

Abstract
Several kinases phosphorylate vimentin, the most common intermediate filament protein, in mitosis. Aurora-B and Rho-kinase regulate vimentin filament separation through the cleavage furrow-specific vimentin phosphorylation. Cdk1 also phosphorylates vimentin from prometaphase to metaphase, but its significance has remained unknown. Here we demonstrated a direct interaction between Plk1 and vimentin-Ser55 phosphorylated by Cdk1, an event that led to Plk1 activation and further vimentin phosphorylation. Plk1 phosphorylated vimentin at ∼1 mol phosphate/mol substrate, which partly inhibited its filament forming ability, in vitro. Plk1 induced the phosphorylation of vimentin-Ser82, which was elevated from metaphase and maintained until the end of mitosis. This elevation followed the Cdk1-induced vimentin-Ser55 phosphorylation, and was impaired by Plk1 depletion. Mutational analyses revealed that Plk1-induced vimentin-Ser82 phosphorylation plays an important role in vimentin filaments segregation, coordinately with Rho-kinase and Aurora-B. Taken together, these results indicated a novel mechanism that Cdk1 regulated mitotic vimentin phosphorylation via not only a direct enzyme reaction but also Plk1 recruitment to vimentin.
Introduction
Mitosis is the fundamental process by which cellular contents are segregated into two daughter cells. Protein phosphorylation is responsible not only for the initiation of mitosis but also for mitotic morphological changes including the rearrangements of three major cytoskeletons (Nigg, 2001). These changes are regulated not only by Cdk1 but also additional kinases such as orthologs of fly Polo (Barr et al., 2004) or Aurora (Carmena and Earnshaw, 2003) or fungus NIMA (Nigg, 2001). For example, Polo-like kinase 1 (Plk1) participates in several events including centrosome maturation, bipolar spindle formation, and chromosome segregation (Barr et al., 2004).
Intermediate filaments (IFs) are one of the three major cytoskeletal networks, together with actin filaments and microtubules. Vimentin is the most abundant IF protein expressed in mesenchymal cells and in a variety of cultured cells and tumors (Fuchs and Weber, 1994; Chang and Goldman, 2004). IFs are dramatically reorganized during mitosis, and this reorganization is considered to be controlled by IF protein phosphorylation (Inagaki et al., 1987, 1996). For example, Cdk1 phosphorylates vimentin from prometaphase to metaphase (Tsujimura et al., 1994). On the other hand, Aurora-B (Goto et al., 2003) and Rho-kinase (Goto et al., 1998) phosphorylate vimentin specifically at the cleavage furrow from anaphase to the end of mitosis. Mutational studies revealed that efficient IF separation largely depends on Aurora-B and Rho-kinase activity (Yasui et al., 2001; Goto et al., 2003). However, there is little information on the significance of vimentin phosphorylation by Cdk1 (Yasui et al., 2001).
In the present study, we present a novel mechanism indicating that Cdk1 governs mitotic vimentin phosphorylation not only by a direct enzyme–substrate reaction but also through Plk1 recruitment to Cdk1 induced vimentin phosphorylation. This event then induces Plk1 activation and further vimentin phosphorylation.
Results and discussion
Plk1 can bind to vimentin phosphorylated by Cdk1
Vimentin phosphorylation is spatiotemporally regulated by several kinases during mitosis (Inagaki et al., 1996). Fig. 1 A shows mitotic vimentin phosphorylation at Ser55, Ser71, and Ser72, which were identified as phosphorylation sites specific to Cdk1 (Chou et al., 1991; Kusubata et al., 1992), Rho-kinase (Goto et al., 1998), and Aurora-B (Goto et al., 2003), respectively. Cdk1 phosphorylates vimentin from prometaphase to metaphase, and this phosphorylation occurs throughout the cytoplasm (Tsujimura et al., 1994). On the other hand, Rho-kinase and Aurora-B coordinately regulate the cleavage furrow-specific vimentin phosphorylation from anaphase to cytokinesis (Goto et al., 1998, 2003; Yasui et al., 2001). Vimentin mutation at Rho-kinase and Aurora-B phosphorylation sites induced the formation of IF bridges (∼50% of postmitotic cells; Fig. 1 B) between two daughter cells (Yasui et al., 2001; Goto et al., 2003). However, vimentin filament segregation appeared to occur successfully in most of the cells expressing vimentin mutated at Cdk1 phosphorylation sites (Fig. 1 B), although 3–5% of postmitotic cells showed the IF bridge phenotype (Yasui et al., 2001). Thus, the significance of vimentin phosphorylation by Cdk1 remained unknown.
Figure 1.
