p53-R175H would allow cells fused by MPMVE to proliferate.
Indeed, fusion of normal Detroit 511 cells (D0P) with cells that were transduced with p53-R175H (Dp53R175HH) produced proliferating hybrids (Fig. 7 A, right). The morphology and the rate of proliferation of hybrids produced with either gene varied widely (Fig. 7 B and not depicted), but p53-R175H was more efficient in producing proliferating hybrids than E1A, and the hybrids were different. Instead of forming well-defined colonies, as did hybrids expressing E1A, the p53-R175H hybrids spread throughout the plate (Fig. 7 A). Given that expression of neither E1A nor p53-R175H affected the rate or extent of cell fusion, we concluded that proliferation of cells produced by fusion is controlled by a mechanism that can be deregulated by mutation of p53 and that a single mutation in one of the fusion partners is sufficient to result in diverse proliferating hybrids.
Fusion produces transformed hybrids from cells destined to die
Cell fusion can add to cell diversity by combining distinct properties of the fusion partners. For example, genes that are insufficient to transform on their own may be brought together by cell fusion to result in a transforming combination. To test whether MPMVE has this effect, we used the observation that expressing E1A together with oncogenic Ras results in transformed cells that grow in soft agar, whereas expressing E1A alone induces apoptosis and expressing Ras senescence (Seger et al., 2002). Indeed, Detroit 511 cells that were freshly transduced with E1A (DEH) or that expressed Ha-Ras (DRasP) alone failed to form colonies in soft agar (Fig. 8). In contrast, hybrids of DEH and DRasP produced by MPMVE grew at least as efficiently in soft agar as did the cells cotransduced with E1A and Ha-Ras (Fig. 5). Hybrids recovered from soft agar also grew efficiently in tissue culture, whereas no parental cells could be recovered, implying that neighboring cells, even if they are otherwise destined to die, could produce transformed proliferating progeny if fused by a virus.
Figure 8.

Fusion produces transformed hybrids from cells destined to die. (A) Detroit 551 cells freshly transduced with E1A and resistant to hygromycin (DEH), transduced with Ha-Ras and resistant to puromycin (DRasP), or cotransduced with both E1A and Ha-Ras and resistant to both antibiotics (DE1ARasHP) were cultured for 16 h alone or in combination, as indicated, with or without MPMVE. The cells were then cultured for 7 d without added virus in medium containing hygromycin (DEH), puromycin (DRasP), or both (DE1ARasHP and the co-cultured DEH and DRasP treated with MPMVE). The cells were then collected, plated into soft agar at equal cell densities as indicated, cultured for 20 d, and visualized by crystal violet. (B) Cell colonies in the corresponding bottom panels in A. The experiment was done four times with similar results. Bars, 100 μm.
Discussion
Overall, we found that a primate virus can produce diverse abnormal proliferating cells by cell fusion and that proliferation of these cells is favored by expression of oncogenes or by a mutated tumor suppressor, p53. Whether proliferating hybrids are produced by viruses in vivo, and whether these hybrids can evolve into cancerous cells, remains to be determined.
At least two sets of observations suggest that viruses produce not only moribund syncytia but also proliferating hybrids. For example, human cancer cells grafted into rodents produce tumors that partially or completely consist of host-human hybrids and host tumor cells that appear to be derived by spontaneous fusion, which is a term used to describe fusion with an unknown cause (Goldenberg et al., 1974; Pathak et al., 1997; Mortensen et al., 2004). Interestingly, mouse cancer cell lines passaged in mice can increase or acquire their metastatic potential by spontaneous fusion to host cells (for review see Duelli and Lazebnik, 2003).
Why these tumor cells are fusogenic is unclear. Our observations suggest an explanation: fusion is caused by viruses or viruslike particles. Indeed, not only are retroviruses ubiquitous in mice, but some viruses were specifically associated with xenografts and the resulting tumors (Bowen et al., 1983; Ristevski et al., 1999). We found that 7 out of the 20 human tumor cell lines that we tested released a particulate fusogen, which is consistent with the possibility that these tumor cell lines released fusogenic viruses (unpublished data).
Another example of cell fusion that remains unexplained is the production of hybrids between bone marrow stem cells and differentiated somatic cells (O'Malley and Scott, 2004; for review see Vignery, 2005). Bone marrow stem cells can give rise not only to somatic differentiated cells but also to gastric cancer by a mechanism that is unclear (Houghton et al., 2004). One possibility that was raised (Marx, 2004) but not tested is that oncogenic transformation in this case was also caused by fusion. Again, viruses may be the fusogen, a possibility that would also explain the puzzlingly low frequency of stem-cell hybrids (10−5; O'Malley and Scott, 2004).
