

Abstract
Ablation of the Raf-1 protein causes fetal liver apoptosis, embryonic lethality, and selective hypersensitivity to Fas-induced cell death. Furthermore, Raf-1–deficient cells show defective migration as a result of the deregulation of the Rho effector kinase Rok-α. In this study, we show that the kinase-independent modulation of Rok-α signaling is also the basis of the antiapoptotic function of Raf-1. Fas activation stimulates the formation of Raf-1–Rok-α complexes, and Rok-α signaling is up-regulated in Raf-1–deficient cells. This leads to increased clustering and membrane expression of Fas, which is rescued both by kinase-dead Raf-1 and by interfering with Rok-α or its substrate ezrin. Increased Fas clustering and membrane expression are also evident in the livers of Raf-1–deficient embryos, and genetically reducing Fas expression counteracts fetal liver apoptosis, embryonic lethality, and the apoptotic defects of embryonic fibroblasts. Thus, Raf-1 has an essential function in regulating Fas expression and setting the threshold of Fas sensitivity during embryonic life.
Introduction
The Raf-1 kinase has been regarded for decades as one of the cornerstones of the Ras–Raf–MEK–ERK cascade, a highly conserved multipurpose signaling module implicated in cell proliferation, activation, survival, and oncogenesis. Genetic ablation of c-raf-1, however, has revealed that the essential function of this protein during embryonic life is not to activate the mitogen and extracellular regulated kinase (ERK) kinase (MEK)–ERK module and promote proliferation, but rather to prevent apoptosis (Huser et al., 2001; Mikula et al., 2001). Raf-1–deficient embryos die in utero between embryonic day (E) 10 and 12.5 with placental defects and increased apoptosis. In cultured embryonic fibroblasts, this prosurvival function is selective in that Raf-1 protects the cells against Fas-induced apoptosis that is induced by Fas activation but not by TNFα. This is surprising, as the two receptors share most of their downstream effector mechanisms. A number of Raf-1 downstream effectors with a potential impact on apoptosis have been described previously, including the MEK–ERK module and the antiapoptotic transcription factor NF-κB but also the proapoptotic BH3-only protein Bad (Baccarini, 2002). Raf-1 also interacts directly with c-FLIP (FADD-like ice–like inhibitory protein; Kataoka et al., 2000), a homologue of caspase-8 that is devoid of catalytic activity. The function of c-FLIP is controversial, as it has been described both as an inhibitor and an activator of caspase-8 downstream of death receptors, depending on the stoichiometry of the complex (Chang et al., 2002; Micheau et al., 2002). Finally, Raf-1 forms a complex with and inhibits the proapoptotic kinases ASK-1 (Chen et al., 2001; Yamaguchi et al., 2004) and MST-2 (O'Neill et al., 2004). In both cases, the interaction results in the inhibition of the proapoptotic kinase, which occurs independently of Raf-1 kinase activity. MST-2 inhibition is selectively relevant in the context of Fas-induced apoptosis (O'Neill et al., 2004).
Fas is the best-characterized member of the TNF receptor (TNFR) superfamily. Like other members of this family, it is expressed as a preassociated trimer that is formed via an extracellular preligand-binding domain (Siegel et al., 2000). Ligand binding stimulates the formation of microaggregates followed by the actin-dependent recruitment of the downstream effectors to a receptor-associated death-inducing signaling complex (DISC). The key components of the DISC are the adaptor protein FADD (Fas-associating death domain–containing protein) and the zymogen of caspase-8, which is the main initiator caspase in Fas signaling (Peter and Krammer, 2003). In addition, the long form of c-FLIP (c-FLIPL) is recruited to the DISC, where it heterodimerizes with caspase-8 (Chang et al., 2002; Micheau et al., 2002). The DISC then induces receptor clustering and internalization. DISC formation and internalization depend on an intact actin cytoskeleton and are essential for Fas signaling (Algeciras-Schimnich et al., 2002). Thus, the interplay between Fas and the actin cytoskeleton is crucial for Fas function. At least in T lymphocytes, the interaction between Fas and the actin cytoskeleton is mediated by the cytolinker ezrin (Parlato et al., 2000; Lozupone et al., 2004). ERM (ezrin–radixin–moesin) proteins link an array of plasma membrane proteins with filamentous actin and play a critical role in the establishment of membrane structures and cell polarity. ERM are activated by a phosphorylation event that relieves intramolecular inhibition and allows their NH2-terminal domain to interact with a target protein, whereas the COOH-terminal domain binds actin (Bretscher et al., 2002). It has been suggested that complex formation among Fas, ezrin, and actin is important for the polarization of Fas and its recruitment to large uropodes, where it appears to concentrate. Notably, the TNFα receptor is homogeneously distributed and does not show any polarization (Parlato et al., 2000).
We have recently shown that Raf-1 plays an essential role in maintaining the organization of the cytoskeleton and, thus, in cell polarity and migration (Ehrenreiter et al., 2005). This function of Raf-1 is kinase independent and consists in restraining Rho downstream signaling. Therefore, the link between the association of Fas with the actin cytoskeleton and the intensity of the apoptotic signal was particularly intriguing in the context of Raf-1 knockout (KO) cells. The goals of this study were to investigate the molecular basis of Fas hypersensitivity in Raf-1 KO cells and to determine the role of this defect in embryonic development.
We show that the molecular basis of Fas hypersensitivity is the lack of Fas internalization as a result of a defect in the regulation of ezrin that results in the accumulation of Fas on the membrane. Increased membrane expression of Fas can be observed in fibroblasts in vitro and in fetal liver cells in situ, and reducing Fas levels rescues fetal liver apoptosis, embryonic lethality, and the apoptotic defects shown by embryonic fibroblasts. Thus, increased membrane expression of Fas is the cause of embryonic death in Raf-1–deficient mice. These results demonstrate that Raf-1, by regulating the cytoskeleton, sets the threshold of Fas sensitivity in fibroblasts in vitro and in the fetal liver in vivo and that this function is essential during embryonic development.
Results
Raf-1–deficient mouse embryonic fibroblasts (MEFs) are selectively hypersensitive to apoptosis induced by Fas activation and showed altered Fas surface expression. 129/SvHsd:Bl6 Raf-1 KO MEFs are more sensitive than wild-type (WT) cells to apoptosis induced by an agonist αFas antibody (Mikula et al., 2001) as well as to the physiological stimulus Fas ligand (FasL; Fig. 1 A, top and middle). In both cases, Fas hypersensitivity could be detected over a broad range of doses. In contrast, Raf-1 KO and WT cells responded in an indistinguishable manner to TNFα over a broad range of concentrations (Fig. 1 A, bottom).
Figure 1.

Selective hypersensitivity of Raf-1–deficient MEFs to Fas activation correlates with increased Fas expression. (A) Raf-1 KO MEFs are hypersensitive toward apoptosis induced by an agonistic Fas antibody or by FasL, but not by TNFα. MEFs were treated either with αFas, with recombinant FLAG-tagged FasL cross-linked with 1 μg/ml α-FLAG M2 antibody, or with recombinant mouse TNFα at the concentrations indicated for 22 h in the presence of 5 μg/ml Chx and 0.5% FCS. Cell death was determined by CytoTox 96 assay. The values represent the mean ± SD (error bars) of three independent cell lines. *, P < 0.02; **, P < 0.01, according to a t test comparing KO with WT cells. (B) The surface expression of Fas but not of TNFRI is altered in Raf-1 KO MEFs. WT and KO cells were stained with FITC-conjugated αFas (left) or with hamster α-mouse TNFRI antibody followed by FITC-conjugated goat α-hamster antibody (right) and were analyzed by flow cytometry. Dashed lines, isotype control (iso). (C and D) Fas is slightly overexpressed in Raf-1–deficient cells. (C) Different amounts of whole cell lysates from WT and KO cells were analyzed by αFas immunoblotting. Ponceau staining of the membrane is shown as a loading control. (D) Fas mRNA levels were determined by RT-PCR. The HPRT gene was used as a normalization control. −, negative control; M, DNA marker. Molecular mass markers (in kilodaltons, C; or bp, D) are shown on the left.
