

Abstract
Emery-Dreifuss muscular dystrophy can be caused by mutations in the nuclear envelope proteins lamin A/C and emerin. We recently demonstrated that A-type lamin-deficient cells have impaired nuclear mechanics and altered mechanotransduction, suggesting two potential disease mechanisms (Lammerding, J., P.C. Schulze, T. Takahashi, S. Kozlov, T. Sullivan, R.D. Kamm, C.L. Stewart, and R.T. Lee. 2004. J. Clin. Invest. 113:370–378). Here, we examined the function of emerin on nuclear mechanics and strain-induced signaling. Emerin-deficient mouse embryo fibroblasts have abnormal nuclear shape, but in contrast to A-type lamin-deficient cells, exhibit nuclear deformations comparable to wild-type cells in cellular strain experiments, and the integrity of emerin-deficient nuclear envelopes appeared normal in a nuclear microinjection assay. Interestingly, expression of mechanosensitive genes in response to mechanical strain was impaired in emerin-deficient cells, and prolonged mechanical stimulation increased apoptosis in emerin-deficient cells. Thus, emerin-deficient mouse embryo fibroblasts have apparently normal nuclear mechanics but impaired expression of mechanosensitive genes in response to strain, suggesting that emerin mutations may act through altered transcriptional regulation and not by increasing nuclear fragility.
Introduction
Emerin is a small (34 kD) nuclear envelope protein with a single transmembrane domain spanning the inner nuclear membrane and a large nucleoplasmic domain that can interact with other nuclear envelope proteins such as lamins (Bengtsson and Wilson, 2004). Emerin is not essential for cell viability, but it contributes to shared vital functions, including nuclear assembly and cell cycle progression. Emerin is encoded by the EMD gene (also known as STA) located on the X-chromosome (Bione et al., 1994). Mutations in this gene can cause the X-linked recessive form of Emery-Dreifuss muscular dystrophy (EDMD), a disease characterized by early onset in childhood with slowly progressive skeletal muscle wasting, contractures of the elbow, neck, and Achilles tendons, a rigid spine, abnormal heart rhythms, heart block, and cardiomyopathy leading to increased risk of cardiac arrest (Bione et al., 1994). The majority of the disease-causing mutations are nonsense mutations that produce soluble forms of the protein, leading to loss of emerin from the nuclear periphery and mislocalization of emerin to the cytoplasm (Ellis et al., 1998; Vaughan et al., 2001). Although X-linked EDMD is associated with a mutation in the EMD gene, the autosomal dominant form of EDMD is caused by missense mutations in the LMNA gene that encodes the nuclear A-type lamins (Bonne et al., 1999). These mutations often result in misfolding or failure of the A-type lamins to correctly assemble, leading to partial or complete loss of function (Burke and Stewart, 2002). Historically, X-linked EDMD was the first disorder to be recognized as a nuclear membrane disease and opened a new area of research on the role of the nuclear envelope in disease. Currently, more than 180 mutations have been identified in the LMNA and EMD genes that are causally linked to at least 10 distinct diseases, including EDMD, dilated cardiomyopathy, familial partial lipodystrophy, and Hutchinson Gilford progeria syndrome (Bonne et al., 1999; Fatkin et al., 1999; Cao and Hegele, 2000; Shackleton et al., 2000; Burke and Stewart, 2002; De Sandre-Giovannoli et al., 2002, 2003; Eriksson et al., 2003).
Although emerin and A-type lamins (predominantly lamins A and/or C) are expressed in most human tissues, EDMD predominantly affects skeletal and cardiac muscles and tendons. The reason for this tissue-specific phenotype is not yet clear, but two alternative hypotheses for the disease mechanism have emerged. The “structural hypothesis” suggests that mutations in genes encoding emerin or A-type lamins lead to increased nuclear fragility and to eventual nuclear disruption in mechanically strained tissues, whereas the “gene regulation hypothesis” is based on the findings that lamin A/C and emerin can bind to a variety of transcriptional regulators that could exert tissue-specific effects. Lamins A/C are a major component of the nuclear lamina, and loss of A-type lamins leads to impaired nuclear mechanics and increased nuclear fragility (Broers et al., 2004; Lammerding et al., 2004). Emerin binds to several structural proteins such as lamin A/C, lamin B, nesprin-1α/2, and nuclear actin, and has recently been demonstrated to promote actin polymerization in vitro (Mislow et al., 2002; Bengtsson and Wilson, 2004; Holaska et al., 2004; Zhang et al., 2005). Loss of emerin from the nuclear envelope could thus interfere with the normal function of these proteins and lead to nuclear structural abnormalities. At the same time, emerin can bind to the transcriptional repressors barrier-to-autointegration factor (BAF), germ cell-less (GCL), and Btf, and to the splicing factor YT521-B, suggesting an important role in gene regulation (Nili et al., 2001; Holaska et al., 2003; Wilkinson et al., 2003; Bengtsson and Wilson, 2004; Haraguchi et al., 2004). The structural hypothesis and the gene regulation hypothesis are not mutually exclusive, and in fact A-type lamin-deficient cells have increased nuclear fragility and abnormal nuclear mechanics as well as impaired signaling responses to mechanical strain or cytokine stimulation, indicating that tissue-specific effects observed in laminopathies could arise from varied degrees of impaired nuclear mechanics and transcriptional activation (Lammerding et al., 2004).
Here, we report independent measures of the structural and gene-regulatory functions of emerin-deficient, A-type lamin-deficient, and wild-type mouse embryo fibroblasts to explore the specific function of emerin on nuclear mechanics and gene regulation. We show that, in contrast to A-type lamin-deficient cells, emerin-deficient fibroblasts have apparently normal nuclear mechanics but display similar, although less profound, deficiencies in strain-induced gene regulation, leading to an increased rate of apoptosis in response to mechanical strain. These data suggest that emerin-associated laminopathies are predominantly caused by an impaired signaling response and not through direct strain-induced injury to the nuclear membrane.
Results
Emerin null fibroblasts
Emerin-deficient mouse embryo fibroblasts were derived from male emerin hemizygous mice (Emd − /y) and have been shown to lack emerin expression (unpublished data). Emerin-deficient mice were derived by deletion of the entire coding region of the Emd gene, which resulted in the complete absence of emerin protein as shown by Western and immunohistochemical analysis of muscle fibroblasts isolated from the null mice (Fig. 1). The Emd null mice (both −/y males and −/− females) showed no overt pathology in either their skeletal or cardiac muscle, even in mice as old as 12–15 mo (unpublished data). Hereafter we have focused our studies on analyzing Emd − /y fibroblasts derived from male embryos to avoid possible heterogeneity due to X-chromosome–associated gene dosage compensation effects in cell lines of female origin.
Figure 1.
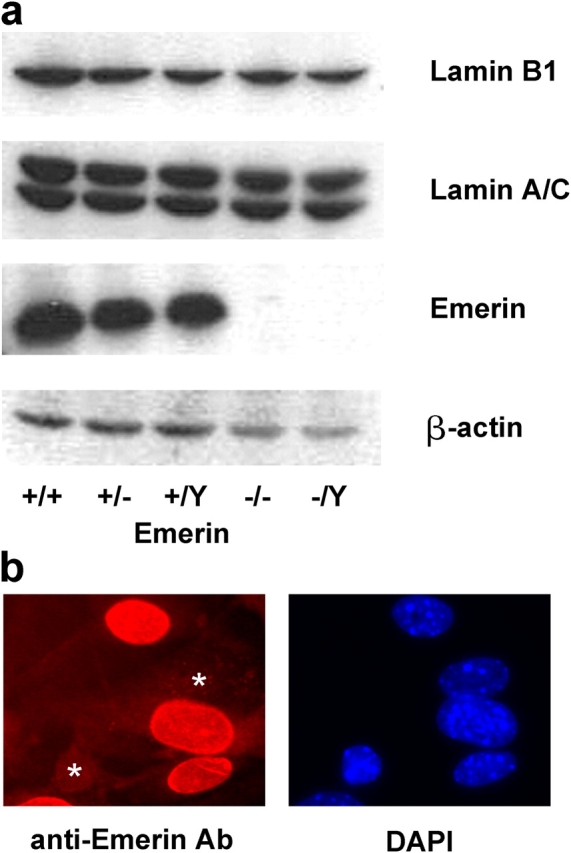
Characterization of emerin null fibroblasts. (a) Western analysis of homozygous (−/− and −/Y) and heterozygous (+/−) emerin null and wild-type (+/+ and +/Y) adult muscle fibroblasts, showing a complete lack of emerin in the homozygous cells. Protein expression of lamin A/C and lamin B1 appeared normal in the emerin null homozygous and heterozygous cells. (b) Female heterozygous (i.e., Emd +/−) muscle fibroblasts stained with anti-emerin antibody (left) and DAPI (right). Two cells (*) clearly lack emerin staining (as visualized by comparing with the DAPI staining), reflecting the differential X-chromosome inactivation in the heterozygous cell population.
