

Abstract
Keratins 8 and 18 (K8/18) are major constituents of Mallory bodies (MBs), which are hepatocyte cytoplasmic inclusions seen in several liver diseases. K18-null but not K8-null or heterozygous mice form MBs, which indicates that K8 is important for MB formation. Early stages in MB genesis include K8/18 hyperphosphorylation and overexpression. We used transgenic mice that overexpress K8, K18, or K8/18 to test the importance of K8 and/or K18 in MB formation. MBs were induced by feeding 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC). Livers of young K8 or K8/K18 overexpressors had no histological abnormalities despite increased keratin protein and phosphorylation. In aging mice, only K8-overexpressing livers spontaneously developed small “pre-MB” aggregates. Only K8-overexpressing young mice are highly susceptible to MB formation after short-term DDC feeding. Thus, the K8 to K18 ratio, rather than K8/18 overexpression by itself, plays an essential role in MB formation. K8 overexpression is sufficient to form pre-MB and primes animals to accumulate MBs upon DDC challenge, which may help explain MB formation in human liver diseases.
Introduction
Intermediate filaments (IFs) are a large family of cytoplasmic and nuclear proteins that are involved in a broad range of diseases that reflect their cell and tissue-specific expression (Herrmann et al., 2003; Coulombe and Wong, 2004; Omary et al., 2004). The nuclear IFs include lamins, whereas examples of cytoplasmic IFs include desmin and neurofilaments that are preferentially expressed in muscle and neuronal cells, respectively. In epithelial cells, cytoplasmic IFs consist of types I and II keratins that are found as obligate noncovalent heteropolymers and are expressed in an epithelial cell–specific manner (Coulombe and Omary, 2002). Type I and II keratins stabilize each other via formation of a coil–coil α helix and are found in cells in a 1:1 type I/II stoichiometric ratio. In digestive organ epithelia, the major IFs are keratin polypeptides 8 and 18 (K8/18), with variable levels of K7, 19, and 20 depending on the tissue/cell type (Ku et al., 1999). Adult hepatocytes are unique in that they express K8/18 exclusively (Omary et al., 2002). The absence of either K8 or K18 results in the rapid degradation of most of the remaining keratin partner by ubiquitinylation in order to maintain the 1:1 type I/II keratin protein ratio (Kulesh et al., 1989; Ku and Omary, 2000).
An important function of K8/18 is to protect hepatocytes from mechanical and nonmechanical stresses, as demonstrated using several transgenic animal models (Omary et al., 2002; Magin et al., 2004; Zatloukal et al., 2004). For example, K8-null or K18 mutant–expressing mice are highly susceptible to liver injury, which led to the identification of K8 and K18 variants in patients with end-stage liver disease (Omary et al., 2004; Ku et al., 2005). K8/18-related liver disease likely occurs by a multi-hit process that includes the keratin mutation and other insults such as viral and toxin-related injuries (Omary et al., 2002; Ku et al., 2005). K8 and K18 are also the major components of Mallory bodies (MBs) and are essential for MB formation (Denk et al., 2000). MBs are typically noted as hyaline, irregular-shaped cytoplasmic aggregates or deposits found particularly but not exclusively in hepatocytes of patients with alcoholic and nonalcoholic steatohepatitis (Denk et al., 2000). Similar cytoplasmic deposits that contain other IFs are seen in other diseases, including astrocyte Rosenthal fibers (glial fibrillary acidic protein deposits) of Alexander disease and neurofilament deposits in several neurodegenerative diseases (Cairns et al., 2004; Omary et al., 2004; Cookson, 2005). These aggregates are hallmarks of several diseases, but the mechanism of their formation is poorly understood, albeit MBs are among the most studied as a result of mouse models that include feeding a diet containing griseofulvin or 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC; Denk et al., 2000).
The accumulation of MBs is a multistep process that includes liver injury–mediated K8/18 hyperphosphorylation and K8/18 mRNA and protein overexpression (Yuan et al., 1996; Cadrin et al., 2000; Denk et al., 2000; Stumptner et al., 2000; Omary et al., 2002). Keratin induction is asymmetric, particularly at the protein level. For example, K8 protein increases nearly 5- and then 3-fold, whereas K18 increases nearly 2- and then 1.5-fold after 1 wk and 2.5 mo of DDC feeding, respectively (Denk et al., 2000). The increases in K8/18 mRNA are somewhat more prominent (K8, nearly sixfold; K18, nearly fivefold after 1 wk of DDC feeding; similar levels were seen after 2.5 mo of feeding; Denk et al., 2000) and are likely mediated by an increase in mRNA stability (Cadrin et al., 2000). Such differences in keratin induction are physiologically relevant because the relative K8 versus K18 levels appear to be important for MB formation. For example, aged K18-null mice (K8 > K18) spontaneously develop abnormal hepatocytes with K8-positive MB-like aggregates (Magin et al., 1998). In contrast, K8-null or heterozygous (+/−) mice (K18 > K8) do not form MBs even after intoxication with DDC (Zatloukal et al., 2000). However, differences between in vivo and cell culture models occur in that transfected K18 is more prone to aggregate than transfected K8 (Nakamichi et al., 2002). Although these studies suggest that keratins are important for MB development, the relationship between single or dual K8 and K18 overexpression and MB formation under basal and stimulated conditions remains unclear. We addressed this issue using transgenic mice that overexpress human (h) K18 (Abe and Oshima, 1990) or mouse (m) K8 or mK8/hK18. A previous study described transgenic mice that overexpressed hK8 and developed progressive chronic pancreatitis, but, although detailed analysis was not performed, no liver abnormalities were described (Casanova et al., 1999).
