

Abstract
Lysosomal storage is the most common cause of neurodegenerative brain disease in preadulthood. However, the underlying cellular mechanisms that lead to neuronal dysfunction are unknown. Here, we report that loss of Drosophila benchwarmer (bnch), a predicted lysosomal sugar carrier, leads to carbohydrate storage in yolk spheres during oogenesis and results in widespread accumulation of enlarged lysosomal and late endosomal inclusions. At the bnch larval neuromuscular junction, we observe similar inclusions and find defects in synaptic vesicle recycling at the level of endocytosis. In addition, loss of bnch slows endosome-to-lysosome trafficking in larval garland cells. In adult bnch flies, we observe age-dependent synaptic dysfunction and neuronal degeneration. Finally, we find that loss of bnch strongly enhances tau neurotoxicity in a dose-dependent manner. We hypothesize that, in bnch, defective lysosomal carbohydrate efflux leads to endocytic defects with functional consequences in synaptic strength, neuronal viability, and tau neurotoxicity.
Introduction
Intact lysosomal function is critical for normal neuronal functioning and survival (Nixon and Cataldo, 1995). In humans, this is most readily illustrated by a group of inherited childhood diseases called lysosomal storage disorders (LSDs) (Neufeld, 1991), in which neuronal brain degeneration is a frequent pathological feature (Walkley, 1998). In preadulthood, lysosomal storage, mainly in the form of lipofuscinosis, is the most common cause of neurodegeneration (Cooper, 2003). In LSDs, lysosomes increase in number and size through the gradual intra-lysosomal build up of storage material. In most cases, substrates accumulate due to the loss of lysosomal hydrolytic enzyme activity, although other causes, such as defective efflux of normally degraded constituents by lysosomal transporters, are also known (Eskelinen et al., 2003). Although studies of LSDs have provided tremendous insights into the biochemical details of lysosomal hydrolytic degradation and although the role of sphingolipid accumulation in endocytic membrane sorting is becoming increasingly recognized (Sillence and Platt, 2003), the cellular mechanism by which lysosomal storage disrupts neuronal viability and membrane trafficking is incompletely understood (Futerman and van Meer, 2004). This is important, however, because the tight association of LSDs with neuronal degeneration suggests that similar mechanisms leading to neurological dysfunction might be shared by at least some of these disorders. Furthermore, understanding the link between lysosomal dysfunction and neurodegeneration is likely to be important for highly prevalent neurodegenerative disorders like Alzheimer's disease. Indeed, endosomal and lysosomal alterations have since long been thought to be an early and prominent feature in Alzheimer's disease (Nixon et al., 2001).
In recent years, Drosophila has become an established model system to study neurodegeneration by using endogenous loss-of-function and heterologous toxic gain-of-function or misexpression approaches (Muqit and Feany, 2002; Bonini and Fortini, 2003). However, no Drosophila models exist that conclusively link a primary lysosomal defect with progressive neurodegeneration, although mutants including eggroll and blue cheese have suggested such a causal relationship (Min and Benzer, 1997; Finley et al., 2003). We report here a neurodegenerative mutant called benchwarmer (bnch), and establish bnch as a model for lysosomal storage-related neurodegeneration. bnch appears most similar to LSDs like sialic acid storage disease and Niemann-Pick disease type C, which are caused by defects in the removal or transport of substrate from lysosomes.
Since we first reported bnch alleles (Kania, 1996), others have isolated allelic Drosophila mutations in screens for abnormal sexual behavior and larval neuromuscular synapse formation, which were assigned the name spinster and diphthong, respectively (Nakano et al., 2001; Sweeney and Davis, 2002). Besides reported defects in courtship behavior, hypomorphic bnch flies have a decreased adult life span, a reduction of neuronal programmed cell death in the pupal central nervous system. and accumulate neuronal autofluorescent pigments (Nakano et al., 2001). In third instar larvae, loss of bnch further causes neuromuscular synaptic overgrowth that is due to increased TGF-β signaling (Sweeney and Davis, 2002). Overexpression of the human homologue in human embryonic kidney cells results in nonapoptotic autophagic/necrotic cell death (Yanagisawa et al., 2003), and loss of the Zebrafish homologue, not really started (nrs), leads to embryonic lethality characterized by accumulation of opaque substances in the yolk (Young et al., 2002). Although these studies have revealed developmental functions of bnch in synaptic growth, decreased programmed cell death, and late endosomal/lysosomal function, its role in the adult nervous system remains uncharacterized.
Here, we demonstrate that loss of bnch leads to lysosomal carbohydrate storage, synaptic defects, subsequent progressive neuronal degeneration, and enhanced tau-mediated toxicity. Ultrastructural, electrophysiological, histological, and endocytic tracer analysis suggest that these phenotypes result from aberrant lysosomal function and defects in endocytic membrane trafficking.
Results
bnch is a semi-lethal locus encoding a highly conserved predicted sugar transporter of the major facilitator superfamily
We identified the bnch mutation in an enhancer detector screen for genes expressed in the nervous system (Kania et al., 1995): P{LacW}[25/11] is inserted 19 bp upstream of the transcription start site of CG8428 (Fig. 1 A) (Kania, 1996). Allelic mutations were recently reported as spinster and diphthong (Nakano et al., 2001; Sweeney and Davis, 2002). Using P{LacW}[25/11], we found LacZ expression in a subset of glial cells in the embryonic, larval, pupal, and adult nervous system (unpublished data), similar to enhancer detector-induced LacZ expression patterns reported previously (Nakano et al., 2001). However, using an anti-Bnch antibody in adult wild-type brains, we observed that Bnch expression is enriched in—but not restricted to—glial cells and also present in neurons (unpublished data).
Figure 1.

bnch loss of function alleles are semi-lethal and disrupt a highly conserved predicted anion/sugar permease of the MFS. (A) The genomic bnch locus (52E, 14 kb) encodes at least five alternatively spliced transcripts. Translation initiation and termination sites are indicated in red. The approximate location of the P{lacW}[25/11] insertion and point mutations (11F5, E14.1, N, P) is shown. The extent of small deletions (Δ29, Δ31, Δ86) generated by imprecise P-element excision of P{LacW}[25/11] is indicated. The alleles affect gene regions that are common to all alternatively spliced isoforms. (B) Predicted membrane topology for Bnch showing the 12 transmembrane domains typical for proteins of the MFS. The position of the anion/cation (ACS) domain signature is indicated in red. EMS mutations result in the introduction of stop codons (for 11F5, E14.1, and P) or a nonconservative amino acid change (for N) and their positions in the protein are indicated by asterisks. The position of the N missense mutation is shown in a partial multiple alignment of eukaryotic Bnch proteins and consensus sequences of the ACS and sugar porter (SP) subfamilies. The percentages identity and similarity of eukaryotic Bnch proteins to Drosophila Bnch are given in the table. Number of identified Bnch homologues per species is between parentheses. (C) Complementation analysis of bnch point mutations (11F5, E14.1, N, P), excision alleles (Δ29, Δ31, Δ86), deletions (Δ2B, Jp8) and P-element insertion (25/11) indicates allelism. The P{LacW}[25/11] chromosome and its derivatives contain an unrelated lethal mutation that is uncovered by the Jp8 deficiency. The frequency of homozygous and transheterozygous escapers (in parentheses) is not absolute and strongly dependent on the genetic background and culture conditions.
Bnch has been reported as a transporter-like protein with 8 putative transmembrane domains (Nakano et al., 2001; Sweeney and Davis, 2002; Young et al., 2002). However, we found a predicted 12-transmembrane domain structure, and homology searches identify significant similarity to transporters of the major facilitator superfamily (MFS) (Fig. 1 B) (Pao et al., 1998). MFS transporters are single polypeptides containing 12 transmembrane domains, capable of transporting a wide range of small solutes, including carbohydrates and amino acids. Bnch has highly conserved homologues in eukaryotic genomes with 42–63% similarity and 26–50% identity (Fig. 1 B). Bnch has similarities with members of the sugar porter subfamily and a Caenorhabditis elegans Bnch homologue has been reported as a member of the anion/cation subfamily (ACS) of MFS transporters (Pao et al., 1998) (Fig. 1 B).
By imprecise P-element excision, small deletions (Δ29, Δ31, Δ86) were generated that disrupt transcription of CG8428 (Fig. 1 A). We have also identified point mutations in common exons of this alternatively spliced gene in four alleles of l(2R)W5, a lethal complementation group in this region (Liu et al., 1999) (Fig. 1, A and B). The four point mutations affect all five predicted alternatively spliced isoforms (Fig. 1 A). In the Bnch protein, these point mutations result in loss of function of bnch through the introduction of stop codons (for bnch 11F5, bnch E14.1, and bnch P) or nonconservative amino acid change, which changes the acidic E to a basic K (for bnch N) (Fig. 1 B). Interestingly, the N mutation affects a consensus residue that is conserved among members of the ACS transporters (Fig. 1 B).