Nuclear shape
Fibroblasts derived from EDMD patients often have irregularly shaped nuclei and show blebbing of the nuclear membrane (Fidzianska et al., 1998; Fidzianska and Hausmanowa-Petrusewicz, 2003). Emerin-deficient mouse embryo fibroblasts had a significantly increased fraction of abnormally shaped nuclei (Fig. 2, a and b) and nuclei with membrane and chromatin protrusions (nuclear blebs) when compared with wild-type fibroblasts. These nuclear shape abnormalities were less pronounced compared with those of A-type lamin-deficient cells (Fig. 2 b). To assess the degree of irregular nuclear shape more quantitatively, we measured nuclear cross-sectional area and perimeter of Hoechst 33342 stained nuclei and computed the nuclear contour ratio (4π × area/perimeter2), which yields a quantitative measure of nuclear roundness. For a circular shape that maximizes the area-to-perimeter ratio the contour ratio has a value of 1, whereas more convoluted outlines lead to smaller values. We found that emerin-deficient cells had a significantly lower mean contour ratio compared with wild-type cells (Fig. 2 c), and the frequency distribution showed that the overall distribution was shifted toward lower values of the contour ratio, indicating a higher prevalence of abnormally shaped nuclei (Fig. 2 d). A-type lamin-deficient cells had an even lower mean contour ratio, with very few cells reaching normal values (∼0.90) and a much wider distribution overall, indicating that both the frequency as well as the extent of irregular nuclear shape is more severe in A-type lamin-deficient cells compared with wild-type and emerin-deficient cells (Fig. 2 d). These findings were confirmed when analyzing the nuclear length/width ratio (unpublished data) that was close to unity for wild-type cells and significantly lower for emerin and especially for A-type lamin-deficient fibroblasts (unpublished data). Fourier shape analysis of nuclear cross-sectional outlines (Diaz et al., 1989) revealed that both emerin and A-type lamin-deficient cells had a significantly higher contribution of higher order harmonics to the nuclear shape, again confirming that these cells have a more irregular nuclear shape compared with wild-type cells (ratio of 1st order harmonics to higher order harmonics was 11.9 ± 0.08 for wild-type, 11.3 ± 0.11 for emerin-deficient, and 7.4 ± 0.07 for A-type lamin-deficient cells; P < 0.001 for wild-type vs. emerin and A-type lamin-deficient cells).
Figure 2.

Emerin and A-type lamin-deficient fibroblasts have abnormal nuclear shape. (a) Fluorescently labeled nuclei of emerin-deficient mouse embryo fibroblasts. Asterisk denotes nucleus with chromatin protruding from the nucleus (nuclear bleb). Arrows indicate nuclei that have mild deviations from the typical round shape. Bar, 20 μm. (b) The fraction of abnormally shaped nuclei and nuclear blebs was significantly increased in emerin and A-type lamin-deficient fibroblasts, with emerin-deficient cells displaying a milder phenotype (cell fractions with abnormal nuclear shape were 0.077 ± 0.006 for wild-type, 0.163 ± 0.009 for emerin-deficient, and 0.378 ± 0.011 for A-type lamin-deficient cells, P < 0.0001 for emerin-deficient and A-type lamin-deficient cells compared with wild-type cells; cell fractions with nuclear blebs were 0.012 ± 0.002, 0.042 ± 0.005, and 0.093 ± 0.006 for wild-type, emerin-deficient, and A-type lamin-deficient cells, respectively, P < 0.0001 for emerin-deficient and A-type lamin-deficient cells compared with wild-type cells; n ∼1,800 for each cell type). (c) Emerin-deficient cells have a significantly decreased contour ratio compared with wild-type cells, but display a milder phenotype compared with A-type lamin-deficient cells (contour ratio = 0.89 ± 0.004 for wild-type, 0.86 ± 0.006 for emerin-deficient, and 0.80 ± 0.007 for A-type lamin-deficient cells, P < 0.001 for emerin-deficient and A-type lamin-deficient cells compared with wild-type; n ∼1500 for each cell type in three independent experiments). (d) Relative frequency distribution of the contour ratio for wild-type, emerin-deficient, and A-type lamin-deficient cells (median values were 0.896, 0.869, and 0.817 for wild-type, emerin-deficient, and A-type lamin-deficient cells, respectively). The difference in the medians was statistically significant (P < 0.001 for emerin-deficient and A-type lamin-deficient vs. wild-type). (e) Emerin and A-type lamin-deficient cells have significantly increased nuclear cross-sectional areas compared with wild-type cells (Nuclear cross-sectional area = 178 ± 4.5 μm2, 252 ± 24.7 μm2, and 259 ± 25.6 μm2 for wild-type, emerin-deficient, and A-type lamin-deficient cells, respectively, P < 0.01 for emerin and A-type lamin-deficient compared with wild-type cells, n ∼1500 for each cell type in three independent experiments). (f) Frequency distribution of the cross-sectional area for wild-type, emerin-deficient, and A-type lamin-deficient cells.
Interestingly, we found that immortalized emerin and A-type lamin-deficient mouse embryo fibroblasts had significantly increased nuclear cross-sectional areas compared with immortalized wild-type cells (Fig. 2, e and f), and DNA content analysis by flow cytometry revealed that the immortalized A-type lamin and emerin-deficient cells had a significantly higher fraction of polyploidal nuclei compared with immortalized wild-type cells. This could be an indication of failure to correctly separate chromatin during mitosis due to abnormal nuclear envelope breakdown or subsequent reassembly. However, DNA content distribution appeared normal in primary fibroblasts, and thus it is possible that these problems in nuclear envelope dynamics only become apparent in the more rapidly proliferating immortalized cells or are an effect of the immortalization process.
Nuclear dynamics
Liu et al. (2000) previously demonstrated that nuclei of lamin-deficient Caenorhabditis elegans cells display significant shape changes over time. To assess dynamic changes in nuclear shape in A-type lamin-deficient and emerin-deficient mouse embryo fibroblasts, we analyzed nuclear shape stability using time-lapse imaging. Phase-contrast images were acquired every 5 min over an 8 h, 20 min period of time for a total of 100 frames. Nuclear motion and deformation were quantified by tracking individual nucleoli within a given nucleus over time (Fig. 3 a) and subsequently computing the translation, rotation, and deformation from these measurements. Cells that underwent mitosis during the observation period were excluded from the analysis. We defined the nuclear deformation as the average deviation from a linear affine transformation, i.e., a change in geometry that can be reduced to a combination of translation, rotation, and scaling in which relative positions to each other are maintained.
Figure 3.
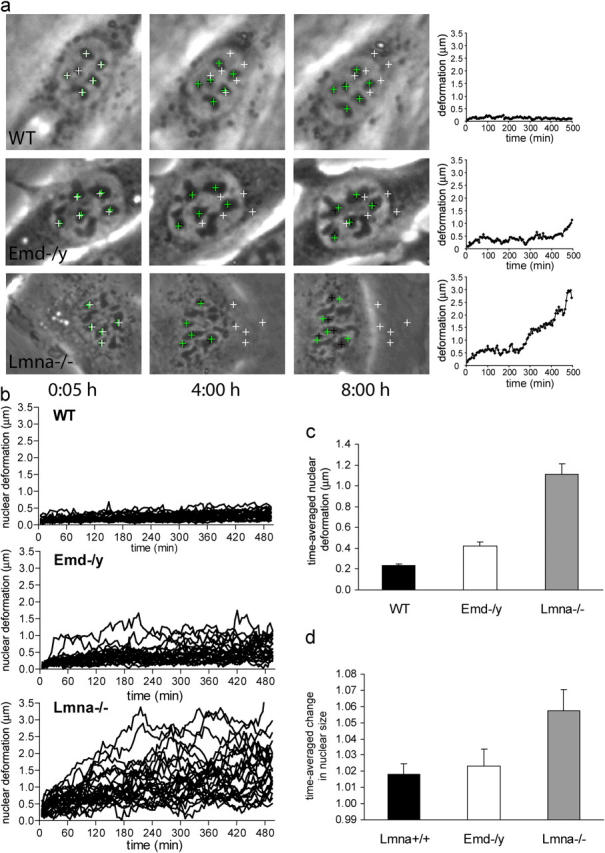
Emerin and A-type lamin-deficient cells have increased nuclear dynamics and decreased nuclear shape stability. (a) Time-lapse series of fibroblasts over an 8 h, 20 min time period. Images shown were acquired at 5 min, 4 h, and 8 h. Wild-type nuclei (top row) appear very stable over time and have only minor deformations, whereas A-type lamin-deficient nuclei (bottom row) show large nuclear deformations over time. Emerin-deficient nuclei (center row) display an intermediate phenotype, with some nuclei appearing very stable and other nuclei undergoing larger deformations. White crosses denote initial positions of nucleoli, green crosses positions according to the least-square fit assuming linear affine transformations (see Materials and methods), and black crosses the actual nucleoli centroid positions. Deviations between the black and green positions indicate nuclear deformations independent of translation, rotation, or uniform changes in nuclear size. Plots on the right show the average deviation between the actual nucleoli positions and the least-square fit. Time-lapse videos are available online at http://www.jcb.org/cgi/content/full/jcb.200502148/DC1. (b) Time courses of the nuclear deformations for wild-type (top), emerin-deficient (center), and A-type lamin-deficient (bottom) fibroblasts, 25 nuclei each. (c) Emerin-deficient cells have significantly increased time-averaged nuclear deformations compared with wild-type cells, but to a much lesser extent than A-type lamin-deficient cells (time-averaged nuclear deformation = 0.23 ± 0.015 μm, 0.42 ± 0.038 μm, and 1.11 ± 0.104 μm for wild-type, emerin-deficient, and A-type lamin-deficient cells, respectively; P < 0.0001 for emerin and A-type lamin-deficient vs. wild-type cells). (d) A-type lamin-deficient cells have significantly increased time-averaged nuclear size changes compared with wild-type and emerin-deficient cells (time-averaged normalized size change = 1.018 ± 0.006, 1.023 ± 0.010, and 1.058 ± 0.013 for wild-type, emerin-deficient, and A-type lamin-deficient cells, respectively; P < 0.01 for A-type lamin-deficient vs. wild-type cells).