The surface expression of Fas and TNFRI, which was determined by FACS analysis, reflected the selective hypersensitivity of Raf-1–deficient MEFs to Fas activation. KO cells showed a 4.5–5-fold increase in Fas surface expression with respect to WT (Fig. 1 B). In contrast, the increase in the total amount of Fas in KO cells was ∼1.5-fold as determined by immunoblotting or RT-PCR (Fig. 1, C and D). The expression of TNFRI was low and indistinguishable in cells of either genotype (Fig. 1 B).
Raf-1 KO MEFs of the 129/SvHsd background were hypersensitive to Fas stimulation and expressed more Fas than their WT counterparts, exactly like Raf-1–deficient MEFs of the 129/SvHsd:Bl6 background, excluding a possible influence of the background on these phenotypes (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200504137/DC1).
Raf-1 is required for Fas internalization and efficient DISC formation
Consistent with the results in Fig. 1, Fas staining was faint, distributed evenly on the cell surface of WT MEFs, and did not appreciably change upon Fas stimulation. In KO cells, the staining was brighter, and Fas was visualized as a rim around the cells and occasionally in patches and clusters that became more prominent upon Fas stimulation (Fig. 2 A). In addition, Fas internalization was significantly reduced in KO fibroblasts (Fig. 2 B). These phenotypes suggested a possible defect in Fas-stimulated cytoskeletal rearrangement, particularly in view of the cytoskeletal anomalies reported in migrating Raf-1 KO cells (Ehrenreiter et al., 2005). Upon Fas stimulation, WT cells produced long protrusions that were brightly stained with an antibody against phosphorylated ezrin (pT567), which was hardly detectable in unstimulated WT cells (Fig. 2 E). These structures are reminiscent of the uropods observed in T lymphocytes—long (at least one third of the whole cell body) and large bulbs transiently protruding from the cell surface—whose formation depends on the phosphorylation of ezrin on T567 (Lee et al., 2004). In T lymphocytes, functionally active Fas colocalizes with ezrin in the uropodes (Parlato et al., 2000); in adherent Raf-1 WT fibroblasts, however, the amount of Fas was too low to be detectable. As previously described, in unstimulated KO fibroblasts, the actin was detected in a rim around the cells, and the vimentin cytoskeleton was disorganized (Ehrenreiter et al., 2005). Upon Fas stimulation, bright patches of actin appeared, which partially colocalized with Fas. In addition, the vimentin cytoskeleton collapsed and was visualized as a dense perinuclear structure and at the tips of the short protrusions induced by Fas in these cells (Fig. 2, C and D). Although full-fledged uropods could not be observed in KO fibroblasts, these small, Fas-induced protrusions may be interpreted as an attempt to form such structures. In contrast to the WT, unstimulated KO fibroblasts contained significant amounts of ezrinpT567 (Figs. 2 E, top; and 4 D, bottom) localized to microvilli, which is in line with previous data (Takeuchi et al., 1994). In the KO, however, ezrinpT567 staining increased and concentrated in large spots, which partially colocalized with Fas (Fig. 2 E). The presence of hyperphosphorylated ezrin in KO cells could be confirmed by immunoblotting (Fig. 3 B).
Figure 2.

Raf-1 modulates cytoskeletal changes during Fas activation and is required for Fas internalization. (A) Fas is concentrated in patches and clusters at the surface of Raf-1 KO MEFs. MEFs were stained with FITC-conjugated αFas for 45 min on ice (0). Cells were then washed, incubated for 30 min at 37°C, and analyzed using a confocal microscope. Fas clustering was observed in 72 ± 4% of KO cells. (B) Defective internalization in Raf-1–deficient MEFs. Cells were treated with 1 μg/ml αFas at 4°C to allow binding, washed, and either kept at 4°C or transferred to 37°C to allow internalization. Internalization was determined by comparing the amount of αFas left on the surface of cells incubated at 37°C with that present on the surface of the cells kept at 4°C. The values represent the mean ± SD (error bars) of three independent experiments. *, P < 0.025; **, P < 0.015, according to a t test comparing KO with WT cells. (C–E) Dramatic cytoskeletal remodeling in Raf-1 KO MEFs. Cells were treated with FITC-conjugated αFas as described in A and were stained with rhodamin-conjugated phalloidin (C), with antibodies against vimentin (D), or with ezrinpT567 (E) followed by the appropriate fluorochrome-conjugated antibodies. As the amount of Fas on the surface of WT cells is too low to be detectable, the merge is shown only for the KO cells. 74 ± 7% untreated KO cells showed the cytoskeletal phenotypes. Fas-induced shrinkage was observed in 70 ± 4% of the KO cells, and uropod formation was observed in 35 ± 3% of WT cells.
Figure 3.

Inefficient DISC formation but increased caspase activation in Raf-1 KO cells. (A) DISC formation is inefficient in Raf-1–deficient MEFs. Cells were either left untreated (0) or were treated with 2 μg/ml αFas plus 5 μg/ml Chx. At the indicated times, the DISC was collected using protein A–Sepharose beads. The presence of Fas, FADD, c-FLIPL, caspase-8, and actin was determined by immunoblotting. Pro-c8, caspase-8 precursor; c8 p43, caspase-8 cleavage product; FlipL p43/p41, c-FLIPL cleavage product; *, unspecific band. (B) Caspase-8, c-FLIPL, and caspase-3 are rapidly cleaved, and ezrin is hyperphosphorylated in KO MEFs treated with αFas. MEFs were treated with αFas/Chx as described in A. Whole cell lysates collected at the indicated times were analyzed by immunoblotting. Molecular mass markers are shown in kilodaltons on the left.
The interaction of Fas with the actin cytoskeleton, which is mediated by ezrin (Lozupone et al., 2004), is required for DISC formation and internalization (Algeciras-Schimnich et al., 2002). DISC formation in KO and WT MEFs was analyzed by exclusively isolating the Fas that bound to excess agonist in vivo. Consistent with the higher surface expression (Figs. 1 B and 2 A), much more agonist-bound Fas was recovered from KO than from WT cells (Fig. 3 A). The amount of DISC components (FADD, procaspase-8, and c-FLIPL) recovered from KO cells was higher than that recovered from WT MEFs. However, considering the amount of agonist-bound receptor isolated, DISC formation on a per-receptor basis was inefficient in KO cells. This suggests either that a smaller pool of receptors are competent to form a DISC in the KO cells or that less DISC components/receptor are recruited. In addition, both procaspase-8 and c-FLIPL cleavage (judged as the ratio between precursors and cleavage products) proceeded less efficiently and with slower kinetics in the KO DISC (Fig. 3 A). Consistent with the defective Fas internalization, much less actin was recruited to the KO than to the WT Fas DISC. Expression of the DISC components was comparable in WT and KO cells (Fig. 3 A, right).
