

Abstract
Two disease-associated missense mutations in the sialin gene (G328E and G409E) have recently been identified in patients with lysosomal free sialic acid storage disease. We have assessed the effect of these mutations and find complete loss of measurable transport activity with both and impaired trafficking of the G409E protein. These results suggest that the two residues are important for proper function of sialin and confirm the association of loss of transport with disease causative mutations.
Keywords: lysosomal storage disorder, sialic acid, Salla disease, infantile sialic acid storage disorders, sialin, transporter
Introduction
Lysosomal storage disorders are marked by an accumulation of organic material within lysosome organelles due to defects either in the degradation of macromolecules within lysosomes or the transport of end products across the lysosomal membrane. Among such disorders are Salla disease and infantile sialic acid storage disease (ISSD), two autosomal recessive neurodegenerative diseases that result from mutations in a gene encoding the lysosomal sialic acid transporter, sialin [1]. The more severe ISSD is typically characterized by neonatal ascites, intrauterine hydrops, dysmorphic features, and death by 2 years of age, but individuals with a less severe phenotype and longer life-spans have been described in the literature. On the other hand, individuals with Salla disease are noted to have cognitive and motor impairment as early as 6-12 months after birth, but typically reach adulthood.
Missense mutations throughout the predicted sialin sequence have been associated with the lysosomal free sialic acid storage disorders [2-5]. Recently disease-associated mutations leading to substitutions of conserved glycines (G328E and G409E) have been identified (figure 1A). The G328E mutation has been found as a homozygous mutation in four relatives in a single family [6]. While one individual presented with congenital ascites and died shortly after birth, others were noted to have a milder phenotype with survival to at least several years of age. The G409E mutation has been identified in a compound heterozygous individual with a phenotype intermediate in severity between ISSD and Salla disease [7]. The second mutation in this individual is predicted to lead to a splicing defect and a subsequent frame-shift leading to a truncated protein of 32 amino acids.
Figure 1.
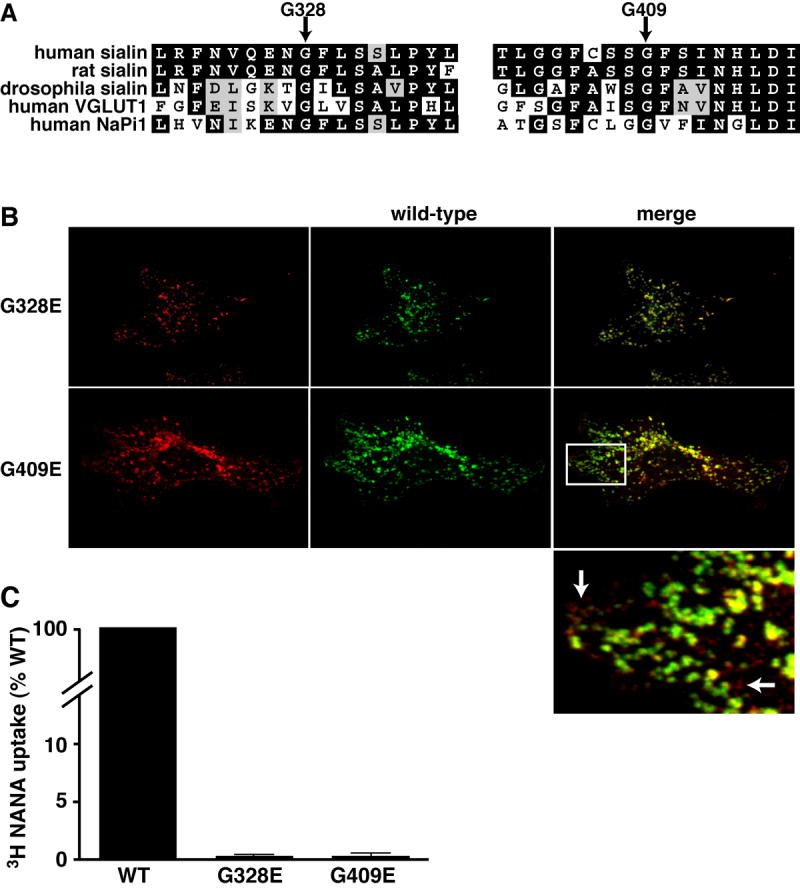
A) Comparison of the sequences corresponding to G328 and G409 and the surrounding residues in human, rat and drosophila sialin and human isoforms of two related proteins, VGLUT1 and NaPi1 demonstrate that these specific residues are completely conserved. B) Immunofluorescence colocalization of HA-tagged G328E and G409E (left panels, red) and myc-tagged wild type sialin (middle panels, green) demonstrate strong colocalization of wild-type and G328E sialin, but less overlap of the wild-type and G409E patterns. In higher magnification view of indicated region (bottom panel right column) arrows indicate G409E puncta that do not colocalize with wild-type sialin. C) Sialic acid transport activity of cells expressing plasma membrane targeted isoforms of G328E and G409E demonstrates less than 1% of the uptake measured in the cells transfected with the wild-type plasma membrane targeted sialin. Uptake by cells transfected with the G328E and G409E isoforms were not statistically different from that measured for LYAAT transfected cells (p=0.48 and p=0.60 respectively, two-tailed unpaired t-test). Duplicate samples were assayed in three separate experiments. The mean values are plotted with the standard deviation.