In wild-type cells, 88 ± 3.7% of the nuclei retained their initial shape throughout the observation period, resulting in only small nuclear deformations over time (Fig. 3 b). In contrast, only 4 ± 3.7% of A-type lamin-deficient nuclei appeared stable while the others displayed large deformations over time, with the deviation from the initial shape increasing rapidly over time (Fig. 3, a and b). Emerin-deficient cells displayed an intermediate phenotype, with 57 ± 11.9% maintaining their shape over time while some cells exhibited large nuclear deformations over time (Fig. 3 b), but rarely to the same degree as the A-type lamin-deficient cells. Comparing the time-averaged nuclear deformations, we found that emerin-deficient cells had significantly increased deformations compared with wild-type cells, but to a much lesser extent than A-type lamin-deficient nuclei (Fig. 3 c). Interestingly, A-type lamin-deficient cells had a significant increase in relative nuclear size compared with wild-type cells, whereas emerin-deficient nuclei had size increases comparable to wild-type cells (Fig. 3 d).
Nuclear mechanics
The nuclear shape changes observed in the time-lapse experiments can be caused by intracellular forces exerted from the cytoskeleton onto the nucleus, from intranuclear processes such as DNA synthesis and chromatin remodeling, or from nuclear envelope dynamics (remodeling) over the 8 h 20 min observation time. To explore the role of emerin on nuclear mechanics independently of intranuclear and cytoskeletal changes that occur over a relatively long time scale, cells were cultured on transparent silicone membranes and subjected to ∼5% biaxial strain. The applied membrane strain is transmitted to the cytoskeleton through membrane receptors such as integrins, resulting in intracellular forces applied to the nucleus (Maniotis et al., 1997; Caille et al., 1998). The induced nuclear deformations were calculated by tracking distinct features in the fluorescently labeled chromatin and normalized to membrane strain to compensate for small variations in the applied membrane strain. This method of strain induction allows quantitative measurements of nuclear stiffness compared with cytoskeletal stiffness in living cells without having to isolate the nuclei (Caille et al., 1998; Lammerding et al., 2004). In wild-type cells, the nucleus is much stiffer than the surrounding cytoskeleton and showed only minor deformations under strain (Fig. 4 a). Emerin-deficient nuclei exhibited deformations comparable to those of wild-type cells, indicating apparently normal nuclear mechanics (Fig. 4 a). In contrast, A-type lamin-deficient nuclei had significantly larger deformations compared with wild-type cells with increased normalized nuclear strain (Fig. 4 a). Experiments were performed in primary and immortalized mouse embryo fibroblasts, and we observed the same trend in all cell lines (data shown are for primary cells), with no significant difference between emerin-deficient and wild-type cells and A-type lamin-deficient cells showing significantly larger deformations.
Figure 4.
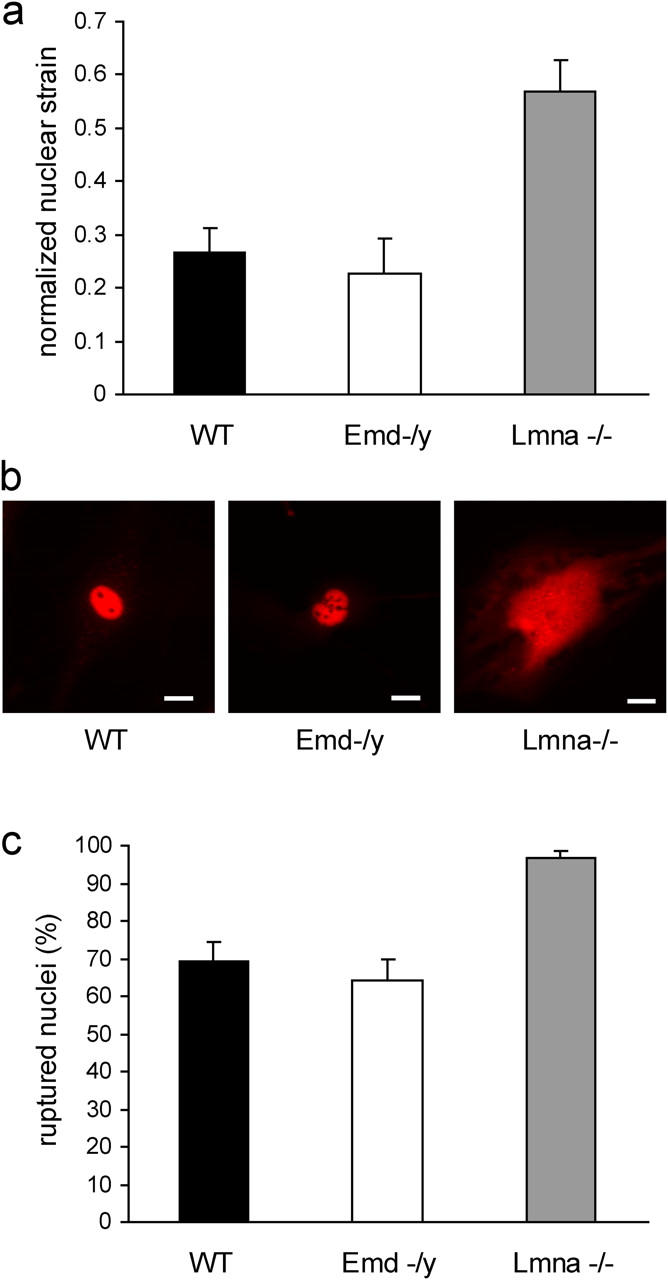
Emerin-deficient cells have apparently normal nuclear mechanics. (a) Emerin-deficient primary mouse embryo fibroblasts have normalized nuclear strain comparable to wild-type cells when subjected to biaxial strain. In contrast, A-type lamin-deficient cells have significantly increased normalized nuclear strain (normalized nuclear strain = 0.27 ± 0.044, 0.23 ± 0.066, and 0.57 ± 0.059 for wild-type, emerin-deficient, and A-type lamin-deficient cells respectively; P < 0.001 for A-type lamin-deficient vs. wild-type cells). (b) Wild-type (left) and emerin-deficient (center) nuclei remain intact when microinjected with fluorescently labeled dextran, whereas A-type lamin-deficient nuclei (right) have more fragile nuclei that allow dextran to leak into the cytoplasm. Bars, 20 μm. (c) Emerin-deficient and wild-type cells have comparable frequency of nuclear rupture for nuclear microinjection at 500 hPa, whereas A-type lamin-deficient cells have a significantly increased fraction of ruptured nuclei (percentage of ruptured nuclei = 69 ± 4.9%, 64 ± 5.5%, and 96 ± 1.9% for wild-type, emerin-deficient, and A-type lamin-deficient cells, respectively).
Strain-induced damage to a mechanically impaired nucleus could provide one explanation for the tissue-specific effects of EDMD. To examine if the observed changes in nuclear stiffness correlate with increased nuclear envelope fragility, we monitored the subcellular localization of fluorescently labeled 70-kD dextran microinjected into emerin-deficient, A-type lamin-deficient, or wild-type fibroblasts. The high molecular weight dextran cannot cross the intact nuclear envelope and is thus excluded from the nucleus when injected into the cytoplasm and retained in the intact nucleus in the case of nuclear injection (Fig. 4 b). We recently demonstrated that the fraction of intact nuclei decreases with increasing injection pressure (Lammerding et al., 2004) and that nuclear microinjection can be effectively used to measure nuclear rupture strength. In this assay, emerin-deficient cells had a similar fraction of ruptured nuclei compared with wild-type cells (Fig. 4 c), again indicating apparently normal nuclear mechanics. In contrast, microinjected A-type lamin-deficient nuclei displayed severely compromised nuclear integrity, and fluorescently labeled dextran escaped into the cytoplasm in a significantly larger fraction of cells. At sufficiently high injection pressures, all nuclei ruptured (unpublished data). Injection of the 70-kD dextran into the cytoplasm showed that dextran was excluded from the nucleus in all three cell types, indicating that nuclear integrity under resting conditions was not impaired in A-type lamin or emerin-deficient cells (unpublished data).
