

Abstract
Despite the fact that the continuity of morphology of fossil specimens of modern humans found in China has repeatedly challenged the Out-of-Africa hypothesis, Chinese populations are underrepresented in genetic studies. Genetic profiles of 28 populations sampled in China supported the distinction between southern and northern populations, while the latter are biphyletic. Linguistic boundaries are often transgressed across language families studied, reflecting substantial gene flow between populations. Nevertheless, genetic evidence does not support an independent origin of Homo sapiens in China. The phylogeny also suggested that it is more likely that ancestors of the populations currently residing in East Asia entered from Southeast Asia.
The majority of China consists of the Han people (93.3%), and 55 official minority nationalities (6.7%), most of which have their own languages, are found predominantly in the peripheral regions. The number of living languages listed for China is 205 (1). Despite the fact that extensive variations among Han Chinese populations and minority populations in China have been observed (2–7), such populations are usually underrepresented in genetic studies of worldwide populations (8–10). The significance of an extensive study of Chinese populations is twofold. First, the distinction between northern and southern Chinese populations (Han and minority alike) has been observed in the analyses of genetic markers (2–4) as well as somatometric and nonmetric features (5–7). Most authors attributed such distinction simply to the presence of geographic barriers (2–7). While it is true that a geographic barrier maintains genetic difference if there is any, but it is irrelevant to a more interesting question: whether southern and northern populations are descendents of the same population or, alternatively, populations that arrived in China from different sources. Furthermore, the understanding of the origin of the populations in East Asia may shed light on the peopling of Siberia and America. Second, the human fossil remains recovered in China have also attracted attention. The regional as well as temporal continuity of fossil records from Homo erectus to Homo sapiens in this region (11–13) repeatedly challenged the Out-of-Africa hypothesis, which suggests a complete replacement of local populations by modern humans originating in Africa. The validity of this analysis (13) has been questioned (14). Genetic evidence became necessary to verify such claims. A systematic genetic study of Chinese populations using contemporary genetic markers therefore was conducted. This report reflects a collaborative effort made by several institutes participating in the Chinese Human Genome Diversity Project (CHGDP).
Microsatellites have been widely used to study the genetic relationship among human populations from different continents (8–10). Simulation results indicated that microsatellite loci generally provide a more reliable phylogenetic relationship among closely related populations than among distantly related ones (15) and therefore have been considered as ideal markers to study closely related populations. However, closely related populations tend to live in the same geographical area and gene flow between neighboring populations can be substantial, which may result in major changes in the original gene frequencies (16). In turn, the reliability of phylogeny inferences in the presence of genetic admixture can be profoundly compromised (17). Nevertheless, the ease of typing of microsatellite alleles and the availability of large numbers of such highly informative loci across the human genome made them the markers of choice in this study.
MATERIALS AND METHODS
Twenty-eight populations speaking the languages that belong to six language families and currently residing in China were studied (see Table 1). The locations of those populations are indicated in Fig. 2 and Table 1. Samples were collected through a coordinated effort of several institutes participating in the Chinese Human Genome Diversity Project. Samples of four Taiwanese Aborigine populations were kindly provided by M. Hsu (Academia Sinica, Taiwan).
Table 1.
Chinese populations sampled in the current study
| Population | Location | Language family | Language subfamily | |
|---|---|---|---|---|
| 1 | Aini | Southwest Yunnan | Sino-Tibetan | Tibeto-Burman |
| 2 | Blang | Southwest Yunnan | Austro-Asiatic | Mon-Khmer |
| 3 | Dai | South Central Yunnan | Daic | Daic |
| 4 | Deang | Southwest Yunnan | Sino-Tibetan | Tibeto-Burman |
| 5 | Dong | Guangxi | Daic | Kam-Sui |
| 6 | Ewenki | Heilongjiang | Altaic | Tungus |
| 7 | Han (Guangdong) | California, U.S. | Sino-Tibetan | Chinese |
| 8 | Han (Henan) | Henan | Sino-Tibetan | Chinese |
| 9 | Han (Northern) | Beijing | Sino-Tibetan | Chinese |
| 10 | Han (Yunnan) | Yunnan | Sino-Tibetan | Chinese |
| 11 | Hui (Muslims) | Ningxia | Sino-Tibetan | Chinese |
| 12 | Jingpo | Western Yunnan | Sino-Tibetan | Tibeto-Burman |
| 13 | Korean | Jilin | Isolate | |
| 14 | Lahu | Southwestern Yunnan | Sino-Tibetan | Tibeto-Burman |
| 15 | Li | Hainan | Daic | Kadai |
| 16 | Manchu | Heilongjiang | Altaic | Tungus |
| 17 | She | Fujian | Hmong-Mien | Ho Nte |
| 18 | Tibetan | Tibet | Sino-Tibetan | Tibeto-Burman |
| 19 | Tujia | Hunan | Sino-Tibetan | Tibeto-Burman |
| 20 | Uyghur | Xinjiang | Altaic | Turkic |
| 21 | Wa | Southwest Yunnan | Austro-Asiatic | Mon-Khmer |
| 22 | Yao (Puno) | Guizhou | Hmong-Mien | Hmongic |
| 23 | Yao (Jinxiu) | Guangxi | Daic | Kam-Sui |
| 24 | Yi | Sichuan | Sino-Tibetan | Tibeto-Burman |
| Taiwan Aboriginies | ||||
| 25 | Ami | Taiwan | Austronesian | Formosan |
| 26 | Atayal | Taiwan | Austronesian | Formosan |
| 27 | Paiwan | Taiwan | Austronesian | Formosan |
| 28 | Yami | Lanyu | Austronesian | Malayo-Polynesian |
Figure 2.