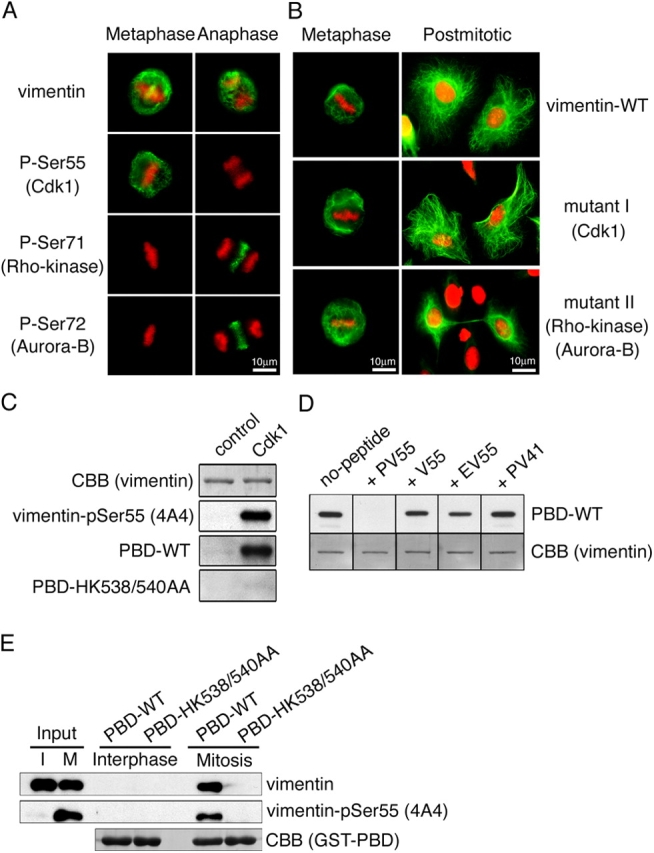
Plk1 binds to vimentin phosphorylated by Cdk1. (A) Site-specific phosphorylation of vimentin during metaphase/anaphase in U251 cells. (B) Metaphase or postmitotic T24 cells expressing vimentin WT or vimentin mutated at Cdk1 (mutant I: Ser41 and Ser55) or Rho-kinase + Aurora-B (mutant II: Ser6, Ser24, Ser38, Ser46, Ser64, Ser65, Ser71, Ser72, and Ser86) phosphorylation sites (Yasui et al., 2001). Green color represents the staining with anti-vimentin (A and B) or the site- and phosphorylation state–specific antibody for Ser55, Ser71, or Ser72 on vimentin (A). DNA was also stained with propidium iodide (red). (C) Specific interaction of Plk1 with vimentin phosphorylated by Cdk1. Vimentin was incubated with (Cdk1) or without (Control) Cdk1. Each sample was then subjected to immunoblotting with 4A4 (P-ser55) or Far Western blotting using GST-PBD. (D) Site specificity of plk1 determined by competition assay. After Far Western blotting (C and D), transferred membrane was stained with Coomassie brilliant blue (CBB). (E) GST pull-down assay using GST-PBD. Each sample was prepared as described in Materials and methods, and then subjected to immunoblotting with 4A4 (P-Ser55) and anti-vimentin antibody.
Plk1 was reported to bind to a phosphoserine (pS)- or phosphothreonine (pT)-containing peptide through its polo-box domain (PBD; Elia et al., 2003a). To examine whether Plk1 interacts with vimentin phosphorylated by Cdk1, vimentin phosphorylated by Cdk1 was subjected to Far Western analysis using GST-PBD proteins. Fig. 1 C shows that GST-PBD wild type (WT) directly interacted with vimentin phosphorylated by Cdk1, but not with nonphosphorylated vimentin. This phenomenon was not observed in the case of GST-PBD-HK538/540AA, which cannot bind phosphoproteins (Elia et al., 2003b), suggesting specific binding between PBD and vimentin phosphorylated by Cdk1 (Fig. 1 C). Next, we examined the phosphorylation site essential for this interaction. Because Ser41 and Ser55 on vimentin were reported to be phosphorylated by Cdk1 (Chou et al., 1991; Kusubata et al., 1992), we did a competition assay using PV55, PV41 (corresponding to vimentin phosphorylated at Ser55 or Ser41, respectively), V55 peptide (corresponding to vimentin unphosphorylated at Ser55; Tsujimura et al., 1994), or EV55 peptide (corresponding to vimentin in which Ser55 is changed to Glu). As shown in Fig. 1 D, binding to vimentin was inhibited by preincubation of GST-PBD-WT with PV55, but not with TBS (control), V55, EV55, or PV41: with regard to Plk1 binding to vimentin, Ser55 mutation to Glu did not mimic Ser55 phosphorylation. PV55 phosphopeptide contains Ser-pSer-Pro reported to be a preferred consensus sequence for a Plk1-binding motif (Elia et al., 2003a). GST pull-down assay revealed that PBD-WT bound to vimentin-Ser55 phosphorylated in mitotic cell extract but not nonphosphorylated vimentin in interphase extract (Fig. 1 E). Thus, these results indicated that Plk1 may directly bind to vimentin-Ser55 phosphorylated by Cdk1.
The binding of vimentin phosphorylated by Cdk1 stimulates the catalytic activity of Plk1, and induces further vimentin phosphorylation
The above specific binding led us to examine its effect on the catalytic activity of Plk1. First, we used casein as a control substrate. As shown in Fig. 2, A and B, the preincubation of Plk1 with PV55 increased its kinase activity toward casein about three times over that without peptide. However, only marginal stimulation was observed in the case of V55 or PV41, confirming the peptide specificity of the Plk1 activation (Fig. 2 A).
Figure 2.