To evaluate the effect of emerin deficiency on cell survival in response to prolonged mechanical stimulation, mouse embryo fibroblasts were subjected to 24 h cyclic biaxial strain (1 Hz; 3, 5, or 10% strain). Total cell death and apoptosis were subsequently measured by propidium iodide uptake and DNA content analysis, respectively. Early apoptotic events can be detected by a characteristic pattern of DNA strand breaks through endonucleolysis, resulting in increased amounts of DNA fragments that are visible by flow cytometry as the sub-G1 phase in the DNA content distribution (Walker et al., 1993). Strain application at the two lowest settings (3 and 5% biaxial strain) had no significant effect on cell death or apoptosis in either cell line, but at the highest strain rate (10% biaxial strain), A-type lamin-deficient fibroblasts had a significantly increased fraction of dead cells compared with nonstretched controls and to strained wild-type cells (Fig. 5 a). Total cell death in emerin-deficient cells was not significantly increased compared with nonstretched controls and to wild-type cells. DNA content analysis of samples from the same experiments revealed a large increase in apoptotic cell fraction in the A-type lamin-deficient cells compared with nonstretched controls and strained wild-type cells. Interestingly, we also found a significantly increased apoptotic cell fraction in the emerin-deficient fibroblasts (Fig. 5 b). Baseline levels of apoptotic cells were comparable for emerin-deficient and wild-type cells, but significantly elevated in the A-type lamin-deficient cells.
Figure 5.
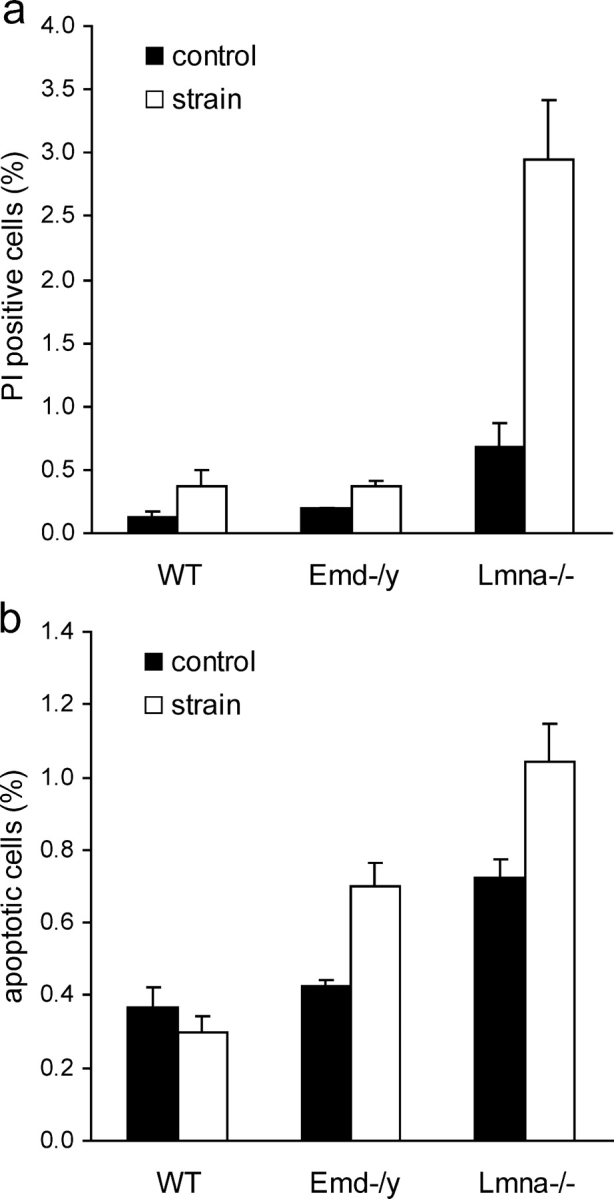
Emerin and A-type lamin-deficient cells are more sensitive to mechanical strain. (a) A-type lamin-deficient cells have a significantly increased number of propidium iodide–positive cells after 24 h of cyclic, 10% biaxial strain application. Emerin-deficient cells are not significantly different from wild-type cells. (Percentage of propidium iodide–positive cells at rest vs. after 24 h strain application = 0.13 ± 0.03% vs. 0.38 ± 0.12%, 0.2 ± 0.00% vs. 0.37 ± 0.04%, and 0.68 ± 0.18% vs. 2.94 ± 0.47% for wild-type, emerin-deficient, and A-type lamin-deficient cells respectively, P < 0.01 for wild-type vs. A-type lamin-deficient strained cells; P < 0.05 for wild-type vs. A-type lamin-deficient controls). (b) Emerin and A-type lamin-deficient cells have increased fractions of apoptotic cells after 24 h of cyclic, 10% biaxial strain application (percentage of apoptotic cells at rest vs. after 24 h of strain application = 0.37 ± 0.05% vs. 0.30 ± 0.04% for wild-type, 0.43 ± 0.02% vs. 0.70 ± 0.06% for emerin-deficient, and 0.72 ± 0.05% vs. 1.04 ± 0.11% for A-type lamin-deficient cells; P < 0.001 for strained emerin and A-type lamin-deficient vs. wild-type cells).
Impaired cellular signaling in response to mechanical stimulation (mechanotransduction) can lead to an altered physiological response and to potentially increased apoptosis in mechanically strained tissue. A-type lamin-deficient cells have impaired mechanotransduction signaling in vivo and in vitro (Lammerding et al., 2004; Nikolova et al., 2004), and insufficient anti-apoptotic signaling could provide one explanation for the increased apoptotic cell fractions in A-type lamin and emerin-deficient cells seen in the 24-h strain experiments. In wild-type cells, expression of the mechanosensitive gene egr-1 and the anti-apoptotic gene iex-1 is rapidly up-regulated in response to mechanical stimulation (Sadoshima et al., 1992; Morawietz et al., 1999; De Keulenaer et al., 2002). To evaluate whether transcriptional activation for these genes was altered in A-type lamin or emerin-deficient cells, fibroblasts plated on fibronectin-coated silicone membranes were subjected to biaxial cyclic strain (4%, 1 Hz) and mRNA levels were subsequently analyzed using Northern analysis and real-time PCR. We found that, in addition to the previously established mechanotransduction deficiency in A-type lamin-deficient cells, emerin-deficient cells also exhibited an attenuated mechanotransduction response. Expression of egr-1 and iex-1 in response to mechanical stimulation was impaired in both A-type lamin and emerin-deficient cells at 2 and 4 h of 4% biaxial cyclic strain application (Fig. 6 a). In contrast, the mechanically unresponsive gene glyceraldehyde 3-phosphate dehydrogenase (gapdh) showed no changes in expression in any cell types, suggesting that transcription was impaired in a specific manner. Impaired mechanosensitive expression of iex-1 in A-type lamin and emerin-deficient cells was confirmed by real-time PCR (Fig. 6 b). Expression levels of iex-1 at baseline (nonstimulated cells) were not significantly different between cell types.
Figure 6.

Emerin and A-type lamin-deficient cells have impaired mechanotransduction. (a) Expression of the mechanosensitive genes iex-1 and egr-1 in response to mechanical strain is drastically reduced in emerin and A-type lamin-deficient fibroblasts compared with wild-type cells. Expression of the nonmechanically inducible gene gapdh is not significantly altered. The elevated baseline expression of iex-1 and egr-1 seen in the emerin-deficient cells in this Northern blot are not representative, and real-time PCR analysis didn't reveal any significant differences in baseline expression between cell types. (b) Real-time PCR analysis confirms the impaired induction of iex-1 in response to strain in emerin and A-type lamin-deficient cells. Results are normalized to β-tubulin expression and presented as percent induction of baseline levels (induction at 2 h: 270 ± 87.1%, 143 ± 41.4%, and 104 ± 35.9% for wild-type, emerin-deficient, and A-type lamin-deficient cells, respectively, P < 0.05 for wild-type vs. A-type lamin-deficient; induction at 4 h: 244 ± 54.8%, 95 ± 20.3%, and 116 ± 39.8% for wild-type, emerin-deficient, and A-type lamin-deficient cells respectively, P < 0.05 for wild-type vs. emerin-deficient and wild-type vs. A-type lamin-deficient cells; differences in baseline expression were not statistically significant between any cell type).
