

Abstract
Huntington disease (HD) is characterized by the preferential loss of striatal medium-sized spiny neurons (MSNs) in the brain. Because MSNs receive abundant glutamatergic input, their vulnerability to excitotoxicity may be largely influenced by the capacity of glial cells to remove extracellular glutamate. However, little is known about the role of glia in HD neuropathology. Here, we report that mutant huntingtin accumulates in glial nuclei in HD brains and decreases the expression of glutamate transporters. As a result, mutant huntingtin (htt) reduces glutamate uptake in cultured astrocytes and HD mouse brains. In a neuron–glia coculture system, wild-type glial cells protected neurons against mutant htt-mediated neurotoxicity, whereas glial cells expressing mutant htt increased neuronal vulnerability. Mutant htt in cultured astrocytes decreased their protection of neurons against glutamate excitotoxicity. These findings suggest that decreased glutamate uptake caused by glial mutant htt may critically contribute to neuronal excitotoxicity in HD.
Introduction
Expansion of a polyglutamine (PolyQ) tract in huntingtin (htt) causes the preferential degeneration of striatal neurons in patients with Huntington's disease (HD) despite the widespread expression of htt in neuronal and nonneuronal cells. The fact that HD transgenic mouse models can develop neurological symptoms without obvious neurodegeneration indicates that early neuronal injury and dysfunction are the major causes of neuropathologic phenotypes in these mice. Consistently, early neuronal injury caused by mutant htt can lead to reactive gliosis in many HD mouse models (Reddy et al., 1998; Lin et al., 2001; Yu et al., 2003) and in postmortem brains of HD patients (Myers et al., 1991; Sapp et al., 2001). Recent studies show that transgenic mice expressing mutant htt only in cortical neurons do not have obvious gliosis and other pathologies, suggesting that cell–cell interactions play a critical role in HD pathology (Gu et al., 2005). However, little is known about the role of glia htt in HD neuropathology, despite findings that htt is also expressed in glial cells (Singhrao et al., 1998; Hebb et al., 1999).
Glial cells constitute 90% of the cells in the brain and provide neurons with nutrition, growth factors, and structural support. They also protect against excitotoxicity by clearing excess excitatory neurotransmitters from the extracellular space (Maragakis and Rothstein, 2001). This protective function may be particularly relevant to the selective degeneration of medium-sized spiny neurons (MSNs) in the striatum in HD and the theory of excitotoxicity for HD pathogenesis (Coyle and Schwarcz, 1976; Beal, 1994). MSNs are innervated by glutamatergic axons, and overstimulation of glutamate receptors induces cell death or excitotoxicity. The involvement of excitotoxicity in HD is supported by considerable evidence. First, administration of NMDA receptor agonists to the striatum of normal animals causes a selective loss of MSNs and neurological symptoms similar to those seen in HD patients (Coyle and Schwarcz, 1976). Second, NMDA receptor antagonists effectively reduce excitotoxicity in HD animal models (Greene et al., 1993). Furthermore, HD transgenic mouse models show increased NMDA receptor activity in neurons (Cepeda et al., 2001; Zeron et al., 2002). The abundant glutamatergic afferents to MSNs and the unique NMDA receptor subunit composition in MSNs (Calabresi et al., 1998; Kuppenbender et al., 2000; Li et al., 2003) may confer their preferential vulnerability in HD, especially when the glutamatergic input is increased or the clearance of extracellular glutamate is decreased.
Clearance of extracellular excitatory neurotransmitters is largely performed by glutamate transporters (GLT-1 and GLAST) in astrocytes, which is the major subtype of glia (Maragakis and Rothstein, 2001). It has been found that mutant htt can reduce the expression level of glutamate transporter-1 (GLT-1) in the brains of HD transgenic mice and Drosophila melanogaster (Lievens et al., 2001, 2005; Behrens et al., 2002). It remains unclear whether mutant htt directly affects glial function and, more important, how glial dysfunction contributes to neuropathology. The present study provides evidence that NH2-terminal mutant htt in glial cells reduces glial glutamate uptake, and this glial dysfunction may critically contribute to neuronal excitotoxicity.
Results
Intranuclear htt aggregates in glial cells
We used an antibody (EM48) to htt and performed immunogold labeling to examine brains from R6/2 mice that express HD exon1 protein with a 115–150-glutamine repeat. EM48 sensitively detects aggregated htt in HD brain (Li et al., 2000), enabling us to identify htt nuclear aggregates in glial cells in the striatum of R6/2 mice (Fig. 1 A). Glia can be classified as microglia, astrocytes, or oligodendrocytes. They are distinguished from neurons by a condensed nuclear envelope, a small and irregular shape, and a limited cytoplasmic area with sparse content. Microglial cells often show highly condensed nuclear membranes. Identification of astrocytes is primarily based on the presence of fibrils within their processes, and oligodendrocytes are often recognized by their association with groups of myelinated nerve fibers. The ultrathin sections used for electron microscopy might not have allowed us to definitively identify astrocytes containing htt aggregates, as a single plane of an ultrathin section could have been too thin to show both htt aggregates and the distinguishing morphological features of the cell. However, some glial cells, which displayed a highly condensed nuclear membrane and a small cytoplasmic space, contained intranuclear aggregates (Fig. 1, A–C). The nuclear htt aggregates in these glial cells are clearly smaller than neuronal nuclear inclusions (Fig. 1 D). Small glial nuclear inclusions were observed in the striatum (Fig. 1 A) and cortex (Fig. 1, B and C) of R6/2 mice 11–12 wk old. We also observed some hypertrophic and dark glial cells with increased electron density in R6/2 mice. These cells did not show visible cytoplasmic organelles and, in many cases, engulfed neuronal bodies or processes (Fig. S1, A and B, available at http://www.jcb.org/cgi/content/full/jcb.200508072/DC1). The hypertrophic glial cells are smaller and darker than the previously identified dark neurons (Turmaine et al., 2000; Yu et al., 2003), which often have long neuronal processes that have begun to degenerate (Fig. S1, C and D). Like dark neurons, the degenerating glial cells were not frequently observed and did not show htt aggregates, suggesting that glial dysfunction rather than degeneration is more important for HD pathology.
Figure 1.
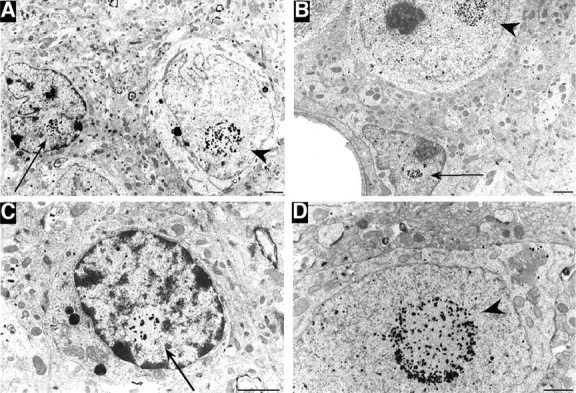
Electron micrographs of HD mouse brains. (A–D) EM48 immunogold labeling of the striatum (A) and cortex (B–D) of R6/2 mice at 12 wk of age. Immunogold-labeled aggregates are present in glial cells (arrows), which show a more condensed nuclear membrane and a smaller sized cytoplasm than do neuronal cells (arrowheads). Bars, 1 μm.
Expression of mutant htt in astrocytes in HD transgenic mouse brains
Because the spectrum of morphological variations among glial cells makes it difficult to define astrocytes containing htt aggregates by electron microscopy, we performed immunofluorescent double labeling on thin sections (8–10 μm) of frozen brain. We first examined white matter that was enriched in astrocytes and was devoid of neurons. Astrocytes are the most abundant glial cell type in the brain and can be specifically labeled by antibody to glial fibrillary acidic protein (GFAP). Although GFAP-labeling was prominent in white matter, only a few cells displayed both strong GFAP staining and mutant htt (Fig. 2 A). This is perhaps because strong GFAP staining is only seen in reactive astrocytes that may have an increased capacity to clear mutant htt. We also performed confocal imaging analysis of white matter and confirmed that mutant htt forms small aggregates in GFAP-positive glial cells (Fig. 2 B). In addition, groups of GFAP-positive astrocytes were often found in the striatum (Fig. 2 C) and were not labeled by antibodies to other glial markers (myelin basic protein for oligodendrocytes and F4/80 for microglia) or the neuronal marker NeuN (not depicted), suggesting that they are mainly astrocytes. Glial nuclei, when stained with the Hoechst dye, are smaller and denser than neuronal nuclei. The small htt aggregates in glial nuclei as well as GFAP staining of glial processes allowed us to distinguish htt-containing glial cells from neurons that show larger and more intense EM48-labeled aggregates in their nuclei (Fig. 2, A and C). Based on these characteristics, we counted htt-containing glial cells in the HD mouse brains.
Figure 2.

Immunofluorescent labeling of htt-containing glia in R6/2 transgenic mouse brains. (A) Immunofluorescent labeling of brain sections containing white matter (WM) and the central region of the cerebellar cortex (Ctx) in an R6/2 mouse 8 wk old. Mouse antibody to GFAP labeled astrocytes (green) and rabbit EM48 labeled mutant htt (red). The nuclei were labeled with Hoechst dye (blue). (B) Confocal imaging of white matter showing that the nuclei of GFAP-positive glial cells contain htt aggregates (Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200508072/DC1). (C) The striatal section of an R6/2 mouse at 8 wk of age was stained with antibody to GFAP and the nuclear dye Hoechst (top). EM48 immunostaining for htt and merged image are also presented (middle). Bottom panels shows merged images of striatal regions from 12- or 4-wk old R6/2 mice. Arrows indicate glial nuclei that contain EM48 staining. Small puncta represent neuropil aggregates. (D) The percentage of glial cells containing nuclear EM48 staining in the brain striatal sections of R6/2 mice at the age of 4, 8, or 12 wk. (E) The percentage of glial cells with intranuclear htt in the striatum (Str), cortex (Ctx), hippocampus (Hipp), and WM in R6/2 mice at the age of 8–10 wk. The data (mean ± SD) were obtained from three to five mice per group. **, P < 0.01 compared with the samples of 8 or 12 wk old R6/2 mice. Bars: (A) 5 μm; (B) 2.5 μm; (C)10 μm.