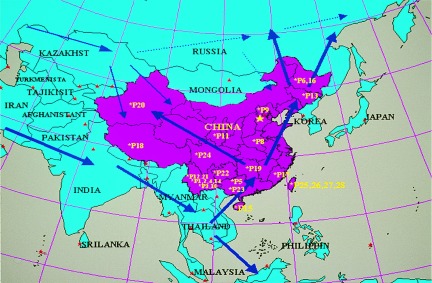
Hypothetical ancestral migration routes to the Far East. Refer to Table 1 for names of the numbered populations.
DNA samples were extracted either directly from lymphocytes or from immortalized cell lines. Some primers were purchased from Perkin–Elmer Applied Biosystems Division and some were kindly provided by Sequana Therapeutics (La Jolla, CA). Selected microsatellite loci were co-amplified in a single 5-μl PCR. TaqStart antibody (CLONTECH) was used to provide a hot-start mechanism. Following the PCR, a 1-μl aliquot of PCR product was loaded on a standard denaturing 6% polyacrylamide sequencing gel. Electrophoresis was conducted using an ABI373A sequencer configured with the B filter wheel during collection of fluorescence signal. GeneScan (Perkin–Elmer, Foster City, CA) was used to collect data, track lanes, measure fragment sizes, and to check the internal size standard. Genotypes were called by Genotyper (Perkin–Elmer, Foster City, CA). A binning method was used to convert raw data to allele frequency distribution.
Phylogenies presented in Fig. 1 were constructed by using the neighbor-joining method (18). Genetic distance proposed by Cavalli-Sforza and Edwards was used to estimate genetic distance between populations (19). A population was selected for phylogeny analysis only when the allele frequency distributions of the population for all microsatellite loci were available. A program, dsw, written by T. Ota, was used to reconstruct phylogeny. Bootstrap values were obtained based on 500 replications. African population lineage was used to root the phylogeny based on the result of Bowcock et al. (8).
Figure 1.

Phylogenies constructed by using the neighbor-joining method based on 30 microsatellites (A) and 15 microsatellites (B), respectively (12–14). Numbers on the branches are bootstrap values based on 500 replications. See text for discussion of clusters S1, etc. indicated on the right.
In Fig. 1A, microsatellites analyzed are D1S484, D2S434, D3S1768, D6S1009, D7S493, D10S537, D12S101, D12S373, D13S126, D15S101, D15S102, D15S230, D16S508, D16S667, D17S1824, D18S465, D19S152, D19S210, D19S414, D19S420, D19S601, D20S100, D20S115, D20S118, D20S171, D20S471, D21S1435, D22S1158, HLIP, and UTSW1523. In this phylogeny, populations and loci were selected to maximize the number of loci in the analysis. Eight Chinese populations were included in Fig. 1A. They are Han from Yunnan, Han from Guangdong, Manchurian, Jingpo, Deang, Atayal, Yami, and Paiwan.
In Fig. 1B, microsatellites analyzed are D1S484, D2S434, D7S493, D10S537, D12S373, D16S667, D17S1824, D19S152, D19S210, D19S414, D19S420, D20S100, D20S115, D20S171, and D21S1435. In this phylogeny, most representative populations in each region were selected and the loci are selected whenever their allele frequency information is available across those populations. Sixteen more Chinese populations were added for the analysis presented in Fig. 1B. They are Uyghur, Han (Northern) from Beijing, Wa, Tujia, Tibetan, Hui, Ewenki, Yao speaking Punu, Yi, She, Yao from Jinxiu, Han from Henan, Dong, Li, Lahu, Dai, Blang, Aini, and Ami.