Complementation analysis with our excision alleles, point mutations, and the reported bnch[Δ2B] null allele (Sweeney and Davis, 2002) reveals that our bnch alleles are null, or at least strong loss-of-function alleles (Fig. 1 C). Transheterozygous combinations of null alleles of bnch are pupal to pharate adult lethal. However, strongly dependent on the culture conditions, a small fraction of adult escapers survive to adulthood in some transheterozygote combinations. Adult escapers exhibit progressive locomotor defects, such as difficulty in righting after a fall and an overall lower level of locomotor activity, hence the name benchwarmer, to honor athletes who play rarely. These behavioral defects worsen during the days after emergence and result in death within 5–12 d. Morphologically bnch escapers appear largely normal, although a subtle and incompletely penetrant extra wing vein phenotype can be observed.
bnch germline clones reveal oogenesis defects associated with defective mobilization of carbohydrates from yolk β-spheres
Developmental gene expression of bnch suggests an important maternal component during early stages of embryonic development (http://genome.med.yale.edu/Lifecycle/) (Arbeitman et al., 2002) that is possibly responsible for the late lethal phase associated with zygotic loss of bnch. Moreover, in zebrafish, loss of the bnch homologue nrs results in early embryonic lethality and defective yolk metabolism (Young et al., 2002). To uncover early developmental consequences of loss of bnch in Drosophila, we generated germline clones.
In the absence of bnch in the germline, few eggs are laid and none become fertilized. The eggs are small, varying from 1/3 to 2/3 of wild-type size (Fig. 2, A and D). The dorsal appendages are malformed and often positioned laterally compared with the wild type. These results indicate that bnch is required for oogenesis.
Figure 2.
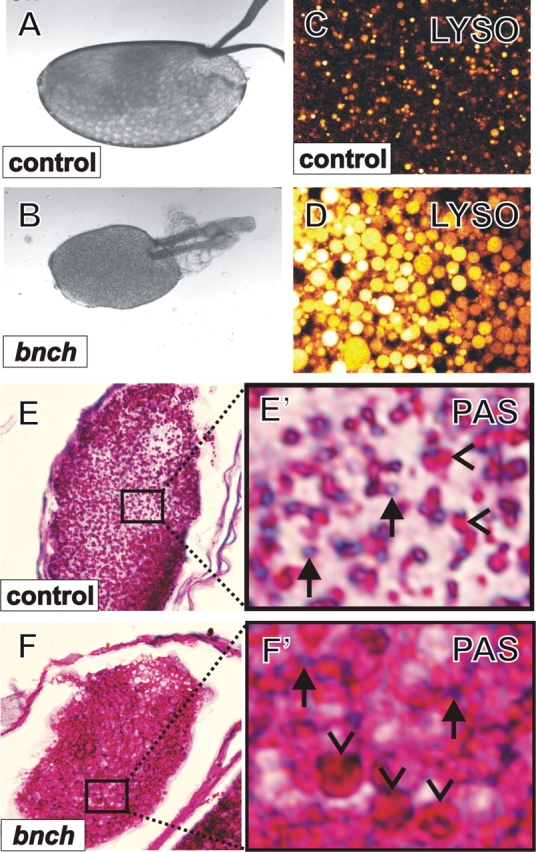
bnch is required for oogenesis and carbohydrate mobilization from yolk spheres. (A and B) Germline clones of bnch E14.1 give rise to malformed oocytes (B) compared with wt (A). The eggs are of significantly reduced size (∼1/3 to 2/3 of wild-type length). Few are laid and none are fertilized. The dorsal appendages are abnormally formed and their distance is somewhat widened. (C and D) Lysotracker staining reveals dramatic expansion of the acidic ooplasmic compartment in bnch 11F5 (D) compared with wild-type oocytes (C). (E, E′, F, and F′) In bnch Δ 2b mutant ooplasm PAS staining reveals dramatic expansion of red PAS positive β-spheres (F′, arrowheads) compared with the control (E′, arrowheads), whereas blue PAS-negative α-spheres are similar in size in bnch (F′, arrows) and control (E′, arrows) ooplasm.
Because bnch is implicated in late-endosomal/lysosomal function and because degradation of yolk protein and carbohydrates in the lysosome-related yolk spheres is crucial for Drosophila oogenesis (DiMario and Mahowald, 1987), we labeled bnch mutant mature oocytes using Lysotracker. Interestingly, we found that the size of acidic vesicles in the bnch-deficient ooplasm is dramatically increased compared with wild type (Fig. 2, B and C). Because the ooplasm of Drosophila contains large amounts of modified lysosomes in the form of yolk spheres (Fagotto, 1995), the acidic compartments likely correspond to yolk spheres that function as nutritional storage organelles of proteins and carbohydrates.
During the last stages of Drosophila oogenesis, protein and carbohydrate reserves are compartmentalized in two different types of yolk spheres and can be differentiated histologically using the periodic acid Schiff (PAS) method (Gutzeit et al., 1994). We therefore performed PAS staining in bnch mutant and control mature oocytes. Although control ooplasm contained similarly sized PAS-negative protein containing α-spheres and PAS-positive carbohydrate containing β-spheres (Fig. 2, E and E′), the bnch mutant ooplasm showed dramatically enlarged β-spheres but α-spheres with similar sizes as in controls (Fig. 2, F and F′). This selective enlargement of β-spheres in the bnch ooplasm suggests that during oogenesis bnch is required for mobilizing carbohydrate but not protein stores from yolk spheres.
Together, our germline clonal analyses indicate that bnch is required for oogenesis and female fertility. At a subcellular level, bnch functions in this process in a subtype of ooplasmic yolk spheres that is specialized in regulated degradation of yolk carbohydrates, but not proteins. Given the lysosomal localization of Bnch (Sweeney and Davis, 2002) and in agreement with its predicted role as a possible sugar transporter, we hypothesize that the oogenesis and fertility defects caused by maternal loss of bnch are related to defective efflux of nutritional carbohydrates from yolk spheres.
bnch mutant eyes accumulate membranous inclusions corresponding to late endosomal, autophagic, and lysosomal structures
Previous studies have suggested a role for bnch in late endosomal/lysosomal membrane trafficking and neuromuscular synapse growth (Nakano et al., 2001; Sweeney and Davis, 2002). To examine the subcellular and developmental consequences of zygotic loss of bnch in the visual system, we performed transmission electron microscopy (TEM) studies of adult homozygous eyes. Because bnch escaper frequencies are generally low and escapers have a short life span, we used the eyFLP system to generate clones in the retina and the lamina (Fig. 3). In the retina we observed a large number of abnormal membranous inclusions in the cell bodies of mutant photoreceptors, while rhabdomere morphology was intact (Fig. 3, A and B). Compared with controls, bnch mutant photoreceptors displayed a 6–10-fold increase in the number of membranous cytoplasmic inclusions (Fig. 3 C).
Figure 3.
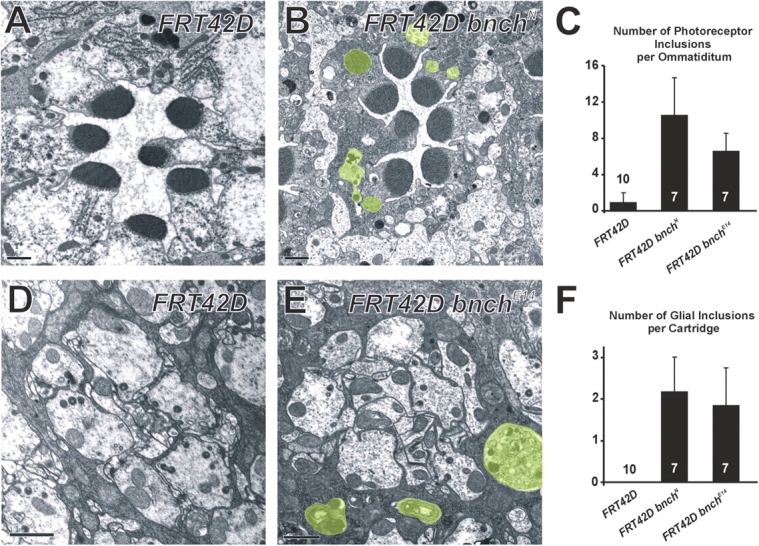
Cytoplasmic membranous inclusions accumulate in the bnch mutant visual system. (A–C) Retinal TEM sections. Compared with controls (A) bnch mutant photoreceptors (B) accumulate cytoplasmic inclusions. Inclusions are highlighted in transparent green (B) and quantified (C). Error bars indicate SEM. (D–F) Laminar TEM sections. Compared with controls (D) bnch mutant laminar cartridges (E) accumulate cytoplasmic inclusions. Inclusions are highlighted in green (E) and quantified (F). Error bars indicate SEM. Bars, 1 μm.
In the lamina, bnch mutant cartridges also accumulated large membranous inclusions, mainly in the glial cell bodies (Fig. 3, D–F). We did not observe abnormalities in laminar architecture, number of photoreceptor terminals per cartridge, or number of glial invaginations per photoreceptor terminal. In addition, labeling photoreceptor cell membranes using mAb 24B10 showed normal photoreceptor projection patterns in the lamina and medulla (unpublished data).
The membranous cytoplasmic inclusions are morphologically highly heterogeneous and vary in size from 0.2 to 2.5 μm (Fig. 4). One population of inclusions contains multilayered membranes often together with partially degraded organelles or other cytoplasmic constituents (Fig. 4 A). Most likely these structures correspond to secondary lysosomes in the phase of digesting endosomal and/or autophagic cargo. This heterogeneous population of secondary lysosomes was observed in retinal photoreceptor cell bodies (Fig. 3 B) and laminar glial cells (Fig. 3 E and Fig. 4 A). In the bnch mutant lamina, these multilamellar structures were also found in presynaptic photoreceptor projections as well as postsynaptic monopolar cells (Fig. 4, C and D).
Figure 4.