Results and discussion
Keratin mRNA and protein expression were analyzed in four transgenic mouse lines: single (K8 or K18) or double overexpressors (K8/18) and control wild-type (WT) nonoverexpressors. These lines were generated by mating mK8 overexpressors (EA5 +/−) together or with hK18 overexpressors (TG2 +/+; Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200507093/DC1). Analysis of keratin transcription by real-time PCR demonstrated a 3.2-fold increase of mK8 RNA in the K8 overexpressors and a similar increase (2.5-fold) in mice that overexpressed K8/18 as compared with WT mice (Fig. 1 A). K8 overexpression had a slightly more prominent effect on K18 mRNA induction (1.8 ± 0.2) as compared with the effect of K18 overexpression on K8 (1.3 ± 0.3). As expected, hK18 mRNA was detected in the K18 and K8/18 overexpressors (Fig. 1 A). Keratin protein overexpression paralleled the mRNA changes (Fig. 1 B). For example, mK8 protein increased ∼2.5-fold in the K8 and K8/18 overexpressors (see Protein analysis). K8 overexpression has a more profound impact on K18 protein induction as compared with K18 overexpression on K8 protein induction, and K8 + K18 protein levels are highest in the K8/18 double and K8 overexpressors (Fig. 1 B). Therefore, of the K8, 18, or 8/18 lines, the normal 1:1 K8 to K18 liver protein ratio is best approximated in the K8/18 overexpressors while yielding a nearly 2.5-fold increase in overall keratin protein.
Figure 1.
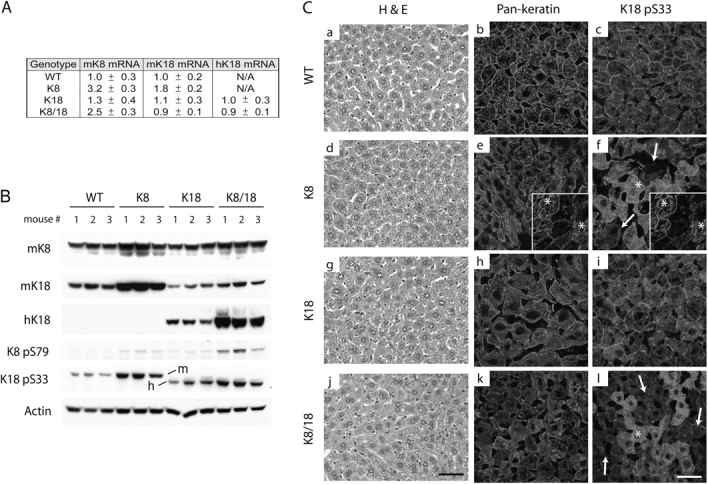
Liver histology and keratin mRNA, protein levels, and immune staining in young WT, K8, K18, and K8/18 mice. (A) Real-time PCR of the indicated keratin mRNA (normalized to ribosomal L7 mRNA) in livers of the four genotypes. For mK8 and mK18 mRNA, the mRNA levels are normalized to those in WT mice (mean ± SD; n = 3/genotype). For mice that overexpress hK18, the levels are normalized to hK18 overexpressors (N/A, not applicable; because WT and K8 mice do not express hK18). (B) Total liver homogenates were prepared, and equal amounts of protein were separated by SDS-PAGE followed by blotting using Abs to the indicated keratin or phosphokeratin epitopes. Actin is included as a loading control. H, human; m, mouse. (C) Livers from 10-wk-old sex-matched mice were fixed, sectioned, and stained with hematoxylin (H) and eosin (E; a, d, g, and j). Alternatively, liver keratins were visualized by immunofluorescence staining using Abs to K8/18 (pankeratin; b, e, h, and k) or to K18 pS33 (c, f, i, and l). Arrows (f and l) highlight basal K18 pS33 staining to contrast with the surrounding enhanced staining (asterisks). Insets show separately performed double staining using anti-mK18 (e) and anti-K18 pS33 (f) to highlight some cells (asterisks) with increased K18 and K18 pS33. Bars, 50 μm.
We examined hepatocyte keratin phosphorylation in the transgenic mouse lines given that keratin dynamics are highly regulated by phosphorylation and that keratin phosphorylation serves as a marker of hepatocyte injury and occurs early during MB formation (Stumptner et al., 2000; Toivola et al., 2004). Keratin phosphorylation was assessed using phosphospecific antibodies (Abs) to the well-characterized phosphorylation sites mK8 phospho- (p)S79 and K18 pS33 (Toivola et al., 2004). These two sites were selected because changes in their phosphorylation correlates with liver injury, apoptosis, or mitosis depending on the biologic context (Toivola et al., 2004). There was a limited increase in the specific activity of K8 pS79 in K8 or K18 livers but a more prominent increase in K8/18 livers (Fig. 1 B). The specific activity of K18 pS33 increased particularly in the K8 transgenic mice as determined by immunoblotting (Fig. 1 B, compare K8 with WT vs. K8/18 with K18) and immunofluorescence staining (Fig. 1 C). Aside from an increased mosaic staining of the filamentous K8 pS33, there was no obvious filament reorganization, and sections of livers from the four genotypes of 10-wk-old mice did not reveal any MBs or other histologic abnormalities (Fig. 1 C). The mosaic increase of K8 pS33 staining in some cells correlated with an increase in K18 staining (Fig. 1 C, e and f; insets). Serum testing of aminotransferases, alkaline phosphatase, total bilirubin, creatinine, and amylase were normal in all four genotypes (not depicted). Therefore, keratin overexpression did not induce MBs in young mice even though K8 and K8/18 mice have abnormal increased hepatocyte K8 and K18 phosphorylation. Because keratin phosphorylation is a hallmark of cell stress (Omary et al., 1998; Coulombe and Omary, 2002; Toivola et al., 2004), the increased keratin phosphorylation in the K8 and K8/18 young mice may point to an underlying early stress response that precedes any obvious liver damage.