Plk1 increases its catalytic activity via its binding to vimentin phosphorylated at Ser55 by Cdk1. (A) After GST-Plk1-WT was preincubated with PV55, PV41, or V55 (Tsujimura et al., 1994) for 5 min at RT, casein was phosphorylated by above Plk1. The SDS-PAGE gel was stained with CBB and then subjected to autoradiography (32P). (B) Time course of Plk1 kinase activity with or without PV55. (C) After vimentin was preincubated with or without Cdk1, each sample was further incubated with or without Plk1. The total amount of vimentin was determined by CBB. The phosphorylation of vimentin at Ser55 was detected by immunoblotting using 4A4. The incorporation of radioactivity was analyzed by autoradiography. (D) Quantification of Plk1 kinase activity indicated in C.
To determine if a similar Plk1 activation also occurred in the context of a full-length protein, vimentin was preincubated with or without Cdk1 in the absence of radioactive ATP and then incubated with or without Plk1 in the presence of radioactive ATP. Plk1 phosphorylated vimentin pretreated with Cdk1 approximately five times more than without Cdk1 (Fig. 2, C and D). Our observations suggest that Plk1 binding to phosphorylated vimentin-Ser55 induces Plk1 activation and further vimentin phosphorylation.
Plk1 phosphorylates vimentin at Ser82, which partly inhibits the filament forming ability of vimentin
We next examined whether vimentin is a good substrate for Plk1. Vimentin was phosphorylated by GST-Plk1-WT, but not by -K82R (Fig. 3 A): the phosphorylation level increased in a time-dependent manner and reached ∼1 mol of phosphate/mol of protein at 120 min (Fig. 3 C). Immunological analyses revealed that Plk1 phosphorylated Ser82, but not Ser6, Ser28, Ser33, Ser38, Ser50, Ser55, Ser71, and Ser72 on vimentin in vitro (Fig. 3 A). In addition, Ser82 phosphorylation was observed in COS-7 cells expressing Plk1 T210D (a constitutively active mutant) but not K82R (Fig. 3 B). These observations suggested that Plk1 induced vimentin phosphorylation at Ser82 likely through a direct enzyme–substrate reaction.
Figure 3.
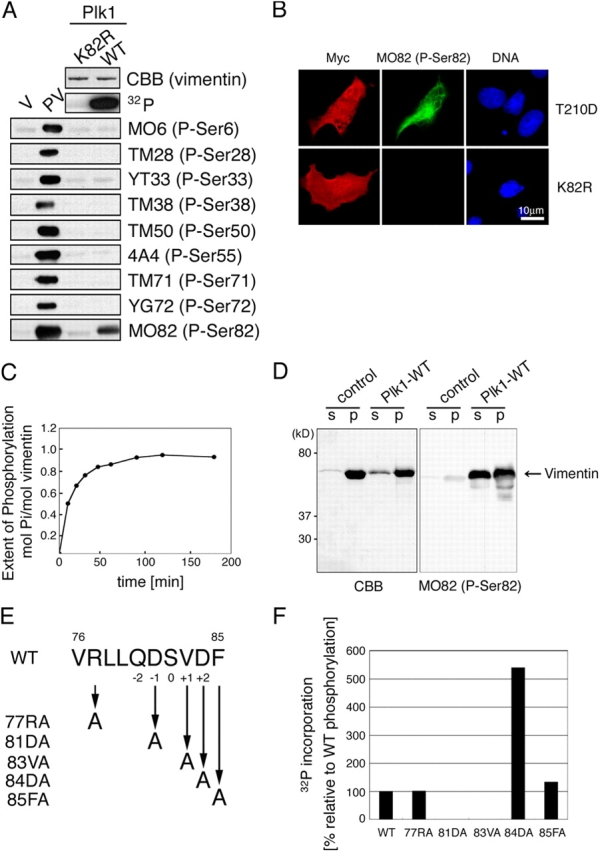
Plk1 phosphorylates vimentin at Ser82. (A) Vimentin was incubated with GST-Plk1-WT or -K82R (a kinase dead mutant). The incorporation of radioactivity into vimentin was analyzed by autoradiography and the total amount of vimentin was determined by CBB. Each sample was also subjected to immunoblotting with each site- and phosphorylation state–specific antibody. The following were used as a positive control for each antibody (phosphorylated vimentin, PV): vimentin phosphorylated by protein kinase A (PKA) for MO6 (P-Ser6) and TM28 (P-Ser28), by PKC for YT33 (P-Ser33) and TM50 (P-Ser50), by Ca2+/calmodulin-dependent protein kinase II for TM38 (P-Ser38) and MO82 (P-Ser82), by Cdk1 for 4A4 (P-Ser55), by Rho-kinase for TM71 (P-Ser71), and by Aurora-B for YG72 (P-Ser72), respectively. As a negative control, nonphosphorylated vimentin (V) was also immunoblotted. (B) COS-7 cells were transfected with myc-Plk1-T210D or -K82R. Green, red, or blue color represents vimentin-phosphoSer82 (MO82), the expression of Plk1 mutants (Myc), or DNA, respectively. (C) Time course of vimentin phosphorylation by Plk1. (D) Vimentin was preincubated with or without Plk1, and then incubated in the polymerization buffer (Goto et al., 1998) at 37°C for 60 min further. After incubation, each sample was centrifuged at 12,000 g. The supernatant (s) and the precipitate (p) fractions were analyzed using SDS-PAGE, then, the gel was subjected to staining with CBB or the immunoblotting with MO82 (P-Ser82). (E) The sequence of residues 76–85 on vimentin and of various vimentin mutant peptides. (F) Plk1 activity toward each vimentin peptide indicated in E was quantified as a percentage of the radioactivity of each peptide relative to that of WT peptide.
