

Summary
Visual snapshots of intracellular kinase activity can be acquired with exquisite temporal control using a light-activatable (caged) sensor, thereby providing a means to interrogate enzymatic activity at any point during the cell division cycle. Robust protein kinase activity transpires just prior to, but not immediately following, nuclear envelope breakdown (NEB). Furthermore, kinase activity is required for progression from prophase into metaphase. Finally, the application of selective protein kinase C (PKC) inhibitors, in combination with the caged sensor, correlates the action of the PKC β isoform with subsequent NEB.
Introduction
Cellular biochemistry is commonly assessed using lysates or cell-free model systems. However, in order to directly correlate enzymatic activity with cellular behavior, new tools must be developed to visualize the action of enzymes in cells, tissues, and ultimately living organisms. Fluorescent microscopy provides the instrumental means to observe intracellular biochemistry, but this methodology is dependent upon the design and construction of fluorescent sensors that can report changes in the catalytic status of the target enzyme. However, live cell microscopy places unique demands on the sensor. In particular, sensors that possess short wavelength fluorophores or exhibit modest enzyme-induced fluorescent changes are insufficient for visualizing enzyme-catalyzed reactions in living cells. The small volume of a typical mammalian cell requires the fluorophore to be both bright and highly responsive to its enzymatic target. Furthermore, the fluorophore must possess the proper photophysical properties (i.e. long wavelength excitation and emission) so that the endogenous fluorophores present in the cell do not interfere with the readout. Finally, the multitude of biochemical transformations transpiring in the cell at any given time requires that the sensor be reasonably selective for its enzymatic target.
In addition to the concerns noted above, live cell enzymology offers a unique challenge that, in general, has no counterpart in experiments that utilize pure enzymes or cell lysates. One of the attractive features associated with observing enzyme action in the natural intracellular environment is the ability to correlate catalytic activity with cellular behavior. However, although the investigator controls the start and stop points of enzyme-catalyzed reactions in a typical cuvette-based experiment, the biochemical apparatus of the cell controls the timing and duration of intracellular enzymatic activity. The loss of investigator control in live cell enzymology has important ramifications. For example, if the intracellular enzyme of interest is constitutively active, then the time required for loading the sensor into the cell could preclude the acquisition of well-defined kinetics. In addition, since intracellular enzymatic activity can cycle on and off, the absence of a fluorescent response in live cell assays may not necessarily be due to the absence of enzymatic activity, but could be a consequence of sensor consumption at an earlier stage. Finally, loading any unnatural molecular entity into a cell, particularly via microinjection or various cell permeablizing delivery systems, can stress the cell and thus generate an artificial response. As a consequence, it is common practice to allow the cell to recover following the introduction of proteins, peptides, nucleic acids, etc. For these reasons, as well as others, it would prove advantageous to devise reagents (inhibitors, sensors, substrates, etc.) that can be delivered to the cell in an inert form, yet sensitive to subsequent activation upon demand. In this regard, light-activatable species offer the possibility of precise temporal control over sensor activity even after the reagent has entered the cell (1).
Protein kinase activity is critical for the G2/M transition (2 - 6), particularly at or around the time of nuclear envelope breakdown (NEB). However, it is unknown if activity is present prior to, during, or after NEB in living cells. We recently described the PKC sensor 1 (Fig. 1), an efficient substrate for the conventional protein kinase C (PKC) isoforms (α, β, and γ) (7). This peptide exhibits a readily observable fluorescent change upon phosphorylation. However, we found that, using sensor 1, PKC is constitutively active in interphase cells unless these cells have been serum starved (7). Consequently, the issue of sensor consumption (i. e. complete phosphorylation of peptide 1) prior to the key biological event (e.g. NEB) represents a significant concern. By contrast, the corresponding caged version 2 (Fig. 1) is not susceptible to phosphorylation until photolyzed, which then furnishes the active sensor 1 (8). In short, the caged derivative 2 can be loaded into cells and subsequently used to visualize kinase activity at any time point relative to NEB, without having to resort to artificial constraints (e.g. serum starvation). We have employed compound 2 to assess intracellular protein kinase activity at precise time intervals just prior to, during, and immediately following NEB. In addition, in combination with known protein kinase inhibitors, we have identified the kinase responsible for both the phosphorylation of the active sensor 1 as well as mitotic progression from prophase to metaphase.
Figure 1.