Given that MBs form spontaneously in old but not in young K18-null mice (Magin et al., 1998), we asked whether keratin deposits form in old mouse livers isolated from WT, K8, K18, and K8/18 mice. Notably, livers from 2-yr-old K8 mice develop keratin aggregates of varied sizes, whereas livers of similarly aged WT, K18, or K8/18 mice do not (Fig. 2). The keratin aggregates consist of small dots located primarily near the cell periphery and of larger, MB-like cytoplasmic aggregates, which also stained positive for K18 pS33 (Fig. 2, C and D) even though clear MB formation was not seen by histochemical staining (not depicted). The larger keratin-containing aggregates uniformly included ubiquitin, whereas the smaller dots did not (Fig. 2, C and D; insets). Some hepatocytes from K8/18 mice had brighter keratin staining, but keratin aggregates were rarely found (Fig. 2, G and H). The K8 aggregates excluded γ-tubulin and, thus, were not related to centrosomes nor to apoptosis, as they did not stain with an Ab specific to an apoptotic K18 fragment, and most did not colocalize with autophagosomes (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200507093/DC1). Altogether, we hypothesize that the keratin aggregates noted in the K8-overexpressing old mice represent pre-MBs as defined by the formation of K8/18-ubiquitin aggregates that are not easily seen by histochemical staining.
Figure 2.
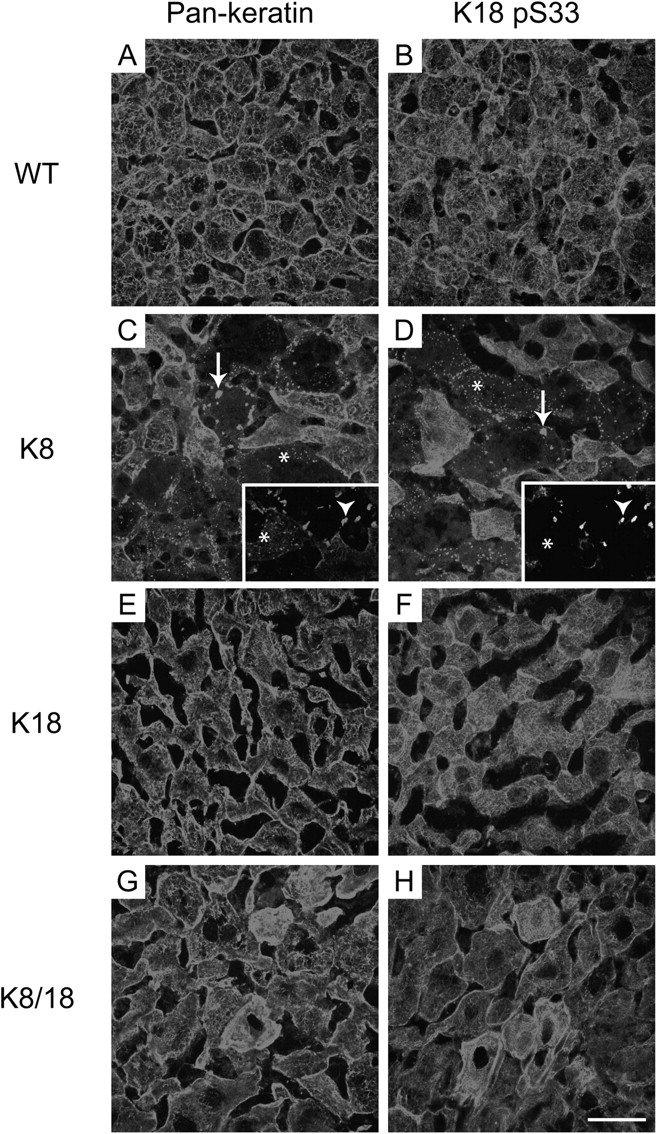
Spontaneous pre-MB formation in livers of old K8 mice. Immunofluorescence staining was performed on livers of 2-yr-old WT, K8, K18, and K8/18 mice. Livers were stained with Abs to K8/18 (pankeratin; A, C, E, and G) or to K18 pS33 (B, D, F, and H). Arrows (C and D) highlight keratin aggregates that are seen only in K8-overexpressing mice, whereas the asterisks highlight fine keratin dots that are likely precursors to the larger aggregates. Bar, 50 μm. Insets show double staining of K8/18 (C) and ubiquitin (D) and illustrate that the larger keratin aggregates are ubiquitin positive (arrowheads), whereas the smaller keratin dots are ubiquitin negative (asterisks).