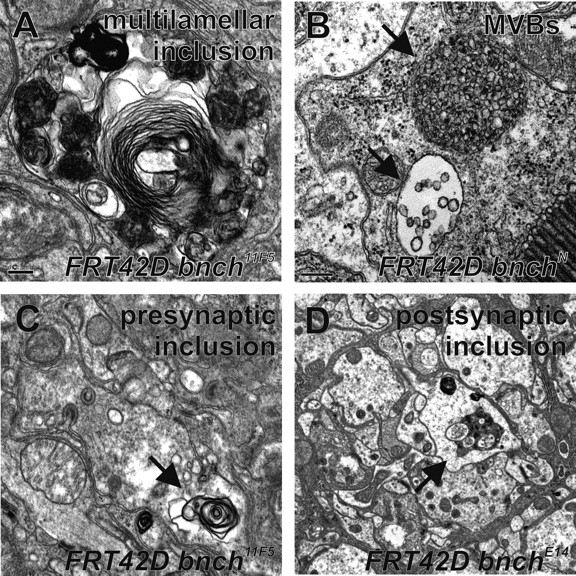
Lysosomes and MVBs in the bnch mutant visual system. (A–D) TEM sections through bnch mutant eyes. (A) Multilammellar onion-like structure with morphologically heterogeneous content in a laminar bnch mutant glial cell (bar, 0.2 μm). (B) MVBs (arrows) in a bnch mutant retinal photoreceptor (bar, 0.2 μm). (C and D) Multilammellar bodies (arrows) are present in a presynaptic photoreceptor terminal (C) as well as a postsynaptic laminar monopolar cell (D).
A second population of inclusions, found in the photoreceptors but not in the laminar glia, consists of organelles with a single limiting membrane surrounding many small regularly shaped internal vesicles (Fig. 3 B and Fig. 4 B). These correspond to enlarged multivesicular bodies (MVBs), which typically represent late endosomes.
These data suggest that loss of bnch in eyFLP clones does not cause striking developmental defects in the visual system. However, the accumulation of enlarged lysosomes with partially degraded contents suggests important defects in lysosomal degradation. In the endocytotically highly active photoreceptors (Stark et al., 1988), this primary lysosomal defect is accompanied by a dramatic expansion and accumulation of MVBs.
Abnormal membranous inclusions and decreased synaptic vesicle endocytosis at the bnch larval neuromuscular junction
In neurons, late endosomes and lysosomes are predominantly located in the soma (Parton et al., 1992). However, the nerve terminal produces significant amounts of endocytic and autophagic cargo destined for degradation (Hollenbeck, 1993). It is assumed that these prelysosomal organelles (Parton et al., 1992; Hollenbeck, 1993) undergo retrograde axonal transport and that their cargo is degraded in lysosomal compartments along the axon and in the soma (Overly et al., 1995; Overly and Hollenbeck, 1996).
At the larval neuromuscular junction (NMJ) of bnch mutants, however, Sweeney and Davis (2002) reported an expanded presynaptic vesicular compartment that is acidic as evidenced by Lysotracker staining. Interestingly, at the larval NMJ we observe abnormal ultrastructural membrane compartments in the cytoplasm of bnch mutant boutons (Fig. 5, B–D), but not in wild-type controls (Fig. 5 A). Given their size and absence in wild-type boutons, these structures likely represent the reported presynaptic acidic compartments (Sweeney and Davis, 2002). These multilamellar structures range in size from 0.3 to 2 μm and resemble the multilayered lysosomal membrane structures present in the visual system.
Figure 5.
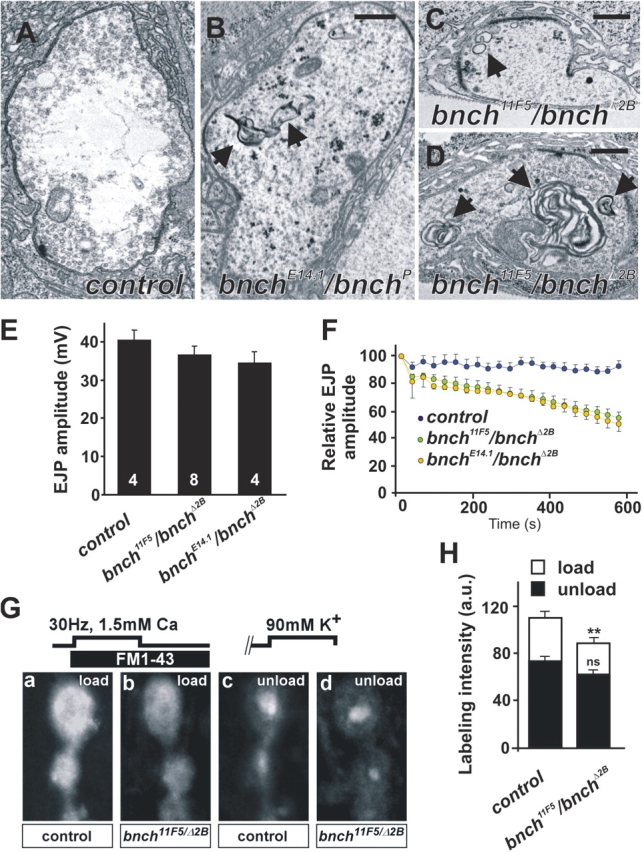
Abnormal membranous structures and presynaptic endocytic defects at the bnch NMJ. (A–D) Ultrastructure of yw (A, control), yw; bnch E14.1/bnchP (B) and yw; bnch 11F5/bnchΔ2B (C and D) NMJ boutons at rest. Bars represent 0.5 μm. Membranous inclusions in bnch mutant boutons range from smaller (0.2 μm) vesicular structures with a single limiting membrane (C, arrow) to larger multilamellar bodies (0.3–1.2 μm) (B and D, arrows) that are absent in controls (A). (E and F) EJP recordings in 1 mM Ca2+ from muscle 6 in yw (control), yw; bnch 11F5/bnchΔ2B, and bnch 11F5/bnchΔ2B third instar larvae. Quantification of EJP amplitudes shows no significant differences between bnch mutants and controls (E). (F) High frequency (10 Hz) stimulation during 10 min reveals a gradual rundown of the EJP amplitudes in bnch mutants (green and yellow) but not controls (blue). (G and H) FM1-43 dye loading and unloading experiments in yw (control) and yw; bnch 11F5/bnchΔ2B third instar larval NMJs preparations. FM1-43 dye was loaded during 5 min 30 Hz stimulation in 1.5 mM Ca2+ and 5 min rest (G, a and b). Unloading of the RRP of synaptic vesicles was achieved by 90 mM K+ stimulation during 5 min (G, c and d). Note the subtle decrease in FM1-43 uptake in bnch mutants (G, b) compared with controls (G, a). Unloading of the RRP is similar in bnch mutants (G, d) and controls (G, c). Quantification of these results is shown (H).
To determine if the observed ultrastructural defects in late endosomal/lysosomal compartments functionally impair exocytosis and/or endocytosis at the bnch larval NMJ, we stimulated the motor neurons and recorded excitatory junctional potentials (EJPs) of abdominal muscles. In 1 mM Ca2+ the amplitude of EJPs in bnch transheterozygotes is slightly but statistically not significantly decreased compared with controls. Hence, bnch barely affects exocytosis in this paradigm (Fig. 5 E). Next, to reveal defects in the synaptic vesicle cycle, we repetitively stimulated motor neurons and monitored the amplitude of the EJP over time. At high stimulation frequency (10 Hz), the amplitude of the EJP measured in bnch animals declines to 55–60% of the original response after 10 min, whereas such decline was absent in control animals (Fig. 5 F). The magnitude of this effect is similar to the defects seen with mutations in dap160, a protein that stabilizes a macromolecular complex implicated in synaptic vesicle endocytosis (Koh et al., 2004). Hence, these results are consistent with a defect in synaptic vesicle recycling, most likely at the level of vesicle endocytosis.
To test the possibility of impaired synaptic vesicle recycling and endocytosis more directly, we incubated third instar fillets with FM1-43, a dye that is internalized into vesicles during clathrin-mediated endocytosis (Betz et al., 1992). To distinguish between the readily releasable pool (RRP) and reserve pool (RP) of synaptic vesicles, we used an FM1-43 labeling protocol to first load the total recycling pool (RRP plus RP) and subsequently selectively unload the RRP (Kuromi and Kidokoro, 2002). Interestingly, in bnch mutant boutons, we observed a significant nearly 20% decrease in uptake of FM1-43 compared with controls, whereas unloading of the RRP was similar in bnch mutants and controls (Fig. 5, G and H). These results suggest that bnch mutations lead to a subtle defect in synaptic vesicle endocytosis but not exocytosis. Together, the ultrastructural, electrophysiological, and FM1-43 uptake data suggest the existence of a presynaptic lysosomal compartment that is important for efficient synaptic vesicle recycling.
bnch mutations affect endosome-to-lysosome trafficking in garland cells
Our ultrastructural analysis in the visual system reveals a dramatic accumulation of abnormal inclusions suggestive of a primary lysosomal defect. Moreover, the accumulation of MVBs in mutant photoreceptors is consistent with a decreased rate of endosome-to-lysosome trafficking, and at bnch mutant neuromuscular synapses we find defects consistent with impaired synaptic vesicle recycling at the level of endocytosis. To further test if bnch mutations affect dynamic aspects of endocytosis, we performed endocytic tracer uptake experiments in live larval garland cells (Fig. 6). Garland cells are similar to nephrocytes, have a rapid rate of fluid phase endocytosis, and are an established system to study endocytic dynamics (Kosaka and Ikeda, 1983). Endocytic uptake of Alexa 488–conjugated avidin (A-avidin) in garland cells labeled with Lysotracker was monitored over time. Although strong colabeling of A-avidin–positive endosomes and Lysotracker was already observed after 20 min in wild-type garland cells, A-avidin–positive endosomes were still easily detectable outside the Lysotracker-positive compartments in bnch mutant cells. After 90 min, A-avidin– and Lysotracker-positive compartments nearly completely overlapped in both bnch and wild-type cells. These results suggest that, although endosome-to-lysosome trafficking is not inhibited, the rate of this process is reduced in bnch-deficient larval garland cells, in agreement with the previous observations.