The aforementioned data indicate that DISC formation is less efficient in Raf-1 KO cells. Regardless of this, however, the analysis of whole cell lysates revealed that the overall Fas-induced caspase activation, as detected by the cleavage of caspase-8, 3, and c-FLIPL, was more rapid and effective in Raf-1 KO than in WT cells (Fig. 3 B). Taking into account the defect in Fas internalization and the recovery of DISC components even at late time points (120 min after stimulation), these data suggest that hypersensitivity to Fas-induced apoptosis results from less efficient but prolonged signaling by surface receptors in Raf-1 KO cells.
Raf-1 associates with Rok-α upon Fas stimulation and modulates Rok-α downstream signaling, Fas surface expression, and sensitivity to Fas-induced apoptosis
We have previously shown that in unstimulated and migrating fibroblasts, the formation of a Raf-1–Rok-α complex restrains the activation of Rok-α independently of Raf-1 kinase activity (Ehrenreiter et al., 2005). Among the consequences of Rok-α hyperactivation in Raf-1 KO cells are the disorganization of the vimentin cytoskeleton, which results in the relocalization of Rok-α to the plasma membrane, and the constitutive phosphorylation of ezrin on T567 (Ehrenreiter et al., 2005). The rapid collapse of vimentin structures and the hyperphosphorylation of ezrin in Fas-stimulated KO cells, which are hallmarks of Rok-α activation, suggested that Raf-1 might serve a similar function during Fas stimulation. Indeed, Rok-α was quickly translocated to the membrane of KO cells upon Fas stimulation, where it often resided in structures similar to small blebs (Fig. 4 A, arrowheads). In contrast, in WT cells, Fas stimulation resulted in an even more defined localization of Rok-α to the vimentin cytoskeleton (Fig. 4 A). Consistent with a role of Raf-1 in the regulation of Rok-α activity and localization, increasing amounts of Rok-α were detectable in endogenous Raf-1 immunoprecipitates (IPs) from Fas-induced WT cells (Fig. 4 B). As described for migration, the stable expression of either kinase-competent (KC) or kinase-dead (KD) Raf-1 in KO cells rescued the increase in Fas expression (Fig. 4, C and D) and ezrin phosphorylation/localization (Fig. 4 D) as well as the cytoskeletal defects (Ehrenreiter et al., 2005). Consistently, both KC and KD clones showed normal sensitivity to Fas-mediated apoptosis. TNFα-induced apoptosis was not affected by the reintroduction of KC or KD Raf-1 (Fig. 4 E). Vector-transfected cells and KO MEFs behaved indistinguishably.
Figure 4.

Endogenous Raf-1 coimmunoprecipitates with Rok-α upon Fas stimulation, and Raf-1 kinase activity is dispensable for the regulation of Fas surface expression. (A) Rok-α is mislocalized in unstimulated and αFas-treated KO fibroblasts. WT and KO fibroblasts were treated with αFas as described in Fig. 2 A. The subcellular localization of Rok-α was determined by immunofluorescence. Arrowheads indicate Rok-α staining in the blebs, which was observed in 66 ± 2% of stimulated KO cells. (B) Fas stimulates the formation of a Raf-1–Rok-α complex. WT MEFs were stimulated with 2 μg/ml αFas, and Raf-1 IPs were prepared at the indicated times. The presence of Raf-1 and Rok-α in the IP (top) and in the input (bottom) was detected by immunoblotting. B, lysates incubated with protein A–Sepharose beads only. (C and D) Expression of KC or KD Raf-1 restores normal Fas expression, ezrin phosphorylation/distribution, and sensitivity to Fas-induced apoptosis in Raf-1−/− MEFs. (C) The amount of Fas and Raf-1 in whole cell lysates was determined by immunoblotting. Molecular mass markers are shown in kilodaltons on the left. (D) Fas surface expression and ezrin phosphorylation/distribution were analyzed by immunofluorescence. The pictures shown are representative of 90 ± 1% KC and 87 ± 4% KD cells. (E) Sensitivity to αFas or TNFα-induced apoptosis was determined as described in Fig. 1 A. The values represent the mean ± SD (error bars) of at least three independent clones, each assessed in at least two independent experiments. *, P < 0.01 according to a t test comparing WT, vector (V), KC, or KD cells with KO cells.
Rok-α–dependent ezrin deregulation is responsible for the defects in Fas clustering and internalization in Raf-1–deficient cells. To determine whether hyperactivation of Rok-α was responsible for the increased clustering of Fas and for Fas hypersensitivity in KO cells, we transfected Raf-1–deficient cells with plasmids expressing a dominant-negative (DN) form of this protein. KD Rok-α (eG–Rok-α KD) abrogated both ezrin phosphorylation and Fas clustering (Fig. 5, A and B); however, as a result of the toxicity of the construct, it was not possible to investigate its effects on Fas-induced apoptosis. Therefore, we used small interfering RNA (siRNA) to silence Rok-α in KO and WT fibroblasts. 72 h after transfection with Rok-α siRNA, the expression of Rok-α, but not of the related kinase Rok-β, was radically reduced in cells of either genotype (Fig. 5 C). Silencing Rok-α abrogated the hypersensitivity of KO cells to Fas-induced apoptosis (Fig. 5 D) and dramatically reduced Fas clustering and ezrin phosphorylation (Fig. 5 E). Scrambled (SCR) siRNA had no effect on Rok-α expression, Fas clustering, Fas-induced apoptosis, or ezrin phosphorylation (Fig. 5, C–E). To determine whether ezrin hyperphosphorylation was causally linked to increased Fas clustering and to hypersensitivity to Fas-induced apoptosis, we transfected WT and KO cells with DN ezrin (DN-Ez-eG or ezrin1-310), which interferes with the function of endogenous ERM and microvilli formation (Crepaldi et al., 1997). DN ezrin abrogated Fas clustering (Fig. 5 F) and significantly decreased Fas-induced apoptosis in KO cells while causing a reproducible, albeit not significant, increase in Fas-induced apoptosis in WT cells (Fig. 5 G, top). In addition, DN ezrin had opposite effects on the internalization in WT and KO cells: it decreased internalization in the former and increased it in the latter (Fig. 5 G, bottom). Together with the data in Figs. 2 E, 3 B, and 4 D, these results show that in WT cells, ezrin is a positive regulator of Fas internalization and that it may modulate apoptosis by regulating the amount of Fas available for further stimulation at the cell surface. In contrast, in KO fibroblasts, hyperphosphorylation of ezrin prevents internalization and prolongs the death signal, thereby determining the difference in Fas sensitivity between WT and KO cells.
Figure 5.