We tested whether the occurrence of pre-MB aggregates in old K8 mice could render younger K8 mice more susceptible to MB formation by feeding the four mouse genotypes a DDC-containing diet. Mice fed with DDC or griseofulvin for 3–4 mo are established models of MB formation (Yuan et al., 1996; Cadrin et al., 2000; Denk et al., 2000; Stumptner et al., 2000). However, in our experimental design, we analyzed the livers at an early time point of 6 wk after DDC feeding, when MBs are typically not seen. In DDC-fed mice, there was a significant increase in serum alanine aminotransferase (ALT; an indicator of hepatocyte necrosis) in K8 as compared with WT, K18, or K8/18 mice (Fig. 3 A) but no significant difference in liver damage when assessed by hematoxylin and eosin staining (not depicted). Other serologic tests (see Animal feeding and tissue experiments) were comparable in all four genotypes (not depicted). However, MBs were found almost exclusively in the K8 mice, as confirmed by hematoxylin and eosin staining (Fig. 3, B and C), standard and immune electron microscopy (Fig. 3 D), and immunofluorescence staining using antikeratin and antiubiquitin antibodies (Fig. 4 A). K8 livers had 35 ± 19 hepatocytes with MBs per 10 high power fields compared with 2.6 ± 3.7, 1.5 ± 1.7, and 0.2 ± 0.4 for WT, K18, and K8/18 livers, respectively (Fig. 3 C). Ubiquitin localized in most, if not all, of the keratin aggregates (Fig. 4 A), whereas p62 (which is a component of MBs; Zatloukal et al., 2000, 2004) was seen in some of the aggregates (not depicted). After DDC feeding, keratin protein levels increased dramatically, as expected in all genotypes (Fig. 4 B), in concert with an increase in K8 and K18 mRNA levels (not depicted). In addition, phosphorylation of K8 S79 and K18 S33 increased similarly in all four genotypes (not depicted) as described previously for WT mice (Stumptner et al., 2000). Therefore, K8-overexpressing mice are highly primed to develop MBs after DDC challenge.
Figure 3.
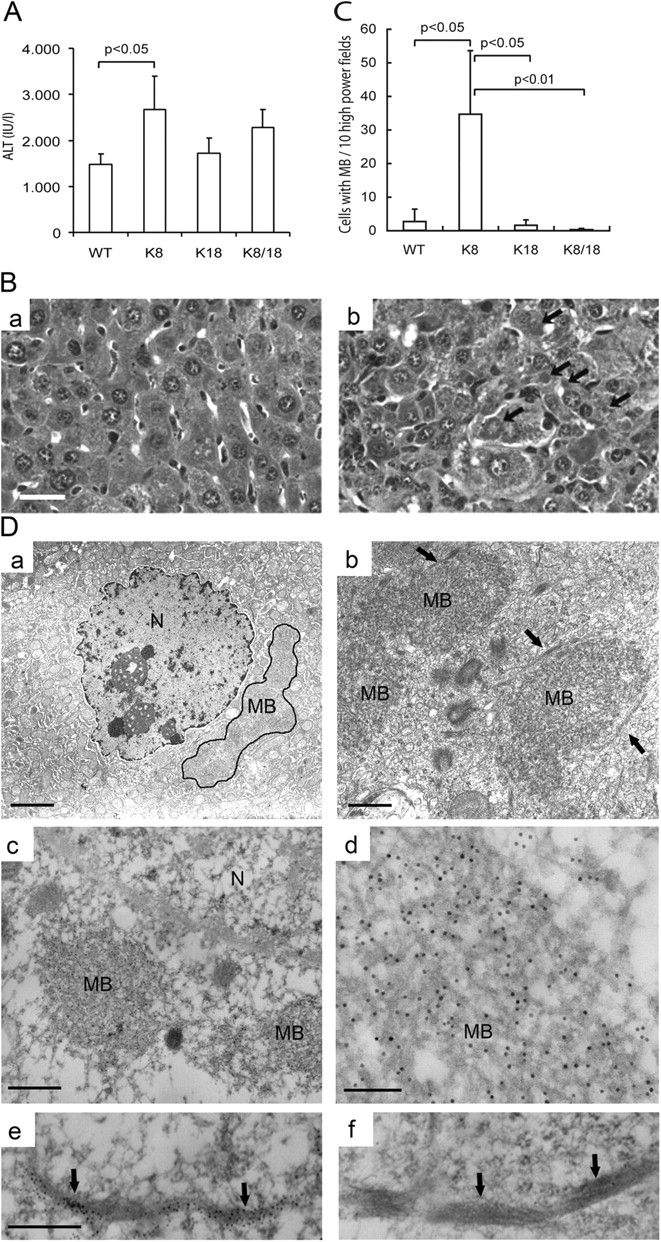
ALT levels and MB formation after DDC feeding. WT, K8, K18, and K8/18 mice (4–5 mice per genotype; 3–4-mo-old sex-matched mice) were fed a DDC-containing diet for 6 wk followed by harvesting of the livers and blood. (A) ALT levels (means ± SD), with a significant increase noted in K8 as compared with WT mice. (B) Livers from mice described in A were fixed and stained with hematoxylin and eosin. Histology of the livers from WT (a) and K8 (b) mice is shown, but K18 and K8/18 liver histology was very similar to WT (not depicted). Note the significant MB formation in K8 livers (arrows). Bar, 200 μm. (C) MBs were quantified (see Materials and methods) using the hematoxylin and eosin–stained liver sections described in B. The P values for comparing the genotypes WT with K8, K8 with K18, and K8 with K8/18 are shown. Error bars represent SD. (D) Conventional (a and b) and immune (c–f) electron microscopy of livers from K8 mice that were fed a DDC-containing diet show typical-appearing MB deposits. In a, the MB is encircled with a black line, and in b, arrows highlight IF bundles that are in close proximity to MBs. For immune electron microscopy, keratins were labeled with 10 nm gold particles (c–e). The particles are difficult to see in c but are clearly evident in the higher magnification images d and e. As a specificity control, the grids were also treated with nonimmune serum (f; note the absence of gold particles as contrasted with e). Arrows in e and f point to keratin IF bundles. Bars (a), 2 μm; (b) 500 nm; (c) 300 nm; (d) 100 nm; (e) 300 nm. N, nucleus.
Figure 4.