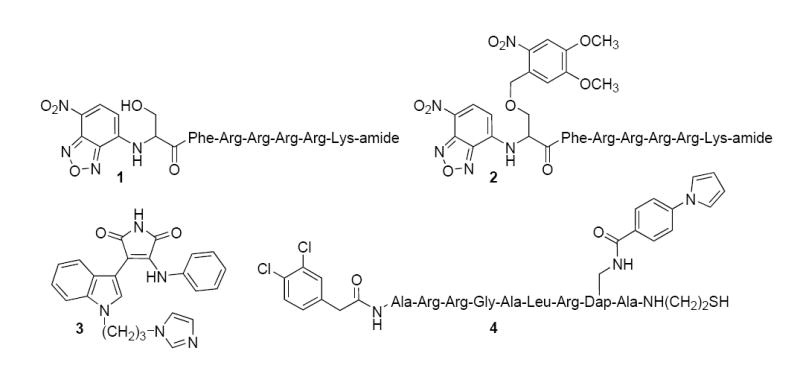
Structures of PKC sensors and inhibitors. Compound 1 responds to PKC-catalyzed phosphorylation in a fluorescently sensitive fashion (7). The nonphosphorylatable analogue 2 is converted to the active sensor 1 by photolysis (8). Compound 3 is a selective PKC ß inhibitor (11), whereas compound 4 serves as a selective PKC α inhibitor (12).
Results and Discussion
Peptide 1 as a Substrate for the PKC Isoforms Present in PtK2 Cells
The PtK2 cell line was employed in this study since these cells remain flat during mitosis and have large chromosomes that allow ready identification of the individual stages of mitosis. Western blot analysis revealed that PtK2 cell express the α, β, ι, μ, θ, and ζ isoforms (Fig. 2). We’ve previously shown that 1 is an outstanding substrate for PKC α, β, and γ (7). The first two isoforms are present in PtK2 cells, whereas PKC γ is not (Fig. 2). In addition, we’ve found that peptide 1 serves as a weak substrate for ι, θ and ζ isoforms. Finally, the fluorescent sensor is not phosphorylated by PKC μ (Fig. 3). These results appear to be consistent with the previously described substrate specificity of some of these isoforms (9). The conventional isoforms, α and β, are known to prefer substrates that possess positively charged residues (in particular Arg) at the +2 and +3 positions relative to the phosphorylatable Ser moiety (i.e. –Ser-Xaa-Arg-Arg). On the other hand, members of the novel and atypical PKC subfamilies generally prefer hydrophobic residues at +2 and +3, although they can tolerate positively charged amino acids at these positions as well. In addition to the PKC family of enzymes, a variety of other protein kinases have been implicated as participants in the onset of mitosis (10). However, these enzymes either fail to (Akt-1, AurB, Cdc-2, Plk1) or only weakly (Nek2) phosphorylate peptide 1.
Figure 2.

Western blots of PKC isoforms present in Ptk2 cells.
Figure 3.
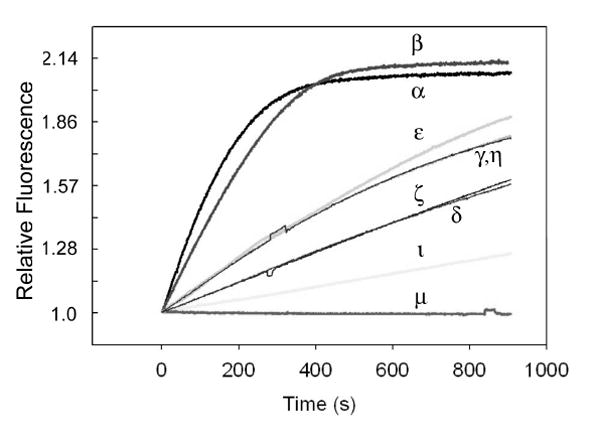
PKC isoform-catalyzed phosphorylation of peptide 1 as a function of time.