To understand how these novel mutations might contribute to disease on a molecular level, we examined the transport activity and localization pattern of mutant recombinant sialin isoforms. Consistent with other disease-associated missense mutations, G328E and G409E mutations both lead to impairment of sialin mediated transport of free sialic acid. In addition the G409E also alters the localization of the protein. These results suggest that these residues may play a crucial role in proper folding and function of the protein, and thus add to our understanding of molecular pathology of the lysosomal free sialic acid storage disorders.
Materials and Methods
All procedures were carried out as previously described [5]. Briefly, disease-associated mutations were generated in wild-type rat sialin background using site-directed mutagenesis with the Quick-Change kit (Stratagene) following the directions of the manufacturer. After sequencing to confirm that desired mutations were generated, cDNAs were expressed in HeLa cells using Effectene (Qiagen) according to the instructions of the manufacturer. For immunofluorescence studies transfected HeLa cells were plated onto glass coverslips. The rabbit anti-HA antibody (Abcam) was used at 1:2,000 and the mouse anti-myc antibody (BD Biosciences) was used at 1:100. Secondary antibodies labeled with Alexa 488 or Alexa 568 (Molecular Probes) were diluted at 1:1,000. A Zeiss Axiovert 100M inverted microscope configured for confocal microscopy was used for imaging. For transport studies HeLa cells were plated on 24 well plates and transfected as described above. After 24-36hours, uptake assays with 3H N-acetyl-neuraminic acid (American Radiolabeled Chemicals) were carried out in an acidic buffer (Krebs-Ringer-MES, pH 5.0). For controls LYAAT1, a lysosomal transporter previously characterized with whole cell uptake assays, was used and the background was subtracted from all values. Quantitation and statistics were performed using the Prism program (GraphPad Software).
Results
The G409E mutation alters the localization of sialin
Previous studies have suggested that mislocalization of disease-associated mutant sialin can lead to the loss of sialic acid transport activity in lysosomes, the presumed site of function [4, 5, 8]. Although sialin has been described as a lysosomal protein [4, 9], our attempts to find another protein that consistently colocalizes with sialin have been unsuccessful. To characterize potential changes in subcellular localization associated with the mutations we therefore compared the expression patterns of the wild-type protein and the mutant proteins differentially tagged with the commonly used epitopes HA and myc. As we have previously demonstrated [5], co-transfection of wild-type HA-tagged and wild-type myc-tagged sialin leads to colocalization of the two epitopes by immunofluorescence staining (data not shown). Comparison of the immunofluorescence patterns for G328E and G409E mutants with the wild-type protein demonstrate overlap in the patterns at 72 hours after transfection (Figure 1B), but careful comparison of the G409E mutant staining and the wild-type staining reveals that there are small punctate structures containing the G409E mutant that do not contain the wild-type sialin.
G328E and G409E are associated with loss of sialic acid transport activity
Although the altered localization of the mutant proteins likely influences function, impaired transport activity might also contribute. To assess this possibility, we directly measured transport activity mediated by the mutant proteins. We have previously demonstrated that disrupting an adaptor protein binding domain in the amino terminal region of the protein targets sialin to the plasma membrane [5]. While the protein is not in its normal subcellular environment, its orientation is such that whole cell uptake corresponds to the physiologically relevant transport of sialic acid out of the lysosome. Cells expressing the plasma membrane targeted wild-type and mutant sialin proteins were assayed for transport of sialic acid in an acidic buffer (pH 5.0) to mimic the lysosomal lumen. Measurements of total uptake at 5 minutes indicate that less than 1% of activity is retained in the G328E (0.4 +/-0.2%) and G409E (0.4+/-0.3%) mutants (figure 1C) despite expression of the proteins on the cell surface (data not shown). These values are not statistically different from those obtained for cells transfected with LYAAT, an unrelated lysosomal amino acid transporter [10].