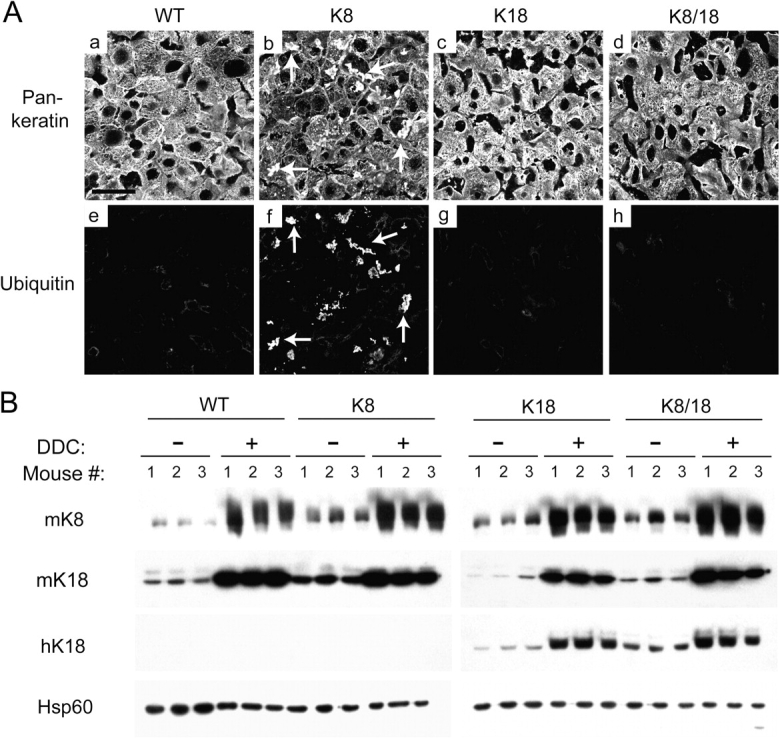
Immunofluorescence staining of keratins and ubiquitin and relative K8/18 protein levels in DDC-fed mouse livers. (A) Mice were fed a DDC-containing diet for 6 wk followed by immunofluorescence double staining of liver sections with anti-K8/18 Ab (pankeratin; a–d) and antiubiquitin Ab. Arrows (b and f) highlight the ubiquitin-positive keratin aggregates. Bar, 50 μm. (B) Total protein was isolated from livers of DDC-fed and control mice (three separate mice/genotype/treatment) followed by blotting with Abs to the indicated epitopes. Because actin increases slightly after DDC treatment (not depicted; Cadrin et al., 2000), heat shock protein 60 (Hsp 60) was used as a loading control.
Our study provides direct evidence that perturbations in individual keratin protein levels, which occur during liver injury, account for MB formation. Overexpression of individual K8 or K18 or combined K8/18 indicated that an increase in the ratio of K8 to K18 is a key ingredient for MB formation. Findings in this study and those previously described for K8 −/− or K8 +/− (which do not form MBs even when challenged) and K18 −/− mice (which form MB-like structures spontaneously) are summarized in Fig. 5 A. Collectively, these data indicate that the availability of “excess” K8 acts as a nidus for hepatocyte MB formation and that excess K18 or K8/18 are well tolerated to the extent that they do not lead to MB formation. Excess K8 requires additional stimuli that ultimately induce MB formation because K18-null (Magin et al., 1998) and K8 mice (Fig. 2) develop MBs only at an old age. Additional stimuli may include aging and its related oxidative damage (Holbrook and Ikeyama, 2002) or chronic toxic liver injury, such as DDC in mice or alcohol or nonalcoholic steatohepatitis in humans (Fig. 5 B). Keratin overexpression is well tolerated under basal conditions (Fig. 1) and does not significantly affect susceptibility to liver injury in DDC-fed mice regardless of whether MBs form in a prominent (K8 mice) or limited (WT, K18, or K8/18 mice) fashion (Figs. 3 and 4). Therefore, in DDC-related injuries, a propensity for MB formation is a reflection of preferential K8 overexpression coupled with additional stimuli, but it does not correlate with the extent of liver injury per se. Interestingly, the formation of K8-containing aggregates also occurs in a physiological context during early mouse embryogenesis when K8 and K7 but not type I keratin proteins are expressed (Lu et al., 2005).
Figure 5.

Summary of MB formation in different mouse genotypes, and a model of MB formation depending on K8 versus K18 protein levels. (A) Summary of MB formation under basal (i.e., spontaneous) and DDC feeding (i.e., induced) conditions in keratin mouse models. (B) A model depicting two types of liver injury: (1) MB-associated liver injury that progressively leads to MB formation in association with keratin overexpression (K8 > K18) and keratin hyperphosphorylation; and (2) non–MB-associated liver injury that results in keratin overexpression (K8 = K18 or K18 > K8) or maintenance of keratin levels coupled with keratin hyperphosphorylation. NASH, nonalcoholic steatohepatitis.