An Assessment of Protein Kinase Activity at the Prophase to Metaphase Transition
The caged peptide 2 (200 μM in needle; 2 – 20 μM final intracellular concentration), and 70 kDa Texas Red-dextran (5 μM in needle) were co-injected into PtK2 cells. The Texas Red-dextran is excluded from the intact nucleus and thus provides an easily visualized readout of NEB, which transpires over a period of approximately 2 min (Fig. 4a, Supplementary Movie 1). In general, photolysis (1 sec) of individual cells during prophase generates a time-dependent increase in fluorescence (Fig. 4a) followed by NEB. Interestingly, the few cells (3 out of 18) that failed to display a fluorescence change in prophase also failed to undergo subsequent nuclear envelope breakdown, suggesting that the former is required for the latter to transpire. As a control, we found that microinjection with buffer alone results in a fraction of cells that do not progress to NEB (8 out of 30). Apparently, the stress of microinjection can have deleterious consequences in a small fraction of cells in terms of mitotic progression. Nevertheless, the majority of cells (>80%) microinjected with sensor share several key enzymological and behavioral properties: (a) a fluorescent enhancement observed up to NEB in all cells that exhibit subsequent NEB, (b) a rate of fluorescence change that levels off as the nuclear envelope collapses, and (c) generally unchanged fluorescence following NEB. These results are consistent with the notion that PKC is active prior to NEB but is rendered inactive following nuclear collapse. However, another interpretation is that all sensor molecules have been phosphorylated by the time the cell reaches the NEB signpost. Consequently, the experiments outlined in Fig. 4a were repeated, but the caged peptide 2 was photoactivated immediately following NEB, as signified by the appearance of Texas Red-dextran within the former nuclear region. Under these conditions, there was no significant change in reporter fluorescence intensity (14 out of 16 cells exhibited no fluorescence change and 2 out of 16 cells displayed <15% fluorescence change) for up to 9 min following NEB (Fig. 4b). These results suggest that kinase activity is biochemically switched off following the collapse of the nuclear envelope. Finally, we also established that the post-NEB phosphorylation state of peptide 1 is not altered by intracellular phosphatases since we failed to observe any change in fluorescence (8 of 10 cells displayed no fluorescence change and 2 out of 10 exhibited <15% fluorescence change) in the presence of the general phosphatase inhibitor okadaic acid (Fig. 4c).
Figure 4.
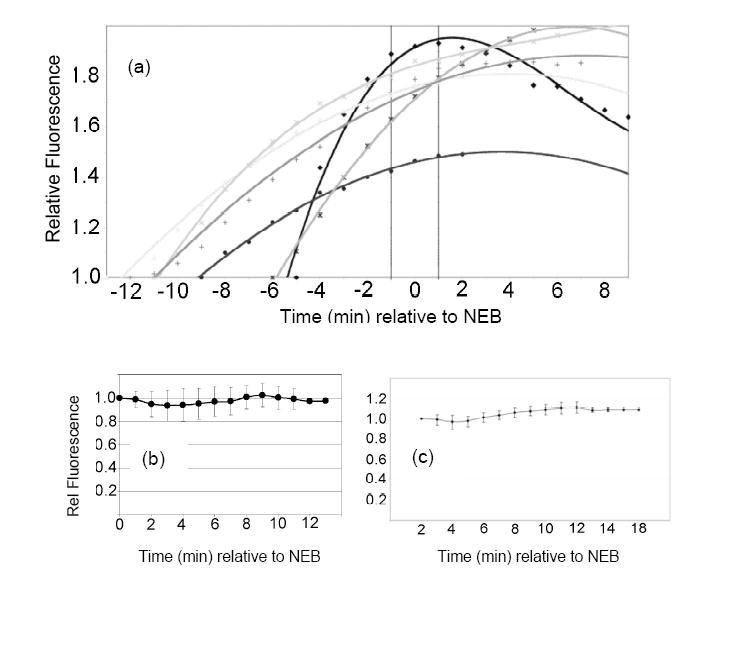
(a) Time course of the phosphorylation of peptide 1 prior to NEB. The nonphosphorylatable analogue 2 and 70 kD Texas Red-dextran were simultaneously microinjected into PtK2 cells. Cells were illuminated (1 sec) during prophase (i.e. prior to NEB) to convert the inert sensor 2 to its active form 1 and the fluorescence change as a function of time recorded with respect to NEB. Each progress curve is derived from a single cell and a total of 18 cells were examined (6 of the progress curves are shown here) (b) Time course of the phosphorylation of peptide 1 after NEB. PtK2 cells were treated as in Figure 2a, except that photolysis was performed following NEB. The progress curve represents the average fluorescence change of 16 cells (each data point represents the mean ± s.d.). (c) Fluorescence change following NEB in the presence of okadaic acid (1.5 μM).
Protein Kinase Inhibitors Establish PKC ß as the Sensor Kinase
Although the sensor experiments expose a clean demarcation in kinase activity prior to and immediately following NEB, they do not reveal which kinase is responsible for sensor phosphorylation nor do they establish a kinase-dependent cause and effect with nuclear collapse. As noted above, Nek-2 as well as the α, ß, ι, μ, θ and ζ isoforms of PKC are capable of catalyzing the phosphorylation of peptide 1. Inhibitors that selectively target each of these individual enzymes would be helpful in assessing which of these kinases is responsible for sensor phosphorylation. In addition, these inhibitors could prove particularly useful in correlating the activity of a specific kinase with the subsequent NEB signpost.