Interfering with Rok-α and ezrin restores normal sensitivity to Fas-induced apoptosis in Raf-1–deficient fibroblasts. (A and B) Transfection with DN Rok-α (eG–Rok-α KD) prevents ezrin hyperphosphorylation (A) and Fas clustering (B) in Raf-1 KO cells. Reduced ezrin phosphorylation and lack of Fas clustering were observed in 89 ± 3% of the cells transfected with eG–Rok-α KD. (C–E) Silencing Rok-α expression reduces Fas sensitivity, Fas clustering, and ezrin hyperphosphorylation in KO cells. (C) Expression of Rok-α was assessed by immunoblotting 72 h after transfection with scrambled (SCR) or Rok-α siRNA. The related kinase Rok-β is shown as a specificity control and tubulin as a loading control. (D) KO and WT MEFs were transfected with Rok-α or SCR siRNA. Apoptosis was induced with 50 ng/ml αFas (5 μg/ml Chx for 22 h) and detected as described in Fig. 1 A. The values represent the mean ± SD (error bars) of triplicates. Two independent transfections are shown. *, P < 0.01 according to a t test comparing Rok-α with SCR siRNA–transfected cells of either genotype. (E) KO cells transfected with Rok-α or SCR siRNA were stained with antibodies against Fas and ezrinpT567. 81 ± 3% of the cells displayed the phenotypes shown. (F and G) DN ezrin restores normal Fas internalization and Fas sensitivity in KO cells. (F) Morphology and Fas staining of KO cells expressing eGFP (eG) or eGFP-ezrin1-310 (DN-Ez-eG). Reduced surface Fas clustering was displayed by 89 ± 4% of the DN-Ez-eG–transfected cells. (G, top) The effect of eG and Ez-eG on Fas-induced apoptosis (200 ng/ml αFas and 5 μg/ml Chx for 16 h) was determined by FACS analysis of Annexin V–positive cells. (bottom) The effect of eG and DN-Ez-eG on Fas internalization was determined as described in Fig. 2 B. The values represent the mean ± SD (error bars) of three independent experiments. *, P < 0.01 (top) and *, P < 0.04 (bottom) according to a t test comparing eG with DN-Ez-eG–expressing cells of either genotype. (A–G) Transfection with the empty vector (eG) or with SCR siRNA did not alter the phenotype of KO cells.
Fas mutation rescues all apoptotic defects of Raf-1–deficient primary MEFs (PEFs)
If the amount of Fas they express is the basis of the hypersensitivity of KO cells to Fas-induced apoptosis, reducing Fas levels should rescue the defects of Raf-1–deficient PEFs in culture. To assay this, we prepared PEFs from 129/SvHsd:Bl6 Raf-1 KO embryos that were heterozygous for the lpr mutation, which functionally inactivates Fas (Watanabe-Fukunaga et al., 1992). In Raf-1 KO PEFs, Fas accumulated at the membrane and was often organized in small patches and clusters (Fig. 6 A and Fig. S2 B, available at http://www.jcb.org/cgi/content/full/jcb.200504137/DC1), as observed in immortalized fibroblasts (Figs. 2 A and 4 D). lpr heterozygosity reduced the amount of total and membrane-associated Fas (Fig. 6, A and B). Raf-1 KO PEFs are hypersensitive to Fas-induced cell death (Fig. 6 F and Fig. S2 A) and fail to accumulate in culture as a result of increased spontaneous apoptosis (Mikula et al., 2001). These defects were rescued in c-raf-1 − / − ;lpr/+ cells (Fig. 6, D–F; KO;lpr/+), indicating that they were caused by alterations in Fas signaling. Because continuously growing KO and WT PEFs constitutively produce indistinguishable amounts of FasL (Fig. 6, B and C), the increase in spontaneous cell death shown by KO PEFs can be ascribed to Fas hypersensitivity (Fig. 6 E). Although KO;lpr/+ PEFs were not hypersensitive to Fas stimulation, Fas internalization was still perturbed in these cells (Fig. 6 G), and some Fas clustering could still be observed (Fig. 6 A). This confirms that Fas hypersensitivity is secondary to the cytoskeletal defects and that the net amount of Fas present on the cell surface is the crucial factor determining the differences between KO and WT cells.
Figure 6.

Heterozygosity at the lpr locus rescues the apoptotic defects of Raf-1 KO PEFs in culture. (A) Fas expression is reduced in the lpr/+ background, but Fas is organized in clusters at the surface of Raf-1 KO PEFs. Cells in suspension were treated with αFas as described in Fig. 2 A. Fas clustering was detectable in 68 ± 3% of KO cells and in 31 ± 2% of c-raf-1 –/– ;lpr/+ cells. (B) Fas expression is reduced in the lpr/+ background. The expression of Raf-1, Fas, and FasL in whole PEF lysates was analyzed by immunoblotting. An ERK immunoblot is shown as a loading control. Molecular mass markers are shown in kilodaltons on the left. (C) FasL mRNA levels were determined by RT-PCR. The HPRT gene was used as a normalization control. −, negative control; M, DNA marker in base pairs. (D) c-raf-1 –/– ;lpr/+ (KO;lpr/+) PEFs successfully accumulate in culture. WT, WT;lpr/+, KO, and KO;lpr/+ PEFs were cultured in DME/10% FCS. Cell numbers were determined at the indicated times. The values are the mean ± SD (error bars) of four individual batches of PEFs/genotype, each assayed in triplicate. *, P < 0.04; **, P < 0.025; ***, P < 0.01 according to a t test, all compared with KO. (E) Spontaneous apoptosis in continuously growing WT, WT;lpr/+, KO, and KO;lpr/+ PEFs. Asynchronous cells were stained with propidium iodide, and their DNA content was determined by FACS analysis. The percentage of apoptotic cells (DNA content < 2n) is indicated. (F) PEFs were treated with 500 ng/ml αFas plus 1 μg/ml Chx for 22 h. Cell death was assessed as in Fig. 1 A. The values in E and F are the mean ± SD (error bars) of at least three individual batches of PEFs/genotype. (E) *, P < 0.01. (F) *, P < 0.025 according to a t test, all compared with KO. (G) Defective internalization in KO and KO;lpr/+ MEFs. Internalization was determined as described in Fig. 2 B and expressed as the percentage of the internalization occurring in WT cells. The values represent the mean ± SD (error bars) of three independent experiments. *, P < 0.025 according to a t test, all compared with WT.
Fas or FasL mutations rescue midgestation lethality caused by c-raf-1 ablation
The major defect of the Raf-1 KO embryos is increased fetal liver apoptosis. Therefore, we investigated whether Fas expression was altered in this organ. At E11.5–12.5, the fetal liver consists of developing hepatocytes and haematopoetic cells, both of which reportedly express Fas (Terada and Nakanuma, 1995; De Maria et al., 1999). Consistent with the results obtained in PEFs and MEFs, Fas was often visualized as a rim around the cells in Raf-1–deficient livers (Fig. 7 A). In addition, FACS analysis showed that KO fetal liver cells expressed higher amounts of surface Fas compared with WT cells (Fig. 7 B). The lpr allele decreased Fas staining in both WT (not depicted) and KO fetal liver (Fig. 7, A and B). Raf-1 ablation is embryonic lethal both on the 129/Ola:Bl6 and on the 129/SvHsd:Bl6 background (Huser et al., 2001; Mikula et al., 2001). Remarkably, lpr heterozygosity rescued the fetal liver apoptosis that is characteristic of 129/SvHsd:Bl6 KO embryos (Fig. 7 A). The lpr allele did not significantly alter the ratio of E11.5 c-raf-1 − / − embryos/litter, which was already submendelian (15%; n = 289) in this background (Huser et al., 2001; Mikula et al., 2001). This indicates the presence of an earlier defect with limited penetrance, and it could be connected with the placental insufficiency caused by Raf-1 ablation (Huser et al., 2001; Mikula et al., 2001). All c-raf-1 − / − ;lpr/+ embryos, however, survived to term, whereas their Raf-1–deficient littermates died within E12.5. Embryonic lethality could similarly be rescued by introducing a mutated fasl allele (Takahashi et al., 1994) in Raf-1 KO animals (unpublished data). Most likely as a consequence of the placental defects that persisted in the Raf-1 KO embryos heterozygous for lpr or gld (not depicted), the rescued Raf-1 KO pups were smaller than their control littermates, but they were not anemic (Fig. 7 C). Their eyes were consistently open at birth, which is a phenotype associated with delayed epithelial cell migration (Zhang et al., 2003) and consistent with the cell-autonomous migration defect of Raf-1 KO cells (Ehrenreiter et al., 2005). The rescued Raf-1 KO animals failed to thrive and died perinatally for reasons presently unknown. Thus, increased Fas surface expression correlates with liver apoptosis and embryonic lethality in Raf-1 KO embryos, and both can be relieved by reducing the amount of Fas. Like in fibroblasts, this does not rescue the defects associated with the cytoskeleton and migration, as the pups are born with their eyes open.