The imbalance in the K8 to K18 ratio may be achieved by transcriptional or posttranscriptional induction in MB-associated mouse injury models (Yuan et al., 1996; Cadrin et al., 2000; Denk et al., 2000; Zatloukal et al., 2000) or by forced expression (this study). Alternate potential mechanisms include differences in keratin protein turnover, particularly because proteasome inhibition occurs as a consequence of protein aggregation (Bence et al., 2001). In this respect, K8/18 degradation in cultured cells occurs by ubiquitination, and K8 and K18 (but less so) phosphorylation protects from degradation (Ku and Omary, 2000). Although keratin hyperphosphorylation is an early event in MB formation (Stumptner et al., 2000), it also correlates with liver injury that is independent of MB formation (Toivola et al., 2004), but its role in MB genesis is unknown. Although phospho–site-specific and mouse strain differences could play an important role in MB formation (Fausther et al., 2004), the inhibitory effect of K8 hyperphosphorylation on keratin degradation (Ku and Omary, 2000) may provide a means to generate a K8 > K18 state after liver injury. In addition, in vivo proteasome inhibition of rats using the compound PS-341 results in keratin accumulation (Bardag-Gorce et al., 2004), and mutant ubiquitin B with likely subsequent interference with MB resorption was found in patient livers that harbored MBs but not in those without MBs (McPhaul et al., 2002). Therefore, alterations in K8/18 turnover may also contribute to a K8 > K18 state. We propose a clinically relevant and testable model whereby MBs form in disease contexts that result in K8 > K18 protein induction but not in situations where there is no induction of K8/18 or where the induction of K8 ≅ K18 or K18 > K8 (Fig. 5 B). The model predicts that human diseases that associate with MB formation, such as alcohol-related liver injury and nonalcoholic steatohepatitis (Fleming and McGee, 1984; Neuschwander-Tetri and Caldwell, 2003; Zatloukal et al., 2004), are more likely to result in a K8 > K18 protein imbalance (K8 > K18). This model may extend to other heteropolymeric IFs, such as neurofilament-containing inclusions (Cairns et al., 2004; Strong et al., 2004), although it is not known whether the stoichiometry of specific neurofilament protein (or inclusion-associated α internexin) induction is critical, as is the case for K8/18.
Materials and methods
Transgenic mice
Transgenic mice that overexpress human WT K18 (called TG2) were previously described and extensively used (Abe and Oshima, 1990; Ku et al., 2003; Zhou et al., 2005). For mouse K8 overexpression, a HindDIII 16-kb fragment of the Balb/c mouse K8 gene (EndoA1α; Vasseur et al., 1985) was injected into zygotes of FVB/n mice. Four independent founder lines (EA2–EA5) were initially selected, from which the EA5 line was propagated further. Double K8/18-overexpressing mice were generated by mating homozygous (+/+) TG2 and EA5 mice. The presence of transgenes was verified by PCR screening of genomic DNA isolated from mouse tails using specific primers (Table S1, available at http://www.jcb.org/cgi/content/full/jcb.200507093/DC1). The primers that were selected to distinguish between BALB/c and endogenous FVB/n utilize an intronic K8 polymorphism that is found in the FVB/n background.
Animal feeding and tissue experiments
Mice were killed by CO2 inhalation, and blood was collected by intracardiac puncture. Livers were rapidly removed and divided into slices or pieces that were immediately fixed in 10% formalin (histology), were embedded in optimal cutting temperature (Miles; immunofluorescence staining), or were snap frozen in liquid N2 (protein/RNA analysis). Fixed tissues were sectioned and stained using hematoxylin and eosin (Histo-tec Laboratory). Immunofluorescence staining was performed as described previously (Ku et al., 2004) using Texas red–conjugated anti–rabbit (Invitrogen) and FITC-conjugated anti–mouse (Biosource International) secondary Abs. Images were taken using 100×/NA 1.40 or 60×/NA 1.40 oil lenses (Nikon) and a microscope (TE300; Nikon) combined with a confocal microscope (MRC1024ES; Bio-Rad Laboratories) and the Lasersharp software (Carl Zeiss MicroImaging, Inc.). Images were compiled using Adobe Illustrator or Adobe Photoshop software. Conventional and immune electron microscopy were performed as described previously (Toivola et al., 2000), and samples were viewed and photographed using an electron microscope (CM-12; Philips). Serologic tests included aspartate aminotransferase and ALT, alkaline phosphatase, total bilirubin, creatinine, and amylase. For MB induction, sex-matched mice (3–4-mo old; three males and two females for each genotype) were fed a powdered Formulab Diet 5008 (Deans Animal Feeds) containing 0.1% DDC (Sigma-Aldrich) for 6 wk (instead of the typical 3-mo feeding; Zatloukal et al., 2000). The diet was replenished every 2 d. Statistical analysis for ALT was performed using Dunnett's method and for MB quantification using the t test. For MB quantification, 10 randomly selected high power fields were used per genotype, and the number of hepatocytes with MBs were counted and presented as means ± SD.
Protein analysis
Total protein tissue lysates were prepared by homogenization in 2% SDS-containing sample buffer and analyzed by SDS-PAGE followed by transfer to membranes for immunoblotting and visualization by enhanced chemiluminescence. The Abs used included (Ku et al., 2004) the following: rabbit Ab 8592 (anti-m/hK8/K18) and Ab 8250 (anti-K18 pS33); rat Troma I (anti-mK8) and Troma II (anti-mK18); mouse Ab L2A1 (anti-hK18) and Ab LJ4 (anti-mK8 pS79); mouse antiactin and anti-hsp60 (NeoMarkers); mouse anti–γ-tubulin (Sigma-Aldrich); mouse Ab M30 (anti-K18 apoptotic fragment; Roche); anti–microtubule-associated protein light chain 3 (a gift from R. Kopito, Stanford University, Palo Alto, CA); and mouse anti-p62 and antiubiquitin (Santa Cruz Biotechnology, Inc.). Keratin protein induction was quantified by densitometric scanning of blots containing serial dilutions of whole liver homogenates from the transgenic lines (analyzed on the same gel).