Compound 3 (Fig. 1) has been reported to be a selective PKC ß inhibitor versus the α and ε isoforms (11). However, its selectivity versus other PKC isoforms has not been explicitly addressed. Consequently, in addition to PKC α and ß, we examined the inhibitory potency of this derivative against the other isoforms present in PtK2 cells, namely ι, μ, θ and ζ. Compound 3 exhibits an IC50 of 11 ± 1 nM for the ßII isoform, which is consistent with the previously reported value of 5 nM (11). By contrast, the IC50 values for the other PKC isoforms are significantly larger. The IC50 for PKCα is 1.3 ± 0.1 μM and there is no observable inhibition for the ι, μ, θ or ζ isoforms or the Nek-2 protein kinase when 3 is present at 50 μM. Consequently, compound 3 serves as a highly selective inhibitor for the ß isoform. With this in mind, we examined the effect of compound 3 (12 μM in the cell media) on mitotic progression. We found that 65% of PtK2 cells (20 out of 31 cells) did not proceed from prophase into metaphase in the presence of 3. These results indicate that passage through NEB is not dependent upon Nek2 nor the ι, μ, θ or ζ PKC isoforms since compound 3 does not inhibit these enzymes at 12 μM. However, the 12 μM concentration of the indolylmaleimide 3 required to establish an NEB block in the majority of the cells is approximately 10-fold above the IC50 value for PKC α and significantly above that for PKC ß obtained under in vitro conditions. Consequently, although compound 3 rules out Nek2 and the ι, μ, θ or ζ PKC isoforms as effectors of peptide 1 phosphorylation and NEB, it does not allow us to conclusively distinguish between the PKC α or ß isoforms as the sensor 1 kinases.
The PKC α inhibitor 4 (Fig. 1, Ki = 800 pM) displays a 385-fold selectivity versus PKC ß and an even greater selectivity versus the other PKC isoforms (12). The caged sensor 2 and the Texas Red-dextran were co-injected along with two different concentrations of the PKC α-selective inhibitor 4, 2 mM (intracellular concentration: 20 – 200 μM) and 1 μM (intracellular concentration: 10 – 100 nM) (13). The cells were photolyzed to convert the caged sensor 2 into the active species 1. Under the first set of conditions (i.e. 2 mM inhibitor 4), both PKC α and ß should be inhibited, whereas under the latter conditions (i.e. 1 μM inhibitor 4), only the activity of PKC α should be compromised. Cells microinjected with 2 mM inhibitor 4 fail to exhibit PKC activity and do not progress through NEB (n = 10 cells). By contrast, cells injected with a 1 μM stock solution of the PKC α-selective inhibitor 4 exhibit both PKC activity and NEB (12 out of 15 cells). In short, compound 3 blocks sensor 1 phosphorylation as well as NEB at a concentration that is only consistent with catalysis by either PKC α or ß. Subsequent studies with compound 4 eliminated PKC α as the kinase sensor, thereby suggesting that PKC ß is the intracellular peptide 1 kinase required for NEB.
Ptk2 cells express both the type I and type II isoforms of PKC ß. These otherwise identical enzymes differ only in their C-terminal 50/52 amino acids (14). Unfortunately, there are no reported inhibitors or substrates that discriminate between these two closely related enzymes. There have been several reports describing the differential translocation of ßI and ßII in response to environmental stimuli (15 - 17). For example, Becker and Hannun have shown that both isoforms, when overexpressed in HeLa cells, are diffusely distributed throughout the cytoplasm and excluded from the nucleus (17). Upon phorbol ester stimulation, however, ßI migrates exclusively to the plasma membrane, whereas ßII is positioned at both the plasma membrane as well as a juxtanuclear location. To the best of our knowledge, the distribution of the two PKC ß isoforms has not been examined as a function of cell cycle. Immunocytochemistry revealed a marked difference in the localization of ßI and ßII in nonmitotic cells. ßI is predominantly associated with the nucleus, both during interphase and in prophase (Fig. 5). Furthermore, since the isoform is excluded from the nucleolus, this implies that ßI is present in the nucleus itself and not merely associated with the nuclear membrane (the latter would generate a uniform distribution about the nucleus). By contrast, ßII is distributed throughout the cytoplasm during interphase (Fig. 5). Upon entry into prophase, the subcellular distribution of ßI remains unchanged, namely this isoform retains its association with the nucleus. By contrast, ßII undergoes a partial subcellular redistribution as it accumulates at or near the nuclear boundary. In short, the subcellular distribution of both isoforms is consistent with a role in promoting nuclear collapse (i.e. via lamin phosphorylation). However, it is tempting to speculate that the cycle-dependent accumulation of ßII at the periphery of the nucleus may be significant, particularly with respect to subsequent NEB.
Figure 5.