An intriguing question is why hybrids of stem cells are capable of proliferating. Our observations suggest that the answer may be in the plastic cell cycle regulation in stem cells, which makes them similar to embryonic or to partially transformed cells (Attar and Scadden, 2004). Therefore, the ability to survive and proliferate after fusion, rather than to fuse, may be a particular characteristic of stem cells that underlies fusion-mediated transdifferentiation, and perhaps cancer, and allows one to detect these hybrids in vivo. Given that fusing somatic cells is being considered as a therapeutic approach (Cowan et al., 2005), it is of practical interest to know for certain that this fusion does not produce malignant progeny.
Whether hybrids observed in animal models occur in humans is unclear, although the reports of premature chromosome condensation (Kovacs, 1985) and hybrids between transplanted and host cells (Chakraborty et al., 2004) are consistent with this possibility. There is little doubt, however, that virus-induced cell fusion in people is common, as syncytia are a feature of many viral infections, from the respiratory syncytial virus to human immunodeficiency virus.
What kind of viruses can produce proliferating hybrids?
Our finding that a virus can produce proliferating hybrids was fortuitous. MPMV is neither cytotoxic nor cytostatic in human cells and in our system primarily produced dikaryons, which are more likely to result in proliferating hybrids than cells with more nuclei. It is unlikely that these characteristics are unique for this virus.
One group are the viruses that, like MPMV, are considered harmless and therefore are studied much less intensely than obvious pathogens. For example, human T cell lymphotropic virus type 1 (HTLV-1) is harmless to most of its carriers, and its association with cancer was uncovered only by epidemiological studies. However, cytotoxic viruses could also produce hybrids by fusion with noninfectious viral particles, which are often more abundant than infectious particles, or if infection is restricted by host factors. In either case, the hybrids may have no traces of the virus, implying that the viral etiology of virus-induced hybrids could be difficult to determine. Because oncogenes can allow proliferation of fused cells, fusogenic viruses carrying such oncogenes (e.g., myc) may not only fuse cells but also allow the hybrids to proliferate. Retroviruses may achieve a similar effect by insertional mutagenesis.
The ability to form dikaryons, which are more likely to have proliferating progeny, may depend not only on the virus but also on how and from what kind of cell it is released. For example, if a virus is released as a part of exosomes, it may carry cellular proteins, such as tetraspanin CD9 and -81, that facilitate fusion of cells. Tetraspanins may also be acquired by the viruses that are released from tetraspanin microdomains of the plasma membrane (Hemler, 2003; Martin et al., 2005).
Endogenous retroviruses (ERVs), whose sequences comprise at least 8% of the human genome (Griffiths, 2001), are also candidates as pathogenic fusogens. At least three Env proteins of human ERV are fusogenic, two of which, Syncytin 1 and 2, are normally expressed only in the placenta, where they mediate fusion required for formation of the syncytiotrophoblast. The third protein, Env(P)b, whose physiological function is unknown, is expressed ubiquitously (Blaise et al., 2005). Interestingly, expression of ERVs is regulated by a variety of factors, including DNA damage, mitogens, and hormones (Taruscio and Mantovani, 2004). Overall, the possibility that viruses produce proliferating hybrids should at least be considered in developing cancer treatments that use fusogenic viruses to kill cells or as vectors for gene therapy.
Are exosomes misidentified viruses?
Because exosomes and some viruses are so similar, at least some particles reported as exosomes may be misidentified viruses. This possibility has practical implications because exosomes are used in cancer therapy (for review see Chaput et al., 2004). Indeed, early reported compositions of exosomes did include retroviral proteins (Thery et al., 1999, 2001). As we found, identifying vesicles as exosomes or viruses by mass spectrometry depends on the protocol used. Defining parameters such as size, shape, and density in sucrose gradients are also conspicuously similar (for review see Fine and Schochetman, 1978; Thery et al., 1999). The morphology of MPMV (Smith et al., 1978), a pleomorphic virus, is difficult to distinguish from that of exosomes.
Cell fusion as a link between viruses and carcinogenesis
A current view is that retroviruses transform cells either by integrating into the cellular genome, thereby affecting normal gene expression or modifying cellular genes, or by introducing oncogenes in the genome of infected cells. However, the transformation induced by viruses is unlikely to be limited by these mechanisms, as indicated by the recent findings that the Env proteins of Jaagsiekte virus and enzootic nasal tumor viruses are causative agents of infectious cancer in animals (Wootton et al., 2005).