Real-time RT-PCR
Total RNA was isolated using an RNeasy Midi Kit (QIAGEN). The RNA was translated into cDNA using oligo-dT primers and Superscript II reverse transcriptase (Invitrogen). Quantitative real-time PCR was performed with a Sequence Detection System (ABI Prism 7900; PE Biosystems) and specific primers (Table S1). Samples were analyzed in quadruplicates, and at least three individual mice were tested for each genotype. L7 ribosomal protein was used as an internal control, and keratin and actin cDNA levels were normalized so that L7 expression was near equal in all tested mice. After confirming that the amplification efficiency was similar for all genes, transcript levels relative to L7 were determined and reported as means ± SD.
Online supplemental material
Fig. S1 shows breeding and screening of WT, K8, K18, and K8/18 lines. Fig. S2 shows characterization of keratin aggregates in aging K8 mice. Table S1 shows sequences of PCR primers used for K8 and K18 transgene screening and sequences of primers used for real-time PCR.
Acknowledgments
We are grateful to Ron Kopito for the antibody gift, Nafisa Ghori for help with electron microscopy, Karin Zeh and Jacqueline Avis for initial work on the K8 mice, Evelyn Resurreccion for assistance with tissue sectioning and immunofluorescence staining, and Kris Morrow for assistance with figure preparation.
This work was supported by the National Institutes of Health (NIH) grant DK52951 and the Department of Veterans Affairs Merit Award (M.B. Omary) as well as the NIH Digestive Disease Center grant DK56339. P. Strnad is supported by a long-term European Molecular Biology Organization postdoctoral fellowship.
I. Nakamichi and D.M. Toivola contributed equally to this paper.
Abbreviations used in this paper: Ab, antibody; ALT, alanine aminotransferase; DDC, 3,5-diethoxycarbonyl-1,4-dihydrocollidine; IF, intermediate filament; MB, Mallory body; WT, wild type.
References
- Abe, M., and R.G. Oshima. 1990. A single human keratin 18 gene is expressed in diverse epithelial cells of transgenic mice. J. Cell Biol. 111:1197–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardag-Gorce, F., N.E. Riley, L. Nan, R.O. Montgomery, J. Li, B.A. French, Y.H. Lue, and S.W. French. 2004. The proteasome inhibitor, PS-341, causes cytokeratin aggresome formation. Exp. Mol. Pathol. 76:9–16. [DOI] [PubMed] [Google Scholar]
- Bence, N.F., R.M. Sampat, and R.R. Kopito. 2001. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 292:1552–1555. [DOI] [PubMed] [Google Scholar]
- Cadrin, M., H. Hovington, N. Marceau, and N. McFarlane-Anderson. 2000. Early perturbations in keratin and actin gene expression and fibrillar organisation in griseofulvin-fed mouse liver. J. Hepatol. 33:199–207. [DOI] [PubMed] [Google Scholar]
- Cairns, N.J., V. Zhukareva, K. Uryu, B. Zhang, E. Bigio, I.R. Mackenzie, M. Gearing, C. Duyckaerts, H. Yokoo, Y. Nakazato, et al. 2004. alpha-internexin is present in the pathological inclusions of neuronal intermediate filament inclusion disease. Am. J. Pathol. 164:2153–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova, M.L., A. Bravo, A. Ramirez, G. Morreale de Escobar, F. Were, G. Merlino, M. Vidal, and J.L. Jorcano. 1999. Exocrine pancreatic disorders in transsgenic mice expressing human keratin 8. J. Clin. Invest. 103:1587–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson, M.R. 2005. The biochemistry of Parkinson's disease. Annu. Rev. Biochem. 74:29–52. [DOI] [PubMed] [Google Scholar]
- Coulombe, P.A., and M.B. Omary. 2002. ‘Hard’ and ‘soft’ principles defining the structure, function and regulation of keratin intermediate filaments. Curr. Opin. Cell Biol. 14:110–122. [DOI] [PubMed] [Google Scholar]
- Coulombe, P.A., and P. Wong. 2004. Cytoplasmic intermediate filaments revealed as dynamic and multipurpose scaffolds. Nat. Cell Biol. 6:699–706. [DOI] [PubMed] [Google Scholar]
- Denk, H., C. Stumptner, and K. Zatloukal. 2000. Mallory bodies revisited. J. Hepatol. 32:689–702. [DOI] [PubMed] [Google Scholar]
- Fausther, M., L. Villeneuve, and M. Cadrin. 2004. Heat shock protein 70 expression, keratin phosphorylation and Mallory body formation in hepatocytes from griseofulvin-intoxicated mice. Comp. Hepatol. 3:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming, K.A., and J.O. McGee. 1984. Alcohol induced liver disease. J. Clin. Pathol. 37:721–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann, H., M. Hesse, M. Reichenzeller, U. Aebi, and T.M. Magin. 2003. Functional complexity of intermediate filament cytoskeletons: from structure to assembly to gene ablation. Int. Rev. Cytol. 223:83–175. [DOI] [PubMed] [Google Scholar]
- Holbrook, N.J., and S. Ikeyama. 2002. Age-related decline in cellular response to oxidative stress: links to growth factor signaling pathways with common defects. Biochem. Pharmacol. 64:999–1005. [DOI] [PubMed] [Google Scholar]