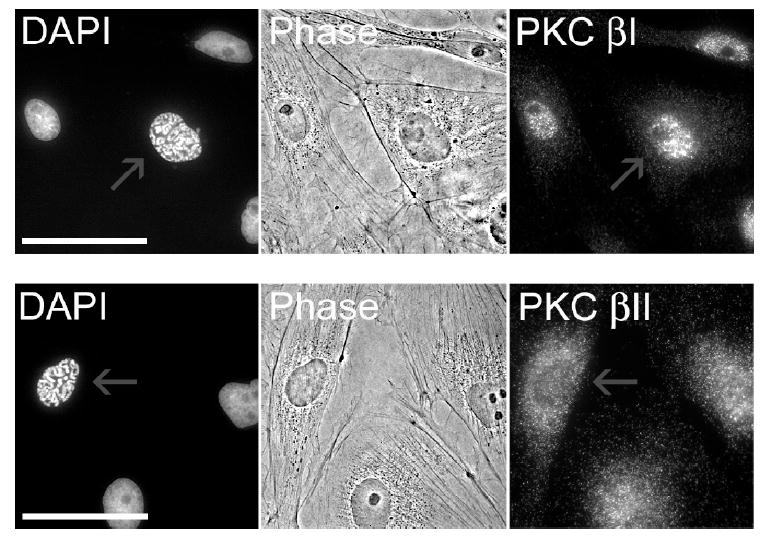
Localization of PKC ß isoforms in prophase (scale bar = 50 μm). Cells stained PKC ßI (upper panel) or PKC ßII (lower panel) antibodies and DAPI. Red arrows highlight cells in prophase. All other cells are in interphase.
Previous studies primarily employed cell free systems to implicate PKC ß as a lamin kinase activated prior to NEB (4). However, these studies employed cell lysates, which were obtained from a large population of chemically-induced synchronized cells. Unfortunately, cells released from aphidicolin or nocodazole blockades rarely progress through the division cycle as a uniform population. Furthermore, some cell lines, such as PtK2 cells, cannot be synchronized. The latter is especially unfortunate given the utility of this particular cell line as the exemplary model system to study mitosis. Perhaps most importantly, however, lysates from large cell populations are simply not ideal for examining short, yet critical time points, which encompass key cellular events. By contrast, the light-activatable sensor 2, provides a temporally precise method to interrogate protein kinase activity in an investigator-controlled fashion. In particular, we found that protein kinase activity is vigorous just prior to NEB, but is abruptly switched off following NEB. Furthermore, in combination with selective protein kinase inhibitors, we identified PKCß as the protein kinase that phosphorylates sensor 1 prior to nuclear envelope collapse.
Significance
Live cell enzymology offers unique challenges that generally do not need to be confronted in studies with pure enzymes or cell lysates. In particular, the cell and not the investigator controls the timing of enzymatic activity. Caged analogs of sensors, inhibitors, substrates, or other reagents offers a means to retain control over the activity of the reagent even after it has entered the cell. In short, light-activatable species allow the investigator to choose, with high temporal precision, when to assess intracellular activity. We have employed this approach to evaluate protein kinase activity in the minutes immediately prior to, during, and following nuclear envelope breakdown.
Experimental Procedures
Spectrofluorometric assays
All spectrofluorometric measurements were carried out on a Photon Technology QM-1 Instrument at 30 °C. The photomultiplier tube setting was held constant at 1000 V during all measurements. The assay volume and pH were maintained at 200 μL and 7.5 respectively. The reaction was initiated by addition of enzyme (10 μL) into the sensor (compound 1) assay buffer solution (190 μL) and the time-dependent change in fluorescence intensity (λex = 520 nm, λem = 560 nm) was continuously monitored. For a direct comparison of relative activities of various PKC isoforms and other protein kinases toward sensor 1, all measurements were carried out under optimum experimental conditions for the enzymes. For (A) PKC α, β, and γ: 62.5 mM HEPES (pH 7.5), MgCl2 (3 mM), CaCl2 (0.3 mM), EGTA (0.1 mM), ATP (1 mM), DTT (1 mM), PS (0.1 mg/mL), DAG (0.1 mg/mL), enzyme (50 nM), and compound 1 (100 μM), (B) PKC ζ and ι: 62.5 mM HEPES (pH 7.5), MgCl2 (10 mM), EGTA (0.5 mM), PS (0.04 mg/mL), ATP (1 mM), DTT (1 mM), enzyme (50 nM), and compound 1 (100 μM), (C) PKC μ, ε, δ, θ and η: 62.5 mM HEPES (pH 7.5), MgCl2 (10 mM), EGTA (0.5 mM), PS (0.04 mg/mL), DAG (0.01 mg/mL), ATP (1 mM), DTT (1 mM), enzyme (50 nM), and compound 1 (100 μM), (D) Nek2: 50 mM Tris-HCl buffer (pH 7.5), MgCl2 (10 mM), DTT (1 mM), ATP (1 mM), enzyme (50 nM) and compound 1 (100 μM), (E) Cdc2: 50 mM Tris-HCl buffer (pH = 7.5), MgCl2 (10 mM), DTT (2 mM), EGTA (1 mM), Brij 35 (0.01%), enzyme (“2 units”), and compound 1 (100 μM), (F) Akt: 20 mM Tris-HCl buffer (pH = 7.5), MgCl2 (10 mM), DTT (5 mM), enzyme (50 nM), and compound 1 (100 μM), (G) Plk1: 50 mM HEPES buffer (pH = 7.5), MgCl2 (10 mM), DTT (2.5 mM), Triton X 100 (0.01%), enzyme (50 nM), and compound 1 (100 μM), (H) Aurora kinase B: 50 mM Tris-HCl buffer (pH = 7.5), MgCl2 (10 mM), enzyme (50 nM), and compound 1 (100 μM).