Unlike neurons, only some glial cells show intranuclear htt labeling, but the prevalence of nuclear htt aggregates in glial cells is increased with age (Fig. 2 C). In the striatum, 17.6% of glial cells showed intranuclear htt staining at 4–5 wk, whereas 42 and 52.6% of glial cells displayed nuclear htt at 8–9 and 11–12 wk, respectively (Fig. 2 D). We also used an antibody to another glial marker, glutamate/aspartate transporter (GLAST), and validated the above finding (unpublished data). Because EM48 is sensitive to aggregated htt and might not label soluble mutant htt, the number of glia expressing mutant htt might be more than what we observed. The progressive increase of glial nuclear htt correlates with disease progression in R6/2 mice, which show significant neurological symptoms at 8 wk and often die after 12 wk (Davies et al., 1997). In white matter, a similar age-dependent increase of intranuclear htt was also seen (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200508072/DC1). Glial htt aggregates seem to be slightly more abundant in the striatum than in the cortex, hippocampus, and white matter (Fig. 2 E), perhaps because of the relatively high density of astrocytes in the striatum (San Jose et al., 2001). However, the accumulation of htt in glial nuclei occurs noticeably later than in neurons (Fig. 2 C), suggesting that glial cells are more capable of clearing misfolded htt than neuronal cells.
Decreased expression of GLT-1 and glutamate uptake in HD mouse brains
Decreased expression of GLT-1 protein was observed in the HD brain by Western blotting (Fig. 3, A and B). The expression of GLT-1 seems to correlate inversely with the age-dependent nuclear accumulation of htt in glial cells, as the brain of HD mice at 11–12 wk showed a greater reduction of GLT-1 than those at 4 wk (Fig. 3, A and B). The expression level of GLAST appeared to be variable in individual mice, but was not significantly decreased in R6/2 mice as compared with littermate controls (Fig. S3, A and B, available at http://www.jcb.org/cgi/content/full/jcb.200508072/DC1). Although Hdh CAG knock-in mice did not show a significant reduction of brain GLT-1 and GLAST even at the age of 9 mo, a slight decrease of GLT-1 expression was observed in the striatum (Fig. S3, C and D). This finding supports the idea that NH2-terminal mutant htt is more toxic than full-length htt in affecting the expression of GLT-1. Consistent with previous studies (Lievens et al., 2001; Behrens et al., 2002), RT-PCR confirmed that GLT-1 transcripts were significantly reduced in HD mouse brains, while GLAST transcripts were slightly decreased (Fig. 3 C).
Figure 3.
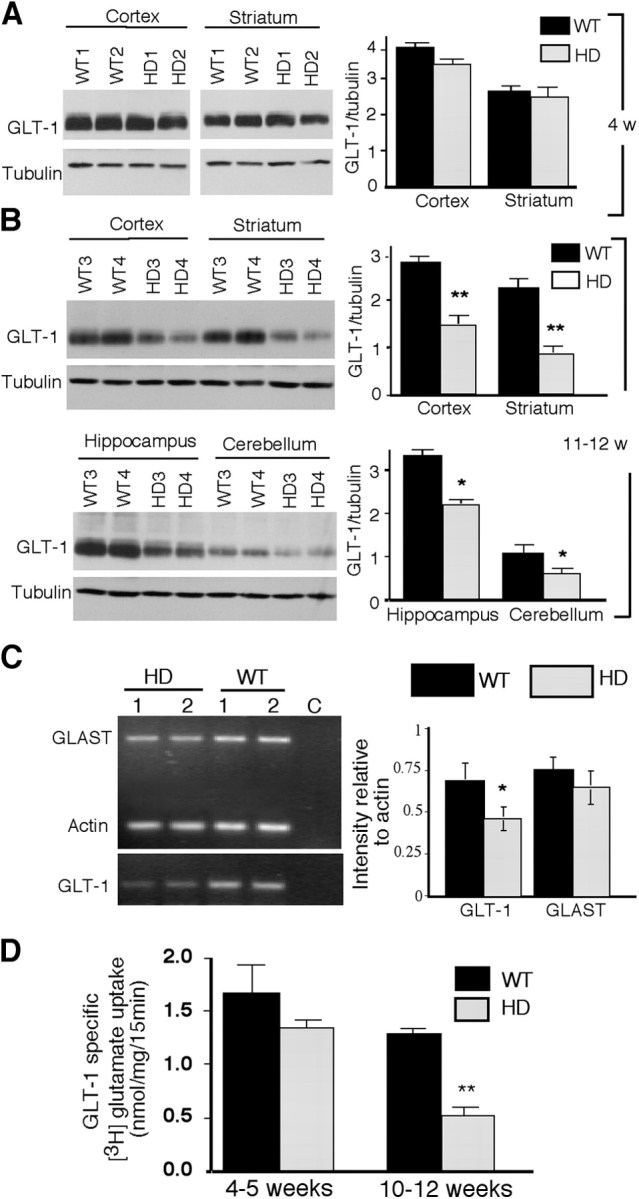
Decreased expression of glutamate transporter GLT-1 and glutamate uptake in HD mouse brain. (A and B) Western blot analysis of the expression of GLT-1 in brain regions of wild-type (W1-W4) or R6/2 (HD1-HD4) mice at 4 wk (A) and 11–12 wk old (B). The same blots were probed with antibody to tubulin. The signal ratios of GLT-1 to tubulin are shown in the right. (C) RT-PCR analysis of the expression of GLT-1 and GLAST in R6/2 (HD) and wild-type (WT) mouse brain cortex. Transcripts for GLAST and actin were amplified in the same reaction (21 cycles), and the same amounts of cDNA were used for GLT-1 PCR (23 cycles). The ratios (mean ± SEM, n = 3) of GLT-1 (*, P = 0.0079, WT vs. HD) and GLAST (P = 0.11, WT vs. HD) to actin were obtained with densitometry. Panel C is the control RT-PCR without reverse transcriptase. (D) GLT-1–specific glutamate uptake in brain slices of littermate control (WT) and R6/2 (HD) mice at the age of 4–5 and 10–12 wk was obtained with DHK (1 mM) treatment. Data are presented as mean ± SEM (n = 4 each group). *, P < 0.05; **, P < 0.01 compared with WT control.
An interesting question is whether decreased glutamate transporters can alter glutamate uptake in astrocytes. The activity of GLT-1 in astrocytes can be specifically inhibited by dihydrokainate (DHK; Kawahara et al., 2002). Although GLT-1 is present in some neurons under certain conditions (Mennerick et al., 1998), a previous study using isolated synaptosomes to examine glutamate uptake into neuronal vesicles did not find a significant difference in DHK-specific glutamate uptake between R6/2 and wild-type mouse brains (Lievens et al., 2001). We decided to use brain slices containing the cortex and striatum to measure their uptake of [3H]glutamate, as the neuron–glia interactions remain intact but the glutamate uptake into astrocytes can be measured with DHK in this assay. Consistent with the age-dependent decrease in the expression of GLT-1 in HD mouse brains, DHK-specific glutamate uptake was more significantly affected in slices from older R6/2 mice (Fig. 3 D), demonstrating a close association between GLT-1 expression and glutamate uptake in HD brain.
Expression of mutant htt in cultured astrocytes from HD mouse brains
Cell culture provides a good system to validate the specific effect of mutant htt on glia. As expected, cultured GFAP-positive astrocytes from R6/2 mice showed intranuclear htt aggregates (Fig. 4 A). Only a fraction of these nuclear aggregates were ubiquitinated (Fig. 4 B), and the formation of these nuclear htt aggregates increased with culturing time (Fig. 4 C). The cells containing htt aggregates were not labeled by antibodies to markers of microglia and oligodendrocytes (not depicted), suggesting that NH2-terminal mutant htt forms aggregates more readily in astrocytic nuclei.
Figure 4.

Age-dependent nuclear accumulation of mutant htt in cultured glial cells. (A) Immunofluorescence double labeling showing that GFAP (green) positive astrocytes display htt aggregates (red) in the nuclei of R6/2 glial cells. (B) Some intranuclear htt aggregates are also labeled by antibody to ubiquitin. (C) Immunofluorescent images of cultured glial cells that were cultured for 2 and 12 wk showing the increase of glial htt aggregates with time. (D) Western blotting of cultured astrocytes that were isolated from the cortex of postnatal Hdh CAG(150) knock-in (KI) and littermate control (WT) mice and had been cultured for 4 wk. The blot was probed with antitubulin (bottom) and 1C2 (top), an antibody that is specific to expanded polyQ tracts and reacts with NH2-terminal htt fragments containing 150Q. Arrow indicates full-length mutant htt. (E) Immunofluorescent images of glial culture from Hdh CAG KI. GFAP (green) positive astrocytes (arrows) contain intranuclear htt (red) aggregates. Some GFAP-negative cells (arrowhead) also show intranuclear htt, suggesting that they might be immature astrocytes or other types of cells. Bars, 5 μm.
To examine whether glial cells expressing full-length mutant htt also display nuclear aggregates, we cultured glial cells from the brain of Hdh CAG(150) knock-in mice that do not show obvious neuropathology and symptoms but have an age-dependent accumulation of NH2-terminal htt in the nuclei of some neurons (Lin et al., 2001). Western blotting verified that NH2-terminal htt fragments were generated in cultured knock-in astrocytes (Fig. 4 D). Some astrocytes contained small aggregates in their nuclei (Fig. 4 E) after 3–4 wk in culture, but relative to R6/2 glial cells, the number of astrocytes containing nuclear aggregates was considerably lower. An antibody (EM121) to the middle region of htt (amino acids 342–456) did not reveal nuclear htt aggregates in knock-in astrocytes (unpublished data). This is consistent with the fact that the cleavage of full-length mutant htt into small NH2-terminal mutant htt fragments and the accumulation of these fragments are required for the formation of nuclear aggregates (DiFiglia et al., 1997; Li et al., 2000).