- Ku, N.O., and M.B. Omary. 2000. Keratins turn over by ubiquitination in a phosphorylation-modulated fashion. J. Cell Biol. 149:547–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku, N.O., X. Zhou, D.M. Toivola, and M.B. Omary. 1999. The cytoskeleton of digestive epithelia in health and disease. Am. J. Physiol. 277:G1108–G1137. [DOI] [PubMed] [Google Scholar]
- Ku, N.O., R.M. Soetikno, and M.B. Omary. 2003. Keratin mutation in transgenic mice predisposes to Fas but not TNF-induced apoptosis and massive liver injury. Hepatology. 37:1006–1014. [DOI] [PubMed] [Google Scholar]
- Ku, N.O., D.M. Toivola, Q. Zhou, G.Z. Tao, B. Zhong, and M.B. Omary. 2004. Studying simple epithelial keratins in cells and tissues. Methods Cell Biol. 78:489–517. [DOI] [PubMed] [Google Scholar]
- Ku, N.O., J.K. Lim, S.M. Krams, C.O. Esquivel, E.B. Keeffe, T.L. Wright, D.A.D. Parry, and M.B. Omary. 2005. Keratins as susceptibility genes for end-stage liver disease. Gastroenterology. 129:885–893. [DOI] [PubMed] [Google Scholar]
- Kulesh, D.A., G. Cecena, Y.M. Darmon, M. Vasseur, and R.G. Oshima. 1989. Posttranslational regulation of keratins: degradation of mouse and human keratins 18 and 8. Mol. Cell. Biol. 9:1553–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, H., M. Hesse, B. Peters, and T.M. Magin. 2005. Type II keratins precede type I keratins during early embryogenic development. Eur. J. Cell Biol. 84:709–718. [DOI] [PubMed] [Google Scholar]
- Magin, T.M., R. Schroder, S. Leitgeb, F. Wanninger, K. Zatloukal, C. Grund, and D.W. Melton. 1998. Lessons from keratin 18 knockout mice: formation of novel keratin filaments, secondary loss of keratin 7 and accumulation of liver-specific keratin 8-positive aggregates. J. Cell Biol. 140:1441–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magin, T.M., J. Reichelt, and M. Hatzfeld. 2004. Emerging functions: diseases and animal models reshape our view of the cytoskeleton. Exp. Cell Res. 301:91–102. [DOI] [PubMed] [Google Scholar]
- McPhaul, L.W., J. Wang, E.M. Hol, M.A. Sonnemans, N. Riley, V. Nguyen, Q.X. Yuan, Y.H. Lue, F.W. Van Leeuwen, and S.W. French. 2002. Molecular misreading of the ubiquitin B gene and hepatic mallory body formation. Gastroenterology. 122:1878–1885. [DOI] [PubMed] [Google Scholar]
- Nakamichi, I., S. Hatakeyama, and K.I. Nakayama. 2002. Formation of Mallory body-like inclusions and cell death induced by deregulated expression of keratin 18. Mol. Biol. Cell. 13:3441–3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuschwander-Tetri, B.A., and S.H. Caldwell. 2003. Nonalcoholic steatohepatitis: summary of an AASLD Single Topic Conference. Hepatology. 37:1202–1219. [DOI] [PubMed]
- Omary, M.B., N.O. Ku, J. Liao, and D. Price. 1998. Keratin modifications and solubility properties in epithelial cells and in vitro. Subcell Biochem. 31:105–140. [PubMed] [Google Scholar]
- Omary, M.B., N.O. Ku, and D.M. Toivola. 2002. Keratins: guardians of the liver. Hepatology. 35:251–257. [DOI] [PubMed] [Google Scholar]
- Omary, M.B., P.A. Coulombe, and W.H. McLean. 2004. Intermediate filament proteins and their associated diseases. N. Engl. J. Med. 351:2087–2100. [DOI] [PubMed] [Google Scholar]
- Strong, M.J., C. Leystra-Lantz, and W.W. Ge. 2004. Intermediate filament steady-state mRNA levels in amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 316:317–322. [DOI] [PubMed] [Google Scholar]
- Stumptner, C., M.B. Omary, P. Fickert, H. Denk, and K. Zatloukal. 2000. Hepatocyte cytokeratins are hyperphosphorylated at multiple sites in human alcoholic hepatitis and in a mallory body mouse model. Am. J. Pathol. 156:77–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toivola, D.M., N.O. Ku, N. Ghori, A.W. Lowe, S.A. Michie, and M.B. Omary. 2000. Effects of keratin filament disruption on exocrine pancreas-stimulated secretion and susceptibility to injury. Exp. Cell Res. 255:156–170. [DOI] [PubMed] [Google Scholar]
- Toivola, D.M., N.O. Ku, E.Z. Resurreccion, D.R. Nelson, T.L. Wright, and M.B. Omary. 2004. Keratin 8 and 18 hyperphosphorylation is a marker of progression of human liver disease. Hepatology. 40:459–466. [DOI] [PubMed] [Google Scholar]
- Vasseur, M., P. Duprey, P. Brulet, and F. Jacob. 1985. One gene and one pseudogene for the cytokeratin endo A. Proc. Natl. Acad. Sci. USA. 82:1155–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, Q.X., N. Marceau, B.A. French, P. Fu, and S.W. French. 1996. Mallory body induction in drug-primed mouse liver. Hepatology. 24:603–612. [DOI] [PubMed] [Google Scholar]
- Zatloukal, K., C. Stumptner, M. Lehner, H. Denk, H. Baribault, L.G. Eshkind, and W.W. Franke. 2000. Cytokeratin 8 protects from hepatotoxicity, and its ratio to cytokeratin 18 determines the ability of hepatocytes to form Mallory bodies. Am. J. Pathol. 156:1263–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatloukal, K., C. Stumptner, A. Fuchsbichler, P. Fickert, C. Lackner, M. Trauner, and H. Denk. 2004. The keratin cytoskeleton in liver diseases. J. Pathol. 204:367–376. [DOI] [PubMed] [Google Scholar]
- Zhou, Q., X. Ji, L. Chen, H.B. Greenberg, S.C. Lu, and M.B. Omary. 2005. Keratin mutation primes mouse liver to oxidative injury. Hepatology. 41:517–525. [DOI] [PubMed] [Google Scholar]