To examine the effect of vimentin phosphorylation by Plk1 on vimentin filament forming ability, soluble vimentin was treated with or without Plk1 and then incubated under conditions favoring polymerization. After incubation, each sample was separated into supernatant and pellet fractions by centrifugation. Plk1 treatment partly inhibited the filament forming ability of vimentin, so that some Plk1-treated vimentin was recovered from the supernatant fraction (Fig. 3 D). Although the amounts of Plk1-treated vimentin in the supernatant fraction were smaller that those in the pellet fraction, there was little difference in the MO82 immunoreactivity between these two fractions (Fig. 3 D), indicating that most of the Ser82-phosphorylated vimentin had lost its filament forming ability in vitro.
Vimentin phosphorylation motif for Plk1
The consensus Plk1 phosphorylation motif was reported to be D/E-X-pS/pT-φ-X-D/E (φ; a hydrophobic amino acid) in which the +3 position is preferred but not essential for kinase recognition (Nakajima et al., 2003). Because the vimentin sequence around Ser82 did not completely match the above consensus (Fig. 3 E), we examined which amino acid(s) surrounding Ser82 are important for Plk1 phosphorylation, using synthetic vimentin peptides (residues 76–85) in which each amino acid was changed to Ala (Fig. 3 E). Compared with WT sequence, the substitution of Asp81 (81DA) or Val83 (83VA) strongly reduced peptide phosphorylation, but the substitution of Asp84 (84DA) increased the phosphorylation (Fig. 3 F). These results suggest that the position of an acidic amino acid is important for vimentin phosphorylation by Plk1: Asp at the −1 position is required for Plk1 phosphorylation, but Asp at the +2 position has an inhibitory effect. In addition, a hydrophobic amino acid at the +1 position is essential for Plk1 recognition, in line with the report by Nakajima et al. (2003).
Plk1 regulates the elevation of vimentin-Ser82 phosphorylation, which follows vimentin-Ser55 phosphorylation by Cdk1, in mitosis
We investigated the temporal and spatial distribution of vimentin-Ser55 and vimentin-Ser82 phosphorylation at each stage of mitosis. As shown in Fig. 4, A and B, Ser55 phosphorylation occurred from prometaphase to metaphase, as reported previously (Tsujimura et al., 1994). Although Ser82 phosphorylation was weakly observed even in interphase, the elevation of Ser82 phosphorylation occurred from metaphase (from prometaphase in some cells) and was maintained until the end of mitosis. These results indicated that the elevation of vimentin-Ser82 phosphorylation occurred just after the initiation of vimentin-Ser55 phosphorylation by Cdk1.
Figure 4.
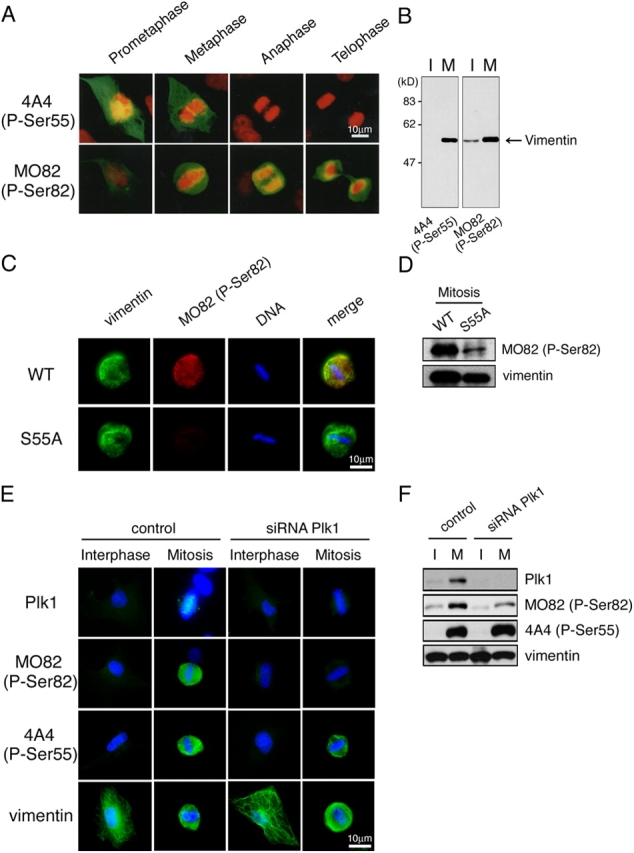
Plk1 depletion impaired the elevation of vimentin-Ser82 phosphorylation in mitosis. (A) Cell cycle–dependent phosphorylation of vimentin-Ser55 and vimentin-Ser82 during mitosis in U251 cells. Green color represents vimentin phosphorylated at Ser55 (4A4) or at Ser82 (MO82). DNA was stained with propidium iodide (red). (B) Interphase (I) or early mitotic (M) U251 cell lysates were subjected to immunoblotting with 4A4 or MO82 antibody. (C) T24 cells were transfected with pDR2 carrying vimentin WT or S55A. 48 h after transfection, cells were stained with anti-vimentin antibody (green) and MO82 (red) and DAPI (blue). (D) 44 h transfection, cells were synchronized at the prometaphase by the addition of nocodazole. After 4 h (48 h after transfection), interphase or early mitotic cell lysate were subjected to immunoblotting analysis with MO82 or anti-vimentin antibody. (E) 48 h after transfection with or without Plk1 siRNA duplex, U251 cells were stained with anti-Plk1, MO82, 4A4, or anti-vimentin antibody (green), and DAPI (blue). (F) 36 h transfection, cells were synchronized at the prometaphase by the addition of nocodazole. After 12 h (48 h after transfection), interphase or early mitotic cell lysate were subjected to immunoblotting analysis with anti-Plk1, MO82, 4A4, or anti-vimentin antibody.