Expression of htt in glial nuclei of HD knock-in mouse and patient brains
Examination of the striatum of Hdh CAG(150) knock-in mice at 14–18 mo of age revealed EM48 labeling in the small and dense nuclei of some GFAP-positive glial cells as compared with intense EM48 labeling in large neuronal nuclei (Fig. 5 A). The presence of mutant htt in glial nuclei was confirmed by both conventional microscopy (Fig. 5 A) and confocal imaging (Fig. 5 B) of white matter, which clearly showed that some GFAP-positive glial nuclei contained small htt aggregates. Younger Hdh CAG(150) knock-in mice (<9 mo) were not found to have obvious EM48 staining in glial nuclei (unpublished data). suggesting that the nuclear accumulation of mutant htt in glia is age dependent. A previous study reported that mutant htt is present in astrocytes of HD patient brains (Singhrao et al., 1998). We also examined postmortem brains from late stage (grade-3) HD patients and confirmed the expression of mutant htt in HD brains by Western blotting (Fig. 5 C). Despite severe degeneration in grade-3 HD brains, some GFAP-positive glial cells still remained in these brains. Small EM48-labeled aggregates (<0.3 μm) were seen in white matter and were much smaller than neuronal nuclear aggregates that often exceeded 1.5 μm and were intensively labeled by EM48 (Fig. 5 D). Immunofluorescent double labeling verified that the nuclei of GFAP-positive glial cells contained EM48 immunoreactive aggregates in HD patient brains (Fig. 5 E), which are similar in size to those in glial cells in Hdh CAG(150) knock-in mice (Fig. 5, A and B) and did not occur in the brain of Alzheimer's disease (AD) patient (Fig. 5 E). We observed that ∼12.3% of GFAP-positive cells contained htt aggregates. Given that EM48 preferentially reacts with aggregated htt and that grade-3 HD brains might have lost some glial cells or their markers, the number of glial cells expressing mutant htt, especially soluble htt, is likely to be higher.
Figure 5.
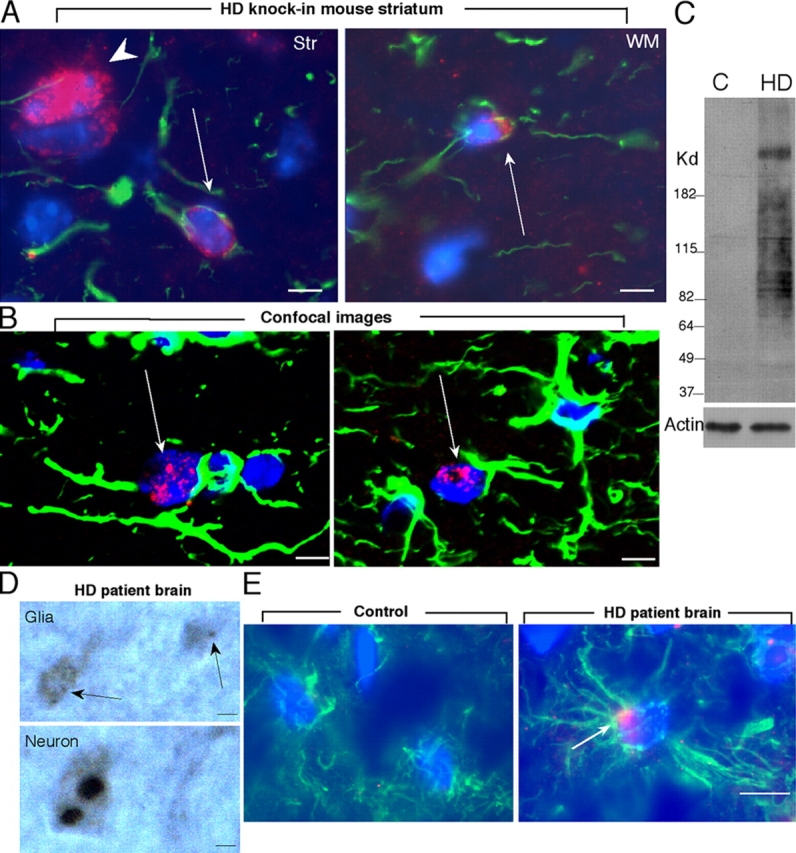
Expression of mutant htt in the brains of Hdh CAG(150) knock-in mice and HD patient. (A) Immunofluorescent double labeling of the striatum of heterozygous Hdh CAG(150) knock-in mice at 14–18 mo old. Arrows indicates the nuclei (blue) of GFAP (green)-positive glial cells containing EM48 labeling (red), and arrowhead indicates a neuronal nucleus, which shows more intense EM48 labeling. (B) High magnification graphs of confocal images of white matter of heterozygous Hdh CAG(150) knock-in mice. Arrows indicate glial nuclei (blue) containing EM48 labeling (red). (C) Western blotting of caudate-putamen tissues from HD and Alzheimer's disease (AD; Control) patients. The blot was probed with antiactin (bottom) and 1C2 antibody (top). (D) Light microscopic graphs of glial cells (arrows in upper panel) in white matter and neurons in the cortex (bottom) in the EM48 stained HD brain sections. (E) Immunofluorescent double labeling of white matter of HD patient brain with rabbit anti-htt (EM48) and mouse anti-GFAP. Mutant htt (red) forms aggregates (arrow) in the nucleus (blue) of an astrocytic cell that shows intense GFAP staining (green) in its processes. The control is the AD brain section. Bars, 2 μm.
Expression of htt in cultured astrocytes by adenoviral infection
To identify the pathological changes that are specifically associated with an expanded repeat, we infected glial cells with adenoviral vectors expressing GFP fusion protein containing the first 208 amino acids of human htt with 23Q (htt-23Q) or 130Q (htt-130Q). Htt-23Q was predominantly diffuse in the cytoplasm, though its overexpression resulted in small puncta or aggregates in the cytoplasm but not in the nucleus. However, expanded polyQ caused htt-130Q to form significantly larger and more abundant inclusions in both the cytoplasm and nucleus (Fig. 6 A). Also, nuclear accumulation and aggregation of htt-130Q were elevated by treatment with the proteasome inhibitor N-acetyl-leucinal-leucinal-norleucinal (Fig. 6 B, ALLN). All of these results indicate a specific nuclear accumulation of htt mediated by a large polyQ tract.
Figure 6.
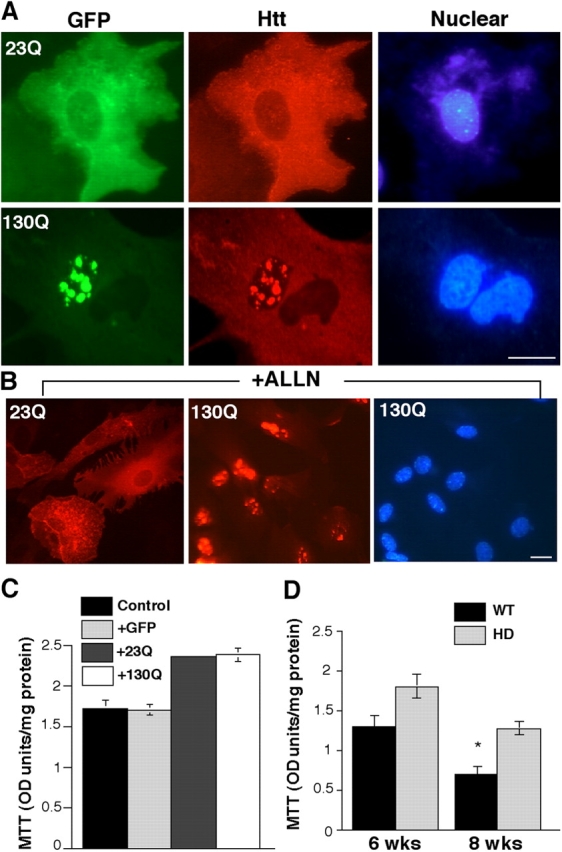
PolyQ-mediated intranuclear htt accumulation in glial cells. (A) Immunofluorescent images of adenovirus-infected astrocytes that express GFP-htt containing a 23- (23Q) or 130- (130Q) glutamine repeat. Immunostaining with EM48 confirms that htt-130Q is located in the nucleus and forms nuclear aggregates. Nuclei are stained with Hoechst. (B) Proteasomal inhibition by ALLN (10 μg/ml) for 5 d significantly increases the formation of nuclear aggregates of htt-130Q, but not htt-23Q, in infected glial cells. (C) MTT assay of cultured glial cells infected by adenoviral-GFP (+GFP), htt-23Q (+23Q), or htt-130Q (+130Q) for 9 d. (D) MTT assay of astrocytes from R6/2 mice (HD) or littermate controls (WT) cultured for 6 or 8 wk. The data are presented as mean ± SEM (n = 3–4) and p values for 6- and 8-wk group comparisons (WT vs. HD) are 0.23 and 0.014, respectively. Bars, 5 μm.
Adenoviral infection resulted in >80% of cultured glial cells expressing mutant htt. Unlike cultured neuronal cells in which NH2-terminal mutant htt can cause nuclear or neuritic fragmentation (Saudou et al., 1998; Li et al., 2000), htt-containing glial cells did not show obvious changes in their morphology at different times after viral infection. We then examined the viability of cultured glial cells using 3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assays. Interestingly, astrocytes infected with either htt-23Q or htt-130Q and HD astrocytes from R6/2 mice showed an increase in MTT value (Fig. 6, C and D). Reactive astrocytes often have activated proliferation and increased mitochondrial respiration, which are reflected by increased MTT values (Setkowicz and Janeczko, 1998; Norton, 1999). Overexpressed or misfolded polyQ proteins might induce reactive astrocytes, resulting in increased proliferation and mitochondrial respiration that lead to a high MTT value. Although electron microscopy revealed degenerated glia in HD mouse brain, this glial pathology may occur in vivo or result from neuronal injury. We could not find any evidence that mutant htt directly kills cultured glial cells in vitro, even in the presence of cocultured neurons (unpublished data), suggesting that mutant htt is more likely to cause glial dysfunction rather than degeneration.