Figure 7.

Heterozygosity at the lpr or gld locus prevents fetal liver apoptosis and embryonic lethality as a result of c-raf-1 ablation. (A) Heterozygosity at the lpr locus prevents fetal liver apoptosis in Raf-1 KO embryos. Parasagittal section of E11.5 livers from WT, Raf-1 KO, and Raf-1 KO;lpr/+ embryos. Note the presence of Fas clusters in Raf-1 KO fetal livers and the reduction in Fas staining in the Raf-1 KO;lpr/+ liver. Apoptotic (TUNEL+) cells are absent in Raf-1 KO;lpr/+. (B) Increased Fas surface expression in KO fetal liver cells. Fas expression (solid lines) was determined by FACS analysis as described in Fig. 1 B. Dashed lines, isotype controls. (C) Comparison of a Raf-1 KO;lpr/+ pup (right), a control littermate (left), and eyes open at birth phenotype of the Raf-1 KO;lpr/+ pup (right). The experiments were performed exclusively with F2 animals.
Discussion
Raf-1 has long been regarded as the downstream effector linking Ras activation to the MEK–ERK module in the context of proliferation, survival, and differentiation. Gene ablation experiments have shown that this function must be redundant and have revealed that the essential role of Raf-1 is to restrain apoptosis in midgestation embryos in vivo as well as spontaneous and Fas-induced apoptosis in cultured MEFs. In addition, Raf-1 is required to modulate Rok-α signaling and the remodeling of the cytoskeleton in migrating cells (Ehrenreiter et al., 2005). In this study, we provide evidence that Raf-1, through its effects on the cytoskeleton, regulates Fas expression and, thereby, sets the threshold of Fas sensitivity in fibroblasts. The hypersensitivity of Raf-1–deficient cells to Fas-induced apoptosis correlates with increased membrane expression and clustering of Fas, which can be observed in Raf-1–deficient fetal liver cells in situ as well as in PEFs and in MEFs of different genetic backgrounds in culture. The increase in Fas density per se does not cause detectable caspase activation or set off apoptosis, but it enhances sensitivity upon Fas activation. This ligand dependence is impressively illustrated in vivo by the fact that c-raf-1 − / − embryos are rescued in a heterozygous FasL background (gld/+); i.e., by the mere reduction of the amount of active ligand produced.
Together, these data imply that Raf-1 is necessary to regulate Fas-dependent apoptosis both upstream and downstream of Fas stimulation. Intriguingly, in both cases, the role of Raf-1 in Fas-induced apoptosis is the same as in migration: namely, restraining signaling by the Rho effector Rok-α. Rok-α is active in unstimulated Raf-1 KO cells (Ehrenreiter et al., 2005), and interfering with Rok-α or with its effector ezrin abrogates Fas clustering in Raf-1–deficient MEFs (Figs. 4 and 5). In these cells, ezrin is hyperphosphorylated and localized in microvilli, and Fas clusters appear to be “trapped” within of these structures (Fig. 2). This, however, does not correlate with enhanced Fas activity in KO MEFs; in contrast, DISC formation on a per-receptor basis is less efficient in these cells, and the DISC formed is less active than the WT DISC. This finding is counterintuitive considering the hypersensitivity of KO cells to Fas-induced apoptosis. In KO cells, however, agonist-induced Fas internalization is defective, and, therefore, many agonist-bound Fas molecules are left on the cell surface (Fig. 2 B). We propose a model in which these receptors continue recruiting DISC components and starting new rounds of membrane-bound Fas signaling, albeit not with maximum efficiency. In WT cells, in contrast, agonist-bound Fas is efficiently removed from the surface (Fig. 2 B) and has to be recycled to the plasma membrane before signaling can proceed. This model is supported by the fact that the amount of FADD, procaspase-8, and c-FLIPL associated with Fas decreases with time in WT but much less so in KO cells (Fig. 3). It is also in line with a previous study demonstrating that efficient DISC formation and activity require internalization (Algeciras-Schimnich et al., 2002).
In agreement with this hypothesis, if we restore Fas internalization, we reduce the sensitivity of KO MEFs to Fas-induced apoptosis. But how does this work? Our experiments clearly indicate a role for Rok-α downstream signaling (more specifically for ezrin) in agonist-induced Fas internalization. In WT fibroblasts, ezrinpT567 can be detected only upon Fas stimulation and is concentrated in uropod-like protrusions (one to two per cell), the site of Fas accumulation in T cells (Parlato et al., 2000). In addition, interfering with ezrin function in WT MEFs results in decreased Fas internalization. The opposite happens in KO cells (Fig. 5), in which ezrin is constitutively phosphorylated and is hyperphosphorylated upon Fas activation. These results indicate that a tight control of ezrin activation is necessary for correct Fas signaling. Together with the evidence in a previous study (Parlato et al., 2000), they suggest a model in which ezrin phosphorylation is necessary for uropod development and Fas clustering and, in turn, for DISC formation. However, if too much ezrin is phosphorylated or if ezrin phosphorylation persists for too long, large Fas clusters that cannot be internalized, ineffective DISC formation, and inefficient substrate cleavage result (Fig. 8). Fas internalization requires both actin and caspase-8 activity (Algeciras-Schimnich et al., 2002), and it has been suggested that DISC internalization may depend on the degradation of substrates involved in cortical actin reorganization by DISC-associated caspase-8. In this situation, an excess of ezrinp567, which acts as an actin filament/plasma membrane cross-linker and may, therefore, prevent cortical actin reorganization, is probably counterproductive (Bretscher et al., 2002). Indeed, Fas-stimulated, caspase-dependent dephosphorylation of ezrin has been shown previously (Kondo et al., 1997). In WT fibroblasts, Fas activation increases the interaction between Raf-1 and Rok-α and, thereby, prevents the hyperactivation of the latter kinase and its substrates, favoring cortical actin rearrangements and Fas internalization. Thus, Rok-α downstream signaling has an impact on both the strength and duration of the apoptotic signal, and restraining Rok-α is the common molecular basis of the essential function of Raf-1 in apoptosis and migration.
Figure 8.

Role of Raf-1 in Fas-mediated apoptosis: a working model. Fas binding to FasL stimulates DISC formation and internalization. Both of these processes depend on the linkage of Fas to the cytoskeleton. Phosphorylation of ezrin on T567 by Rok-α promotes Fas clustering but reduces DISC formation and internalization, generating a prolonged, albeit less efficient, Fas signal. In WT cells, formation of a Raf-1–Rok-α complex restrains Rok-α activity and ezrin phosphorylation. In addition, direct binding to Raf-1 prevents the dimerization and phosphorylation of the proapoptotic kinase MST-2 (O'Neill et al., 2004). In the absence of Raf-1, Rok-α activity and ezrin phosphorylation generate a prolonged Fas signal, boosted by unrestrained MST-2 stimulation. White rods, Fas; gray rods, DISC components.