NF-κB signaling
iex-1 is an NF-κB–dependent survival gene, and we recently demonstrated that NF-κB signaling in response to strain or cytokine stimulation is impaired in A-type lamin-deficient cells (Lammerding et al., 2004). To test if this signaling pathway was also responsible for the impaired mechanotransduction observed in the emerin-deficient cells, we measured NF-κB–regulated luciferase activity in response to cytokine IL-1β stimulation in emerin-deficient and wild-type fibroblasts. Normalized NF-κB luciferase activity at baseline versus after 12 h of IL-1β stimulation was 0.32 ± 0.04 vs. 0.83 ± 0.10 for wild-type and 0.38 ± 0.08 vs. 0.86 ± 0.21 for emerin-deficient cells (n = 9 for each group in three independent experiments), yielding no significant difference between cell types. Consistent with this negative finding, IL-1β–induced expression of NF-κB was not significantly different between wild-type and emerin-deficient cells (unpublished data). Although these experiments do not represent a comprehensive study of the NF-κB signaling pathway, these results suggest that NF-κB signaling is not responsible for the observed impaired mechanotransduction and that emerin may exert its effect through alternative signaling pathways.
Discussion
In this work, we conducted a quantitative comparison between wild-type, emerin-deficient, and A-type lamin-deficient mouse embryo fibroblasts using previously established (Lammerding et al., 2004) and novel experimental methods to independently measure structural and gene-regulatory function of these nuclear envelope proteins. We found that emerin-deficient cells, unlike A-type lamin-deficient cells, had apparently normal nuclear mechanics evaluated by nuclear strain and microinjection experiments, despite their obviously abnormal nuclear architecture. Although it is possible that the sensitivity of our assay was insufficient to detect small differences in nuclear mechanics between emerin-deficient cells and wild-type cells, the data strongly suggest that the effect of A-type lamin deficiency on nuclear mechanics is much greater than the effect of emerin deficiency. The importance of A-type lamins on nuclear structure and mechanics has been demonstrated in vivo (Nikolova et al., 2004) and in vitro (Broers et al., 2004; Lammerding et al., 2004), but the structural function of emerin is still unclear. Emerin binds to a variety of structural proteins such as lamin A, actin, and nesprin-1α/2α/2β (Mislow et al., 2002; Bengtsson and Wilson, 2004; Holaska et al., 2004; Libotte et al., 2005; Zhang et al., 2005). Nuclei from muscle biopsies of patients suffering from X-linked EDMD are characterized by breakdown of the nuclear membrane and the presence of chromatin blebs extruding from the nucleus, whereas nuclei from patients with the autosomal dominant form typically show abnormal distribution of the nuclear matrix and focal loss of chromatin from the nuclear envelope and interior (Fidzianska and Hausmanowa-Petrusewicz, 2003). Several nesprin isoforms (nesprin 1α, 2α, 2β) bind to emerin and are necessary to anchor emerin at the inner nuclear membrane (Libotte et al., 2005; Zhang et al., 2005). Nesprin isoforms containing actin-binding domains are thought to physically connect the nucleus to the cytoskeleton and play an important role in anchoring muscle nuclei at the neuromuscular junction (Starr and Han, 2002, 2003; Padmakumar et al., 2004; Grady et al., 2005; Zhang et al., 2005). Furthermore, emerin can directly bind and stabilize the pointed ends of F-actin in vitro, suggesting that emerin may stabilize actin polymers at the nuclear envelope (Holaska et al., 2004). At the same time, the nucleoplasmic domain of emerin has dominant effects in Xenopus nuclear assembly extracts and overexpression of the full nucleoplasmic domain of emerin extends the mammalian cell cycle by 7 h (Fairley et al., 2002), supporting emerin's involvement in nuclear assembly and cell cycle regulation. Our experimental findings suggest that loss of emerin does not directly affect large scale nuclear mechanics such as overall nuclear stiffness and fragility. Instead, emerin could predominantly affect nuclear envelope assembly and organization at a smaller length scale that is not apparent in global measurements of nuclear strain and rupture. This dynamic and local effect is supported by the more variable nuclear shape of emerin-deficient cells observed in our time-lapse sequences.
In contrast to the apparently normal nuclear mechanics, mechanosensitive gene regulation was deficient in emerin-deficient cells, resulting in a significantly increased rate of apoptosis in response to mechanical stimulation. The absence of changes in total cell death (propidium iodide uptake) in these cells could be due to the fact that the DNA contents assay measures early apoptotic chromatin breakdown while loss of plasma membrane integrity that is required for propidium iodide uptake occurs at a much later stage in apoptosis. Consistent with the apparently normal nuclear mechanics and the impaired activation of anti-apoptotic genes in response to mechanical stimulation, our findings indicate that emerin-deficient cells are more susceptible to strain-induced apoptosis, whereas both necrosis and apoptosis contribute to mechanically induced cell death in A-type lamin-deficient cells. Although the current study was narrowed to two representative mechanosensitive genes (egr-1 and iex-1) whose activation has previously been reported to be impaired in A-type lamin-deficient cells (Lammerding et al., 2004), we expect that the role of emerin (and A-type lamins) is not limited to these particular genes, and a more comprehensive gene array analysis of mechanically stimulated cells will provide more insight into the extent of lamin/emerin-dependent mechanotransduction.
The molecular mechanism that is responsible for the observed mechanotransduction deficiency in A-type lamin and emerin-deficient cells is not clear. We previously reported impaired NF-κB–regulated transcriptional activation in A-type lamin-deficient cells, but emerin-deficient cells had apparently normal NF-κB signaling, indicating that alternative pathways are responsible for the observed mechanotransduction deficiencies. Emerin directly binds to the transcriptional regulators BAF, Btf, GCL, and YT521-B (Nili et al., 2001; Holaska et al., 2003; Wilkinson et al., 2003; Haraguchi et al., 2004). Btf is a transcription repressor that induces cell death when overexpressed (Kasof et al., 1999). Emerin is cleaved during apoptosis in proliferating mouse myoblasts and differentiating myotubes, and emerin may have an anti-apoptotic effect by sequestering Btf (Columbaro et al., 2001). This anti-apoptotic role of emerin is consistent with the increased rate of apoptosis observed in emerin-deficient cells after strain application. GCL is a repressor of E2F-DP–regulated genes. Emerin cannot bind BAF and GCL simultaneously, and presence of BAF at the nuclear envelope might inhibit GCL binding to emerin in vivo (Bengtsson and Wilson, 2004). YT521-B is involved in determining sites for alternate mRNA splicing, and emerin influences splice site selection by YT521-B (Wilkinson et al., 2003). The role of emerin as a modulator for transcriptional regulation is further supported by DNA microarray analysis of fibroblasts from patients with the X-linked form of EDMD, which showed that at least 28 genes are specifically up-regulated in the emerin mutant cells (Tsukahara et al., 2002). The affected genes include both structural (lamin A/C, αII-spectrin, and filamin) and signal transduction–associated proteins such as protein phosphatase 2A (Tsukahara et al., 2002).
In addition to these direct interactions between emerin and transcriptional regulators, we cannot exclude the possibility that the impaired mechanotransduction response is caused by more indirect consequences of emerin deficiency. Abnormal nuclear shape and ultrastructure in emerin-deficient cells could affect force transmission from the cytoskeleton to the nucleus (Maniotis et al., 1997), and interaction of emerin with nuclear actin (Holaska et al., 2004) could affect both nuclear ultrastructure as well as transcription itself, as nuclear actin is emerging as a critical component of polymerase II transcription (Hofmann et al., 2004; Zhu et al., 2004).
In our experiments, emerin-deficient cells generally displayed a milder phenotype compared with the A-type lamin-deficient cells, including fewer and less extensive nuclear shape abnormalities, a less profound increase in apoptosis in response to strain, and less severe mechanotransduction deficiency. This observation is consistent with the milder phenotype found in emerin-deficient mice that don't display overt muscular dystrophy or cardiac problems but show signs of impaired muscle regeneration later on (unpublished data). Loss of function in lamin A/C mutants often leads to mislocalization of emerin from the nuclear envelope to the ER (Sullivan et al., 1999; Östlund et al., 2001; Raharjo et al., 2001; Muchir et al., 2003) and to a subsequent loss of normal emerin function at the nuclear envelope. Therefore, we expect that A-type lamin-deficient cells encompass phenotypes associated with emerin deficiency or loss of function. Loss of emerin, on the other hand, does not appear to grossly affect A-type lamin function, providing a possible explanation for the more severe phenotype in the A-type lamin-deficient cells compared with the emerin-deficient cells.
In conclusion, we found that A-type lamin and emerin deficiencies share common features such as abnormal nuclear shape and impaired mechanotransduction, but also selectively interfere with other structural and gene-regulatory functions. In the case of emerin deficiency, we found that emerin predominantly affects mechanosensitive gene regulation with only small effects on nuclear mechanics. By providing independent tests for measuring structural and gene-regulatory functions, our experiments can help clarify the effects of specific mutations in nuclear envelope proteins. Elucidating the molecular mechanisms will provide new insights into the specific disease mechanisms of X-linked recessive and autosomal dominant EDMD, and might eventually lead to new treatment courses.