Decreased expression of glutamate transporters in astrocytes expressing mutant htt
Next, we examined whether mutant htt in astrocytes directly affects the expression of glutamate transporters. Western blots revealed that the basal level of GLT-1 was lower in cultured astrocytes from R6/2 mouse brains than those from littermate controls (Fig. 7 A), whereas the basal level of GFAP did not differ. We then treated the astrocytes with dibutyryl cyclic adenosine monophosphate (dBcAMP), which can significantly increase the expression of GLT-1 by regulating its transcription (Eng et al., 1997). The difference in the expression of GLT-1, but not GFAP, between control and R6/2 astrocytes was enhanced by dBcAMP treatment (Fig. 7, A and B), suggesting that mutant htt negatively affects gene transcription of GLT-1. The expression of GLAST in HD glial cells and its up-regulation by dBcAMP were also reduced as compared with wild-type cells (Fig. 7, A and B), but this reduction was not as great as the change in GLT-1. Because GLAST expression is not significantly decreased in HD brains (Lievens et al., 2001; Behrens et al., 2002), in vivo neuron–glia interactions may attenuate the inhibitory effect of mutant htt on the protein expression of GLAST in the brain.
Figure 7.
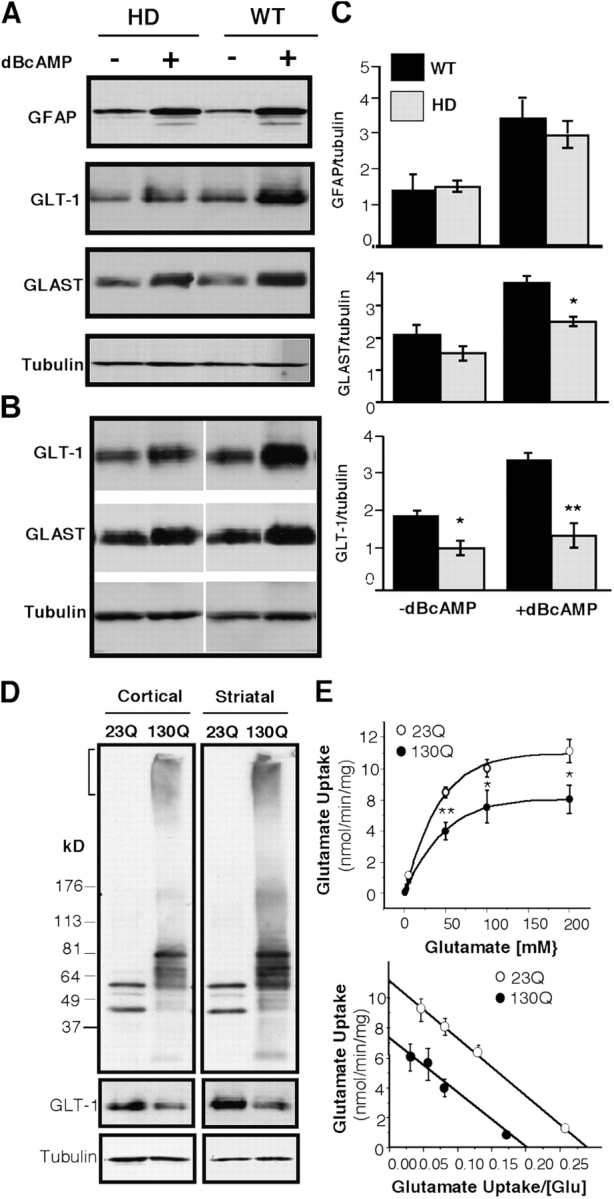
Decreased expression of GLT-1 in glial cells expressing mutant htt. (A and B) Western blots of cultured astrocytes from R6/2 (HD) and littermate control (WT) mice. Astrocytes that had been cultured 4–6 wk and treated with (+) or without (–) 0.25 mM dBcAMP were examined by Western blotting with antibodies to GFAP and GLT-1. The same blots were also probed with an antibody to tubulin. Two blots (a, b) containing different cell samples are presented. (C) Densitometric analysis of signals of immunoreactive bands. The ratios (mean ± SEM, n = 3–4) of GLAST (*, P = 0.045, WT vs. HD), GLT-1 (*, P = 0.013; **, P < 0.001 WT vs. HD), and GFAP to tubulin. (D) Western blot analysis of the expression of htt-23Q and htt-130Q in adenovirus-infected glial cells. Bracket indicates the stacking gel in which aggregated htt-130Q is present. GLT-1 expression is reduced in htt-130Q astrocytes cocultured with cortical or striatal neurons. (E) [3H]Glutamate uptake assays of astrocytes infected with htt-23Q or htt-130Q. Note that the glutamate uptake in htt-130Q–infected cells is lower than in htt-23Q infected cells. The data are presented as mean ± SEM (*, P < 0.05; **, P < 0.01). The Vmax of [3H]glutamate uptake in htt-23Q and htt-130Q infected cells was 11.1 ± 0.84 and 7.3 ± 1.21 nmol/min/mg protein, respectively. There was no significant difference in the apparent glutamate Kms (37.7 ± 3.24 and 38.6 ± 6.3 for htt-23Q and htt-130Q, respectively).
To examine whether the decreased expression of GLT-1 is associated with expanded polyQ and also occurred in the presence of neurons, we cultured adenovirus–htt–infected glial cells with cortical or striatal neurons. Aggregated htt was seen in htt-130Q–infected glial cells in EM48 Western blots (Fig. 7 D). Because EM48 preferentially reacts with mutant and aggregated htt (Li et al., 2000), htt-130Q and htt-20Q might be expressed at a similar level. GLT-1, but not tubulin, was significantly reduced in htt-130Q infected glial cells. Densitometry analysis confirmed that this decrease was associated with expanded polyQ (unpublished data). Furthermore, htt-130Q infected astrocytes showed a significant reduction in uptake of [3H] glutamate as compared with htt-23Q astrocytes (Fig. 7 E). Because the Vmax, but not the Km, of [3H]glutamate uptake was decreased in htt-130Q astrocytes (Fig. 7 E), expanded polyQ appears to reduce the number of glutamate transporters in these glial cells. This finding is consistent with the decreased expression of GLT-1 and GLAST in cultured R6/2 glial cells.
Protection against htt-mediated neuronal toxicity by glial cells
To determine whether glia can protect against neuronal htt cytotoxicity, we infected cultured neurons with adenoviral GFP, htt-23Q, and htt-130Q constructs, and then cocultured them with wild-type astrocytes. In the absence of glial cells, htt-130Q neurons generally showed a decrease in the staining of microtubule-associated protein 2 (MAP2) (Fig. 8 A), a neuron-specific protein whose decrease reflects early neurodegeneration (Matesic and Lin, 1994). When htt-130Q neurons were cocultured with wild-type glial cells, the number of MAP2-positive neurons was significantly increased (Fig. 8 B). The NMDA receptor blocker MK801 (dizocilpine maleate), which increases the survival of cultured neurons (Driscoll et al., 1991), also increased the number of MAP2-positive neurons expressing htt-130Q (Fig. 8 B).
Figure 8.
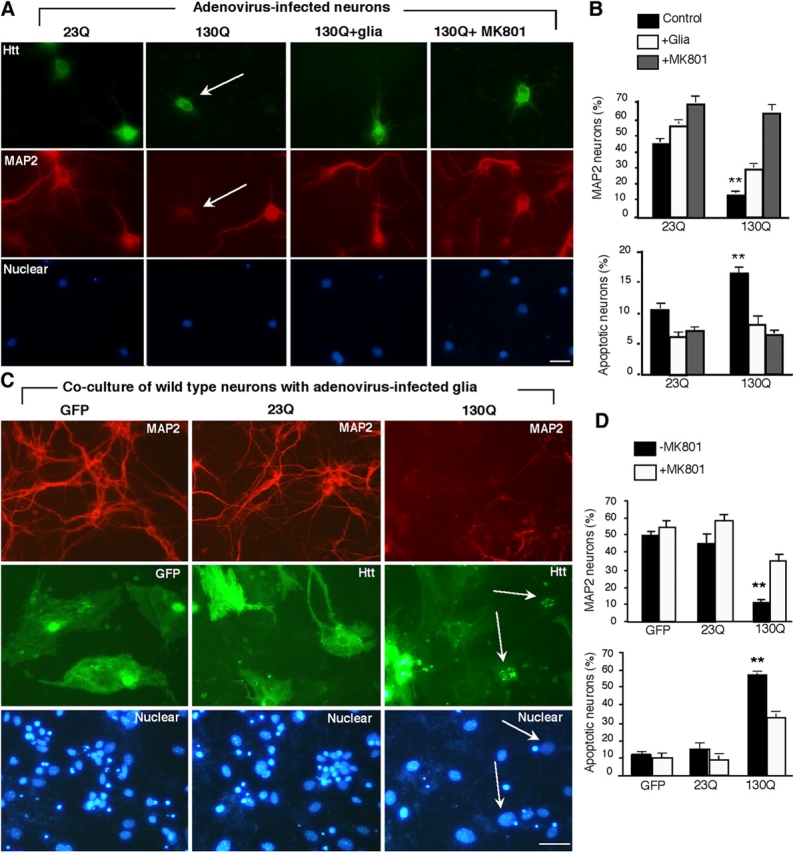
Htt-mediated neurotoxicity in glia–neuron coculture. (A) Cultured cortical neurons were infected with adenoviral htt-23Q or htt-130Q for 24 h. The infected neurons were then cultured with or without wild-type glial cells or MK801 (10 μM) and labeled by antibodies to htt (top), MAP2 (middle), and Hoechst (bottom). Htt-130Q–infected neurons show decreased MAP2-staining (arrows). (B) The percentage of MAP2-positive neurons and apoptotic neurons with nuclear DNA fragmentation in the presence or absence of wild-type glial cells or MK801. (C) Cultured glial cells (4–6 wk) infected with adenoviral htt-23Q or htt-130Q were cocultured with wild-type cortical neurons. EM48 immunofluorescence staining of glia–neuron coculture shows that htt-23Q is distributed in the cytoplasm whereas htt-130Q accumulates in the nuclei (arrows) of infected glial cells. The size of nuclei of cultured glial cells is often larger than that of cultured cortical neurons. There is a decrease in the number of MAP2-positive neurons in the coculture with htt-130Q–infected glial cells. Nuclei were stained with Hoechst (blue). (D) The percentage of MAP2-positive neurons and apoptotic cells in the presence of adenoviral infected glial cells. Neurons were treated with or without MK801 (10 μM). The data (mean ± SEM) were obtained by counting the number of degenerated cells and the total number of nuclei per image. **, P < 0.01 compared with neurons cocultured with glial cells. Bars, 10 μm.