The control of Fas expression and internalization via the modulation of Rok-α is one of three examples of how Raf-1 can counteract apoptosis by inhibiting another kinase. Strikingly, in all three cases, this function of Raf-1 is kinase independent. The two other kinases, ASK-1 (Chen et al., 2001; Yamaguchi et al., 2004) and MST-2 (O'Neill et al., 2004), can both be activated by Fas. Thus, Raf-1 ablation perturbs the Fas cascade at different levels: it lowers the threshold of Fas activation, thereby boosting Fas signaling in general, and it inhibits proaptotic kinases, reducing their specific contribution to Fas-induced cell death (O'Neill et al., 2004). If we correct only the former defect by reducing Fas–FasL interaction in Raf-1–deficient cells (by interfering with Rok-α signaling or by lpr heterozygosity) and embryos (by lpr or gld heterozygosity), we rescue their hypersensitivity to Fas. This indicates that the defective control of Fas expression is the rate-limiting defect in Raf-1–deficient cells and embryos. Consistent with this scenario, mathematical modeling of Fas-induced apoptosis has recently identified the concentration of activated Fas as a critical factor in setting the threshold of Fas-induced apoptosis (Bentele et al., 2004).
Besides rescuing fetal liver apoptosis, a reduction in Fas–FasL expression also appears to restore normal fetal erythropoiesis, as lpr/+ or gld/+ (unpublished data) Raf-1–deficient embryos or pups are not anemic. We have previously demonstrated that the anemia is caused by accelerated erythroblast differentiation, secondary to premature caspase-associated differentiation (Kolbus et al., 2002). To date, the stimulus causing differentiation-associated caspase activation in erythroblasts is entirely unclear. It has, however, been shown that erythroid cells express Fas–FasL, whereby immature erythroblasts express the receptor and react to the ligand, and more mature cells express the ligand. The paracrine interaction between Fas and FasL is hypothesized to regulate erythroid homeostasis (De Maria et al., 1999). In line with this, increased numbers of erythroid progenitor cells have been observed in lpr/lpr or gld/gld mice (Schneider et al., 1999). Indeed, heterozygosity at the lpr or gld locus restores normal caspase activation and erythroid differentiation in Raf-1–deficient erythroblasts in vitro (unpublished data).
The finding that Raf-1 selectively regulates the sensitivity to Fas-mediated apoptosis has important biomedical implications. Fas-mediated apoptosis plays a capital role in liver physiology and disease. Fas up-regulation has been observed in alcohol-induced liver disease and viral hepatitis; in addition, cytotoxic T lymphocytes, which express FasL, may induce apoptosis in the course of viral hepatitis, autoimmune liver disease, and allograft rejection (Kanzler and Galle, 2000). In all of these conditions, Fas activation is undesirable, and knowledge of the processes regulating it might help to find ways of preventing it. On the other hand, apoptosis plays a very relevant role in the progression of hepatocellular carcinoma (Kanzler and Galle, 2000). During progression, these tumors down-regulate Fas expression and are therefore insensitive to FasL produced by T lymphocytes; in addition, the tumor cells express FasL, which enables them to kill the T lymphocytes. Thus, modulating Fas–FasL expression enables the tumors to bypass tumor surveillance. In light of these observations, signal transducers selectively modulating Fas-mediated apoptosis can be considered attractive therapeutic targets in a number of conditions. Mice harboring tissue-restricted or inducible c-raf-1 deletion will be instrumental in defining the role of Raf-1 in the regulation of Fas expression and signaling during tumor progression.
Materials and methods
Mice
c-raf-1 + / − 129/SvHsd mice (Mikula et al., 2001) and lpr/lpr and gld/gld (C57/Bl6; gift of A. Martin-Villalaba, Deutschen Kresbforschungszentrum, Heidelberg, Germany) animals were housed in the animal facility of the Max F. Perutz Laboratories. PCR analysis of offspring was performed as previously described (Mixter et al., 1995; Van Parijs et al., 1998; Mikula et al., 2001).
Cell isolation, culture, infection, and transfection
PEFs were isolated as previously described (Mikula et al., 2001). PEFs and MEFs were cultured in DME supplemented with 10% FCS (GIBCO BRL). Stable cell clones expressing KC and KD Raf-1 have been previously described (Ehrenreiter et al., 2005). For transient transfections, cells were transfected with KD DN Rok-α (pXJ40-eGFP–Rok-α KD; gift of Thomas Leung, Institute of Molecular and Cell Biology, Singapore) and DN ezrin (DN-Ez-eG; gift of R. Lamb, Institute of Cancer Research, London, UK) or the corresponding empty vectors using LipofectAMINE (Invitrogen) according to the manufacturer's instructions. Apoptosis was induced by treatment with either Jo2 (BD Biosciences), recombinant FLAG-tagged FasL (gift of Jürg Tschopp, University of Lausanne, Lausanne, Switzerland) cross-linked with 1 μg/ml αFLAG M2 antibody (Sigma-Aldrich), or murine TNFα (Calbiochem), all in the presence of cycloheximide (Chx; Calbiochem). Chx prevents the expression of NF-κB target genes (Micheau et al., 2001) and was added to sensitize the relatively resistant MEFs to Fas- or TNFα-induced apoptosis. Cell death was measured using the CytoTox 96 NonRadioactive Cytotoxicity Assay (Promega).
RNA isolation and RT-PCR
RNA was prepared using the NucleoSpin Purification Kit (CLONTECH Laboratories, Inc.) according to the manufacturer's instructions. cDNA was generated by reverse transcription using oligo(dT) as primer and M-MuLV Reverse Transcriptase (Fermentas). PCR reactions (94°C for 30 s, 55°C for 30 s, and 72°C for 1 min; 27 cycles) were performed using the following primers: Fas, 5′-CTCCGAGAGTTTAAAGCTGAGG-3′ and 5′-GGAGAATCGCAGTAGAAGTCTGG-3′; FasL, 5′-TCCAGGGTGGGTCTACTTACTAC-3′ and 5′-CCCTCTTACTTCTCCGTTAGGA-3′; and hypoxanthine–guanine phosphoribosyl transferase (HPRT), 5′-GCTGGTGAAAAGGACCTCT-3′ and 5′-CACAGGACTAGAACACCTGC-3′.
Rok-α knockdown
MEFs were transfected with 100 nM siRNA against the mouse Rok-α (siGENOM SMARTpool reagent M-040429-00-05; Dharmacon) or nonspecific control duplex (siCONTROL nontargeting pool D-001206-13-05) using DharmaFECT3 in combination with Neowater-Transfection Enhancer (Do-Coop Ltd.). Cells were analyzed 72 h after transfection.