Materials and methods
Cells
Emerin-deficient (Emd − / − and Emd −/ y) mouse embryo and muscle fibroblasts were derived from Emd null mice. Emd null mice were derived by deleting the entire coding region for the Emd gene, using standard gene targeting procedures in mouse ES cells (Joyner, 1999). Lmna + / +, Lmna − / −, and Emd − /y mouse embryo fibroblasts were maintained in DME (Invitrogen) containing 10% FCS (HyClone) and penicillin/streptomycin (Invitrogen). Experiments were performed on primary mouse embryo fibroblasts and on cells immortalized by repeated passage of primary cells. Unless otherwise specified, cells referred to in the text are immortalized mouse embryo fibroblasts.
Nuclear shape analysis
To quantify the overall fraction of irregularly shaped or blebbing nuclei, cells of various passages were incubated for 15 min with 1 μg/ml Hoechst 33342 (Molecular Probes) and washed with HBSS (Invitrogen). For each passage, fluorescence images of 100–500 randomly selected nuclei were acquired at 20× on a microscope (model IX-70; Olympus) using a CoolSNAP camera (Roper Scientific), and were stored for subsequent image analysis. Nuclei of Lmna + / +, Lmna − / −, and Emd -/y cells were scored as normal, irregularly shaped (deviation from an oval or spherical shape), or as nuclei with nuclear blebs. These determinations were made using an observer blinded for the genotype. To quantify the variation in nuclear morphology, we measured nuclear area and perimeter in midsections of the fluorescent nuclei using custom-written MATLAB software. From these data, we computed the nuclear roundness or contour ratio (4π × area/perimeter2) (Goldman et al., 2004). The contour ratio reaches a maximum value of 1 for a circle and decreases with increasingly convoluted nuclear shapes. Elliptic Fourier analysis of nuclear shape was performed as described previously (Diaz et al., 1989). Custom-written MATLAB software was used to automatically trace the outline of fluorescently labeled nuclei and to compute the first 20 elliptic harmonics. Each single elliptic harmonic can be geometrically visualized as a pair of orthogonal semiaxes, and we used the ratio of the sum of the first major and minor semiaxes over the sum of the higher order semiaxes as indicator of irregular nuclear shape, as the higher order harmonics represent deviations from a purely elliptical shape.
Time-lapse experiments
Cells were plated at subconfluent density in 35-mm polystyrene cell culture dishes (Corning). Cells were grown in DME + 10% FCS at 37°C for at least 24 h before the start of the experiments. Subsequently, lidded culture dishes were sealed with parafilm M (American National Can) and placed at RT on the microscope stage (model IX-70; Olympus). After a brief equilibration period, images were automatically acquired every 5 min for a minimum of 8 h, 20 min (corresponding to 100 frames) using a digital CCD camera (CoolSNAP HQ; Roper Scientific) and were stored on a computer for subsequent analysis. Continued cell viability was confirmed by monitoring the cells for 24 h after the experiments. Nuclear rotation, translation, and shape changes were analyzed by tracking the centroid positions of 3–6 nucleoli for each nucleus using custom-written MATLAB software. For each frame, the linear conformal image transformation was computed that best mapped the current centroid positions to the original positions minimizing the least-square error. The linear conformal transformations can account for a combination of translation, rotation, and scaling, and preserves the relative position of objects to each other. The deviation from the best fit, i.e., the error between the least-square fit transformation and the actual nucleoli positions, was used as a measure of nuclear deformation as it describes the extent of nuclear deformation from its initial shape independent of absolute nuclear movement or uniform changes in size (see time-lapse videos, available at http://www.jcb.org/cgi/content/full/jcb.200502148/DC1). Nucleoli that fused over time or cells that underwent mitosis during or immediately after the observation period were excluded from the analysis. We analyzed 25 nuclei of each cell type and computed for each nucleus the time-averaged deformation, the time-averaged and maximal change in size, translation, and rotation.
Nuclear strain experiments
Experiments were performed as previously described (Lammerding et al., 2004). In brief, cells were plated at subconfluent cell density on fibronectin-coated silicone membranes in DME supplemented with 10% FCS, followed by serum starvation for 48 h in DME containing ITS supplement (Sigma-Aldrich). Preceding the strain experiments, cells were incubated with Hoechst 33342 nuclear stain (final concentration 1 μg/ml; Molecular Probes) in DME + ITS for 20 min. Membranes were placed on a custom-made strain device and uniform biaxial strain was applied to the silicone membrane. Membrane and nuclear strains were computed based on bright field and fluorescence images acquired before, during, and after strain application using a custom-written image analysis algorithm. Normalized nuclear strain was defined as the ratio of nuclear strain to membrane strain to compensate for small variations in applied membrane strain (range 4.5–5.5%).
Microinjection experiments
Cells were plated on fibronectin-coated glass dishes (WillCo Wells) and incubated overnight. Microinjections were performed using a microinjector (Eppendorf) with Femtotips (Eppendorf). In each dish, 30–50 cells were injected with Texas red–labeled 70-kD dextran (dissolved at 10 mg/ml in PBS; Molecular Probes) into the cytoplasm or into the nucleus (injection pressure 500 hPa; injection time 0.6 s). After the microinjection, cells were washed with HBSS (Invitrogen) and intracellular localization of dextran–Texas red was recorded under a fluorescent microscope using a CCD camera (CoolSNAP HQ; Roper Scientific) on a microscope (model IX-70; Olympus). Cells were considered as having defective nuclear envelope integrity in the case of uniform nuclear and cytoplasmic staining, and the fraction of defective nuclei was expressed as the ratio of total fluorescent cells.
Image acquisition and manipulation
Phase-contrast and fluorescence images were acquired as described above using a digital CCD-camera (CoolSNAP HQ; Roper Scientific) mounted on an inverted microscope (model IX-70; Olympus) using ImagePro image acquisition software (Media Cybernetics). Nuclear shape experiments, time-lapse studies, and microinjected cells were imaged using an Olympus LCPlanF 20× phase-contrast objective (NA 0.40), whereas nuclear strain experiments were conducted using an Olympus UApo/340 40× water immersion objective (NA 1.15). Cells were kept in HBSS+ buffer during imaging, except for time-lapse experiments that were performed on cells in full media. All experiments were performed at RT. Radiographs from Northern analysis were digitized on a scanner (Perfection 2450; Epson) using linear intensity settings. Digital images were processed using Adobe Photoshop (ver. 6.0) by adjusting the linear image intensity display range, and fluorescence grayscale images were colorized in Adobe Photoshop by selecting a colorplane (RGB) appropriate for the chromophore (i.e., blue for Hoechst 33342, red for Texas red).
Mechanotransduction experiments
Strain stimulation was performed as described previously (Cheng et al., 1997; Lammerding et al., 2004). In brief, cells were plated on fibronectin-coated silicone membranes (∼3,000 cells/cm2). After 72 h serum starvation, cells were subjected to bi-axial cyclic strain (4%, 1 Hz) for 2 or 4 h. For chemical stimulation, cells were incubated with IL-1β (10 ng/ml; R&D Systems) or PMA (200 ng/ml; Sigma-Aldrich) in DME + ITS. Cellular mRNA of strained and unstrained control samples was isolated using the RNeasy Minikit (QIAGEN), and samples were subsequently analyzed by Northern analysis and real-time PCR.
Cell viability and apoptosis assay
Cells were plated on fibronectin-coated silicone membranes and maintained in full media for 48 h to provide sufficient cell attachment. After 24 h of cyclic biaxial strain application (1 Hz; 3, 5, or 10% strain), cells were incubated with propidium iodide (PI, final concentration 1 μg/ml; Sigma-Aldrich). Cells and culture media were collected, washed once in PBS− and resuspend in HBSS−. Each sample was divided into two equal parts to independently measure cell death (PI uptake) and apoptosis (DNA content analysis). One part was immediately analyzed for PI uptake using a Cytomics FC 500 flow cytometer (Beckman Coulter), counting 10,000–30,000 events in each group. Thresholds for PI incorporation were determined based on negative (no PI staining) and positive (cells permeabilized by 50% ethanol) controls. The other cell fraction from the 24-h strain experiments was fixed in 80% ethanol and stored at −20°C. Samples were then spun down, resuspended in PBS− and treated with Ribonuclease A (Sigma-Aldrich) for 1 h and subsequently stained with PI (final concentration 100 μg/ml). DNA content was measured on a flow cytometer and the apoptotic cell fraction was identified as cells with sub-G1 DNA content, counting 30,000–50,000 events per sample as described previously (Walker et al., 1993).
Northern analysis
For Northern analysis of iex-1, egr-1, and GAPDH, mRNA from unstrained controls and from cells subjected to 2 and 4 h of biaxial cyclic strain was harvested using RNAeasy Mini Kit kit (QIAGEN). 7–12 μg of each collected RNA sample were separated by gel electrophoresis at 110–130 V and mRNA was transferred overnight to a transfer membrane (MAGNA, Nylon, 0.45 micron; Osmonics, Inc.). Expression of iex-,1egr-1, and GAPDH mRNA was assessed by Northern analysis as described previously (De Keulenaer et al., 2002).