To examine the relationship between Ser55 and Ser82 phosphorylation, we transfected T24 cells with vimentin WT or its mutant in which Ser55 is changed into Ala (S55A). 48 h after transfection, the level of Ser82 phosphorylation significantly reduced in cells expressing S55A, compared with WT (Fig. 4, C and D). These results suggested that Ser55 phosphorylation plays an important role in mitotic elevation of Ser82 phosphorylation.
To examine whether Plk1 regulates vimentin-Ser82 phosphorylation, we depleted endogenous Plk1 using small interference RNA (siRNA), as reported previously (Liu and Erikson, 2002). 48 h after transfection, Plk1 expression was significantly reduced in Plk1 siRNA–treated cells compared with control cells, although no significant difference was observed in vimentin expression (Fig. 4 F). Plk1 depletion diminished vimentin-Ser82 phosphorylation in mitosis but not in interphase, whereas no significant reduction was observed in the level of vimentin-Ser55 phosphorylation (Fig. 4, E and F). These results suggested that Plk1 regulates the mitotic elevation of vimentin-Ser82 phosphorylation.
Effects of vimentin-Ser82 phosphorylation by Plk1 on cytokinesis
We constructed a mutant vimentin in which Ser82 is changed to Glu (S82E), which is assumed to mimic Ser82 phosphorylation. Exogenous WT expression induced the formation of a filamentous IF network in T24 cells (expressing no endogenous vimentin), similar to the network observed in cells expressing endogenous vimentin (Fig. 5 A). On the other hand, S82E mutant no longer formed a clear filamentous IF network: instead, the dot-like IF structures and short filaments were distributed diffusely in the cytoplasm. Together with in vitro data (Fig. 3 D), these results indicated that Plk1, as well as Aurora-B and Rho-kinase, may participate in the reorganization of vimentin filaments through vimentin phosphorylation.
Figure 5.
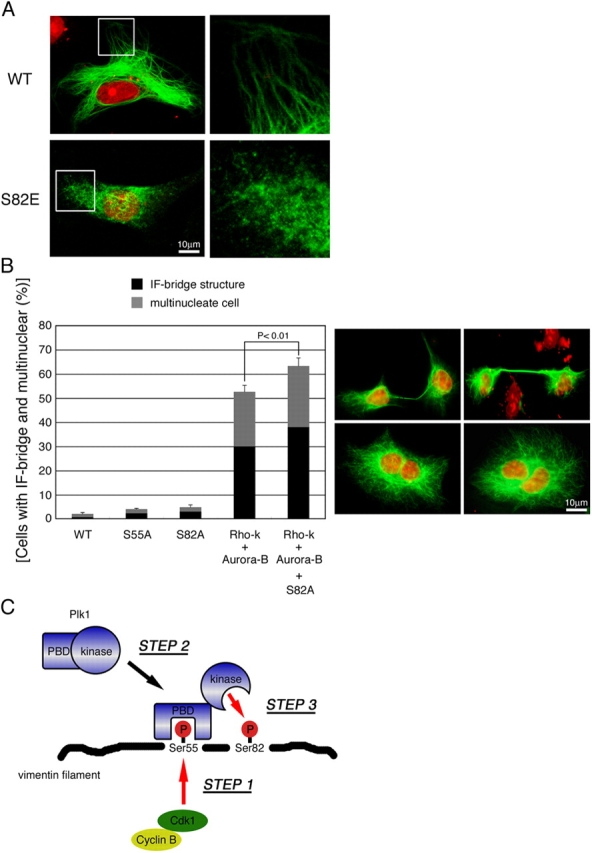
The significance of Plk1-induced vimentin phosphorylation on vimentin filament segregation. (A) T24 cells were transfected with pDR2 carrying vimentin WT or S82E. 48 h after transfection, cells were stained with anti-vimentin antibody (green) and propidium iodide (red). Magnified images were also indicated in the right panel. Cells were analyzed by laser-scanning confocal microscopy. (B) Vimentin IF bridge formation and multinucleate cell in T24 cells expressing vimentin mutants (right). Cells were stained with anti-vimentin antibody (green) and propidium iodide (red). The percentage of vimentin IF-bridge or multinucleate cells were scored as described (Yasui et al., 2001). Data represents means ± SEM in at least three independent experiments. (C) Blue, green, yellow, or red color represents Plk1, Cdk1, cyclin B, and phosphates within residues on vimentin, respectively. The red arrow indicates phosphorylation reaction.