RESULTS
The phylogeny based on 30 microsatellites (Fig. 1A) revealed a clear distinction between southern and northern Chinese populations, although the number of Chinese populations included in this phylogeny is small. Three northern Chinese populations clustered with the Japanese and Korean as expected. The southern populations in this phylogeny are not representative because three of the five southern populations are Taiwanese Aborigines speaking Austronesian languages. However, this phylogeny provides validation for our current approach, given the fact that the relationship among worldwide populations is identical to that presented in Bowcock et al. (8). The latter was derived by using a completely different set of markers, but some populations analyzed in this study were included in Bowcock et al. (Cambodian, Karitiana, Mayan, Australian, New Guinean, Italian, Zaire Pygmy, Central Republic Pygmy, and Lissongo). Populations from East Asia form a distinctive cluster indicating a common ancestry shared among those groups. Taiwanese Aborigines populations derived from the southern population cluster from the continent, indicating the probable origin of those populations and probably Polynesians.
The distinction between southern populations and northern populations was noticeable but far less clear when 16 more Chinese populations were added, producing the phylogeny presented in Fig. 1B. The number of loci was reduced to 15 due to incomplete data for some loci. Again, the populations from East Asia were derived from the same lineage.
In Fig. 1B, two clusters for the northern populations are discernible. Altaic language-speaking Buryat, Yakut, Uyghur, and Manchu clustered with the Korean and Japanese, two language isolates but closely related to Altaic. Two Han populations, one from north China and the other from Yunnan, also contributed to this cluster (cluster N1). Another Altaic language-speaking population, Ewenki, formed a cluster (cluster N2) with Tibetan, Tujia, and Hui, all of which were originally derived from the northern populations though currently living in the western part of China (21).
Populations of southern origin formed three clusters. In the first south cluster (S1), Blang, an Austro-Asiatic population, grouped with Deang, Aini, Lahu, and Dai, all sampled from the southwest part of Yunnan. This lineage then clustered with three populations from Taiwan (Paiwan, Atayal, and Yami), probably reflecting the origin of Taiwanese Aborigines and thus Polynesians from Southeast Asia. The fourth Taiwanese aboriginal population, Ami, forms a separate cluster with Han Chinese of southern origin living in the U.S. before they joined the previous cluster to form cluster S1. The second southern group consists of three Daic populations (Li, Dong, and Yao from Jinxiu) all from Guangxi or Hainan, two Hmong-Mien populations (She and Yao speaking Punu), Cambodian (a Austro-Asiatic population), Yi and Han from Henan (cluster S2). The second northern lineage (cluster N2) consists of mostly western populations derived from this southern group except Ewenki. Jingpo and Wa formed the third southern lineage (cluster S3). In this phylogeny, populations in East Asia can be divided into two groups: a northern group consisting of populations in cluster N1 and a southern group including all southern populations (clusters S1, S2, and S3) and the second cluster of northern origin (cluster N2). This relationship was not strongly supported by the bootstrap values among major clusters most of which were small. However, a phylogeny with 17 Chinese populations and 8 worldwide populations based on 26 loci presented a topology very similar to that of Fig. 1B, and the bootstrap value supporting the separation of the first northern cluster and the southern clusters being 13% and the bootstrap value supporting the second northern lineage being 19% (data not shown).
The measure of genetic distance, Dc (19), was used in this study because it generally outperformed other measures in obtaining correct topology for microsatellite markers in an extensive simulation study (15). The neighbor-joining method tends to be less affected by the presence of admixture occurring among populations in recovering the correct topology compared with the unweighted pair-group method of averages (UPGMA) and therefore became the method of choice in this analysis (17). Phylogenies using UPGMA were also constructed but not included because the relationships of worldwide populations are different from those in Bowcock et al. and other studies using microsatellites (8–10). Other measures of genetic distance such as Dsw, Rst, and (Δμ)2 were also used in the analysis (20–23), but they lead to less sensible results inconsistent with known ethnohistory of the populations studied (15–17).
CONCLUSIONS AND DISCUSSION
Validation of the utility of microsatellites in reconstructing evolutionary history of human populations has been made not only theoretically (20–23) but also empirically; the relationships based on microsatellites are generally consistent with morphological and paleontological evidence and other types of genetic markers (8–10). However, many of such studies used distantly related populations and, therefore, the utility of such markers in the study of closely related populations is yet to be explored. The current study reflects, to some extend, a lack of resolution of microsatellites in the reconstruction of closely related populations, probably because of an insufficient number of loci and a large number of populations studied but less likely because of the insufficient number of samples for each population as demonstrated by Shriver et al. (20). This is so because the variance of the genetic distance between loci is much larger than the variance due to sampling error (20) in the estimation of genetic distance. Small bootstrap values reflect insufficient amount of information available to resolve the genetic relationship among closely related populations in the presence of strong gene flow among those populations. But the employment of a much larger number of microsatellite loci in the current analysis may not guarantee a better resolution under such a scenario. Nevertheless, it is not our primary intention to reveal the detailed genetic relationship among those closely related populations, rather we are interested in exploring the major pattern of evolutionary history of the human populations currently residing in East Asia.