Whether retroviruses cause human cancer is a subject of discussion. The claim that they do is based on epidemiological data (for review see Mant et al., 2004), whereas the claim that they do not is based on the argument that viruses fail the requirements imposed by Koch's postulates (Duesberg, 1987; Blaho and Aaronson, 1999; Talbot and Crawford, 2004). These postulates, as applied to cancer, argue that a candidate virus must be present in cancer but not in healthy cells, must be isolated from the cancer cells, must cause oncogenic transformation if introduced into normal cells, and must be present in these cells once they are transformed. Indeed, if these standards apply, the proof that viruses are etiological agents of human cancer falls short (for review see Mant et al., 2004). For example, only 1% of the people infected with HTLV-1 develop cancer with no apparent correlation between carcinogenesis and the virus integration sites (Hanai et al., 2004), whereas another small fraction of the carriers develop a disease unrelated to cancer.
However, Koch's postulates are valid only if the causal relationship between viruses and oncogenic transformation is as direct as that between viruses and infectious diseases, which is an assumption that is consistent with the current view of viral oncogenesis. If the causal relationship between viruses and oncogenic transformation includes events with random outcomes in respect to carcinogenesis, such as cell fusion and abnormal mitoses, and the virus may even be absent in the cells it produced, then the cause–effect relationship between viruses and cancer is intrinsically stochastic (Fig. 9), which would mean that Koch's postulates do not apply. Therefore, the observed correlations between viral infections and human cancer may be more than coincidental, even though because of its random nature it may be impossible, in principle, to establish a mechanistic link by analyzing only cancer cells.
Figure 9.

Cell fusion as a link between viruses and carcinogenesis. Potential implications of our findings to carcinogenesis could be summarized by the following speculative model. Although illicit cell fusion induced by viruses may be a frequent and common event, it usually has no consequences for carcinogenesis because, as a rule, the resulting cells either die or do not proliferate. However, if the cells have a deregulated cell cycle and if the virus tends to produce dikaryons rather than syncytia, the fused cells may proliferate. The majority of these cells, however, die within a few cell divisions because of chromosomal aberrations associated with abnormal mitoses or other, yet unrecognized, consequences of cell fusion. However, the few cells that survive are abnormal in that they have a deregulated cell cycle, lack normal response to this deregulation, are aneuploid, and have epigenetic regulation that is an unpredictable result of the merger between fusion partners. A combination of these properties in a minute fraction of the fused cells may be sufficient to make them cancerous. Considering that a single cell can give rise to a cancer, the frequency with which cell fusion produces such cells does not need to be high to have pathological consequences.
Irrespective to the origin of cancer cells, cell fusion has the potential to promote diversity of transformed cells, as our results and previous studies have demonstrated. In this case, the frequency with which proliferating hybrids are produced would be even higher because at least one of the fusion partners has a deregulated cell cycle and, perhaps, greater survival capabilities.
Overall, the evidence presented in this study suggests a mechanistic link between viral infections and the genetic and epigenetic instability model of carcinogenesis. Whether this link contributes to carcinogenesis or cancer progression remains to be determined.
Materials and methods
Cell lines and tissue culture
Normal human diploid embryonic lung fibroblast cell line IMR90 and normal human diploid skin fibroblast cell lines Detroit 551, HSF43, and Hs68, as well as transformed monkey cell lines COS-1 and CMMT cell lines were obtained from American Type Culture Collection and cultured in 90% DME and 10% FBS and in the absence of antibiotics. Normal human diploid skin fibroblast cell line BJ and normal human diploid lung fibroblasts WI-38 were cultured in 90% MEM and 10% FBS and nonessential amino acids. Renal proximal tubule cells were obtained from Cambrex and cultured in REGM renal epithelial cell medium from the same manufacturer. Normal human umbilical vein endothelial cells were obtained from Cambrex and were maintained in Ham's F12K medium with 2 mM l-glutamine. All cells were routinely monitored for mycoplasma contamination using the Mycoplasma detection kit 2 (American Type Culture Collection).
Gene transduction
Retroviral vectors pMarxIVpuro, pMarxIVhygro, pBABEpuro, and pWZLhygro or the vectors containing 12S E1A, Ha-Ras, or p53 dominant-negative mutant were transfected (all constructs were gifts from G. Hannon, the Hannon Laboratory, Cold Spring Harbor, NY) into the packaging cell line Phoenix-ampho (a gift from G. Nolan, Stanford University, Stanford, CA) using fugene (Roche). 96 h later, culture supernatant was filtered through a 0.45-μm filter, supplemented with 8 μg/ml polybrene, and added to freshly plated 70% confluent recipient cells that were centrifuged for 1 h at 500 g, replated after reaching confluency, and selected with appropriate antibiotics.