To investigate the significance of vimentin-Ser82 phosphorylation in mitosis, we constructed a series of mutant vimentin in which phosphorylation site(s) are changed to Ala, and transfected then with T24 (type III IF–deficient) cells (Fig. 5 B). IF bridge and multinuclear phenotype were frequently observed in cells expressing vimentin with mutations in Rho-kinase and Aurora-B sites (52.6 ± 2.6%) as compared with WT (2.1 ± 0.5%): this observation was consistent with our previous reports (Goto et al., 2003). Additional Ser82 mutation showed synergistic effects (63.3 ± 3.3%) in the IF bridge formation and multinucleate cell, although single mutation at Ser82 showed a little effect (S82A; 4.86 ± 0.9%, Fig. 5 B). These mutational analyses revealed that Plk1 might play important roles in the correct separation of vimentin filaments, together with Rho-kinase and Aurora-B.
We propose that Cdk1 phosphorylation induces Plk1-mediated reorganization of vimentin filaments (Fig. 5 C). According to our model, Cdk1 phosphorylates vimentin at Ser55 from prometaphase to metaphase (step 1), then, Plk1 is recruited to phosphoSer55 on vimentin via its PBD (step 2). The binding of the PBD to phosphorylated vimentin-Ser55 serves to increase not only the catalytic activity of Plk1 but also the accessibility of the activated Plk1 to vimentin (Fig. 2). This mechanism facilitates vimentin phosphorylation at Ser82 by Plk1 (step 3), and explains why Ser82 phosphorylation is elevated just after the initiation of Ser55 phosphorylation (Fig. 4 A). At anaphase, Plk1 binding to vimentin may be diminished by the reduction of vimentin-Ser55 phosphorylation (Fig. 4 A). However, the elevation of Ser82 phosphorylation appears to continue until the end of mitosis (Fig. 4 A). We consider that this retention may relate to resistance to the activity of protein phosphatase 1 (PP1), which was reported to be a vimentin phosphatase (Inada et al., 1999). Interestingly, phosphoSer82 on vimentin was relatively insensitive to PP1 in vitro, and the stimulation of a calcium signaling pathway in primary glia cells resulted in a sustained increase of vimentin-Ser82 phosphorylation (unpublished data). So, phosphoSer82 on vimentin may hardly be dephosphorylated by PP1 in mitosis. This sustained Ser82 phosphorylation by Plk1 may play some roles in the efficient segregation of vimentin filaments, although Rho-kinase and Aurora-B may play greater roles.
This study demonstrates a novel mechanism indicating that Cdk1 controls vimentin phosphorylation not only by a direct reaction but also through Plk1 recruitment to vimentin. The crosstalk between Cdk1 and Plk1 signaling opens a new field to study mechanisms of mitotic regulation.
Materials and methods
Materials
Recombinant mouse vimentin (Ogawara et al., 1995; NCBI accession no. P20152) and mouse Cdk1 (Kusubata et al., 1992) were purified from Escherichia coli and FM3A cells, respectively. For Plk1 purification, Sf9 cells were infected with baculovirus carrying His-tagged or GST-tagged human Plk1-WT or K82R (a kinase dead mutant) for 2 d and then each protein was purified. In some experiments (Fig. 3), Sf9 cells were further treated with okadaic acid for 3 h after a 2 d-infection: this method having the advantage that Plk1 is already active in Sf9 cells (Kumagai and Dunphy, 1996). GST-PBD WT or HK538/540AA was purified from BL21(DE3)pLysS transformed with pGEX-6P-3 carrying residues 326–603 of human Plk1 WT or a mutant in which His538 and Lys540 were changed into Ala, respectively. Anti-vimentin and site- and phosphorylation state–specific antibodies of vimentin were prepared as described elsewhere (Yasui et al., 2001; Goto et al., 2003). Mammalian expression vectors of mouse vimentin WT or phosphorylation site mutants were prepared as described previously (Yasui et al., 2001). Interphase and mitotic U251 cell lysates were prepared as reported by Goto et al. (1998). Plk1-targeted siRNA duplexes (Liu and Erikson, 2002) were obtained from QIAGEN. Vimentin peptides (residues 76–85) as shown in Fig. 3 E were chemically synthesized by Greiner JAPAN.
Production of mouse monoclonal anti-Plk1 antibody
For the production of the anti-Plk1 mouse monoclonal antibody 36–298, BALB/c mice were then immunized by repeated injections of 100–150 μg of His-tagged human Plk1, using either Freund's or aluminium hydroxide as an adjuvant. Spleen cells were fused with PAIB3Ag81 mouse myeloma cells. Supernatant screening was performed using ELISA and dot-blot assays with His–tagged human Plk1 as an antigen, and by Western blots on HeLa S3 extracts. Primary hybridoma clones were subcloned by limiting dilution. Isotyping revealed that the mouse monoclonal antibody 36–298 is an IgG1.
Transfection
U251, COS-7, or EBNA-expressing T24 cells (Yasui et al., 2001) were transfected using Lipofectamine (Invitrogen) or FuGENE (Roche). For siRNA transfection, U251 cells were transfected using Oligofectamine (Invitrogen) according to the manufacturer's instructions. 24 or 48 h after transfection, cells were subjected to immunocytochemical or immunoblotting studies.