Discussion
The data we present here for two sialin missense mutations recently associated with the human lysosomal free sialic acid storage disease are consistent with the results obtained for the previously studied mutations [4, 5]. Our studies confirm that disease-associated mutations can impair the transport activity and/or localization of sialin and support the hypothesis that these effects underlie the pathophysiology of the lysosomal free sialic acid storage disorders.
The mechanisms by which the G328E and G409E missense mutations impair function remain unclear. Glycine residues tend to disrupt α-helical structures and are important in formation of appropriate secondary structures, particularly in regions where there is a transition from membranous to extramembranous environment. The membrane topology of sialin has yet to be determined by molecular methods, but there is considerable homology between sialin and the Drosophila vesicular glutamate transporter (DVGLUT) for which the structure has been partially determined [11]. Interestingly, the residues in DVGLUT that correspond to G328 and G409 are predicted to reside near the interface between transmembrane domains and extramembranous portions of the protein. This suggests that the influence of these glycine residues on secondary structure explains their conservation among related proteins and possibly the effect of the missense mutations on protein function. It is worth noting, however, that recently solved crystal structures for other transporter proteins indicate that glycine residues can be important in helix packing and substrate binding [12, 13]. Thus, further studies on the structure of sialin and related proteins will be necessary for a fuller understanding of how these mutations lead to impaired function and trafficking of sialin and ultimately the disease phenotype.
Acknowledgments
The authors would like to acknowledge members of the Meyer lab and members of the Garner lab for assistance and guidance. This work was supported by grants awarded to R.J.R. from the March of Dimes and the National Institute of Neurological Disorders and Stroke (1R01NS050417).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aula P, Gahl WA. Disorders of free sialic acid storage. In: Scriver CR, Sly WS, Childs B, et al., editors. The Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw-Hill; 2001. pp. 5109–20. [Google Scholar]
- 2.Verheijen FW, Verbeek E, Aula N, et al. A new gene, encoding an anion transporter, is mutated in sialic acid storage diseases. Nature Genetics. 1999;23:462–5. doi: 10.1038/70585. [DOI] [PubMed] [Google Scholar]
- 3.Aula N, Salomaki P, Timonen R, et al. The spectrum of SLC17A5-gene mutations resulting in free sialic acid-storage diseases indicates some genotype-phenotype correlation. Am J Hum Genet. 2000;67:832–40. doi: 10.1086/303077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morin P, Sagne C, Gasnier B. Functional characterization of wild-type and mutant human sialin. Embo J. 2004;23:4560–70. doi: 10.1038/sj.emboj.7600464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wreden CC, Wlizla M, Reimer RJ. Varied mechanisms underlie the free sialic acid storage disorders. J Biol Chem. 2005;280:1408–16. doi: 10.1074/jbc.M411295200. [DOI] [PubMed] [Google Scholar]
- 6.Landau D, Cohen D, Shalev H, et al. A novel mutation in the SLC17A5 gene causing both severe and mild phenotypes of free sialic acid storage disease in one inbred Bedouin kindred. Mol Genet Metab. 2004;82:167–72. doi: 10.1016/j.ymgme.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 7.Kleta R, Morse RP, Orvisky E, et al. Clinical, biochemical, and molecular diagnosis of a free sialic acid storage disease patient of moderate severity. Mol Genet Metab. 2004;82:137–43. doi: 10.1016/j.ymgme.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 8.Aula N, Jalanko A, Aula P, Peltonen L. Unraveling the molecular pathogenesis of free sialic acid storage disorders: altered targeting of mutant sialin. Mol Genet Metab. 2002;77:99–107. doi: 10.1016/s1096-7192(02)00124-5. [DOI] [PubMed] [Google Scholar]
- 9.Aula N, Kopra O, Jalanko A, Peltonen L. Sialin expression in the CNS implicates extralysosomal function in neurons. Neurobiol Dis. 2004;15:251–61. doi: 10.1016/j.nbd.2003.11.017. [DOI] [PubMed] [Google Scholar]
- 10.Wreden CC, Johnson J, Tran C, et al. The H+-coupled electrogenic lysosomal amino acid transporter LYAAT1 localizes to the axon and plasma membrane of hippocampal neurons. J Neurosci. 2003;23:1265–75. doi: 10.1523/JNEUROSCI.23-04-01265.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fei H, Karnezis T, Reimer RJ, Krantz DE. Membrane topology of the Drosophila vesicular glutamate transporter. J Neurochem. 2007;101:1662–71. doi: 10.1111/j.1471-4159.2007.04518.x. [DOI] [PubMed] [Google Scholar]
- 12.Boudker O, Ryan RM, Yernool D, Shimamoto K, Gouaux E. Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature. 2007;445:387–93. doi: 10.1038/nature05455. [DOI] [PubMed] [Google Scholar]
- 13.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters. Nature. 2005;437:215–23. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]