Antibodies
Mouse mAbs to human CD9, -63, and -81 were purchased from BD Biosciences and IgG of murine serum from Sigma-Aldrich. The biotinylated antibody to human CD81 was obtained from Ancell. The HC-10 antibody to human MHCI was a gift from H. Ploegh (Harvard Medical School, Cambridge, MA). The mAb F2-1 to Pr86, gp20, and gp22 of MPMV (Sommerfelt et al., 2003) was a gift from J. Yee and N. Pedersen (California National Primate Research Center, Davis, CA). Goat antiserum to MPMV p27Ca was provided by D. Ott (Science Applications International Corporation–Frederick and National Cancer Institute–Frederick, Frederick, MD). Secondary antibodies used for immunofluorescence were goat anti–mouse IgG and donkey anti–goat labeled with AlexaFluor 594 (Invitrogen) and for immunoblotting anti-mouse HRP (GE Healthcare) and anti-goat HRP (Santa Cruz Biotechnology, Inc.).
Fusion assay
All fusion experiments were performed in media with no antibiotics, fungicides, or fungistats, as such compounds influence the mode or efficiency of fusion. Assays were performed in Falcon Multiwell 24-well plates (BD Biosciences) as described previously (Duelli and Lazebnik, 2000). In brief, 3 × 104 to 4 × 104 of one fusion partner that had been dyed with CellTracker Green CMFDA (5-chloromethylfluorescein diacetate; Invitrogen) were plated with the same number of cells of another fusion partner that had been dyed with CellTracker Blue CMAC (7-amino-4-chloromethylcoumarin; Invitrogen) and cultured for 12–16 h, unless otherwise indicated, in 500–1,000 μl of medium or in medium supplemented with a tested fusogen, as indicated in the figure legends. The fusion rate was assessed by immunofluorescence by measuring the fraction of green cells that became part of a heterokaryon. At least 600 cells were counted for each data point, and each experiment was performed at least twice, unless otherwise indicated.
Exosome purification
To prepare exosomes, cells were grown in DME that was supplemented with the serum replacement TM-235 (MP Biomedicals), Nutridoma HU (Roche; for mass spectrometry/mass spectrometry [MS/MS] analyses), or FBS, as indicated in the figure legends. FBS was clarified of particulate matter by centrifugation at 110,000 g for 2 h, and the medium was filtered through 0.2-μm filters after FBS was added. The resulting medium contained no exosome-like, or other, particles detectable by EM. The medium was used to culture 1–20 15-cm culture dishes containing I0 or IEH cells to near confluency. The conditioned tissue culture medium was collected and clarified by centrifugation at 500 g and filtered through Acrodisc 0.2-μm HT Tuffryn filters (Pall Corporation), and the filtrate was subjected to successive centrifugation at 12,000, 70,000, and 110,00 g. The 70,000-g pellet (P70) was resuspended in PBS at 1% of the initial volume of tissue culture medium and used for further applications immediately or stored at −70°C.
Exosomes were purified by flotation in sucrose gradients using a published protocol (Thery et al., 1999). In brief, 100 μl of freshly obtained P70 was applied under a 12-ml 0.25–2 M gradient of sucrose in PBS. The resulting samples were centrifuged at 100,000 g for 24 h and collected as 1-ml fractions. The fractions were diluted with PBS, their content was pelleted at 70,000 g, and the pellets were assessed for fusogenicity. Purification on iodixanol gradients, which provided a better yield of fusogenic activity, was performed by overlaying 100 μl of concentrated P70 onto a 10-ml PBS gradient of 0.6–30% OptiPrep (Axis-Shield PoC). To test for fusogenicity, the fractions were directly added to 1 ml of medium to achieve a final concentration of no more than 3% OptiPrep.
Capture assay
The assay was performed using μMACS streptavidin microbeads (Miltenyi Biotec) according to the manufacturer's instructions. 50 μl of P70 preparation was incubated for 30 min with 0.5 μg of the biotinylated anti-human CD81 antibody, complemented with 20 μl of μMACS streptavidin microbeads, and incubated for 10 min. The suspension was then applied to magnetized μ columns. The unbound material was collected, and the bound material was washed four times with PBS supplemented with 2% FBS and released into SDS sample buffer.
Mass spectrometry
Exosomes purified on iodixanol gradients were precipitated in TCA, washed in acetone, and lysed in hot SDS buffer. The lysates were fractionated by electrophoresis on precast NuPage 4–12% BisTris gels (Invitrogen), and the proteins were visualized with zinc staining (Bio-Rad Laboratories) and excised and processed for identification by mass spectrometry. Spectra resulting from matrix-assisted laser desorption ionization–time of flight/time of flight mass spectrometry were analyzed using an ABI4700 (Applied Biosystems) and the MASCOT search engine (Matrix Science, Inc.).