Figure 6.
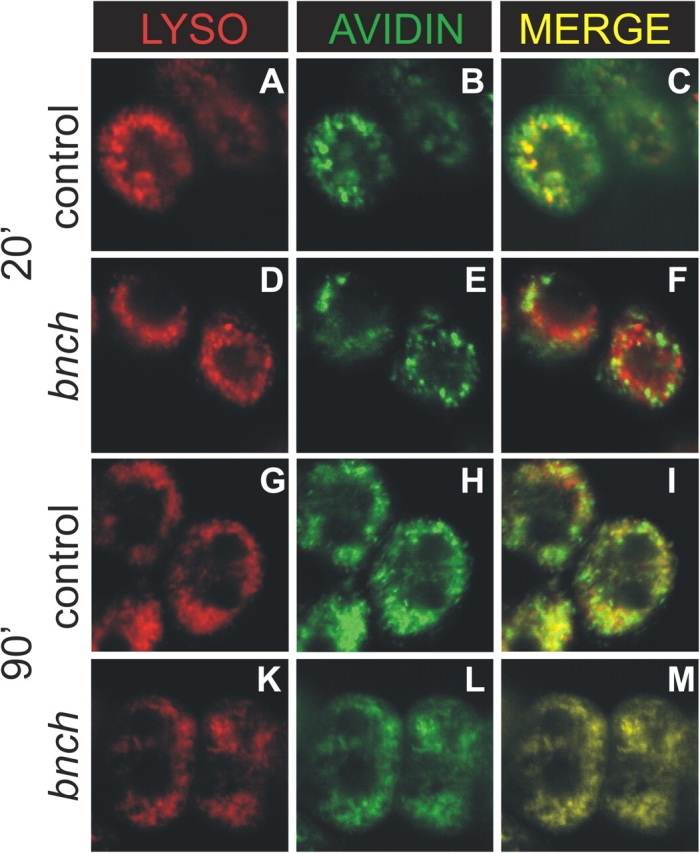
Endosome-to-lysosome trafficking is reduced in bnch mutant garland cells. Endocytosed A-avidin is labeled in green and Lysotracker in red. After 20 min, strong colabeling of avidin positive endosomes and Lysotracker was observed in wild-type cells (A–C), whereas in bnch mutant cells the vast majority of A-avidin positive endosomal compartments was detected outside Lysotracker-positive compartments (D–F). After 90 min A-avidin– and Lysotracker-positive compartments nearly completely overlapped in both wild-type (G–I) and bnch cells (K–M).
Synaptic function is impaired in the visual system of bnch escapers and age-dependently deteriorates in bnch eye mosaics
To examine the functional consequences of loss of bnch at visual synapses, we performed electroretinogram (ERG) recordings (Fig. 7). We found that 1-d-old mosaic animals show a normal depolarization in response to light and have normal on/off transients (Fig. 7 A). In agreement, they behave normally during phototaxis assays (unpublished data). However, in 40-d-old bnch mosaic flies but not control flies, ERG recordings consistently displayed reduced depolarization amplitudes and smaller or absent on/off transients (Fig. 7 A). These results show that, although normal at eclosion, phototransduction and visual synaptic transmission progressively deteriorate over a 40-d period in bnch mutant eyes.
Figure 7.
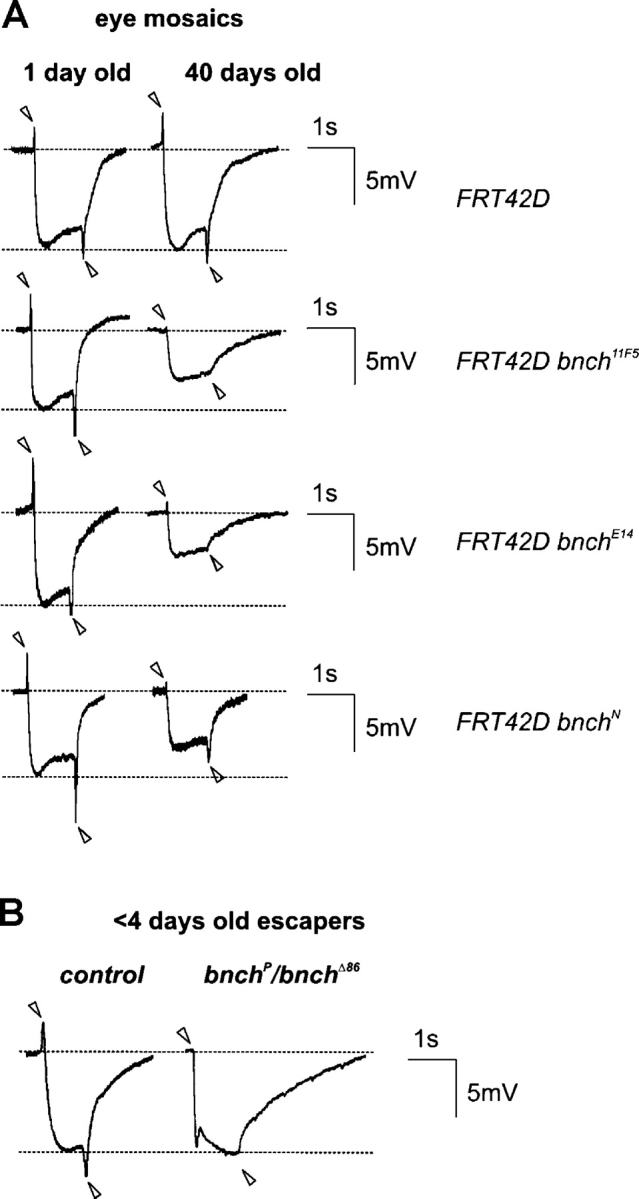
Impaired visual synaptic function in bnch escapers and aged eye mosaics. (A) ERG recordings in 1-d-old flies having mosaic bnch eyes are similar to wild type. In 40-d-old bnch eye mosaic flies, ERG recordings reveal reduced depolarization in response to light and smaller to absent on/off transients (arrowheads) with stronger phenotypes for bnch alleles with premature stop codons (11F5 and E14.1) compared with bnch missense allele (N). (B) ERGs of bnch adult escapers show a depolarization in response to light similar to controls but lack on/off transients (arrowheads) and exhibit slowed repolarization.
In adult bnch escapers, ERGs recorded <4 d after eclosion are also abnormal (Fig. 7 B). In such animals depolarization is normal, but the repolarization is slowed and the on/off transients are completely missing. Hence, in whole mutant animals visual synaptic transmission, but not phototransduction, is severely impaired.
Together, these results demonstrate that loss of bnch leads to defects in visual synaptic transmission that deteriorate in an age-dependent manner.
bnch mutations cause age-dependent neurodegeneration associated with carbohydrate-containing granules
Because adult homozygous bnch escapers exhibit progressive behavioral defects and because bnch mutant eyes show progressive visual synaptic defects, we studied the morphology of their central nervous system. Previously, loss of bnch has been reported to cause a longer abdominal ganglion in adult hypomorphic flies due to decreased developmental programmed cell death (Nakano et al., 2001). Similar to Nakano et al. (2001), we also observe a longer abdominal ganglion in 4-d-old bnch escapers (unpublished data). However, sagittal histological sections through the abdominal ganglion reveals vacuolization of the tissue (see online supplemental data, available at http://www.jcb.org/cgi/content/full/jcb.200412001/DC1), suggesting that neuronal degeneration occurs after the developmental apoptotic defect responsible for the abnormally long ventral ganglion.
In agreement, we observe severe neuronal loss in histological sections of brains of bnch null pharate adults and escapers. This neuronal loss is characterized by a significant decrease in the number of neuronal cell bodies and severe vacuolization in cortical brain layers (Fig. 8, A and B). In addition, the layered architecture of the visual lobe is disrupted (Fig. 8, A and B). The medulla of bnch mutants does not undergo its normal rotation with respect to the antero-posterior axis.
Figure 8.