Glial htt promotes neuronal vulnerability
The preceding experiments examined glial protection for neurons in which mutant htt was overexpressed. Overexpressed mutant htt in neurons activates NMDA receptors and triggers many other pathological pathways, such as caspase cascades (Cepeda et al., 2001; Zeron et al., 2002; Li and Li, 2004), which could counteract glial protection. Thus, a more important issue is whether mutant htt, when expressed in glia, can reduce their ability to protect neurons. To address this issue, we placed wild-type neurons on glia that had been infected with adenoviral htt. After >3–4 wk in culture, astrocytes displayed larger nuclei than neuronal nuclei and lacked MAP2 immunostaining, allowing us to definitively distinguish them from neurons in the coculture. We did not observe a significant change in neuronal morphology after coculturing with adenoviral htt-infected astrocytes for 4–5 d. However, after >7–8 d, a significant decrease in the number of MAP2-positive neurons was seen in the presence of htt-130Q infected astrocytes (Fig. 8 C). Because cultured neurons normally start to express NMDA receptors at 7–8 d in vitro (DIV), we added MK801 to block glutamate receptors. MK801 (10 μM) significantly increased the number of MAP2-positive neurons (Fig. 8 D and Fig. S4 [available at http://www.jcb.org/cgi/content/full/jcb.200508072/DC1]), also suggesting that mutant htt in cultured glia can promote neuronal glutamate excitotoxicity in vitro.
To assess the specific protective effect of astrocytes in the removal of extracellular glutamate from the medium, we placed astrocytes, which were cultured on coverslips, on the top of neuronal cells without direct contact during 1 h stimulation with glutamate or NMDA. The astrocytes were removed after stimulation, and neurons were cultured in the absence of astrocytes for 24 h. Stimulation of 15–17 DIV striatal neurons with 0.1 mM glutamate in the absence of astrocytes led to a marked reduction in MAP2-positive neurons, whereas the presence of wild-type astrocytes significantly increased the number of MAP2-positive neurons (Fig. 9, A, B, and D). This glial protection was diminished by the glutamate transporter blockers (Fig. S5, available at http://www.jcb.org/cgi/content/full/jcb.200508072/DC1). NMDA (0.4 mM), whose neuronal toxicity is independent of glutamate transporters, elicited a similar reduction of MAP2-positive neurons in the absence or presence of astrocytes (Fig. 9, B and E). Also, cultured neurons were more sensitive to glutamate (0.1 mM) than to NMDA (0.4 mM; Fig. 9 A). Thus, this coculture system allowed us to specifically examine the ability of glial cells to remove extracellular glutamate and to protect neurons from glutamate excitotoxicity.
Figure 9.
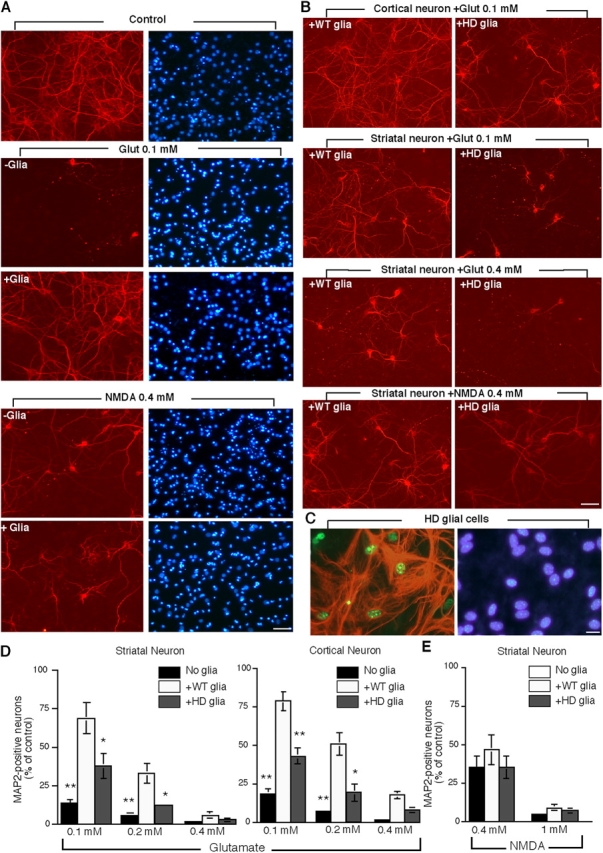
Reduced glial protection against glutamate excitotoxicity by mutant htt in R6/2 astrocytes. (A) Cultured rat striatal neurons (15–17 DIV) were stimulated for 1 h with glutamate (0.1 mM) or NMDA (0.4 mM) in the absence (−glia) or presence (+glia) of wild-type rat cortical astrocytes. The astrocytes were removed after the stimulation. Glutamate stimulation caused neurons to degenerate and lose MAP2 staining. Wild-type astrocytes increased MAP2-positive neurons following glutamate (0.1 mM), but not NMDA (0.4 mM) stimulation, suggesting a specific protection resulting from their glutamate uptake. (B) Cultured astrocytes from littermate control (+WT glia) mouse cortex also protected against glutamate neuronal toxicity. Cultured astrocytes from R6/2 (+HD glia) mouse cortex, however, showed a decrease in protection against glutamate (0.1 mM), but not NMDA (0.4 mM), toxicity. (C) Cultured R6/2 astrocytes contained GFAP (green) in the cytoplasm and mutant htt (red) in their nuclei but had normal nuclear morphological appearance (blue). (D) The percentage of MAP2-positive striatal or cortical neurons after glutamate stimulation in the absence (No glia) or presence of wile type (+WT glia) or R6/2 (+HD glia) astrocytes. (E) The percentage of MAP2-positive striatal neurons after NMDA stimulation. The control is the number of cells without excitotoxin stimulation. The data (mean ± SEM) were obtained from four to five independent coculture experiments. *, P < 0.05; *, P < 0.01 as compared with WT astrocytes. Bars: (A and B) 20 μm; (C) 5 μm.
We observed that wild-type astrocytes could protect ∼78% of cortical neurons from glutamate toxicity, whereas HD astrocytes from R6/2 mice provided significantly less protection (43%). Similar results were also seen in cultured striatal neurons, though striatal neurons were more sensitive than cortical neurons to glutamate toxicity (Fig. 9 D). The increased neuronal excitotoxicity by R6/2 astrocytes is apparently related to glutamate transporters, as NMDA did not cause a significant difference in the number of MAP2-positive neurons between cocultures with wild-type or R6/2 astrocytes (Fig. 9, B and E). Examination of cocultured astrocytes verified that they expressed intranuclear aggregated htt but retained a normal nuclear appearance (Fig. 9 C). These results support the idea that mutant htt in glial nuclei affects the expression of glutamate transporters, leading to reduced protection against glutamate neuronal excitotoxicity.
Discussion
Expression of mutant htt in glial cells
The present findings suggest that mutant htt in glial cells can contribute to neuronal dysfunction and excitotoxicity in HD brains. Detection of the expression of mutant htt in glial cells seems to depend largely on the antibodies used, as the previous study with different antibodies did not identify glial htt aggregates (Davies et al., 1997). Also, the small size and irregular shape of glial cells made it difficult to identify nuclear htt in these cells. EM48 sensitively detects mutant htt and its aggregates (Li et al., 2000), allowing us to demonstrate the presence of nuclear htt in glial cells in HD mouse brains. We also demonstrated that cultured astrocytes isolated from R6/2 mouse brains contain nuclear aggregates, consistent with a previous report that cultured glia from a different exon1 htt transgenic mouse model express nuclear htt (Martin-Aparicio et al., 2002). Because HD mice (R6/2) expressing small NH2-terminal htt fragments show more glial cells (up to 52.6% in white matter at 12 wk) containing nuclear htt aggregates than Hdh CAG knock-in mice, NH2-terminal mutant htt seems to be able to accumulate in the nuclei of glial cells. HD mice display some early HD neuropathology in the absence of obvious neurodegeneration. Thus, study of the expression of mutant htt in glial cells in HD mice is important for understanding the role of glial htt in early HD pathology. We also observed htt aggregates in the nuclei of ∼12.3% of GFAP-positive cells in postmortem brains of patients with late-stage HD. This number might be underestimated because it did not include cells that express soluble mutant htt and was obtained from the limited number of grade-3 HD patient brains. Further quantitative investigation of the expression of mutant htt in glial cells in HD patient brains, especially those at early disease stages, remains to be performed.