Analysis of peptides released after treatment of the P70 with trypsin (Roche) for 12 h was performed after removing particulate matter from the digest by centrifugation. Liquid chromatography-MS/MS was performed using 75-μm × 5-cm picofrit columns (New Objective) coupled directly to an LCQdeca (Thermo Savant). Spectra resulting from liquid chromatography-MS/MS were analyzed using the SONARS software package (Genomic Solutions).
Plating assay to visualize proliferating hybrids
Cell lines, as indicated in the figure legends, were plated at equal ratios in 10-cm dishes at 60% confluency and treated with P70 or medium alone. 24 h after the treatment, the medium was replaced with that containing puromycin and hygromycin and cultured until the cell colonies became visible (for 1 wk or longer), and it was replaced with fresh medium every 5 d. To obtain the hybrids by PEG, we cultured I0P and IEH cells as a mixture and fused them as described previously (Duelli and Lazebnik, 2000).
Effect of antibodies to CD9 and -81 on fusion
Mouse IgG and the antibodies to CD9 and -81 were diluted to 5 ng/ml. The HC-10 ascites to MHCI and goat sera were diluted at 1:100, and F2-1 tissue culture supernatant to Env was diluted at 1:10 in medium containing clarified 10% FBS. All antibodies were then clarified by centrifugation. Aliquots of P70 were incubated with these antibodies for 2 h at RT and washed three times by centrifugation at 16,000 g with PBS. No antibodies were detected by immunoblotting in the supernatant of the third wash. The final pellets were resuspended in PBS to use in the fusion assay or in SDS sample buffer to analyze by immunoblotting. The amounts of the antibody–MPMVE complexes were normalized using MPMV reverse transcriptase activity.
Reverse transcriptase assay
The colorimetric reverse transcriptase assay (Roche) was performed according to the manufacturer's recommendations.
EM
Purified exosomes were directly adsorbed to butvar-coated grids followed by negative staining/embedding in 1% aqueous uranyl acetate in 1% methyl cellulose (Raposo et al., 1996). Micrographs were taken using a transmission electron microscope (H7000T; Hitachi).
Routine transmission EM of purified exosomes was done by fixation in suspension with 2% PFA and 0.2% glutaraldehyde followed by pelleting and postfixation with 1% osmium tetroxide and embedding in epon-araldite resin (Electron Microscopy Sciences). Thin sections were counterstained with uranyl acetate and lead citrate.
Immunogold labeling for CD81 was done on unfixed, suspended exosomes using 5 nm of colloidal gold conjugated to goat anti–mouse IgG (GE Healthcare) followed by PFA fixation and adsorption and staining as described in the previous paragraph. Postembedding immunogold labeling for MPMV p27CA was done on sections of lowicryl K4M–embedded exosomes. Primary antibody bound to exosomes was detected with a two-step protocol: incubation in rabbit anti–goat IgG (The Jackson Laboratory) followed by 5 nm of colloidal gold conjugated to goat anti–rabbit IgG (GE Healthcare). Labeled thin sections were counterstained with uranyl acetate. Negative control sections for immunogold labeling using irrelevant primary antibodies followed by detection steps showed an absence of gold particles.
Image processing
Digital images of immunoblots were acquired by scanning the autoradiograms on a flatbed scanner, and digital images of plates with cell colonies were acquired at RT with an inverted microscope (Axiovert 405M; Carl Zeiss MicroImaging, Inc.) equipped with an Achroplan LD 20× lens, with NA 0.4 (Carl Zeiss MicroImaging, Inc.), a Photometrics CH250 camera (Roper Scientific), and OncorImage analysis software v. 2.0.5d. Immunofluorescence images were acquired at RT using Axioskop 2 plus (Carl Zeiss MicroImaging, Inc.) equipped with Plan Neo-Fluar 40× lens with NA 0.75, AxioCam digital camera, and OpenLab 4.0.2 imaging software. Images for figures were formatted using Adobe Photoshop, which included change of contrast or brightness in some images, in which case each image was processed as a whole. If contrast and brightness were changed in multipanel images, the changes were performed on the entire image. The figures were assembled using PowerPoint.
Online supplemental material
Table S1 shows the composition of exosomes, and Table S2 shows identification of viral peptides from fusogenic exosomes. Fig. S1 demonstrates that the fusogen is infectious, and Fig. S2 gives evidence that productive infection is not required for fusion and that normal cells fused by MPMV survive but do not proliferate. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200507069/DC1.
Acknowledgments
We thank our student helpers Jovana Drinjakovic, David Rubenstein, Marie Mizuno, and Carolyn Hirschman for their contributions at various stages of the study; Eckard Wimmer for guidance with experiments on viruses; and Vadim Agol, Eli Hatchwell, Senthil Muthuswamy, Michelle Hastings, and the Lazebnik Laboratory for critically evaluating the manuscript. We also thank Greg Hannon, Niels Pedersen, David Ott, Hidde Ploegh, and JoAnn Yee for their kind gifts of antibodies and plasmids.