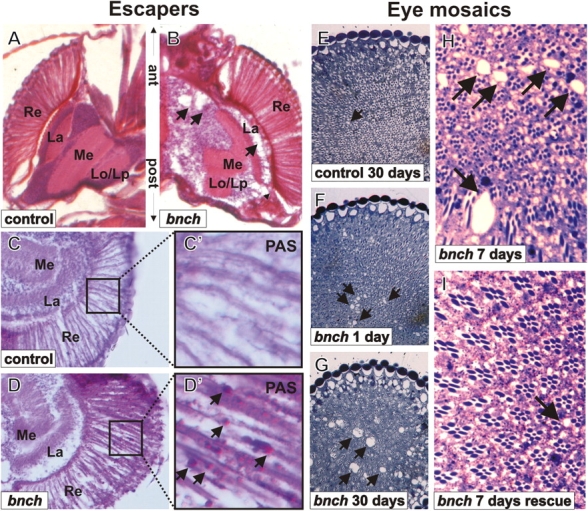
Loss of bnch leads to severe progressive neurodegeneration. (A–D) Brain and retinal morphology of bnch adult escapers. Eosin and hematoxylin stained horizontal sections of adult heads (A and B). Compared with wild type (A), a severe decrease in the cortical neuronal density is observed in transheterozygous bnch mutant escapers (B). Arrows indicate cortical vacuoles. In mutants (B), the external surface of the medulla is not entirely parallel to the face of the lamina, indicating that the medulla has not rotated properly during development. PAS staining in horizontal sections of the adult retina (C, C′, D, and D′). In the control, normal ommatidial organization is seen with lenses, cone cells, pigment cells, and photoreceptors, but no PAS-positive reaction (C and C′). In a bnch transheterozygous escaper (D), the ommatidia as seen in C are disrupted and is accompanied by a marked accumulation of PAS-positive granules (arrows in D′ compared with C′). Names of optic lobe regions are abbreviated as Re (retina), La (lamina), Me (medulla), Lo (lobula), and Lp (lobula plate). (E–I) Retinal light microscopical sections of bnch mutant eye clones stained with toluidine blue. Homozygous bnch mutant eye tissue of 1-d-old flies (F) displays vacuolar degeneration (arrows), swelling of the cytoplasm and cytoplasmic inclusions. Compared with the lesions of 1-d-old flies (F), there is significant increase in size and number of vacuoles after 30 d (G). Such lesions are never found in 30-d-old control flies (E). 7-d-old bnch mutant eyes consistently demonstrate a significant number of vacuoles (>1/2 size of a single ommatidium) (H). In the presence of Spin-GFP (expressed under control of ey-Gal4) these vacuoles are largely absent (I). An occasional small vacuole is present (<1/2 size of a single ommatidium). The occasional ommatidial loss seen in both bnch mutant eyes and controls is an artifact of the GMR-hid insertion present on the FRT chromosome used to induce clones.
Because we found a role for Bnch in mobilizing carbohydrates from yolk spheres during oogenesis and because bnch mutant neuronal tissue accumulates abnormal lysosomal inclusions, we also stained the nervous system of 4-d-old bnch escapers using the PAS method for carbohydrates. Interestingly, although no obvious defects were observed in 1-d-old flies (unpublished data), 4-d-old flies showed a loss of retinal structural integrity associated with the accumulation of PAS-positive granules (Fig. 8, C and D).
To further address whether the observed neuronal loss has a progressive character, we tested if the lesions worsen in an age-dependent manner in adult flies. Because bnch adult escapers eclose at low frequencies and die after a few days, we generated bnch eye mosaics (Newsome et al., 2000) and studied the progressive nature of bnch-associated neuronal loss in adult eye clones. Although the external morphology of bnch eyes appears macroscopically normal, histological light microscopy sections reveal vacuoles throughout the retina in 1-d-old flies (Fig. 8 F) not present in controls (Fig. 8 E). Vacuolization of neuronal tissues is the major hallmark of neurodegeneration in flies (Muqit and Feany, 2002). Although these results demonstrate that vacuolar lesions are already present at eclosion, comparison of 1- to 30-d-old mosaic flies reveals that the extent of these defects is progressive, as illustrated by the clear increase in number and size of retinal vacuoles (Fig. 8, F and G). In bnch mutant retinas the cytoplasm becomes progressively swollen, which leads to an increase in the distance between neighboring rhabdomere clusters. The development of vacuolar defects is almost completely corrected when a Bnch transgene (Spin-GFP fusion protein) (Sweeney and Davis, 2002) is expressed under control of ey-Gal4 (Fig. 8, H and I). These data demonstrate that in bnch null neuronal tissues, the neurodegeneration starts during development and continues in an age-dependent progressive manner during adulthood. These data provide a morphological correlate to the progressive behavioral defects seen in homozygous and transheterozygous escapers and to the progressive ERG defects in bnch eye clones (Fig. 7 A).
Together with the ultrastructural demonstration of abnormal multilamellar inclusions, these data suggest that loss of bnch leads to progressive neurodegeneration associated with the accumulation of carbohydrate-storing lysosomes.
Loss of bnch dose-dependently enhances tau toxicity
Our phenotypic analysis and the possible role of bnch as a sugar carrier suggest strong similarities between bnch and a subclass of neurodegenerative LSDs that are caused by defective efflux of lysosomal substrates. In humans, sialic acid storage disorder and Niemann-Pick disease type C are caused by defective efflux of acidic sugars and cholesterol, respectively (Walkley, 2001), and in both diseases the pathological hallmarks of LSD are accompanied by tauopathy in the form of neurofibrillary tangles (Autio-Harmainen et al., 1988; Auer et al., 1995). However, the question of whether neurofibrillary tangles constitute a pathogenic lesion in such disorders is controversial (German et al., 2001; Treiber-Held et al., 2003).
To test the possibility of whether defective lysosomal function due to loss of bnch is able to trigger tauopathy, we used a reported tauopathy fly model that expresses the longest four repeat isoform of human wild-type tau in the eye (GMR-htau4R) (Jackson et al., 2002). This model behaves as a genetically sensitized system that allows the identification of genetic modifiers of tau-mediated neurodegeneration by assessing roughening of the eye as a quantitative readout of tau toxicity. We combined this model with the eyFLP system to generate homozygous bnch eyes that simultaneously express one copy of the GMR-htau4R transgene and compared such eyes with several controls. As shown in Fig. 9, eyes that express either one copy of GMR-htau4R (Fig. 9 A) or that are homozygous bnch mutant (Fig. 9 B) display no obvious roughening. However, removal of one (Fig. 9 C) or two copies (Fig. 9 D) of bnch in eyes expressing one copy of GMR-htau4R induced a moderate and very severe rough eye phenotype, respectively. These results indicate that loss of bnch can dose-dependently enhance tau toxicity and suggest that lysosomal dysfunction is a potent trigger of tau toxicity in Drosophila.
Figure 9.
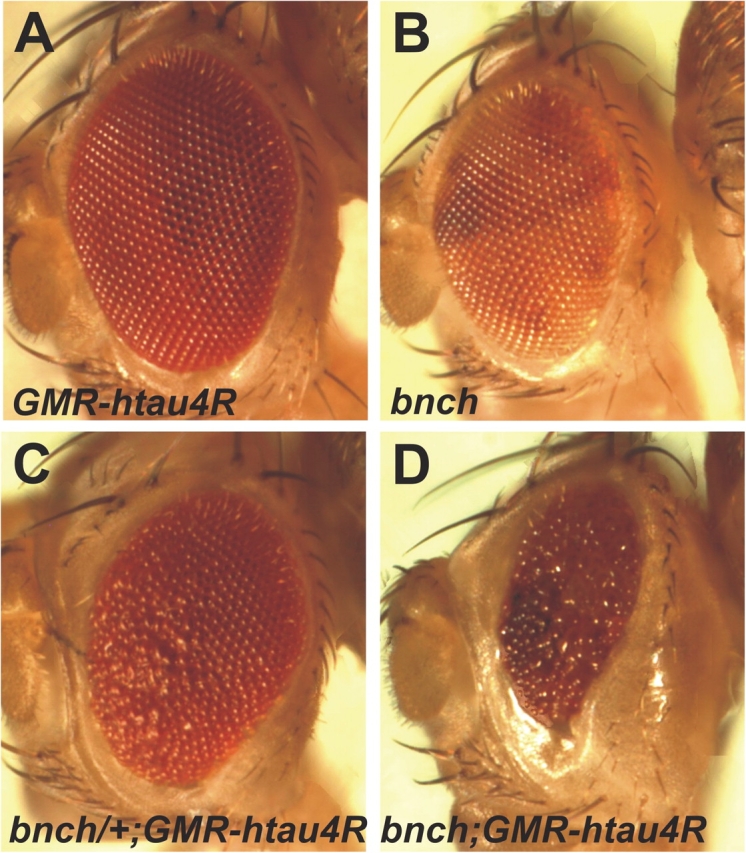
Loss of bnch enhances tau neurotoxicity in a dose-dependent manner. (A and B) Eyes that express one copy of GMR-htau4R (A) or lack bnch (B) appear morphologically normal. (C and D) Removal of one copy of bnch in eyes expressing one copy of GMR-htau4R induces a mild rough eye phenotype (C), whereas overexpression of one of GMR-htau4R copy in a bnch null background results in a small and rough eye (D).
Discussion
A Drosophila model for human neurodegenerative lysosomal efflux disorders
Although mutations in the human homologues of bnch are not a known cause of human disease, the present study suggests that the bnch mutant phenotype is very closely related to the human neurodegenerative LSDs. Together with the reported subcellular lysosomal localization of Bnch (Sweeney and Davis, 2002), the present demonstration of progressive neurodegeneration characterized by dramatic ultrastructural lysosomal abnormalities links a primary lysosomal defect to neurodegeneration in Drosophila. Not only are the ultrastructural characteristics highly similar to the lesions observed in human LSDs, but the predicted molecular nature of the Bnch protein as a lysosomal transporter protein further suggests similarity with a subset of LSDs that are caused by defective efflux of substrates from lysosomes. Interestingly, the human protein sialin, which is defective in sialic acid storage disease (Verheijen et al., 1999), is very similar to bnch as it is an MFS transporter of the ACS subfamily (Pao et al., 1998).