IC50 Determination of PKC β inhibitor 3 against PKC isoforms
All inhibition studies were performed using the [γ−33P]ATP radioactive method in a 96 multiwell plate format at 30 °C. Optimal conditions (including specific peptide substrates) where chosen for the classical, novel, and atypical PKC isoforms as follows: (A) PKC α and β II: An assay volume of 50 μL/well was used. Thus, the assay involving 100 μM ATP was performed as follows. A 20 μl aliquot of inhibitor 3, appropriately diluted in 1:4 DMSO:H2O, was added (final concentration = 3310, 1655, 827.5, 413.75, 206.87, 103.4, 51.7, 25.6, 12.9 and 0 nM) to 20 μL of assay buffer to give a final concentration of 62.5 mM HEPES (pH 7.5), MgCl2 (10 mM), EGTA (0.5 mM), CaCl2 (0.8 mM), DAG (17.6 μg/mL), PS (100 μg/mL), peptide substrate Ac-Ser-Phe-Arg-Arg-Arg-Arg-NH2. (75 μM) and cold ATP (100 μM or 1 mM), supplemented with 1.5 μCi/well [γ−33P] ATP for radioactive detection. The reaction was initiated with the addition of 10 μL of enzyme buffer containing 20 mM Tris (pH 7.5), PKC (final concentration = 0.1 ng/μL), BSA (final concentration = 0.15 mg/mL), EDTA (final concentration = 0.2 mM), and DTT (final concentration = 0.2 mM). The reaction was allowed to progress for 15 min at 30 °C and quenched subsequently with 6% phosphoric acid (100 μl). An aliquot of 75 μL of quenched reaction mixture was transferred to a P81 cellulose phosphate paper unifilter plates and the solution was allowed to stand for 12 min. The solution was then filtered off and the membrane was washed with 0.1% phosphoric acid (4x), followed by water. The scintillation fluids (100 μL) were then added and counting was performed on a 1420 MicroBeta™ counter (Perkin Elmer). The rate of reaction was assessed by the incorporation of 33P into the peptide substrate. (B) PKC μ: performed as described above except that CaCl2 was not added and the peptide substrate Ala-Ala-Leu-Val-Arg-Gln-Met-Ser-Val-Ala-Phe-Phe-Phe-Lys-OH (EMD Biosciences) wasemployed at a final concentration of 60 μM. (C) PKC ι and ζ: performed as described above except that CaCl2 and DAG were not added and the peptide substrate Ac-Pro-Arg-Lys-Arg-Gln-Gly-Ser-Val-Arg-Arg-Arg-Val-NH2 was employed at a final concentration of 75 μM. SigmaPlot, Enzyme Kinetics 1.1, was used for data analyses.
Western blots
Cells were lysed (Laemmli sample buffer containing chymostatin, leupeptin, and pepstatin), proteins separated on a 4 - 20% Tris-HCl SDS-PAGE gel and subsequently transferred overnight onto a Immuno-Blot™ PVDF membrane at 4 °C. The membrane was incubated with 5% non-fat dry milk solution in Tris Buffer Saline with Tween (0.05%) for 1 hr and the membrane incubated with primary antibodies in 5% non-fat dry milk solution in TBS-T for 3 hr at RT. The following dilutions of primary antibodies were used, as recommended by the manufacturers: PKC α: (1:500); β1: (1:500); βII: (1:500); θ: (1:200); μ: (1:1000); ζ: (1:1000); ι: (1:200); γ: (1:1000); δ: (1:500); ε: (1:1000); η: (1:1000); λ: (1:1000). After incubation with primary antibody, the membrane was washed with TBS-T for 15 min (2x), and treated for 1 hr at RT with secondary antibodies (1:2000 dilution) conjugated to horseradish peroxidase (HRP). The membrane was subsequently washed for 15 min (2x) with TBS-T, it treated with HRP substrate (Pierce Supersignal West Pico Chemiluminescent Substrate) for 1 min and the chemiluminescent signal detected on film.