Mutant htt and glial dysfunction
Whereas nuclear htt aggregates reflect the accumulation of mutant htt in cells, soluble mutant htt can also affect cellular function. An important question is whether mutant htt affects glial function. Because glutamate excitotoxicity is thought to be an important mechanism in HD pathology and astrocytes can prevent this excitotoxicity, we focused on the effect of mutant htt on glutamate uptake in astrocytes. Among the five known glutamate transporters (GLT-1, GLAST, EAAC1, EAAT4, and EAAT5), GLT-1 and GLAST are primarily expressed in astrocytes and seem to be the predominant glutamate transporters in the brain (Maragakis and Rothstein, 2001). Although previous studies have revealed that GLT-1 is decreased in HD mouse brains (Lievens et al., 2001; Behrens et al., 2002), it remains unclear how GLT-1 expression is altered and whether glial htt affects neuronal viability. Using cultured astrocytes, we have provided direct evidence that mutant htt in glial cells reduces the expression of GLT-1 and GLAST. Also, dBcAMP-mediated expression of GLT-1 and, to a lesser extent, GLAST was impaired by mutant htt. This reduction does not appear to be caused by an altered dBcAMP stimulation by mutant htt, as dBcAMP-mediated up-regulation of GFAP is not impaired. The promoters of the GLT-1 and GLAST genes contain Sp1-binding sites (Su et al., 2003). Considering that mutant htt binds Sp1 (Dunah et al., 2002; Li et al., 2002), it is possible that Sp1-mediated expression of GLT-1 and GLAST is affected by intranuclear htt. However, in vivo glia–neuron interaction may counteract the inhibitory effect of mutant htt on the expression of GLAST and thereby minimize the reduction of GLAST in HD brains.
Although electron microscopy in the present study identified degenerating glial cells in HD mouse brains, the number of degenerating glial cells is not substantive. Also, overexpression of NH2-terminal mutant htt did not cause obvious death of cultured glial cells in vitro. Thus, glial dysfunction may be more important for neuronal toxicity than glial degeneration. In fact, the expression of glutamate transporters and glutamate uptake are reduced in HD mouse brains and cultured astrocytes without degeneration.
Glial htt and neuropathology
Unlike cultured glial cells, cultured neurons are vulnerable to transfected NH2-terminal mutant htt (Saudou et al., 1998). However, when neurons that express full-length mutant htt are cultured with glial cells, they do not show increased vulnerability to glutamate excitotoxicity (Snider et al., 2003). The intracellular accumulation of NH2-terminal mutant htt fragments may be required for neuronal htt toxicity, and the neurons may be protected from this toxicity by glial and other cells. Transgenic mice that express exon-1 mutant htt in many types of cells in the brain show obvious neuropathology and neurological symptoms (Davies et al., 1997; Gu et al., 2005). In contrast, transgenic mice expressing the same mutant htt only in cortical neurons do not have obvious neuropathology and phenotypes, leading to the idea that cell–cell interactions play an important role in HD pathology (Gu et al., 2005). The present study shows that neurodegeneration caused by overexpressed NH2-terminal mutant htt in vitro is reduced in the presence of glial cells.
A more interesting question is whether mutant htt affects glial function to contribute to neuropathology. We found that NH2-terminal mutant htt in glia promoted the death of cultured neurons that did not express mutant htt. Mutant htt may affect various functions of glial cells, including their production of chemokines and neurotrophic factors. While this possibility remains to be explored, the coculture system allowed us to detect the specific effect of removal of extracellular glutamate by glia on neurotoxicity (Fig. 9).
Glial htt and the selective vulnerability of MSNs
MSNs constitute 70–80% of the total number of human striatal neurons and receive glutamatergic afferents from all areas of the neocortex, thalamus, and limbic system (Parent and Hazrati, 1995). The specific composition of NMDA receptor subunits could also underlie the hypersensitivity of MSNs to glutamate excitotoxicity (Landwehrmeyer et al., 1995; Kuppenbender et al., 2000; Li et al., 2003). In addition, mutant htt in MSNs is found to increase their sensitivity to NMDA in brain slices by modulating the activity or expression of glutamate receptor subunits (Cepeda et al., 2001; Li et al., 2003). All of these characteristics make MSNs highly vulnerable to glutamate stimulation. Thus, although NH2-terminal mutant htt may affect GLT-1 expression and glutamate uptake in various brain regions, the impairment of extracellular glutamate removal in the striatum could particularly increase the vulnerability of MSNs to glutamate excitotoxicity. Impaired glial glutamate uptake in R6/2 mice may also significantly increase extracellular glutamate concentration in the brain (Lievens et al., 2001; Behrens et al., 2002), which, in turn, would raise the threshold of neuronal response to excitotoxins so that neurons in R6/2 mouse brain are less responsive to exogenously administered NMDA agonists (Hansson et al., 2001).
Conclusion
Glial dysfunction is known to contribute to other neurodegenerative diseases, and wild-type nonneuronal cells can ameliorate motor neuron degeneration in a mouse model of amyotrophic lateral sclerosis (Clement et al., 2003). In addition to its effect on glial nuclei, mutant htt in neurons can cause multiple dysfunctions (Li and Li, 2004) including transcriptional dysregulation and impaired transport (Gunawardena et al., 2003). Mutant htt in neurons and glia can independently or synergistically affect neuronal function depending on its subcellular accumulation in these cells during disease progression. This study underscores the importance of glial protection against neurodegeneration caused by misfolded polyQ proteins and also provides a new avenue for studying the pathogenesis of other polyQ diseases in which the disease proteins may also be expressed in glial cells.
Materials and methods
Reagents
R6/2 mice and Hdh CAG(150) knock-in mice, which express a 150Q repeat, were bred and maintained in the animal facility at Emory University under specific pathogen-free conditions in accordance with institutional guidelines. Postmortem brain samples of patients with HD or AD were provided by the Harvard Brain Tissue Resource Center. The brain samples were from three female HD patients. Their ages of death were 57 (B5295), 65 (B5297), and 39 (B5299). The postmortem intervals were 18, 25, and 24 h, respectively. The pathological severity of these cases is grade-3, and expanded CAG repeats are 41–47 units. AD patients were female cases (B4842 and B5092) at 61 and 67 yr of age with postmortem interval 6.75 and 27 h, respectively.
Rabbit polyclonal antibody (EM48) and mouse monoclonal antibodies (mEM48) against the NH2-terminal region (amino acids 1–256) of human huntingtin were described in our previous study (Li et al., 2002). Rabbit antibody against GLT-1 (cGLT T88) was provided by J. Rothstein (Johns Hopkins University, Baltimore, MD). Rabbit antibody to GLAST was provided by K. Tanaka (Tokyo Medical and Dental University, Tokyo, Japan) and Gi. Bonvento (CEA CNRS, Orsay, France). Other antibodies used included mouse monoclonal antibodies against polyglutamine (1C2) and GFAP (Chemicon), MAP2 (AP20; BD Biosciences), monoclonal γ-tubulin (Sigma Aldrich), GLAST (Chemicon), and anti–mouse or –rabbit antibodies conjugated with Alexa Fluor 488 (Invitrogen). Antibodies to myelin basic protein (1:500; Chemicon) and a microglial protein (F4/80 1:100, Serotec/Biozol) were also used.
Immunocytochemistry and Western blotting
Electron microscopy was performed as described previously (Li et al., 2000). For Immunofluorescence light microscopy, brains of R6/2 and littermate control mice were rapidly isolated and cut to sections (8–10 μm) with a cryostat at −20°C. The brain sections were placed on gelatin-precoated glass slides. Mouse and human brain sections were examined with immunofluorescence labeling as described previously (Li et al., 2000; Zhou et al., 2003). Light micrographs were taken using a microscope (Axiovert 200 MOT; Carl Zeiss MicroImaging, Inc.) and a 63× lens (LD-Achroplan 63×/0.75) with a digital camera (Orca-100; Hamamatsu) and the Openlab software (Improvision Inc). To count glial cells containing htt aggregates in HD mouse brains, random images (five to eight) of white matter or other brain regions in each brain section were captured using LD plan-neofluar lens (20×/0.4 or 40×/0.6) and stored in a computer for counting. Glial cells (15–60) in each image were counted. To examine HD patient brains, we studied three cases of HD patient brains and counted 1,004 GFAP-positive cells in four independent immunocytochemical experiments. AD patient brain samples were used as the control. Confocal image analysis was performed with a confocal microscope system (LSM 510 NLO; Carl Zeiss MicroImaging, Inc.).
For Western blots, cultured cells or brain tissues were solubilized in 1% SDS, and then resuspended in SDS sample buffer and sonicated for 10 s. The total lysate was used for Western blotting with the ECL kit (Amersham). RT-PCR to examine the transcripts of GLT-1 and GLAST was performed using the same oligonucleotide primers and methods as described previously (Tortarolo et al., 2004). Oligonucleotide primers for GLAST cDNA and actin were included in the same reaction for amplification with 21 cycles, whereas GLT-1 cDNA was amplified alone with 23 cycles because its PCR products had a molecular weight similar to that of actin.
Adenoviral vector construction and preparation
Recombinant adenoviruses were generated according to a previously reported protocol (He et al., 1998). Human htt cDNAs coding the first 208 amino acid plus an additional 23 or 130 glutamine repeats were fused in-frame to GFP COOH-terminal cDNA, resulting in GFP-htt fusion proteins containing a 23- (htt-23Q) or 130 (htt-130Q)-glutamine repeat that were expressed under the control of a cytomegalovirus (CMV) promoter. The viral titer was determined by measuring the number of infected HEK293 cells expressing GFP. All viral stocks were adjusted to 109 VP/ml before their use.
Glial and neuronal cultures
Enriched glial cultures were prepared from 1 to 2 d postnatal rat or mouse pups. Microglia cells were dissociated from the culture after shaking cultured glial cells or enriched for culture. Immunostaining with antibodies to specific makers (GFAP for astrocytes, F4/80 for microglia, and myelin basic protein for oligodendrocytes) was used to identify different types of glial cells. Cultured astrocytes were treated with 0.25 mM dBcAMP for 7–10 d to increase the expression of glutamate transporters (Eng et al., 1997).