Based mainly on the accumulation of autofluorescent pigments, previous studies have emphasized the similarities between bnch phenotypes and LSDs called neuronal ceroid lipofuscinoses (Nakano et al., 2001; Sanyal and Ramaswami, 2002; Sweeney and Davis, 2002). In addition, here we point out the striking similarities between bnch and human lysosomal efflux disorders. More specifically, from both a molecular mechanistic and phenotypic point of view, bnch appears similar to sialic acid storage disease, a lysosomal acid sugar efflux disorder with severe neurodegeneration characterized by neuronal accumulation of abnormal lysosomes including lipofuscin pigments and neurofibrillary tangles (Autio-Harmainen et al., 1988). Given our data suggesting a role of bnch in lysosomal carbohydrate metabolism, it is tempting to propose bnch as a candidate gene for either the molecularly undefined lysosomal neutral hexose or N-acetylhexosamine transporter systems (Mancini et al., 1990).
Lysosomes are present at the presynaptic terminal and function in synaptic vesicle recycling
Despite their high rates of membrane turnover, presynaptic terminals are not known to contain active lysosomal-degradative compartments (Nixon and Cataldo, 1995). Nevertheless, the local regulation of endocytosis at the nerve terminal is crucial for the function of synapses in the nervous system due to its role in synaptic vesicle recycling (Murthy and De Camilli, 2003). However, the extent to which endosomal compartments impinge on the synaptic vesicle cycle is not completely understood and controversial.
Together with the previously reported observation that bnch mutant NMJ boutons accumulate expanded acidic organelles (Sweeney and Davis, 2002), our ultrastructural and physiological data indicate that nerve terminals contain degradative compartments that are important for normal synaptic function.
Aberrant lysosomal sugar storage accompanies endocytic defects, neurodegeneration, and enhanced tau neurotoxicity
Besides severe neurodegeneration and alterations in lysosomal morphology, our phenotypic analysis of bnch mutants indicates morphological and functional alterations in endocytic compartments. The evidence comes mainly from cell types that have high rates of membrane trafficking. First, in the photoreceptors, known for their high rate of membrane turnover and endocytosis (Stark et al., 1988), loss of bnch induces a dramatic increase in number and size of enlarged late endosomal MVBs, suggestive of a block downstream of the late endosomal compartment. Second, we observed reduced endosome-to-lysosome trafficking in bnch mutant garland cells, a cell type with high rates of fluid phase endocytosis. Third, at bnch mutant NMJ synapses, we observed defects in endocytosis and synaptic vesicle recycling, further suggesting functional abnormalities in extralysosomal membrane compartments.
In contrast with hrs mutants, in which a specific functional endosomal defect leads to phenotypes suggestive of increased receptor tyrosine kinase signaling (Lloyd et al., 2002), loss of bnch does not seem to result in phenotypes indicative of a downstream defect in a specific pathway. However, as bnch most likely functions at the level of lysosomal degradation, the cellular consequences of loss of bnch are expected to be highly pleiotropic, simultaneously affecting upstream autophagic and endocytic pathways that converge at the level of the lysosome. In addition, because Bnch, as a potential mobilizer of lysosomal carbohydrate reserves, is also highly expressed in glial cells, it is possible that the neurodegeneration seen in the bnch nervous system can be partially caused by lack of trophic sugar support of neurons by glia.
Our observation that loss of bnch is a potent enhancer of tau neurotoxicity gives some clues as to how lysosomal dysfunction might lead to neurodegeneration. It is well established that tau is on the final common pathway in a wide range of human neurodegenerative diseases (Hardy et al., 1998; Lee et al., 2001), and our results give experimental in vivo evidence that alterations in lysosomal membrane compartments are able to induce tauopathy. Together with the fact that tauopathy has been reported in sialic acid storage disease (Autio-Harmainen et al., 1988) and is a highly consistent finding in Niemann-Pick disease type C (Auer et al., 1995), our results suggest a link between lysosomal function and tau pathology.
Materials and methods
Fly stocks, mitotic recombination, and phenotypic rescue
The P{LacW}[25/11] insertion was obtained from I. Kiss (Kania et al., 1995). The P{LacW} [Δ29], [Δ31] and [Δ86] alleles were generated through imprecise excision and mapped by Southern blot. The l(2R)W5[11F5, N, E14.1, and P] alleles were obtained from Ruth Steward (Liu et al., 1999). For the studies in the eye, l(2R)W5[11F5, N, and E14.1] alleles and bnch[Δ2B] (provided by G. Davis and S. Sweeney) were recombined onto a P{ry[+t7.2]=neoFRT}42D chromosome and crossed to yw P{ry[+t7.2]=ey-FLP.N}2 P{GMR-lacZ.C(38.1)}TPN1; P{ry[+t7.2]=neoFRT}42D P{w[+t*ry[+t*]=white-un1}47A l(2)cl-R11[1]/CyO-KrGFP (obtained from Barry Dickson) (Newsome et al., 2000). For rescue of the degenerative defects in the eye the phenotypes of yw; P{ry[+t7.2]=neoFRT}42D bnch[N]/P{ry[+t7.2]=neoFRT}42D P{y[+t7.7] ry[+t7.2]=Car20y}44B, P{w[+mC]=GMR-hid}SS2, l(2)cl-R[1]; ey-Gal4, UAS-FLP/UAS-Spin-GFP and yw; P{ry[+t7.2]=neoFRT}42D bnch[N]/P{ry[+t7.2]=neoFRT}42D P{y[+t7.7] ry[+t7.2]=Car20y}44B, P{w[+mC]=GMR-hid}SS2, l(2)cl-R[1]; ey-Gal4, UAS-FLP/+ were compared. UAS-Spin-EGFP was given by S. Sweeney (Sweeney and Davis, 2002).
For generation of germ line clones we used the ovoD technique and recombined the alleles l(2R)W5[11F5, N, P, and E14.1] and bnch[Δ2B] onto a P{w[+mW.hs]=FRT(w[hs])}G13(42B) chromosome (Chou and Perrimon, 1996).
The gl-tau2.1 line (Jackson et al., 2002) expressing human 4-repeat tau in the eye was provided by George Jackson and is referred to as GMR-htau4R throughout the manuscript. For the tau interaction study yw; P{ry[+t7.2]=neoFRT}42D spin[Δ2B]/CyO; gl-tau2.1/TM6b males were crossed to yw P{ry[+t7.2]=ey-FLP.N}2 P{GMR-lacZ.C(38.1)}TPN1; P{ry[+t7.2]=neoFRT}42D P{w[+t* ry[+t*]=white-un1}47A l(2)cl-R11[1]/CyO-KrGFP females and eye morphology of the progeny was compared. Photographs of the eyes are shown as a composite image from a Z-stack captured using a microscope (model MZ16; Leica) with a camera (Magnafire; Optronics) driven by InFocus v1.6 software (Meyer Instruments). The composite image was generated using the maximum local contrast algorithm in the ImagePro Plus v4.5.1 software (MediaCybernetics) and refined using Lucis Pro v5.0 software (Image Content Technology).
DNA sequencing
Genomic DNA was prepared from various alleles of l(2R)W5 in trans to another l(2R)W chromosome using Puregene (Gentra). PCR primers were designed using a modified Primer3 algorithm (http://flypush.imgen.bcm.tmc.edu/primer/) (Zhai et al., 2003). PCR reactions were performed using HotstarTaq (QIAGEN). Sequences were analyzed using Seqman II (DNAStar).
Topology analysis
The membrane topology of Bnch was predicted using the combined TMHMM/SignalP predictor Phobius (Kall et al., 2004).
Lysotracker and PAS staining of ovaries
After feeding flies on grape juice agar and fresh yeast paste for several days, ovaries were dissected in Schneider's Drosophila medium (SDM; GIBCO BRL). Ovarioles were gently separated and incubated for 15 min at RT in SDM with added Lysotracker (Molecular Probes, Inc.), diluted according to the manufacturer's instructions. After washing, ovarioles were mounted in SDM for confocal microscopy. Images were captured with a confocal microscope (Bio-Rad Laboratories) and processed with Amira 2.2 software and Adobe Photoshop 7. For PAS staining, ovaries were dissected and then fixed, dehydrated, and embedded as the adult flies (see below). Images were obtained using a light microscope (model BX61; Olympus) and AnalySIS 3.2. imaging software.
ERG recordings
For ERG recordings, flies were immobilized with nail polish. A sharp glass reference electrode was inserted in the thorax, while a sharp recording electrode, filled with 3 M NaCl, was placed on the eye (Alawi and Pak, 1971). Light flashes of 1 s or 500 ms were delivered using a halogen lamp. We performed recordings on at least seven flies per genotype. Data were digitized and stored on a PC with pClamp and analyzed with Clampfit and Microsoft Excel.
TEM
TEM preparation of adult eye retinae and laminae were performed as described previously (Fabian-Fine et al., 2003). 1-d-old flies were dissected and fixed at 4°C in 2% PFA; 2% glutaraldehyde; 0.1 M sodium cacodylate; 0.005% CaCl2, pH 7.2, and postfixed in 2% OsO4. 50-nm thin sections were stained in 4% uranyl acetate and 2.5% lead nitrate. Larvae were dissected in fixative and prepared for TEM as described previously (Verstreken et al., 2002). Data were captured with an electron microscope (model JEM-1010; JEOL), and images were processed in Photoshop 7 (Adobe) and assembled in CorelDRAW 12 (Corel).
Larval NMJ electrophysiology and FM1-43 dye uptake
Third instar larval muscle recordings were performed as described in the online supplemental data (Verstreken et al., 2002). Larvae were dissected and recordings were performed in modified HL3 (mM): NaCl (110), KCl (5), NaHCO3 (10), Hepes (5), sucrose (30), trehalose (5), CaCl2 (1), and MgCl2 (20) (pH 7.2).