Live cell enzymology
PtK2 cells were cultured at 37 °C for 2 - 3 days (70 - 80% confluence) on 50 mm MatTek glass bottom dishes (MatTek Corp, Ashland, MA) containing photo-etched gridded coverslips (thickness 0.19 - 0.22 mm) in Dulbeco’s MEM with non-essential amino acids and 10% FBS in a humidified atmosphere containing 5% CO2. Prior to microinjection, the dishes were washed three times with PBS and then 4 mL of L-15 medium (without phenol red) supplemented with 10% FBS was added. The caged sensor 2 [synthesized and characterized as previously described (8)] was dissolved in PBS at a concentration of 200 μM and pre-filtered through a 0.22 μm centrifuge filter. Cells were microinjected with the 200 μM caged sensor and 1 μM of 70K dextran tagged with Texas Red (Molecular Probes, Eugene, OR) using a commercial microinjection system (Transjectors 5246, Eppendorf, Westbury, NY) at an estimated final concentration of 2 - 20 μM for the caged peptide and 10 – 100 nM Texas Red-dextran. Cells were exposed to UV illumination from a 100 W Hg-Arc lamp (using a set of 2 KG5 heat filters in the light path and a narrow band pass DAPI filter 355 ± 10 nm) through a PlanApo 40X N.A. 0.75 objective for 1 sec to activate the sensor (i.e. only cells in the immediate microscopic field were illuminated). Immediately after UV treatment, time-lapse images were collected with 2 × 2 binning using a Cooke Sensicam QE cooled CCD camera (driven by IPlab software) mounted on an Olympus 1X70 inverted microscope (Melville, NY) with a PlanApo 40X, 0.75 N.A. objective in conjunction with a 1.5X “booster” slider, and a FITC filter set with an excitation wavelength of 460 - 500 nm and an emission wavelength of 510 - 560 nm. A long working distance condenser (N. A. 0.55) was used. Images were collected every minute for 9 - 30 min (1000 ms exposure time). The digitized images were analyzed using ImageJ to measure the mean intensity of the whole cell. Images and fluorescence intensity measurements were also obtained from control cells that were injected with the same caged sensor in the presence of 200 μM staorosporine to make photobleaching adjustments. For experiments involving the PKCβ inhibitor 3 or the phosphatase inhibitor okadaic acid, cells were incubated with the relevant inhibitor for at least 1 hr prior to microinjection.
Immunofluorescence
To examine the distribution of PKC ß I and II isoforms throughout the cell cycle, PtK2 cells growing on glass coverslips were fixed in freshly prepared 3.7% formaldehyde in PBS for 15 min, permeabilized for 20 sec with −20 °C acetone, immediately rehydrated in PBS containing 0.02% azide, and blocked in PBS containing 1% bovine serum albumin (BSA). Cells were then incubated with primary PKC ß I (C-16) sc-209 or PKC ß II (C-18) sc-210 rabbit polyclonal antibodies (Santa Cruz Biotechnology) at a dilution of 1:100 in PBS containing 1% BSA for 1 h at RT. After washing with 1% BSA in PBS, cells were incubated with a Cy3-conjugated donkey anti-rabbit antibody (Jackson ImmunoResearch Lab) at a dilution of 1:500 in 1% BSA in PBS for 40 min at RT and rinsed again. Coverslips were incubated with DAPI (Sigma) at 0.1 μg/mL in 1% BSA in PBS for 10 min before mounting in Pro-Long Antifade (Molecular Probes). Immunofluorescence images were acquired using IPLab Spectrum software (Scanalytics, Inc.) and a CoolSNAP HQ interline 12-bit, cooled CCD camera (Roper Scientific) mounted on an Olympus IX70 microscope with a PlanApo 60X, 1.4 N.A. objective (Olympus) and HiQ bandpass filters (Chroma Technology Corp.). Images were processed using Photoshop (Adobe Systems).
Supplementary Material
Supplemental movies are available online.