For neuronal culture, mixed neuronal–glial cultures were prepared from the cerebral cortex and striatum of rat fetuses at embryonic day 17–18. Viable cells were plated at 1 × 106 cells/ml on poly-d-lysine–coated plastic culture plates (Corning Costar) in B27-supplemented Neurobasal medium (Invitrogen). To reduce glial proliferation, we added cytosine arabinoside to the cultures 3 d after plating (5 × 106 M final concentration). At 7–8 DIV, the majority of cells were mature neurons with elongated processes, and they were used for viral infection. In some experiments, neurons were treated with cytosine arabinoside for 6–7 d, and then MK801 (10 μM) for 30 min. Cultured neurons were then infected with or without adenoviral vectors for 24 h. After washing, infected neurons were cocultured with wild-type glial cells that were on coverslips for 3 d.
For glia–neuron coculture, we first infected astrocytes (4–6 wk) from rat brain cortex with adenoviral GFP, htt-23Q, or htt-130Q for 24 h. After washing, we then isolated cortical or striatal neurons and plated them on the top of infected astrocytes and cultured these neurons in B27-supplemented neurobasal medium. To inhibit the proliferation of newly added glial cells, 2.5 × 106 M cytosine arabinoside was added 1 d after plating neurons. The newly isolated glial cells were unable to grow because serum free media and cytosine arabinoside suppressed their growth. Neurons cocultured with infected astrocytes for more than 4 d were then examined.
For neuronal–glial coculture treated with glutamate or NMDA, cortical or striatal neurons (DIV 14–17) were cocultured with glial cells that were attached to coverslips. The glial cells were facing the cultured neurons without direct contact. The coverslips were placed over the cultured neurons during stimulation with glutamate or NMDA in B27-supplemented neurobasal medium for 1 h at 37°C. This glial protection was verified by using the glutamate transporter blockers DHK and threo-β-benzyloxyaspartate. After stimulation, the glial cells on the coverslips were removed, and the medium was changed to fresh drug-free medium. The treated neurons were cultured for 24 h before immunofluorescent examination.
Glutamate uptake assay
Cortico-striatal brain slices (400 μm, three pieces/brain) from R6/2 and littermate controls at the age of 4–5 or 10–12 wk (four animals per group) were prepared with vibratome. The slices were placed in ice-cold artificial CSF (aCSF) ([mM] 118 NaCl, 4.8 KCl, 2.6 CaCl2, 1.2 MgSO4, 25 NaHCO3, and 1.2 KH2PO4, 11 glucose, 0.6 ascorbic acid) with aeration of 95% O2 5% CO2. Half of a slice was preincubated with 1 mM dihydrokainate (DHK; Sigma Aldrich) for 1 h at 37°C, and the other half was preincubated without DHK treatment. After preincubation, [3H]L-glutamate was added into solution at a final concentration of 25 nM and incubated for 15 min. The incubation was terminated by rapidly removing the solution, and the slices were washed with 4 ml of ice-cold aCSF buffer three times. Slices were lysed in 0.1% NaOH with sonication, and the radioactivity was determined using a liquid scintillation counter. Protein amount of brain slices was measured to normalize counting results. The differences between DHK treated and nontreated samples were obtained to reflect GLT-1–specific glutamate uptake (nmol/mg protein/15 min).
For glutamate uptake assay of cultured astrocytes, glial cells preincubated with 10 mM unlabeled glutamate served as a control to obtain a background value. Vmax and Km were determined from Eadie-Hofstee plots.
Neuronal degeneration and cell viability assays
Degenerated neurons were identified by their disrupted or reduced neurites and condensed or fragmented nuclear DNA using the same method as described previously (Saudou et al., 1998). Cultured neurons were fixed and stained with antibody to MAP2 antibody for neuronal cells and Hoechst for nuclear DNA. Fluorescent images were captured and neurons with normal or degenerating morphology were counted. To count cells after immunocytochemistry, 10–20 pictures of each experimental group were taken with a 20× objective (Carl Zeiss MicroImaging, Inc.). The imaged areas were chosen randomly from at least three different wells per experimental group. The controls were neurons that had not been treated with excitotoxins. The numbers of MAP2-positive neurons and apoptotic neurons were expressed as the percentage of total neurons counted. Data were obtained from 4–5 independent coculture experiments.
The viability of glial cells was determined by a modified MTT assay using a microplate reader (SPECTRAmax Plus; Li et al., 2002). The absorbance at 490 nm was normalized with the amount of cellular protein used for the assay in a 100-μl reaction.
Statistical analysis
Statistical significance was assessed by using Student's t test or two-way ANOVA test followed by Bonferroni post-hoc analysis. Calculations were performed by SigmaPlot 4.11 and Prism (version 4) software. Statistical significance was taken to be P < 0.05.
Online supplemental material
Fig. S1 shows electron micrographs of R6/2 mouse brain containing degenerated neurons and glial cells. Fig. S2 shows an age-dependent increase of glial cells containing intracellular mutant htt in R6/2 mice. Fig. S3 shows Western blot analysis of glutamate transporters in R6/2 and HD knock-in mouse brains. Fig. S4 shows that MK801 reduced neurotoxicity in the presence of glial cells expressing mutant htt. Fig. S5 shows that glial protection was inhibited by glutamate transporter blockers. Video 1 shows three-dimensional images of a GFAP-positive astrocyte containing htt aggregates in its nucleus. Online supplemental material is available at available at http://www.jcb.org/cgi/content/full/jcb.200508072/DC1.
Acknowledgments
We are grateful to H. Rizvi and A. Orr for their technical assistance and the members of the Li laboratory for their comments. We thank J. Rothstein for providing antibody to GLT-1, K. Tanaka and G. Bonvento for antibody to GLAST, R. Wen for adenoviral vectors, W. Chen and S. Warren for advice, and M. Friedman for critical reading of the manuscript. We also thank Harvard Brain Tissue Resource Center for providing HD patient and control brain tissues.
This work was supported by grants (AG19206 and NS36232) from the National Institutes of Health.
Abbreviations used in this paper: AD, Alzheimer's disease; DHK, dihydrokainate; DIV, days in vitro; GFAP, glial fibrillary acidic protein; GLT-1, glutamate transporter-1; GLAST, glutamate/aspartate transporter; HD, Huntington's disease; htt, huntingtin; MAP2, microtubule-associated protein 2; MSNs, medium-sized spiny neurons; MTT, 3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide; PolyQ, polyglutamine.
References
- Beal, M.F. 1994. Huntington's disease, energy, and excitotoxicity. Neurobiol. Aging. 15:275–276. [DOI] [PubMed] [Google Scholar]
- Behrens, P.F., P. Franz, B. Woodman, K.S. Lindenberg, and G.B. Landwehrmeyer. 2002. Impaired glutamate transport and glutamate-glutamine cycling: downstream effects of the Huntington mutation. Brain. 125:1908–1922. [DOI] [PubMed] [Google Scholar]
- Calabresi, P., D. Centonze, A. Pisani, G. Sancesario, P. Gubellini, G.A. Marfia, and G. Bernardi. 1998. Striatal spiny neurons and cholinergic interneurons express differential ionotropic glutamatergic responses and vulnerability: implications for ischemia and Huntington's disease. Ann. Neurol. 43:586–597. [DOI] [PubMed] [Google Scholar]
- Cepeda, C., M.A. Ariano, C.R. Calvert, J. Flores-Hernandez, S.H. Chandler, B.R. Leavitt, M.R. Hayden, and M.S. Levine. 2001. NMDA receptor function in mouse models of Huntington disease. J. Neurosci. Res. 66:525–539. [DOI] [PubMed] [Google Scholar]
- Clement, A.M., M.D. Nguyen, E.A. Roberts, M.L. Garcia, S. Boillee, M. Rule, A.P. McMahon, W. Doucette, D. Siwek, R.J. Ferrante, et al. 2003. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 302:113–117. [DOI] [PubMed] [Google Scholar]
- Coyle, J.T., and R. Schwarcz. 1976. Lesion of striatal neurones with kainic acid provides a model for Huntington's chorea. Nature. 263:244–246. [DOI] [PubMed] [Google Scholar]
- Davies, S.W., M. Turmaine, B.A. Cozens, M. DiFiglia, A.H. Sharp, C.A. Ross, E. Scherzinger, E.E. Wanker, L. Mangiarini, and G.P. Bates. 1997. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 90:537–548. [DOI] [PubMed] [Google Scholar]
- DiFiglia, M., E. Sapp, K.O. Chase, S.W. Davies, G.P. Bates, J.P. Vonsattel, and N. Aronin. 1997. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 277:1990–1993. [DOI] [PubMed] [Google Scholar]
- Driscoll, B.F., M.J. Law, and A.M. Crane. 1991. Cell damage associated with changing the medium of mesencephalic cultures in serum-free medium is mediated via N-methyl-D-aspartate receptors. J. Neurochem. 56:1201–1206. [DOI] [PubMed] [Google Scholar]
- Dunah, A.W., H. Jeong, A. Griffin, Y.M. Kim, D.G. Standaert, S.M. Hersch, M.M. Mouradian, A.B. Young, N. Tanese, and D. Krainc. 2002. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science. 296:2238–2243. [DOI] [PubMed] [Google Scholar]