Immunofluorescence analysis
Immunofluorescence analyses were performed as Goto et al. (1998) with slight modification. In brief, fluorescence images were visualized using BX60 microscope (Olympus) controlled by Viewerfinder Lite software (Pixera Corp.) or using Radiance2100 laser confocal microscope controlled by LaserSharp2000 software (Bio-Rad Laboratories). Confocal images (captured at 100×) represents projections of Z series scan (Fig. 5 A). Other images were captured at 60× (by using objective lenses) and recorded using a CCD camera (DP50 camera; Olympus).
Phosphorylation of proteins and peptides
The phosphorylation reaction for vimentin was done in the mixture (25 mM Tris-Cl, pH 7.5, 1.5 mM MgCl2, 0.1 mM ATP, 0.1 μM calyculin A, and 150 μg/ml vimentin) with or without 20 μg/ml of GST-Plk1 at 25°C for 60 min. Vimentin phosphorylation by Cdk1 was done as described previously (Kusubata et al., 1992). Some reactions were done in the presence of [γ-32P] ATP. The phosphorylation reaction for casein was done as follows: after 20 μg/ml GST-Plk1 WT was preincubated with or without 250 μM of each peptide in 25 mM Hepes-NaOH, pH 7.5, buffer at RT for 5 min, phosphorylation reaction was performed in 20 μl of 25 mM Hepes-NaOH, pH 7.5, 5 mM MgCl2, 0.1 mM [γ-32P] ATP, 0.1 μM calyculin A, 200 μg/ml casein, and the above preincubation mixture (5 μl) at 30°C. The reaction was stopped by the addition of SDS sample buffer and boiled. The phosphorylation reaction for peptides was performed in the mixture (25 mM Hepes-NaOH, pH 7.5, 10 mM MgCl2, 50 μM ATP, 0.1 μM calyculin A, 100 μM [γ-32P] ATP, 500 μg/ml each vimentin peptide, and 60 μg/ml of purified Plk1) at 25°C for 60 min. After phosphorylation, each radioactive peptide was separated by reverse phase-HPLC as described (Goto et al., 1998).
Far Western analysis
Vimentin was treated with or without Cdk1 at 25°C for 60 min. Each reaction mixture was separated by SDS-PAGE and transferred onto membranes. Transferred membranes were incubated with 6 μg/ml of GST-PBD-WT or -HK538/540AA in TBS containing 5% skim milk at 4°C for 6 h. For peptide competition assay, GST-PBD-WT was preincubated with 100 μg/ml of phosphopeptide PV55 (CSLYASpSPGGVY) or PV41 (CTYSLGpSALRPS), unphosphosphorylated peptide V55 (CSLYASSPGGVY), or peptide EV55 (CSLYASEPGGVY; in which Ser55 is changed into Glu) at RT for 30 min. As a control, GST-PBD-WT was preincubated only with TBS (no peptide). After washing with TBS, these membranes were incubated with anti-GST rabbit polyclonal antibody (MBL) and then with HRP-conjugated anti–rabbit antibody.
GST pull-down assay
U251 interphase or mitotic cells were treated with the following buffer (1 ml): 50 mM Tris-Cl, pH 7.5, 100 mM NaCl, 1 mM EGTA, 1 mM EDTA, 50 mM NaF, 20 mM β-glycerophosphate, 1 mM Na3VO4, 1% Triton X-100, and protease inhibitor cocktail (Sigma-Aldrich). After centrifugation, each cell extract (1 mg total protein) was incubated with 50 μg of GST-PBD at 4°C for 1 h. GST–PBD protein complex were immobilized on 20 μl of glutathione-sepharose beads, eluted by adding with 20 mM glutathione, and then subjected to immunoblotting with 4A4 (P-Ser55) and anti-vimentin antibody.
Plk1 protein kinase activity toward vimentin pretreated with or without Cdk1
Vimentin was preincubated with or without Cdk1 at 25°C for 3 h. Each reaction mixture was denatured with 7 M urea buffer (7 M urea, 25 mM Tris-Cl, pH 8.8) at 4°C for 16 h and then dialyzed with renaturing buffer (10 mM Tris, 1.7 mM EGTA, 50 mM 2-mercaptoethanol, 1 mM PMSF). Phosphorylation assay for Plk1 was done as described above.
Acknowledgments
We thank Ms. M. Ohara for assistance with the manuscript; Y. Hayashi for preparing materials and technical assistance; Y. Yasui for helpful discussion.
This work supported in part by Grants-in-Aid for Scientific Research and Cancer Research from the Ministry of Education, Science, Technology, Sports, and Culture of Japan, by a grant-in-aid for the Second Term Comprehensive 10-Year Strategy for Cancer Control from the Ministry of Health and Welfare, Japan, by The Naito Foundation, and by the Uehara Memorial Foundation.
Abbreviations used in this paper: CBB, Coomassie brilliant blue; IF, intermediate filament; PBD, Polo-box domain; Plk1, Polo-like kinase 1; PP1, protein phosphatase 1; pS, phosphoserine; pT, phosphothreonine; siRNA, small interference RNA; WT, wild type.