For FM1-43 experiments, larvae were incubated in modified HL3 with 1.5 mM CaCl2 and 10 μm FM1-43FX. Preparations were nerve stimulated at 2× threshold at 30 Hz for 5 min; the FM1-43 dye was left in the bath for an additional 5 min. Such a labeling protocol is assumed to label vesicles in the RRP (exo/endo recycling pool) as well as in the RP (Kuromi and Kidokoro, 2002). Preparations were then extensively washed with 0 Ca2+ solution and boutons in the stimulated segment were imaged with a confocal microscope (model 510; Carl Zeiss MicroImaging, Inc.). To unload the RRP (but not the RP), preparations were subsequently incubated in 90-mM KCl solution with 1.5 mM CaCl2 (in modified HL3 with 25 mM NaCl to preserve osmolarity) for 5 min. After extensive washing in 0 Ca2+ solution, the same synapses were imaged again. Quantification of FM1-43 fluorescence after loading or unloading was performed as described previously (Verstreken et al., 2003; Koh et al., 2004). FM1-43 data were captured with an upright confocal microscope (model 510; Carl Zeiss MicroImaging, Inc.), processed with Amira 2.2 using the projection view module with bilinear interpolation (Template Graphics Software, Inc.), and images were processed in Photoshop 7 (Adobe) and assembled in CorelDRAW 12 (Corel).
Histological stainings and sections of the adult nervous system
Fly heads were fixed in 1.25% glutaraldehyde in PBT for 1–2 h on ice and dehydrated. After equilibration in xylene-paraffin, the heads were incubated in paraffin at 58°C for 3–4 h. The heads were then reoriented in melted paraffin blocks and allowed to solidify. The blocks were cooled down on ice and sliced into 10-μm sections with a microtome. The sections were then rinsed in distilled water and stained with hematoxylin (Gill's formulation #1; Fisher Scientific) for 3 min and de-stained in tap water for 5 min. The stain was fixed in a 0.25% HCl solution in 70% ethanol. After a series of tap water and distilled water rinses, the sections were counterstained in eosin (Y solution; Sigma-Aldrich) for 1 min and de-stained in ethanol. Slides were then transferred into xylene and mounted in Permount (Fisher Scientific). Images were captured using a microscope (Axiophot; Carl Zeiss MicroImaging, Inc.).
For PAS staining, adult flies were fixed overnight in 4% formaldehyde in PBS. To facilitate entry of fixative and subsequent solvents during dehydration and embedding, several openings were made in the head, thorax, and abdomen. Flies were dehydrated through ethanol series, methylbenzoate and paraffin embedded. Sections of 10 μm were made on a microtome (model 5040; Bright Instruments). PAS staining was performed as described previously (Gutzeit et al., 1994).
For light microscopy of adult retina, specimens were dissected, fixed, mounted, and stained with toluidine blue as described previously (Tomlinson and Ready, 1987). Images were captured using a microscope (Axiophot; Carl Zeiss MicroImaging, Inc.).
Endocytic tracer studies
Garland cell uptake studies were performed by incubating cells in Schneider's medium containing 0.2 mg/ml Alexa 488–conjugated avidin (Sigma-Aldrich) and 50 nM Lysotracker. Live cells were imaged and data were captured with an upright confocal microscope (model 510; Carl Zeiss MicroImaging, Inc.), processed with Amira 2.2 using the projection view module with bilinear interpolation (Template Graphics Software, Inc.), and images were processed in Photoshop 7 (Adobe) and assembled in CorelDRAW 12 (Corel).
Online supplemental material
The supplemental figure contains histological sagittal sections stained with the PAS method of the ventral ganglion of a bnch transheterozygous escaper (B) at the same level as the control (shown in A). Numerous vacuoles typical for neurodegeneration can be observed in the bnch ventral ganglion (arrows). Online supplemental material available at http://www.jcb.org/cgi/content/full/jcb.200412001/DC1.
Acknowledgments
We thank Ruth Steward, George Jackson, Sean Sweeney, and Graeme Davis for sharing reagents. We thank Peter Robin Hiesinger, Hubert Carchon, Eva Beke, and Jason Clements for technical help and discussions.
This work was supported by the Pediatric Brain Tumor Foundation and the National Institutes of Health to H.J. Bellen. H.J. Bellen is an Investigator and K.K. Norga was a Physician Postdoctoral Associate of the Howard Hughes Medical Institute and is now Senior Clinical Investigator of the Fund for Scientific Research, Flanders. B. Dermaut is a Long-term Postdoctoral Fellow of the Human Frontier Science Program.
B. Dermaut and K.K. Norga contributed equally to this paper.
A. Kania's present address is Institut de Recherches Cliniques de Montreal, Avenue des Pins Ouest 110, Montréal H2W 1R7, Québec, Canada.
Abbreviations used in this paper: ACS, anion/cation subfamily; bnch, benchwarmer gene; EJP, excitatory junctional potential; ERG, electroretinogram; LSD, lysosomal storage disorder; MFS, major facilitator superfamily; MVB, multivesicular bodies; NMJ, neuromuscular junction; RP, reserve pool; RRP, readily releasable pool; TEM, transmission electron microscopy; PAS, periodic acid Schiff.
References
- Alawi, A., and W.L. Pak. 1971. On-transient of insect electroretinograms: its cellular origin. Science. 172:1055–1057. [DOI] [PubMed] [Google Scholar]
- Arbeitman, M.N., E.E. Furlong, F. Imam, E. Johnson, B.H. Null, B.S. Baker, M.A. Krasnow, M.P. Scott, R.W. Davis, and K.P. White. 2002. Gene expression during the life cycle of Drosophila melanogaster. Science. 297:2270–2275. [DOI] [PubMed] [Google Scholar]
- Auer, I.A., M.L. Schmidt, V.M. Lee, B. Curry, K. Suzuki, R.W. Shin, P.G. Pentchev, E.D. Carstea, and J.Q. Trojanowski. 1995. Paired helical filament tau (PHFtau) in Niemann-Pick type C disease is similar to PHFtau in Alzheimer's disease. Acta Neuropathol. (Berl.). 90:547–551. [DOI] [PubMed] [Google Scholar]
- Autio-Harmainen, H., A. Oldfors, P. Sourander, M. Renlund, K. Dammert, and S. Simila. 1988. Neuropathology of Salla disease. Acta Neuropathol. (Berl.). 75:481–490. [DOI] [PubMed] [Google Scholar]
- Betz, W.J., F. Mao, and G.S. Bewick. 1992. Activity-dependent fluorescent staining and destaining of living vertebrate motor-nerve terminals. J. Neurosci. 12:363–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonini, N.M., and M.E. Fortini. 2003. Human neurodegenerative disease modeling using Drosophila. Annu. Rev. Neurosci. 26:627–656. [DOI] [PubMed] [Google Scholar]
- Chou, T.B., and N. Perrimon. 1996. The autosomal FLP-DFS technique for generating germline mosaics in Drosophila melanogaster. Genetics. 144:1673–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, J.D. 2003. Progress towards understanding the neurobiology of Batten disease or neuronal ceroid lipofuscinosis. Curr. Opin. Neurol. 16:121–128. [DOI] [PubMed] [Google Scholar]
- DiMario, P.J., and A.P. Mahowald. 1987. Female sterile (1) yolkless: a recessive female sterile mutation in Drosophila melanogaster with depressed numbers of coated pits and coated vesicles within the developing oocytes. J. Cell Biol. 105:199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskelinen, E.L., Y. Tanaka, and P. Saftig. 2003. At the acidic edge: emerging functions for lysosomal membrane proteins. Trends Cell Biol. 13:137–145. [DOI] [PubMed] [Google Scholar]
- Fabian-Fine, R., P. Verstreken, P.R. Hiesinger, J.A. Horne, R. Kostyleva, Y. Zhou, H.J. Bellen, and I.A. Meinertzhagen. 2003. Endophilin promotes a late step in endocytosis at glial invaginations in Drosophila photoreceptor terminals. J. Neurosci. 23:10732–10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagotto, F. 1995. Regulation of yolk degradation, or how to make sleepy lysosomes. J. Cell Sci. 108(Pt 12):3645–3647. [DOI] [PubMed] [Google Scholar]
- Finley, K.D., P.T. Edeen, R.C. Cumming, M.D. Mardahl-Dumesnil, B.J. Taylor, M.H. Rodriguez, C.E. Hwang, M. Benedetti, and M. McKeown. 2003. blue cheese mutations define a novel, conserved gene involved in progressive neural degeneration. J. Neurosci. 23:1254–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futerman, A.H., and G. van Meer. 2004. The cell biology of lysosomal storage disorders. Nat. Rev. Mol. Cell Biol. 5:554–565. [DOI] [PubMed] [Google Scholar]
- German, D.C., E.M. Quintero, C.L. Liang, B. Ng, S. Punia, C. Xie, and J.M. Dietschy. 2001. Selective neurodegeneration, without neurofibrillary tangles, in a mouse model of Niemann-Pick C disease. J. Comp. Neurol. 433:415–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutzeit, H.O., D. Zissler, V. Grau, M. Liphardt, and U.R. Heinrich. 1994. Glycogen stores in mature ovarian follicles and young embryos of Drosophila: ultrastructural changes and some biochemical correlates. Eur. J. Cell Biol. 63:52–60. [PubMed] [Google Scholar]
- Hardy, J., K. Duff, K.G. Hardy, J. Perez-Tur, and M. Hutton. 1998. Genetic dissection of Alzheimer's disease and related dementias: amyloid and its relationship to tau. Nat. Neurosci. 1:355–358. [DOI] [PubMed] [Google Scholar]
- Hollenbeck, P.J. 1993. Products of endocytosis and autophagy are retrieved from axons by regulated retrograde organelle transport. J. Cell Biol. 121:305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, G.R., M. Wiedau-Pazos, T.K. Sang, N. Wagle, C.A. Brown, S. Massachi, and D.H. Geschwind. 2002. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron. 34:509–519. [DOI] [PubMed] [Google Scholar]
- Kall, L., A. Krogh, and E.L. Sonnhammer. 2004. A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 338:1027–1036. [DOI] [PubMed] [Google Scholar]
- Kania, A. 1996. Mutations in neuromusculin and benchwarmer affect early nervous system development and neuronal survival. Ph.D. thesis. Baylor College of Medicine, Houston, TX. 122 pp.