Flow cytometric analysis
Cells were stained with either FITC-conjugated Jo2 or αTNFRI antibody (hamster α-mouse; BD Biosciences) followed by a FITC-conjugated secondary antibody (Jackson ImmunoResearch Laboratories) in PBS, 0.5% BSA, and 2 mM EDTA (all from Sigma-Aldrich) for 20 min at 4°C. Unrelated isotype-matched immunoglobulins (BD Biosciences) were used as a specificity control. Cells were analyzed using a FACScalibur. To assess the relative intensity of Fas surface staining, we used the CellquestPro set-up that converts the channel value to a linear value corresponding to fluorescence intensity. The relative surface levels of Fas were calculated by subtracting the relative mean fluorescence of the isotype control from the αFas values and determining the ratio between KO and WT cells. For Fas internalization, cells were stained with unconjugated Jo2 and washed to remove unbound antibody before a 30-min incubation at either 37 or 4°C (to prevent internalization) in DME supplemented with 0.5% FCS. Cells were then placed at 4°C, stained with a phycoerythrin-conjugated α-hamster antibody (Jackson ImmunoResearch Laboratories), and analyzed by flow cytometry. The percentage of Fas that was internalized was calculated as follows:
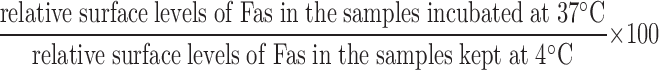 |
Apoptosis was determined by propidium iodide staining of methanol-fixed, RNase A–treated cells followed by FACS analysis or, in transient transfections, using the Annexin V (allophycocyanin) assay (BD Biosciences) according to the manufacturer's protocol. In the transient transfection experiments, at least 10,000 eGFP-positive cells/sample were gated and analyzed.
Histological, immunofluorescence, and TUNEL analysis
Histological and TUNEL analysis were performed as described previously (Mikula et al., 2001). Actin, vimentin cytoskeleton, and Rok-α were visualized as previously described (Ehrenreiter et al., 2005). For Fas immunofluorescence analysis, 4-μm–thick paraffin sections were stained with 1 μg/ml FITC-conjugated Jo2 (1 h at 37°C). Cultured fibroblasts that were adherent or in suspension were treated with 1 μg/ml αFas antibody (FITC- or phycoerythrin-conjugated or unconjugated, as indicated) for 45 min at 4°C. Unconjugated αFas antibody was visualized by staining with rhodamine-conjugated α-hamster antibody (45 min at 4°C; Jackson ImmunoResearch Laboratories). Unbound antibodies were removed by washing, and cells were warmed and kept at 37°C for 30 min to trigger Fas stimulation or were kept on ice (time = 0). After stimulation, cells were fixed in cold methanol/5 mM EDTA or 4% PFA (Figs. 2 C and 5, B and F) for 10 min at room temperature. Unrelated isotype-matched immunoglobulin (BD Biosciences) were used as a specificity control. For ezrinpT567 localization, cells were fixed in cold methanol/5 mM EDTA or 4% PFA (Fig. 5 A) and blocked (10% goat serum/1% BSA) before incubation with αpERM (pT567; Cell Signaling) antibody followed by the appropriate AlexaFluor-conjugated secondary antibody. ProLong Antifade (Invitrogen) was used as a mounting medium. Fas and ezrinpT567 staining are suboptimal in cells fixed in PFA. Hence, this fixation was used only in conjunction with stainings that cannot be visualized in methanol-fixed cells (actin and eGFP). Confocal microscopy was performed with a microscope (model DMRE; Leica) equipped with a TCS-SP confocal scan head (Leica) and a PL-fluotar 40×/1.00–0.50 oil objective or with a microscope (Axiovert 100M; Carl Zeiss MicroImaging, Inc.) fitted with a plan-Apochromat 63×/1.40 oil objective and equipped with the confocal laser scanning module (LSM 510; Carl Zeiss MicroImaging, Inc.). Immersol 518 (Carl Zeiss MicroImaging, Inc.) was used as imaging medium. The confocal images were acquired using the Leica TCS-NT or the LSM 510 2.3 software. 300 cells/slide were counted by two independent investigators. The pictures show representative Z-stacks.
Immunoprecipitation and immunoblotting
Cell lysis and immunoblotting were performed as previously described (Mikula et al., 2001) using the following primary antibodies: αFas, α-actin, α–caspase-3, and α–caspase-8 (Santa Cruz Biotechnology, Inc); αFasL, α–Raf-1, α–Rok-α, α–Rok-β, and α–panERK (BD Biosciences); α–FADD (BioVision); α–FLIP (Q Biogene); α-tubulin (Sigma-Aldrich); and α-ERM and α-pERM (Cell Signaling). Raf-1 IPs were prepared in TBS Tween-20 lysis buffer (200 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, and protease inhibitors) as previously described (Ehrenreiter et al., 2005).
DISC isolation
WT and KO MEFs were stimulated with 2 μg/ml αFas plus 5 μg/ml Chx at 37°C. At the indicated time points, cells were lysed in ice-cold TBS Tween-20 lysis buffer. Insoluble material was removed by centrifugation, and the Fas DISC was collected using protein A–Sepharose beads. The IPs were washed four times and eluted by boiling in SDS-PAGE sample buffer. Protein A–Sepharose beads incubated with lysates from unstimulated cells were used as a negative control.
Statistical analysis
All values are expressed as means ± SD. P values were calculated using the unpaired two-tailed t test assuming unequal variances. A P value ≤ 0.05 is considered statistically significant.
Online supplemental material
Fig. S1 shows that 129/SvHsd Raf-1–deficient MEFs are selectively hypersensitive to Fas stimulation and express increased amounts of surface Fas. Fig. S2 shows hypersensitivity to Fas-induced apoptosis, Fas clustering, and ezrin hyperphosphorylation in Raf-1–deficient 129/SvHsd PEFs.
Acknowledgments
We thank K. Bartalska, C. Khrla, D. Haiderer, and M. Hamerl for technical support, P. Bauer and S. Zehetmayer of the Institute for Statistics of the Vienna Medical University for statistical analysis of the data, and T. Decker for critical reading of the manuscript.
This work was supported by Fond zur Förderung Wissenschaftlicher Forschung grant P15784-B07, European Community grant QLRT-CT-2000-02278, and by a Boehringer Ingelheim grant to M. Baccarini.
D. Piazzolla and K. Meissl contributed equally to this work.
Abbreviations used in this paper: DISC, death-inducing signaling complex; DN, dominant negative; ERK, extracellular regulated kinase; ERM, ezrin–radixin–moesin; FADD, Fas-associating death domain–containing protein; FLIP, FADD-like ice–like inhibitory protein; IP, immunoprecipitate; KC, kinase competent; KD, kinase dead; KO, knockout; MEF, mouse embryonic fibroblast; MEK, mitogen and ERK kinase; PEF, primary MEF; SCR, scrambled; siRNA, small interfering RNA; TNFR, TNF receptor; WT, wild type.