Real-time polymerase chain reaction
For further analysis of iex-1 expression, cells were prepared for strain experiments with 2 and 4 h biaxial strain. Cells were harvested using the QIAGEN RNAeasy kit. For each sample, 1 μl of collected RNA was added to RT Mixes of Stratagene Light Cycler kit with iex-1 and β-tubulin primers from Integrated DNA Technologies, Inc. (De Keulenaer et al., 2002). The polymerase reaction was conducted in Roche Molecular Biochemicals Light Cycler Version 5.32 with 45 cycles. Results were normalized with β-tubulin expression and expressed as percent increase from baseline (unstrained controls).
Luciferase experiments
Cells were transfected with plasmids for NF-κB–controlled luciferase expression and SV40-regulated β-galactosidase (Promega) using FuGENE6 (Roche). After transfection, cells were serum starved in DME + ITS medium for 48 h, followed by overnight stimulation with 200 ng/ml PMA or 10 ng/ml IL-1β. Luciferase assays were quantified in a Victor2 Multilabel Counter (PerkinElmer). Results were normalized for β-galactosidase activity and expressed as percentage of wild-type control.
Statistical analysis
All experiments were performed at least three independent times. Data are expressed as mean ± SEM. Statistical analysis was performed using the PRISM 3.0 and INSTAT software (GraphPad). The data were analyzed by unpaired t test (allowing different SD), one-way ANOVA (followed by Tukey's multiple comparison test) or, in case of non-Gaussian distribution, the Mann-Whitney or Kruskal Wallis tests (the latter when comparing more than two groups, followed by Dunn's multiple comparison test). For nuclear shape analysis, two-way ANOVA was performed on datasets from three different passages to evaluate the source of variation. In contrast to cell type, passage number was not a significant source of variation, and thus datasets from different passages were pooled and analyzed using the Kruskal-Wallis test for non-Gaussian distributions with Dunn's post test. Real-time PCR data were analyzed using a paired test and comparing induction of iex-1 (as percentage of baseline levels) at 2 and 4 h to wild-type induction. A two-tailed P-value of <0.05 was considered significant.
Online supplemental material
Full-length time-lapse sequences of A-type lamin-deficient, emerin-deficient, and wild-type mouse embryo fibroblasts corresponding to the still images presented in Fig. 3 can be found in the supplemental material. Each video consists of a sequence of images acquired every 5 min over an 8 h, 20 min period for a total of 100 frames. Online supplemental material available at http://www.jcb.org/cgi/content/full/jcb.200502148/DC1.
Acknowledgments
The authors would like to thank Julie Ji for her help in the nuclear morphology analysis. Microinjection was performed at the W.M. Keck Microscopy Facility at the Whitehead Institute.
This work was supported by grants from National Heart, Lung, and Blood Institute (HL073809, HL64858) and a post-doctoral fellowship from the American Heart Association for J. Lammerding.
Abbreviations used in this paper: BAF, barrier-to-autointegration factor; EDMD, Emery-Dreifuss muscular dystrophy; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; GCL, germ cell-less.
References
- Bengtsson, L., and K.L. Wilson. 2004. Multiple and surprising new functions for emerin, a nuclear membrane protein. Curr. Opin. Cell Biol. 16:73–79. [DOI] [PubMed] [Google Scholar]
- Bione, S., E. Maestrini, S. Rivella, M. Mancini, S. Regis, G. Romeo, and D. Toniolo. 1994. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat. Genet. 8:323–327. [DOI] [PubMed] [Google Scholar]
- Bonne, G., M.R. Di Barletta, S. Varnous, H.M. Becane, E.H. Hammouda, L. Merlini, F. Muntoni, C.R. Greenberg, F. Gary, J.A. Urtizberea, et al. 1999. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet. 21:285–288. [DOI] [PubMed] [Google Scholar]
- Broers, J.L., E.A. Peeters, H.J. Kuijpers, J. Endert, C.V. Bouten, C.W. Oomens, F.P. Baaijens, and F.C. Ramaekers. 2004. Decreased mechanical stiffness in LMNA−/− cells is caused by defective nucleo-cytoskeletal integrity: implications for the development of laminopathies. Hum. Mol. Genet. 13:2567–2580. [DOI] [PubMed] [Google Scholar]
- Burke, B., and C.L. Stewart. 2002. Life at the edge: the nuclear envelope and human disease. Nat. Rev. Mol. Cell Biol. 3:575–585. [DOI] [PubMed] [Google Scholar]
- Caille, N., Y. Tardy, and J.J. Meister. 1998. Assessment of strain field in endothelial cells subjected to uniaxial deformation of their substrate. Ann. Biomed. Eng. 26:409–416. [DOI] [PubMed] [Google Scholar]
- Cao, H., and R.A. Hegele. 2000. Nuclear lamin A/C R482Q mutation in Canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet. 9:109–112. [DOI] [PubMed] [Google Scholar]
- Cheng, G.C., W.H. Briggs, D.S. Gerson, P. Libby, A.J. Grodzinsky, M.L. Gray, and R.T. Lee. 1997. Mechanical strain tightly controls fibroblast growth factor-2 release from cultured human vascular smooth muscle cells. Circ. Res. 80:28–36. [DOI] [PubMed] [Google Scholar]
- Columbaro, M., E. Mattioli, G. Lattanzi, C. Rutigliano, A. Ognibene, N.M. Maraldi, and S. Squarzoni. 2001. Staurosporine treatment and serum starvation promote the cleavage of emerin in cultured mouse myoblasts: involvement of a caspase-dependent mechanism. FEBS Lett. 509:423–429. [DOI] [PubMed] [Google Scholar]
- De Keulenaer, G.W., Y. Wang, Y. Feng, S. Muangman, K. Yamamoto, J.F. Thompson, T.G. Turi, K. Landschutz, and R.T. Lee. 2002. Identification of IEX-1 as a biomechanically controlled nuclear factor-κB target gene that inhibits cardiomyocyte hypertrophy. Circ. Res. 90:690–696. [DOI] [PubMed] [Google Scholar]
- De Sandre-Giovannoli, A., M. Chaouch, S. Kozlov, J.M. Vallat, M. Tazir, N. Kassouri, P. Szepetowski, T. Hammadouche, A. Vandenberghe, C.L. Stewart, et al. 2002. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am. J. Hum. Genet. 70:726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sandre-Giovannoli, A., R. Bernard, P. Cau, C. Navarro, J. Amiel, I. Boccaccio, S. Lyonnet, C.L. Stewart, A. Munnich, M. Le Merrer, and N. Levy. 2003. Lamin A truncation in Hutchinson-Gilford progeria. Science. 300:2055. [DOI] [PubMed] [Google Scholar]
- Diaz, G., A. Zuccarelli, I. Pelligra, and A. Ghiani. 1989. Elliptic fourier analysis of cell and nuclear shapes. Comput. Biomed. Res. 22:405–414. [DOI] [PubMed] [Google Scholar]
- Ellis, J.A., M. Craxton, J.R. Yates, and J. Kendrick-Jones. 1998. Aberrant intracellular targeting and cell cycle-dependent phosphorylation of emerin contribute to the Emery-Dreifuss muscular dystrophy phenotype. J. Cell Sci. 111:781–792. [DOI] [PubMed] [Google Scholar]
- Eriksson, M., W.T. Brown, L.B. Gordon, M.W. Glynn, J. Singer, L. Scott, M.R. Erdos, C.M. Robbins, T.Y. Moses, P. Berglund, et al. 2003. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 423:293–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairley, E.A., A. Riddell, J.A. Ellis, and J. Kendrick-Jones. 2002. The cell cycle dependent mislocalisation of emerin may contribute to the Emery-Dreifuss muscular dystrophy phenotype. J. Cell Sci. 115:341–354. [DOI] [PubMed] [Google Scholar]
- Fatkin, D., C. MacRae, T. Sasaki, M.R. Wolff, M. Porcu, M. Frenneaux, J. Atherton, H.J. Vidaillet Jr., S. Spudich, U. De Girolami, et al. 1999. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med. 341:1715–1724. [DOI] [PubMed] [Google Scholar]
- Fidzianska, A., and I. Hausmanowa-Petrusewicz. 2003. Architectural abnormalities in muscle nuclei. Ultrastructural differences between X-linked and autosomal dominant forms of EDMD. J. Neurol. Sci. 210:47–51. [DOI] [PubMed] [Google Scholar]
- Fidzianska, A., D. Toniolo, and I. Hausmanowa-Petrusewicz. 1998. Ultrastructural abnormality of sarcolemmal nuclei in Emery-Dreifuss muscular dystrophy (EDMD). J. Neurol. Sci. 159:88–93. [DOI] [PubMed] [Google Scholar]
- Goldman, R.D., D.K. Shumaker, M.R. Erdos, M. Eriksson, A.E. Goldman, L.B. Gordon, Y. Gruenbaum, S. Khuon, M. Mendez, R. Varga, and F.S. Collins. 2004. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA. 101:8963–8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady, R.M., D.A. Starr, G.L. Ackerman, J.R. Sanes, and M. Han. 2005. Syne proteins anchor muscle nuclei at the neuromuscular junction. Proc. Natl. Acad. Sci. USA. 102:4359–4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraguchi, T., J.M. Holaska, M. Yamane, T. Koujin, N. Hashiguchi, C. Mori, K.L. Wilson, and Y. Hiraoka. 2004. Emerin binding to Btf, a death-promoting transcriptional repressor, is disrupted by a missense mutation that causes Emery-Dreifuss muscular dystrophy. Eur. J. Biochem. 271:1035–1045. [DOI] [PubMed] [Google Scholar]
- Hofmann, W.A., L. Stojiljkovic, B. Fuchsova, G.M. Vargas, E. Mavrommatis, V. Philimonenko, K. Kysela, J.A. Goodrich, J.L. Lessard, T.J. Hope, et al. 2004. Actin is part of pre-initiation complexes and is necessary for transcription by RNA polymerase II. Nat. Cell Biol. 6:1094–1101. [DOI] [PubMed] [Google Scholar]
- Holaska, J.M., K.K. Lee, A.K. Kowalski, and K.L. Wilson. 2003. Transcriptional repressor germ cell-less (GCL) and barrier to autointegration factor (BAF) compete for binding to emerin in vitro. J. Biol. Chem. 278:6969–6975. [DOI] [PubMed] [Google Scholar]
- Holaska, J.M., A.K. Kowalski, and K.L. Wilson. 2004. Emerin caps the pointed end of actin filaments: evidence for an actin cortical network at the nuclear inner membrane. PLoS Biol. 2:E231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyner, A. 1999. Gene Targeting: A Practical Approach. Oxford University Press, Oxford, UK. 312 pp.