- Kania, A., A. Salzberg, M. Bhat, D. D'Evelyn, Y. He, I. Kiss, and H.J. Bellen. 1995. P-element mutations affecting embryonic peripheral nervous system development in Drosophila melanogaster. Genetics. 139:1663–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh, T.W., P. Verstreken, and H.J. Bellen. 2004. Dap160/intersectin acts as a stabilizing scaffold required for synaptic development and vesicle endocytosis. Neuron. 43:193–205. [DOI] [PubMed] [Google Scholar]
- Kosaka, T., and K. Ikeda. 1983. Reversible blockage of membrane retrieval and endocytosis in the garland cell of the temperature-sensitive mutant of Drosophila melanogaster, shibirets1. J. Cell Biol. 97:499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuromi, H., and Y. Kidokoro. 2002. Selective replenishment of two vesicle pools depends on the source of Ca2+ at the Drosophila synapse. Neuron. 35:333–343. [DOI] [PubMed] [Google Scholar]
- Lee, V.M., M. Goedert, and J.Q. Trojanowski. 2001. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 24:1121–1159. [DOI] [PubMed] [Google Scholar]
- Liu, Z., T. Xie, and R. Steward. 1999. Lis1, the Drosophila homolog of a human lissencephaly disease gene, is required for germline cell division and oocyte differentiation. Development. 126:4477–4488. [DOI] [PubMed] [Google Scholar]
- Lloyd, T.E., R. Atkinson, M.N. Wu, Y. Zhou, G. Pennetta, and H.J. Bellen. 2002. Hrs regulates endosome membrane invagination and tyrosine kinase receptor signaling in Drosophila. Cell. 108:261–269. [DOI] [PubMed] [Google Scholar]
- Mancini, G.M., C.E. Beerens, and F.W. Verheijen. 1990. Glucose transport in lysosomal membrane vesicles. Kinetic demonstration of a carrier for neutral hexoses. J. Biol. Chem. 265:12380–12387. [PubMed] [Google Scholar]
- Min, K.T., and S. Benzer. 1997. Spongecake and eggroll: two hereditary diseases in Drosophila resemble patterns of human brain degeneration. Curr. Biol. 7:885–888. [DOI] [PubMed] [Google Scholar]
- Muqit, M.M., and M.B. Feany. 2002. Modelling neurodegenerative diseases in Drosophila: a fruitful approach? Nat. Rev. Neurosci. 3:237–243. [DOI] [PubMed] [Google Scholar]
- Murthy, V.N., and P. De Camilli. 2003. Cell biology of the presynaptic terminal. Annu. Rev. Neurosci. 26:701–728. [DOI] [PubMed] [Google Scholar]
- Nakano, Y., K. Fujitani, J. Kurihara, J. Ragan, K. Usui-Aoki, L. Shimoda, T. Lukacsovich, K. Suzuki, M. Sezaki, Y. Sano, et al. 2001. Mutations in the novel membrane protein spinster interfere with programmed cell death and cause neural degeneration in Drosophila melanogaster. Mol. Cell. Biol. 21:3775–3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld, E.F. 1991. Lysosomal storage diseases. Annu. Rev. Biochem. 60:257–280. [DOI] [PubMed] [Google Scholar]
- Newsome, T.P., B. Asling, and B.J. Dickson. 2000. Analysis of Drosophila photoreceptor axon guidance in eye-specific mosaics. Development. 127:851–860. [DOI] [PubMed] [Google Scholar]
- Nixon, R.A., and A.M. Cataldo. 1995. The endosomal-lysosomal system of neurons: new roles. Trends Neurosci. 18:489–496. [DOI] [PubMed] [Google Scholar]
- Nixon, R.A., P.M. Mathews, and A.M. Cataldo. 2001. The neuronal endosomal-lysosomal system in Alzheimer's disease. J. Alzheimers Dis. 3:97–107. [DOI] [PubMed] [Google Scholar]
- Overly, C.C., and P.J. Hollenbeck. 1996. Dynamic organization of endocytic pathways in axons of cultured sympathetic neurons. J. Neurosci. 16:6056–6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overly, C.C., K.D. Lee, E. Berthiaume, and P.J. Hollenbeck. 1995. Quantitative measurement of intraorganelle pH in the endosomal-lysosomal pathway in neurons by using ratiometric imaging with pyranine. Proc. Natl. Acad. Sci. USA. 92:3156–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao, S.S., I.T. Paulsen, and M.H. Saier Jr. 1998. Major facilitator superfamily. Microbiol. Mol. Biol. Rev. 62:1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton, R.G., K. Simons, and C.G. Dotti. 1992. Axonal and dendritic endocytic pathways in cultured neurons. J. Cell Biol. 119:123–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal, S., and M. Ramaswami. 2002. Spinsters, synaptic defects, and amaurotic idiocy. Neuron. 36:335–338. [DOI] [PubMed] [Google Scholar]
- Sillence, D.J., and F.M. Platt. 2003. Storage diseases: new insights into sphingolipid functions. Trends Cell Biol. 13:195–203. [DOI] [PubMed] [Google Scholar]
- Stark, W.S., R. Sapp, and D. Schilly. 1988. Rhabdomere turnover and rhodopsin cycle: maintenance of retinula cells in Drosophila melanogaster. J. Neurocytol. 17:499–509. [DOI] [PubMed] [Google Scholar]
- Sweeney, S.T., and G.W. Davis. 2002. Unrestricted synaptic growth in spinster-a late endosomal protein implicated in TGF-beta-mediated synaptic growth regulation. Neuron. 36:403–416. [DOI] [PubMed] [Google Scholar]
- Tomlinson, A., and D.F. Ready. 1987. Cell fate in the Drosophila ommatidium. Dev. Biol. 123:264–275. [DOI] [PubMed] [Google Scholar]
- Treiber-Held, S., R. Distl, V. Meske, F. Albert, and T.G. Ohm. 2003. Spatial and temporal distribution of intracellular free cholesterol in brains of a Niemann-Pick type C mouse model showing hyperphosphorylated tau protein. Implications for Alzheimer's disease. J. Pathol. 200:95–103. [DOI] [PubMed] [Google Scholar]
- Verheijen, F.W., E. Verbeek, N. Aula, C.E. Beerens, A.C. Havelaar, M. Joosse, L. Peltonen, P. Aula, H. Galjaard, P.J. van der Spek, and G.M. Mancini. 1999. A new gene, encoding an anion transporter, is mutated in sialic acid storage diseases. Nat. Genet. 23:462–465. [DOI] [PubMed] [Google Scholar]
- Verstreken, P., O. Kjaerulff, T.E. Lloyd, R. Atkinson, Y. Zhou, I.A. Meinertzhagen, and H.J. Bellen. 2002. Endophilin mutations block clathrin-mediated endocytosis but not neurotransmitter release. Cell. 109:101–112. [DOI] [PubMed] [Google Scholar]
- Verstreken, P., T.W. Koh, K.L. Schulze, R.G. Zhai, P.R. Hiesinger, Y. Zhou, S.Q. Mehta, Y. Cao, J. Roos, and H.J. Bellen. 2003. Synaptojanin is recruited by Endophilin to promote synaptic vesicle uncoating. Neuron. 40:733–748. [DOI] [PubMed] [Google Scholar]
- Walkley, S.U. 1998. Cellular pathology of lysosomal storage disorders. Brain Pathol. 8:175–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkley, S.U. 2001. New proteins from old diseases provide novel insights in cell biology. Curr. Opin. Neurol. 14:805–810. [DOI] [PubMed] [Google Scholar]
- Yanagisawa, H., T. Miyashita, Y. Nakano, and D. Yamamoto. 2003. HSpin1, a transmembrane protein interacting with Bcl-2/Bcl-xL, induces a caspase-independent autophagic cell death. Cell Death Differ. 10:798–807. [DOI] [PubMed] [Google Scholar]
- Young, R.M., S. Marty, Y. Nakano, H. Wang, D. Yamamoto, S. Lin, and M.L. Allende. 2002. Zebrafish yolk-specific not really started (nrs) gene is a vertebrate homolog of the Drosophila spinster gene and is essential for embryogenesis. Dev. Dyn. 223:298–305. [DOI] [PubMed] [Google Scholar]
- Zhai, R.G., P.R. Hiesinger, T.W. Koh, P. Verstreken, K.L. Schulze, Y. Cao, H. Jafar-Nejad, K.K. Norga, H. Pan, V. Bayat, et al. 2003. Mapping Drosophila mutations with molecularly defined P element insertions. Proc. Natl. Acad. Sci. USA. 100:10860–10865. [DOI] [PMC free article] [PubMed] [Google Scholar]