References
- Algeciras-Schimnich, A., L. Shen, B.C. Barnhart, A.E. Murmann, J.K. Burkhardt, and M.E. Peter. 2002. Molecular ordering of the initial signaling events of CD95. Mol. Cell. Biol. 22:207–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccarini, M. 2002. An old kinase on a new path: Raf and apoptosis. Cell Death Differ. 9:783–785. [DOI] [PubMed] [Google Scholar]
- Bentele, M., I. Lavrik, M. Ulrich, S. Stosser, D.W. Heermann, H. Kalthoff, P.H. Krammer, and R. Eils. 2004. Mathematical modeling reveals threshold mechanism in CD95-induced apoptosis. J. Cell Biol. 166:839–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher, A., K. Edwards, and R.G. Fehon. 2002. ERM proteins and merlin: integrators at the cell cortex. Nat. Rev. Mol. Cell Biol. 3:586–599. [DOI] [PubMed] [Google Scholar]
- Chang, D.W., Z. Xing, Y. Pan, A. Algeciras-Schimnich, B.C. Barnhart, S. Yaish-Ohad, M.E. Peter, and X. Yang. 2002. c-FLIP(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J. 21:3704–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J., K. Fujii, L. Zhang, T. Roberts, and H. Fu. 2001. Raf-1 promotes cell survival by antagonizing apoptosis signal- regulating kinase 1 through a MEK-ERK independent mechanism. Proc. Natl. Acad. Sci. USA. 98:7783–7788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crepaldi, T., A. Gautreau, P.M. Comoglio, D. Louvard, and M. Arpin. 1997. Ezrin is an effector of hepatocyte growth factor-mediated migration and morphogenesis in epithelial cells. J. Cell Biol. 138:423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Maria, R., U. Testa, L. Luchetti, A. Zeuner, G. Stassi, E. Pelosi, R. Riccioni, N. Felli, P. Samoggia, and C. Peschle. 1999. Apoptotic role of Fas/Fas ligand system in the regulation of erythropoiesis. Blood. 93:796–803. [PubMed] [Google Scholar]
- Ehrenreiter, K., D. Piazzolla, V. Velamoor, I. Sobczak, J.V. Small, J. Takeda, T. Leung, and M. Baccarini. 2005. Raf-1 regulates Rho signaling and cell migration. J. Cell Biol. 168:955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huser, M., J. Luckett, A. Chiloeches, K. Mercer, M. Iwobi, S. Giblett, X.M. Sun, J. Brown, R. Marais, and C. Pritchard. 2001. MEK kinase activity is not necessary for Raf-1 function. EMBO J. 20:1940–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzler, S., and P.R. Galle. 2000. Apoptosis and the liver. Semin. Cancer Biol. 10:173–184. [DOI] [PubMed] [Google Scholar]
- Kataoka, T., R.C. Budd, N. Holler, M. Thome, F. Martinon, M. Irmler, K. Burns, M. Hahne, N. Kennedy, M. Kovacsovics, and J. Tschopp. 2000. The caspase-8 inhibitor FLIP promotes activation of NF-κB and Erk signaling pathways. Curr. Biol. 10:640–648. [DOI] [PubMed] [Google Scholar]
- Kolbus, A., S. Pilat, Z. Husak, E.M. Deiner, G. Stengl, H. Beug, and M. Baccarini. 2002. Raf-1 antagonizes erythroid differentiation by restraining caspase activation. J. Exp. Med. 196:1347–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo, T., K. Takeuchi, Y. Doi, S. Yonemura, S. Nagata, and S. Tsukita. 1997. ERM (ezrin/radixin/moesin)-based molecular mechanism of microvillar breakdown at an early stage of apoptosis. J. Cell Biol. 139:749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J.-H., T. Katakai, T. Hara, H. Gonda, M. Sugai, and A. Shimizu. 2004. Roles of p-ERM and Rho-ROCK signaling in lymphocyte polarity and uropod formation. J. Cell Biol. 167:327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone, F., L. Lugini, P. Matarrese, F. Luciani, C. Federici, E. Iessi, P. Margutti, G. Stassi, W. Malorni, and S. Fais. 2004. Identification and relevance of the CD95-binding domain in the N-terminal region of ezrin. J. Biol. Chem. 279:9199–9207. [DOI] [PubMed] [Google Scholar]
- Micheau, O., S. Lens, O. Gaide, K. Alevizopoulos, and J. Tschopp. 2001. NF-kappaB signals induce the expression of c-FLIP. Mol. Cell. Biol. 21:5299–5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheau, O., M. Thome, P. Schneider, N. Holler, J. Tschopp, D.W. Nicholson, C. Briand, and M.G. Grutter. 2002. The long form of FLIP is an activator of caspase-8 at the Fas death- inducing signaling complex. J. Biol. Chem. 277:45162–45171. [DOI] [PubMed] [Google Scholar]
- Mikula, M., M. Schreiber, Z. Husak, L. Kucerova, J. Ruth, R. Wieser, K. Zatloukal, H. Beug, E.F. Wagner, and M. Baccarini. 2001. Embryonic lethality and fetal liver apoptosis in mice lacking the c-raf-1 gene. EMBO J. 20:1952–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mixter, P.F., J.Q. Russell, F.H. Durie, and R.C. Budd. 1995. Decreased CD4-CD8- TCR-alpha beta + cells in lpr/lpr mice lacking beta 2-microglobulin. J. Immunol. 154:2063–2074. [PubMed] [Google Scholar]
- O'Neill, E., L. Rushworth, M. Baccarini, and W. Kolch. 2004. Role of the kinase MST2 in suppression of apoptosis by the proto-oncogene product Raf-1. Science. 306:2267–2270. [DOI] [PubMed] [Google Scholar]
- Parlato, S., A.M. Giammarioli, M. Logozzi, F. Lozupone, P. Matarrese, F. Luciani, M. Falchi, W. Malorni, and S. Fais. 2000. CD95 (APO-1/Fas) linkage to the actin cytoskeleton through ezrin in human T lymphocytes: a novel regulatory mechanism of the CD95 apoptotic pathway. EMBO J. 19:5123–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter, M.E., and P.H. Krammer. 2003. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 10:26–35. [DOI] [PubMed] [Google Scholar]
- Schneider, E., G. Moreau, A. Arnould, F. Vasseur, N. Khodabaccus, M. Dy, and S. Ezine. 1999. Increased fetal and extramedullary hematopoiesis in Fas-deficient C57BL/6-lpr/lpr mice. Blood. 94:2613–2621. [PubMed] [Google Scholar]
- Siegel, R.M., J.K. Frederiksen, D.A. Zacharias, F.K. Chan, M. Johnson, D. Lynch, R.Y. Tsien, and M.J. Lenardo. 2000. Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science. 288:2354–2357. [DOI] [PubMed] [Google Scholar]
- Takahashi, T., M. Tanaka, C.I. Brannan, N.A. Jenkins, N.G. Copeland, T. Suda, and S. Nagata. 1994. Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell. 76:969–976. [DOI] [PubMed] [Google Scholar]
- Takeuchi, K., N. Sato, H. Kasahara, N. Funayama, A. Nagafuchi, S. Yonemura, and S. Tsukita. 1994. Perturbation of cell adhesion and microvilli formation by antisense oligonucleotides to ERM family members. J. Cell Biol. 125:1371–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada, T., and Y. Nakanuma. 1995. Detection of apoptosis and expression of apoptosis-related proteins during human intrahepatic bile duct development. Am. J. Pathol. 146:67–74. [PMC free article] [PubMed] [Google Scholar]
- Van Parijs, L., D.A. Peterson, and A.K. Abbas. 1998. The Fas/Fas ligand pathway and Bcl-2 regulate T cell responses to model self and foreign antigens. Immunity. 8:265–274. [DOI] [PubMed] [Google Scholar]
- Watanabe-Fukunaga, R., C.I. Brannan, N.G. Copeland, N.A. Jenkins, and S. Nagata. 1992. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 356:314–317. [DOI] [PubMed] [Google Scholar]
- Yamaguchi, O., T. Watanabe, K. Nishida, K. Kashiwase, Y. Higuchi, T. Takeda, S. Hikoso, S. Hirotani, M. Asahi, M. Taniike, et al. 2004. Cardiac-specific disruption of the c-raf-1 gene induces cardiac dysfunction and apoptosis. J. Clin. Invest. 114:937–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L., W. Wang, Y. Hayashi, J.V. Jester, D.E. Birk, M. Gao, C.-Y. Liu, W.W.-Y. Kao, M. Karin, and Y. Xia. 2003. A role for MEK kinase 1 in TGF-{beta}/activin-induced epithelium movement and embryonic eyelid closure. EMBO J. 22:4443–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]