- Eng, D.L., Y.L. Lee, and P.G. Lal. 1997. Expression of glutamate uptake transporters after dibutyryl cyclic AMP differentiation and traumatic injury in cultured astrocytes. Brain Res. 778:215–221. [DOI] [PubMed] [Google Scholar]
- Greene, J.G., R.H. Porter, R.V. Eller, and J.T. Greenamyre. 1993. Inhibition of succinate dehydrogenase by malonic acid produces an “excitotoxic” lesion in rat striatum. J. Neurochem. 61:1151–1154. [DOI] [PubMed] [Google Scholar]
- Gu, X., C. Li, W. Wei, V. Lo, S. Gong, S.H. Li, T. Iwasato, S. Itohara, X.J. Li, I. Mody, et al. 2005. Pathological cell-cell interactions elicited by a neuropathogenic form of mutant huntingtin contribute to cortical pathogenesis in HD mice. Neuron. 46:433–444. [DOI] [PubMed] [Google Scholar]
- Gunawardena, S., L.S. Her, R.G. Brusch, R.A. Laymon, I.R. Niesman, B. Gordesky-Gold, L. Sintasath, N.M. Bonini, and L.S. Goldstein. 2003. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 40:25–40. [DOI] [PubMed] [Google Scholar]
- Hansson, O., E. Guatteo, N.B. Mercuri, G. Bernardi, X.J. Li, R.F. Castilho, and P. Brundin. 2001. Resistance to NMDA toxicity correlates with appearance of nuclear inclusions, behavioural deficits and changes in calcium homeostasis in mice transgenic for exon 1 of the huntington gene. Eur. J. Neurosci. 14:1492–1504. [DOI] [PubMed] [Google Scholar]
- He, T.C., S. Zhou, L.T. da Costa, J. Yu, K.W. Kinzler, and B. Vogelstein. 1998. A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA. 95:2509–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebb, M.O., E.M. Denovan-Wright, and H.A. Robertson. 1999. Expression of the Huntington's disease gene is regulated in astrocytes in the arcuate nucleus of the hypothalamus of postpartum rats. FASEB J. 13:1099–1106. [DOI] [PubMed] [Google Scholar]
- Kawahara, K., R. Hosoya, H. Sato, M. Tanaka, T. Nakajima, and S. Iwabuchi. 2002. Selective blockade of astrocytic glutamate transporter GLT-1 with dihydrokainate prevents neuronal death during ouabain treatment of astrocyte/neuron cocultures. Glia. 40:337–349. [DOI] [PubMed] [Google Scholar]
- Kuppenbender, K.D., D.G. Standaert, T.J. Feuerstein, J.B. Penney Jr., A.B. Young, and G.B. Landwehrmeyer. 2000. Expression of NMDA receptor subunit mRNAs in neurochemically identified projection and interneurons in the human striatum. J. Comp. Neurol. 419:407–421. [DOI] [PubMed] [Google Scholar]
- Landwehrmeyer, G.B., D.G. Standaert, C.M. Testa, J.B. Penney Jr., and A.B. Young. 1995. NMDA receptor subunit mRNA expression by projection neurons and interneurons in rat striatum. J. Neurosci. 15:5297–5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H., S.H. Li, H. Johnston, P.F. Shelbourne, and X.J. Li. 2000. Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat. Genet. 25:385–389. [DOI] [PubMed] [Google Scholar]
- Li, L., M. Fan, C.D. Icton, N. Chen, B.R. Leavitt, M.R. Hayden, T.H. Murphy, and L.A. Raymond. 2003. Role of NR2B-type NMDA receptors in selective neurodegeneration in Huntington disease. Neurobiol. Aging. 24:1113–1121. [DOI] [PubMed] [Google Scholar]
- Li, S.H., and X.J. Li. 2004. Huntingtin-protein interactions and the pathogenesis of Huntington's disease. Trends Genet. 20:146–154. [DOI] [PubMed] [Google Scholar]
- Li, S.H., A.L. Cheng, H. Zhou, S. Lam, M. Rao, H. Li, and X.J. Li. 2002. Interaction of Huntington disease protein with transcriptional activator Sp1. Mol. Cell. Biol. 22:1277–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lievens, J.C., B. Woodman, A. Mahal, O. Spasic-Boscovic, D. Samuel, L. Kerkerian-Le Goff, and G.P. Bates. 2001. Impaired glutamate uptake in the R6 Huntington's disease transgenic mice. Neurobiol. Dis. 8:807–821. [DOI] [PubMed] [Google Scholar]
- Lievens, J.C., T. Rival, M. Iche, H. Chneiweiss, and S. Birman. 2005. Expanded polyglutamine peptides disrupt EGF receptor signaling and glutamate transporter expression in Drosophila. Hum. Mol. Genet. 14:713–724. [DOI] [PubMed] [Google Scholar]
- Lin, C.H., S. Tallaksen-Greene, W.M. Chien, J.A. Cearley, W.S. Jackson, A.B. Crouse, S. Ren, X.J. Li, R.L. Albin, and P.J. Detloff. 2001. Neurological abnormalities in a knock-in mouse model of Huntington's disease. Hum. Mol. Genet. 10:137–144. [DOI] [PubMed] [Google Scholar]
- Maragakis, N.J., and J.D. Rothstein. 2001. Glutamate transporters in neurologic disease. Arch. Neurol. 58:365–370. [DOI] [PubMed] [Google Scholar]
- Martin-Aparicio, E., J. Avila, and J.J. Lucas. 2002. Nuclear localization of N-terminal mutant huntingtin is cell cycle dependent. Eur. J. Neurosci. 16:355–359. [DOI] [PubMed] [Google Scholar]
- Matesic, D.F., and R.C. Lin. 1994. Microtubule-associated protein 2 as an early indicator of ischemia-induced neurodegeneration in the gerbil forebrain. J. Neurochem. 63:1012–1020. [DOI] [PubMed] [Google Scholar]
- Mennerick, S., R.P. Dhond, A. Benz, W. Xu, J.D. Rothstein, N.C. Danbolt, K.E. Isenberg, and C.F. Zorumski. 1998. Neuronal expression of the glutamate transporter GLT-1 in hippocampal microcultures. J. Neurosci. 18:4490–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers, R.H., J.P. Vonsattel, P.A. Paskevich, D.K. Kiely, T.J. Stevens, L.A. Cupples, E.P. Richardson Jr., and E.D. Bird. 1991. Decreased neuronal and increased oligodendroglial densities in Huntington's disease caudate nucleus. J. Neuropathol. Exp. Neurol. 50:729–742. [DOI] [PubMed] [Google Scholar]
- Norton, W.T. 1999. Cell reactions following acute brain injury: a review. Neurochem. Res. 24:213–218. [DOI] [PubMed] [Google Scholar]
- Parent, A., and L.N. Hazrati. 1995. Functional anatomy of the basal ganglia. I. The cortico-basal ganglia-thalamo-cortical loop. Brain Res. Brain Res. Rev. 20:91–127. [DOI] [PubMed] [Google Scholar]
- Reddy, P.H., M. Williams, V. Charles, L. Garrett, L. Pike-Buchanan, W.O. Whetsell Jr., G. Miller, and D.A. Tagle. 1998. Behavioural abnormalities and selective neuronal loss in HD transgenic mice expressing mutated full-length HD cDNA. Nat. Genet. 20:198–202. [DOI] [PubMed] [Google Scholar]
- San Jose, I., N. Garcia-Atares, B. Pelaez, R. Cabo, I. Esteban, J.A. Vega, and J. Represa. 2001. Reduction of glial fibrillary acidic protein-immunoreactive astrocytes in some brain areas of old hairless rhino-j mice (hr-rh-j). Neurosci. Lett. 309:81–84. [DOI] [PubMed] [Google Scholar]
- Sapp, E., K.B. Kegel, N. Aronin, T. Hashikawa, Y. Uchiyama, K. Tohyama, P.G. Bhide, J.P. Vonsattel, and M. DiFiglia. 2001. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J. Neuropathol. Exp. Neurol. 60:161–172. [DOI] [PubMed] [Google Scholar]
- Saudou, F., S. Finkbeiner, D. Devys, and M.E. Greenberg. 1998. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 95:55–66. [DOI] [PubMed] [Google Scholar]
- Setkowicz, Z., and K. Janeczko. 1998. Effects of prenatal gamma-irradiation on the astrocyte proliferation in response to injury in the brain of 6-day-old rat. Brain Res. 803:122–128. [DOI] [PubMed] [Google Scholar]
- Singhrao, S.K., P. Thomas, J.D. Wood, J.C. MacMillan, J.W. Neal, P.S. Harper, and A.L. Jones. 1998. Huntingtin protein colocalizes with lesions of neurodegenerative diseases: An investigation in Huntington's, Alzheimer's, and Pick's diseases. Exp. Neurol. 150:213–222. [DOI] [PubMed] [Google Scholar]
- Snider, B.J., J.L. Moss, F.J. Revilla, C. Lee, V.C. Wheeler, M.E. Macdonald, and D.W. Choi. 2003. Neocortical neurons cultured from mice with expanded cag repeats in the huntingtin gene: unaltered vulnerability to excitotoxins and other insults. Neuroscience. 120:617–625. [DOI] [PubMed] [Google Scholar]
- Su, Z.Z., M. Leszczyniecka, D.C. Kang, D. Sarkar, W. Chao, D.J. Volsky, and P.B. Fisher. 2003. Insights into glutamate transport regulation in human astrocytes: cloning of the promoter for excitatory amino acid transporter 2 (EAAT2). Proc. Natl. Acad. Sci. USA. 100:1955–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortarolo, M., A.J. Crossthwaite, L. Conforti, J.P. Spencer, R.J. Williams, C. Bendotti, and M. Rattray. 2004. Expression of SOD1 G93A or wild-type SOD1 in primary cultures of astrocytes down-regulates the glutamate transporter GLT-1: lack of involvement of oxidative stress. J. Neurochem. 88:481–493. [DOI] [PubMed] [Google Scholar]
- Turmaine, M., A. Raza, A. Mahal, L. Mangiarini, G.P. Bates, and S.W. Davies. 2000. Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington's disease. Proc. Natl. Acad. Sci. USA. 97:8093–8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, Z.X., S.H. Li, J. Evans, A. Pillarisetti, H. Li, and X.J. Li. 2003. Mutant huntingtin causes context-dependent neurodegeneration in mice with Huntington's disease. J. Neurosci. 23:2193–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeron, M.M., O. Hansson, N. Chen, C.L. Wellington, B.R. Leavitt, P. Brundin, M.R. Hayden, and L.A. Raymond. 2002. Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron. 33:849–860. [DOI] [PubMed] [Google Scholar]
- Zhou, H., F. Cao, Z. Wang, Z.X. Yu, H.P. Nguyen, J. Evans, S.H. Li, and X.J. Li. 2003. Huntingtin forms toxic NH2-terminal fragment complexes that are promoted by the age-dependent decrease in proteasome activity. J. Cell Biol. 163:109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]