This work was funded by National Institutes of Health grant GM069357-01 and by the Seraph foundation to Y. Lazebnik and an American Leukemia and Lymphoma Society fellowship 5958-01 to D.M. Duelli.
Abbreviations used in this paper: ERV, endogenous retrovirus; MCHI, major histocompatibility complex class I; MPMV, Mason-Pfizer monkey virus; PEG, polyethylene glycol.
References
- Aichel, O. 1911. Über Zellverschmelzung mit qualitativ abnormer Chromosomenverteilung als Ursache der Geschwulstbildung. Vorträge und Aufsätze über Entwicklungsmechanik der Organismen. Vol 13. Leipzig, Germany: Verlag von Wilhelm Engelmann.
- Attar, E.C., and D.T. Scadden. 2004. Regulation of hematopoietic stem cell growth. Leukemia. 18:1760–1768. [DOI] [PubMed] [Google Scholar]
- Blaho, J.A., and S.A. Aaronson. 1999. Convicting a human tumor virus: guilt by association? Proc. Natl. Acad. Sci. USA. 96:7619–7621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaise, S., N. de Parseval, and T. Heidmann. 2005. Functional characterization of two newly identified Human Endogenous Retrovirus coding envelope genes. Retrovirology. 2:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohannon, R.C., L.A. Donehower, and R.J. Ford. 1991. Isolation of a type D retrovirus from B-cell lymphomas of a patient with AIDS. J. Virol. 65:5663–5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveri, T. 1929. The Origin of Malignant Tumors. The Williams & Wilkins Company, Baltimore, MD. 119 pp.
- Bowen, J.M., R. Cailleau, B.C. Giovanella, S. Pathak, and M.J. Siciliano. 1983. A retrovirus-producing transformed mouse cell line derived from a human breast adenocarcinoma transplanted in a nude mouse. In Vitro. 19:635–641. [DOI] [PubMed] [Google Scholar]
- Chakraborty, A., R. Lazova, S. Davies, H. Backvall, F. Ponten, D. Brash, and J. Pawelek. 2004. Donor DNA in a renal cell carcinoma metastasis from a bone marrow transplant recipient. Bone Marrow Transplant. 34:183–186. [DOI] [PubMed] [Google Scholar]
- Chaput, N., J. Taieb, N.E. Schartz, F. Andre, E. Angevin, and L. Zitvogel. 2004. Exosome-based immunotherapy. Cancer Immunol. Immunother. 53:234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charrin, S., F. Le Naour, M. Oualid, M. Billard, G. Faure, S.M. Hanash, C. Boucheix, and E. Rubinstein. 2001. The major CD9 and CD81 molecular partner. Identification and characterization of the complexes. J. Biol. Chem. 276:14329–14337. [DOI] [PubMed] [Google Scholar]
- Chen, E.H., and E.N. Olson. 2005. Unveiling the mechanisms of cell-cell fusion. Science. 308:369–373. [DOI] [PubMed] [Google Scholar]
- Clayton, A., J. Court, H. Navabi, M. Adams, M.D. Mason, J.A. Hobot, G.R. Newman, and B. Jasani. 2001. Analysis of antigen presenting cell derived exosomes, based on immuno-magnetic isolation and flow cytometry. J. Immunol. Methods. 247:163–174. [DOI] [PubMed] [Google Scholar]
- Couzin, J. 2005. Cell biology: the ins and outs of exosomes. Science. 308:1862–1863. [DOI] [PubMed] [Google Scholar]
- Cowan, C.A., J. Atienza, D.A. Melton, and K. Eggan. 2005. Nuclear reprogramming of somatic cells after fusion with human embryonic stem cells. Science. 309:1369–1373. [DOI] [PubMed] [Google Scholar]
- Duelli, D.M., and Y.A. Lazebnik. 2000. Primary cells suppress oncogene-dependent apoptosis. Nat. Cell Biol. 2:859–862. [DOI] [PubMed] [Google Scholar]
- Duelli, D., and Y. Lazebnik. 2003. Cell fusion: a hidden enemy? Cancer Cell. 3:445–448. [DOI] [PubMed] [Google Scholar]
- Duesberg, P.H. 1987. Retroviruses as carcinogens and pathogens: expectations and reality. Cancer Res. 47:1199–1220. [PubMed] [Google Scholar]
- Duesberg, P., R. Li, and D. Rasnick. 2004. Aneuploidy approaching a perfect score in predicting and preventing cancer: highlights from a conference held in Oakland, CA in January, 2004. Cell Cycle. 3:823–828. [PubMed] [Google Scholar]
- Fevrier, B., and G. Raposo. 2004. Exosomes: endosomal-derived vesicles shipping extracellular messages. Curr. Opin. Cell Biol. 16:415–421. [DOI] [PubMed] [Google Scholar]
- Fine, D., and G. Schochetman. 1978. Type D primate retroviruses: a review. Cancer Res. 38:3123–3139. [PubMed] [Google Scholar]
- Fine, D.L., G.C. Clarke, and L.O. Arthur. 1979. Characterization of infection and replication of Mason-Pfizer monkey virus in human cell cultures. J. Gen. Virol. 44:457–469. [DOI] [PubMed] [Google Scholar]