- Kasof, G.M., L. Goyal, and E. White. 1999. Btf, a novel death-promoting transcriptional repressor that interacts with Bcl-2-related proteins. Mol. Cell. Biol. 19:4390–4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammerding, J., P.C. Schulze, T. Takahashi, S. Kozlov, T. Sullivan, R.D. Kamm, C.L. Stewart, and R.T. Lee. 2004. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J. Clin. Invest. 113:370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libotte, T., H. Zaim, S. Abraham, V.C. Padmakumar, M. Schneider, W. Lu, M. Munck, C. Hutchison, M. Wehnert, B. Fahrenkrog, et al. 2005. Lamin A/C dependent localization of nesprin-2, a giant scaffolder at the nuclear envelope. Mol Biol Cell. 16:3411–3424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J., T.R. Ben-Shahar, D. Riemer, M. Treinin, P. Spann, K. Weber, A. Fire, and Y. Gruenbaum. 2000. Essential roles for Caenorhabditis elegans lamin gene in nuclear organization, cell cycle progression, and spatial organization of nuclear pore complexes. Mol. Biol. Cell. 11:3937–3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniotis, A.J., C.S. Chen, and D.E. Ingber. 1997. Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc. Natl. Acad. Sci. USA. 94:849–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mislow, J.M., J.M. Holaska, M.S. Kim, K.K. Lee, M. Segura-Totten, K.L. Wilson, and E.M. McNally. 2002. Nesprin-1α self-associates and binds directly to emerin and lamin A in vitro. FEBS Lett. 525:135–140. [DOI] [PubMed] [Google Scholar]
- Morawietz, H., Y.H. Ma, F. Vives, E. Wilson, V.P. Sukhatme, J. Holtz, and H.E. Ives. 1999. Rapid induction and translocation of Egr-1 in response to mechanical strain in vascular smooth muscle cells. Circ. Res. 84:678–687. [DOI] [PubMed] [Google Scholar]
- Muchir, A., B.G. van Engelen, M. Lammens, J.M. Mislow, E. McNally, K. Schwartz, and G. Bonne. 2003. Nuclear envelope alterations in fibroblasts from LGMD1B patients carrying nonsense Y259X heterozygous or homozygous mutation in lamin A/C gene. Exp. Cell Res. 291:352–362. [DOI] [PubMed] [Google Scholar]
- Nikolova, V., C. Leimena, A.C. McMahon, J.C. Tan, S. Chandar, D. Jogia, S.H. Kesteven, J. Michalicek, R. Otway, F. Verheyen, et al. 2004. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J. Clin. Invest. 113:357–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nili, E., G.S. Cojocaru, Y. Kalma, D. Ginsberg, N.G. Copeland, D.J. Gilbert, N.A. Jenkins, R. Berger, S. Shaklai, N. Amariglio, et al. 2001. Nuclear membrane protein LAP2β mediates transcriptional repression alone and together with its binding partner GCL (germ-cell-less). J. Cell Sci. 114:3297–3307. [DOI] [PubMed] [Google Scholar]
- Östlund, C., G. Bonne, K. Schwartz, and H.J. Worman. 2001. Properties of lamin A mutants found in Emery-Dreifuss muscular dystrophy, cardiomyopathy and Dunnigan-type partial lipodystrophy. J. Cell Sci. 114:4435–4445. [DOI] [PubMed] [Google Scholar]
- Padmakumar, V.C., S. Abraham, S. Braune, A.A. Noegel, B. Tunggal, I. Karakesisoglou, and E. Korenbaum. 2004. Enaptin, a giant actin-binding protein, is an element of the nuclear membrane and the actin cytoskeleton. Exp. Cell Res. 295:330–339. [DOI] [PubMed] [Google Scholar]
- Raharjo, W.H., P. Enarson, T. Sullivan, C.L. Stewart, and B. Burke. 2001. Nuclear envelope defects associated with LMNA mutations cause dilated cardiomyopathy and Emery-Dreifuss muscular dystrophy. J. Cell Sci. 114:4447–4457. [DOI] [PubMed] [Google Scholar]
- Sadoshima, J., L. Jahn, T. Takahashi, T.J. Kulik, and S. Izumo. 1992. Molecular characterization of the stretch-induced adaptation of cultured cardiac cells. An in vitro model of load-induced cardiac hypertrophy. J. Biol. Chem. 267:10551–10560. [PubMed] [Google Scholar]
- Shackleton, S., D.J. Lloyd, S.N. Jackson, R. Evans, M.F. Niermeijer, B.M. Singh, H. Schmidt, G. Brabant, S. Kumar, P.N. Durrington, et al. 2000. LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat. Genet. 24:153–156. [DOI] [PubMed] [Google Scholar]
- Starr, D.A., and M. Han. 2002. Role of ANC-1 in tethering nuclei to the actin cytoskeleton. Science. 298:406–409. [DOI] [PubMed] [Google Scholar]
- Starr, D.A., and M. Han. 2003. ANChors away: an actin based mechanism of nuclear positioning. J. Cell Sci. 116:211–216. [DOI] [PubMed] [Google Scholar]
- Sullivan, T., D. Escalante-Alcalde, H. Bhatt, M. Anver, N. Bhat, K. Nagashima, C.L. Stewart, and B. Burke. 1999. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 147:913–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukahara, T., S. Tsujino, and K. Arahata. 2002. CDNA microarray analysis of gene expression in fibroblasts of patients with X-linked Emery-Dreifuss muscular dystrophy. Muscle Nerve. 25:898–901. [DOI] [PubMed] [Google Scholar]
- Vaughan, A., M. Alvarez-Reyes, J.M. Bridger, J.L. Broers, F.C. Ramaekers, M. Wehnert, G.E. Morris, W.G.F. Whitfield, and C.J. Hutchison. 2001. Both emerin and lamin C depend on lamin A for localization at the nuclear envelope. J. Cell Sci. 114:2577–2590. [DOI] [PubMed] [Google Scholar]
- Walker, P.R., J. Kwast-Welfeld, H. Gourdeau, J. Leblanc, W. Neugebauer, and M. Sikorska. 1993. Relationship between apoptosis and the cell cycle in lymphocytes: roles of protein kinase C, tyrosine phosphorylation, and AP1. Exp. Cell Res. 207:142–151. [DOI] [PubMed] [Google Scholar]
- Wilkinson, F.L., J.M. Holaska, Z. Zhang, A. Sharma, S. Manilal, I. Holt, S. Stamm, K.L. Wilson, and G.E. Morris. 2003. Emerin interacts in vitro with the splicing-associated factor, YT521-B. Eur. J. Biochem. 270:2459–2466. [DOI] [PubMed] [Google Scholar]
- Zhang, Q., C.D. Ragnauth, J.N. Skepper, N.F. Worth, D.T. Warren, R.G. Roberts, P.L. Weissberg, J.A. Ellis, and C.M. Shanahan. 2005. Nesprin-2 is a multi-isomeric protein that binds lamin and emerin at the nuclear envelope and forms a subcellular network in skeletal muscle. J. Cell Sci. 118:673–687. [DOI] [PubMed] [Google Scholar]
- Zhu, X., X. Zeng, B. Huang, and S. Hao. 2004. Actin is closely associated with RNA polymerase II and involved in activation of gene transcription. Biochem. Biophys. Res. Commun. 321:623–630. [DOI] [PubMed] [Google Scholar]