References
- Barr, F.A., H.H. Silljé, and E.A. Nigg. 2004. Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol. 5:429–440. [DOI] [PubMed] [Google Scholar]
- Carmena, M., and W.C. Earnshaw. 2003. The cellular geography of aurora kinases. Nat. Rev. Mol. Cell Biol. 4:842–854. [DOI] [PubMed] [Google Scholar]
- Chang, L., and R.D. Goldman. 2004. Intermediate filaments mediate cytoskeletal crosstalk. Nat. Rev. Mol. Cell Biol. 5:601–613. [DOI] [PubMed] [Google Scholar]
- Chou, Y.H., K.L. Ngai, and R. Goldman. 1991. The regulation of intermediate filament reorganization in mitosis. p34cdc2 phosphorylates vimentin at a unique N-terminal site. J. Biol. Chem. 266:7325–7328. [PubMed] [Google Scholar]
- Elia, A.E., L.C. Cantley, and M.B. Yaffe. 2003. a. Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science. 299:1228–1231. [DOI] [PubMed] [Google Scholar]
- Elia, A.E., P. Rellos, L.F. Haire, J.W. Chao, F.J. Ivins, K. Hoepker, D. Mohammad, L.C. Cantley, S.J. Smerdon, and M.B. Yaffe. 2003. b. The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell. 115:83–95. [DOI] [PubMed] [Google Scholar]
- Fuchs, E., and K. Weber. 1994. Intermediate filaments: structure, dynamics, function, and disease. Annu. Rev. Biochem. 63:345–382. [DOI] [PubMed] [Google Scholar]
- Goto, H., H. Kosako, K. Tanabe, M. Yanagida, M. Sakurai, M. Amano, K. Kaibuchi, and M. Inagaki. 1998. Phosphorylation of vimentin by Rho-associated kinase at a unique amino-terminal site that is specifically phosphorylated during cytokinesis. J. Biol. Chem. 273:11728–11736. [DOI] [PubMed] [Google Scholar]
- Goto, H., Y. Yasui, A. Kawajiri, E.A. Nigg, Y. Terada, M. Tatsuka, K. Nagata, and M. Inagaki. 2003. Aurora-B regulates the cleavage furrow-specific vimentin phosphorylation in the cytokinetic process. J. Biol. Chem. 278:8526–8530. [DOI] [PubMed] [Google Scholar]
- Inada, H., H. Togashi, Y. Nakamura, K. Kaibuchi, K. Nagata, and M. Inagaki. 1999. Balance between activities of Rho kinase and type 1 protein phosphatase modulates turnover of phosphorylation and dynamics of desmin/vimentin filaments. J. Biol. Chem. 274:34932–34939. [DOI] [PubMed] [Google Scholar]
- Inagaki, M., Y. Nishi, K. Nishizawa, M. Matsuyama, and C. Sato. 1987. Site-specific phosphorylation induces disassembly of vimentin filaments in vitro. Nature. 328:649–652. [DOI] [PubMed] [Google Scholar]
- Inagaki, M., Y. Matsuoka, K. Tsujimura, S. Ando, T. Tokui, T. Takahashi, and N. Inagaki. 1996. Dynamic property of intermediate filaments: regulation by phosphorylation. Bioessays. 18:481–487. [Google Scholar]
- Kumagai, A., and W.G. Dunphy. 1996. Purification and molecular cloning of Plx1, a Cdc25-regulatory kinase from Xenopus egg extracts. Science. 273:1377–1380. [DOI] [PubMed] [Google Scholar]
- Kusubata, M., T. Tokui, Y. Matsuoka, E. Okumura, K. Tachibana, S. Hisanaga, T. Kishimoto, H. Yasuda, M. Kamijo, Y. Ohba, et al. 1992. p13suc1 suppresses the catalytic function of p34cdc2 kinase for intermediate filament proteins, in vitro. J. Biol. Chem. 267:20937–20942. [PubMed] [Google Scholar]
- Liu, X., and R.L. Erikson. 2002. Activation of Cdc2/cyclin B and inhibition of centrosome amplification in cells depleted of Plk1 by siRNA. Proc. Natl. Acad. Sci. USA. 99:8672–8676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima, H., F. Toyoshima-Morimoto, E. Taniguchi, and E. Nishida. 2003. Identification of a consensus motif for Plk (Polo-like kinase) phosphorylation reveals Myt1 as a Plk1 substrate. J. Biol. Chem. 278:25277–25280. [DOI] [PubMed] [Google Scholar]
- Nigg, E.A. 2001. Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell Biol. 2:21–32. [DOI] [PubMed] [Google Scholar]
- Ogawara, M., N. Inagaki, K. Tsujimura, Y. Takai, M. Sekimata, M.H. Ha, S. Imajoh-Ohmi, S. Hirai, S. Ohno, H. Sugiura, et al. 1995. Differential targeting of protein kinase C and CaM kinase II signalings to vimentin. J. Cell Biol. 131:1055–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimura, K., M. Ogawara, Y. Takeuchi, S. Imajoh-Ohmi, M.H. Ha, and M. Inagaki. 1994. Visualization and function of vimentin phosphorylation by cdc2 kinase during mitosis. J. Biol. Chem. 269:31097–31106. [PubMed] [Google Scholar]
- Yasui, Y., H. Goto, S. Matsui, E. Manser, L. Lim, K. Nagata, and M. Inagaki. 2001. Protein kinases required for segregation of vimentin filaments in mitotic process. Oncogene. 20:2868–2876. [DOI] [PubMed] [Google Scholar]