- Ford, R.J., L.A. Donehower, and R.C. Bohannon. 1992. Studies on a type D retrovirus isolated from an AIDS patient lymphoma. AIDS Res. Hum. Retroviruses. 8:742–751. [PubMed] [Google Scholar]
- Goldenberg, D.M., R.A. Pavia, and M.C. Tsao. 1974. In vivo hybridisation of human tumour and normal hamster cells. Nature. 250:649–651. [DOI] [PubMed] [Google Scholar]
- Gould, S.J., A.M. Booth, and J.E. Hildreth. 2003. The Trojan exosome hypothesis. Proc. Natl. Acad. Sci. USA. 100:10592–10597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths, D.J. 2001. Endogenous retroviruses in the human genome sequence. Genome Biol. 2:Reviews1017.1–Reviews1017.5. [DOI] [PMC free article] [PubMed]
- Hanai, S., T. Nitta, M. Shoda, M. Tanaka, N. Iso, I. Mizoguchi, S. Yashiki, S. Sonoda, Y. Hasegawa, T. Nagasawa, and M. Miwa. 2004. Integration of human T-cell leukemia virus type 1 in genes of leukemia cells of patients with adult T-cell leukemia. Cancer Sci. 95:306–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansemann, D.P.v. 1890. Ueber asymmetrische Zelltheilung in Epithelkrebsen und deren biologische Bedeutung. Virchow's Archiv fuer pathologische Anatomie und fuer klinische Medicin. 119:299–326. [Google Scholar]
- Hemler, M.E. 2003. Tetraspanin proteins mediate cellular penetration, invasion, and fusion events and define a novel type of membrane microdomain. Annu. Rev. Cell Dev. Biol. 19:397–422. [DOI] [PubMed] [Google Scholar]
- Hingorani, S.R., L. Wang, A.S. Multani, C. Combs, T.B. Deramaudt, R.H. Hruban, A.K. Rustgi, S. Chang, and D.A. Tuveson. 2005. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 7:469–483. [DOI] [PubMed] [Google Scholar]
- Houghton, J., C. Stoicov, S. Nomura, A.B. Rogers, J. Carlson, H. Li, X. Cai, J.G. Fox, J.R. Goldenring, and T.C. Wang. 2004. Gastric cancer originating from bone marrow-derived cells. Science. 306:1568–1571. [DOI] [PubMed] [Google Scholar]
- Klebs, E. 1887. Die Allgemeine Pathologie. Vol. 1. Verlag von Gustav Fischer, Jena, Germany. 499 pp.
- Kovacs, G. 1985. Premature chromosome condensation: evidence for in vivo cell fusion in human malignant tumours. Int. J. Cancer. 36:637–641. [DOI] [PubMed] [Google Scholar]
- Mant, C., S. Hodgson, R. Hobday, C. D'Arrigo, and J. Cason. 2004. A viral aetiology for breast cancer: time to re-examine the postulate. Intervirology. 47:2–13. [DOI] [PubMed] [Google Scholar]
- Margolis, R.L., O.D. Lohez, and P.R. Andreassen. 2003. G1 tetraploidy checkpoint and the suppression of tumorigenesis. J. Cell. Biochem. 88:673–683. [DOI] [PubMed] [Google Scholar]
- Martin, F., D.M. Roth, D.A. Jans, C.W. Pouton, L.J. Partridge, P.N. Monk, and G.W. Moseley. 2005. Tetraspanins in viral infections: a fundamental role in viral biology? J. Virol. 79:10839–10851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx, J. 2004. Medicine. Bone marrow cells: the source of gastric cancer? Science. 306:1455–1457. [DOI] [PubMed] [Google Scholar]
- Mortensen, K., J. Lichtenberg, P.D. Thomsen, and L.I. Larsson. 2004. Spontaneous fusion between cancer cells and endothelial cells. Cell. Mol. Life Sci. 61:2125–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Malley, K., and E.W. Scott. 2004. Stem cell fusion confusion. Exp. Hematol. 32:131–134. [DOI] [PubMed] [Google Scholar]
- Pathak, S., M.A. Nemeth, A.S. Multani, G.N. Thalmann, A.C. von Eschenbach, and L.W. Chung. 1997. Can cancer cells transform normal host cells into malignant cells? Br. J. Cancer. 76:1134–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelchen-Matthews, A., B. Kramer, and M. Marsh. 2003. Infectious HIV-1 assembles in late endosomes in primary macrophages. J. Cell Biol. 162:443–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, K.W., C.J. TenEyck, E. Galanis, K.R. Kalli, L.C. Hartmann, and S.J. Russell. 2002. Intraperitoneal therapy of ovarian cancer using an engineered measles virus. Cancer Res. 62:4656–4662. [PubMed] [Google Scholar]
- Rajagopalan, H., and C. Lengauer. 2004. Aneuploidy and cancer. Nature. 432:338–341. [DOI] [PubMed] [Google Scholar]
- Raposo, G., H.W. Nijman, W. Stoorvogel, R. Liejendekker, C.V. Harding, C.J. Melief, and H.J. Geuze. 1996. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 183:1161–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringertz, N.R., and R.E. Savage. 1976. Cell Hybrids. Academic Press, New York. 366 pp.