Acknowledgments
We thank the NIH [CA095019 (DSL) and GM069945 (ARB)] for financial support. DSL is a member of the scientific advisory board of Panomics, the company to which this work has been licensed.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lawrence DS. The preparation and in vivo applications of caged peptides and proteins. Curr Opin Chem Biol. 2005;9:570–575. doi: 10.1016/j.cbpa.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 2.Black JD. Protein kinase C-mediated regulation of the cell cycle. Front Biosci. 2000;5:406–423. doi: 10.2741/black. [DOI] [PubMed] [Google Scholar]
- 3.Murray NR, Burns DJ, Fields AP. Presence of a ßII protein kinase C-selective nuclear membrane activation factor in human leukemia cells. J Biol Chem. 1994;269:21385–21390. [PubMed] [Google Scholar]
- 4.Hocevar BA, Burns DJ, Fields AP. Identification of protein kinase C (PKC) phosphorylation sites on human lamin B. J Biol Chem. 1993;268:7545–7552. [PubMed] [Google Scholar]
- 5.Goss VL, Hocevar BA, Thompson LJ, Stratton CA, Burns DJ, Fields AP. Identification of nuclear beta II protein kinase C as a mitotic lamin kinase. J Biol Chem. 1994;269:19074–19080. [PubMed] [Google Scholar]
- 6.Thompson LJ, Fields AP. ßII protein kinase C is required for the G2/M phase transition of cell cycle. J Biol Chem. 1996;271:15045–15053. doi: 10.1074/jbc.271.25.15045. [DOI] [PubMed] [Google Scholar]
- 7.Yeh R-H, Yan X, Cammer M, Bresnick AR, Lawrence DS. Real Time Visualization of Protein Kinase Activity in Living Cells. J Biol Chem. 2002;277:11527–11532. doi: 10.1074/jbc.M111300200. [DOI] [PubMed] [Google Scholar]
- 8.Veldhuyzen WF, Nguyen Q, McMaster G, Lawrence DS. A light-activated probe of intracellular protein kinase activity. J Am Chem Soc. 2003;125:13358–13359. doi: 10.1021/ja037801x. [DOI] [PubMed] [Google Scholar]
- 9.Nishikawa K, Toker A, Johannes FJ, Songyang Z, Cantley LC. Determination of the specific substrate sequence motifs of protein kinase C isozymes. J Biol Chem. 1997;272:952–960. doi: 10.1074/jbc.272.2.952. [DOI] [PubMed] [Google Scholar]
- 10.Ferrari S. Protein kinase controlling the onset of mitosis. Cell Mol Life Sci. 2006;63:781–795. doi: 10.1007/s00018-005-5515-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tanaka M, Sagawa S, Hoshi J, Shimoma F, Matsuda I, Sakoda K, Sasase T, Shindo M, Inaba T. Synthesis of anilino-monoindolylmaleimides as potent and selective PKCß inhibitors. Bioorg Med Chem Lett. 2004;14:5171–5174. doi: 10.1016/j.bmcl.2004.07.061. [DOI] [PubMed] [Google Scholar]
- 12.Lee JH, Nandy SK, Lawrence DS. A highly potent and selective PKCα inhibitor generated via combinatorial modification of a peptide scaffold. J Am Chem Soc. 2004;126:3394–3395. doi: 10.1021/ja037300b. [DOI] [PubMed] [Google Scholar]
- 13.The 10- to 100-fold dilution upon microinjection is estimated from the equation for volume (V) flow through a capillary tube where p is the difference in pressure at the ends of the tube (290 hPa), r the radius (0.05 μm) and l the length (10 μm) of the tube, η the viscosity of the injected solution (0.69 × 10-2 g/cm-sec), and t the total injection time. The average volume of a fibroblast is 2 pL Kislauski EH, Zhu X-C, Singer R. J Cell Biol. 1997;136:1263–70. doi: 10.1083/jcb.136.6.1263.
- 14.Ono Y, Kikkawa U, Ogita K, Fujii T, Kurokawa T, Asaoka Y, Sekiguchi K, Ase K, Igarashi K, Nishizuka Y. Expression and properties of two types of protein kinase C: alternative splicing from a single gene. Science. 1987;236:1116–120. doi: 10.1126/science.3576226. [DOI] [PubMed] [Google Scholar]
- 15.Becker KP, Hannun YA. Isoenzyme-specific translocation of protein kinase C (PKC) ßII and not PKCßI to a juxtanuclear subset of recycling endosomes: involvement of phospholipase D. J Biol Chem. 2004;279:28251–28256. doi: 10.1074/jbc.M400770200. [DOI] [PubMed] [Google Scholar]
- 16.Goodnight JA, Mischak H, Kolch W, Mushinski JF. Immunocytochemical localization of eight protein kinase C isozymes overexpressed in NIH 3T3 fibroblasts. Isoform-specific association with microfilaments, Golgi, endoplasmic reticulum, and nuclear and cell membranes. J Biol Chem. 1995;270:9991–10001. doi: 10.1074/jbc.270.17.9991. [DOI] [PubMed] [Google Scholar]
- 17.Disatnik MH, Winnier AR, Mochly-Rosen D, Arteaga CL. Distinct responses of protein kinase C isozymes to c-erbB-2 activation in SKBR-3 human breast carcinoma cells. Cell Growth Differ. 1994;5:873–880. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental movies are available online.