- Ristevski, S., D.F. Purcell, J. Marshall, D. Campagna, S. Nouri, S.P. Fenton, D.A. McPhee, and G. Kannourakis. 1999. Novel endogenous type D retroviral particles expressed at high levels in a SCID mouse thymic lymphoma. J. Virol. 73:4662–4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert-Guroff, M., T.L. Stern, E.S. Richardson, B.C. Giovanella, and F.H. Michaels. 1996. Presence of Mason-Pfizer monkey virus in some stocks of the human HBL-100 mammary epithelial cell line. J. Natl. Cancer Inst. 88:372–374. [DOI] [PubMed] [Google Scholar]
- Schmid, E., A. Zurbriggen, U. Gassen, B. Rima, V. ter Meulen, and J. Schneider-Schaulies. 2000. Antibodies to CD9, a tetraspan transmembrane protein, inhibit canine distemper virus-induced cell-cell fusion but not virus-cell fusion. J. Virol. 74:7554–7561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seger, Y.R., M. Garcia-Cao, S. Piccinin, C.L. Cunsolo, C. Doglioni, M.A. Blasco, G.J. Hannon, and R. Maestro. 2002. Transformation of normal human cells in the absence of telomerase activation. Cancer Cell. 2:401–413. [DOI] [PubMed] [Google Scholar]
- Smith, G.C., R.L. Heberling, and S.S. Kalter. 1978. Comparison of Mason-Pfizer monkey virus and squirrel monkey (Saimiri sciureus) retrovirus by immunoelectron microscopy. J. Natl. Cancer Inst. 61:411–413. [PubMed] [Google Scholar]
- Sommerfelt, M.A., N. Harkestad, and E. Hunter. 2003. The endogenous langur type D retrovirus PO-1-Lu and its exogenous counterparts in macaque and langur monkeys. Virology. 315:275–282. [DOI] [PubMed] [Google Scholar]
- Song, C., and E. Hunter. 2003. Variable sensitivity to substitutions in the N-terminal heptad repeat of Mason-Pfizer monkey virus transmembrane protein. J. Virol. 77:7779–7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot, S.J., and D.H. Crawford. 2004. Viruses and tumours—an update. Eur. J. Cancer. 40:1998–2005. [DOI] [PubMed] [Google Scholar]
- Taruscio, D., and A. Mantovani. 2004. Factors regulating endogenous retroviral sequences in human and mouse. Cytogenet. Genome Res. 105:351–362. [DOI] [PubMed] [Google Scholar]
- Thery, C., A. Regnault, J. Garin, J. Wolfers, L. Zitvogel, P. Ricciardi-Castagnoli, G. Raposo, and S. Amigorena. 1999. Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. J. Cell Biol. 147:599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thery, C., M. Boussac, P. Veron, P. Ricciardi-Castagnoli, G. Raposo, J. Garin, and S. Amigorena. 2001. Proteomic analysis of dendritic cell-derived exosomes: a secreted subcellular compartment distinct from apoptotic vesicles. J. Immunol. 166:7309–7318. [DOI] [PubMed] [Google Scholar]
- Vignery, A. 2005. Macrophage fusion: are somatic and cancer cells possible partners? Trends Cell Biol. 15:188–193. [DOI] [PubMed] [Google Scholar]
- Wootton, S.K., C.L. Halbert, and A.D. Miller. 2005. Sheep retrovirus structural protein induces lung tumours. Nature. 434:904–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhivotovsky, B., and G. Kroemer. 2004. Apoptosis and genomic instability. Nat. Rev. Mol. Cell Biol. 5:752–762. [DOI] [PubMed] [Google Scholar]