In both phylogenies with different loci and populations, populations from East Asia always derived from a single lineage, indicating the single origin of those populations. It does not preclude the possibility of an independent origin of modern humans in East Asia, but its contribution to the extant populations is not detectable in this analysis. It is now probably safe to conclude that modern humans originating in Africa constitute the majority of the current gene pool in East Asia. A phylogeny with very different topological structure would have been expected if an independent Asian origin of modern human had made a major contribution to the current gene pool in Asian populations. Since the methods employed in this analysis can detect only major genetic contribution from particular sources, a haplotype-based analysis will probably detect minor contribution from an independent origin of modern humans in East Asia (24, 25).
In contrast with previous studies (2–4) where distinction between southern and northern populations was clear, our current analysis showed that northern populations belong to two different groups, although statistical support was still weak. One noticeable difference in our study is the employment in the phylogeny reconstruction of the neighbor-joining method, which is supposedly more robust in the presence of genetic admixture. The use of microsatellites, a different type of genetic markers from previous studies, and the measures of genetic distance introduced further complication. However, the northern populations in cluster N2 were sampled from the southwestern part of China, except for Ewenki, where genetic admixture with the southern population was more likely to occur. This might explain why this group of northern populations clustered with southern populations.
Another noticeable feature from this analysis is that the linguistic boundaries are often transgressed across the six language families studied (Sino-Tibetan, Daic, Hmong-Mien, Austro-Asiatic, Altaic, and Austronesian). Such a phenomenon is even more pronounced among southern populations, where populations from the same geographic regions tend to cluster in the phylogeny (see Fig. 1B). This observation is consistent with the history of Chinese populations, where population migrations were substantial.
The current analysis suggests that the southern populations in East Asia may be derived from the populations in Southeast Asia that originally migrated from Africa, possibly via mid-Asia, and the northern populations were under strong genetic influences from Altaic populations from the north. But it is unclear how Altaic populations migrated to Northeast Asia. It is possible that ancestral Altaic populations arrived there from middle Asia, or alternatively they may have originated from East Asia.
The analyses of metric and nonmetric cranial traits of modern and prehistoric Siberian and Chinese populations showed that Siberians are closer to Northern Chinese and Mongolian than European (26, 27). The same notion holds for the facial flatness (26–28). European populations did not appear in Siberia, western Mongolia, and China until the Neolithic and Bronze Age (26, 27, 29, 30). Furthermore, cranial and dental analyses have linked the Arctic peoples, Buryat and east Asians with American Indians (31–35), which arrived through Beringia (Bering land bridge) somewhere between 15,000 and 30,000 years ago (36). These observations are generally consistent with the genetic evidence based on this research and mitochondrial DNA data (37–40). Therefore, it is more likely that ancestors of Altaic-speaking populations originated from an East Asian population that was originally derived from Southeast Asia, although the current Altaic-speaking populations undeniably admixed with later arrivers from mid-Asia and Europe (see Fig. 2, thin solid lines). The possibility of early northern route migration from mid-Asia to Siberia is doubtful, given the fact that the last glacier started to recede only 15,000 years ago (see Fig. 2, dashed lines).
This conclusion can be tested by using simple inductive logic. If the ancestral Altaic-speaking population was of northern origin, the genetic relationship of extant populations should follow the phylogeny presented in the bottom of Fig. 3. The phylogeny generated in the current study apparently supports the upper phylogeny of Fig. 3. In this analysis, Altaic populations are represented by Buryat and Yakut. Southern Chinese populations are those populations from Yunnan and Taiwan that reportedly did not have any admixture with Altaic populations. Populations from Middle Asia were not available to this study.
Figure 3.

Phylogenetic relationships of worldwide populations under two hypotheses; see text for discussion.
Now that we have established that populations in East Asia were subjected to genetic contributions from multiple sources: Southeast Asia, Altaic from northeast Asia, and mid-Asia or Europe. It would be interesting to estimate relative contributions from each source. Unfortunately, the current study involved only mostly minority populations. A study involving populations across the country is necessary to reveal such a picture.
Acknowledgments
We thank the people whose DNA was provided by L. L. Cavalli-Sforza, J. Kidd, M. Hsu, S. Q. Mehdi, and J. Bertranpetit. Informed consent was obtained for the newly collected Chinese samples. This project was completed under the organization of Z. Chen and B. Q. Qiang and funded by the National Natural Sciences Foundation of China. We also thank P. Watkin and P. Morin from Sequana Therapeutics, Inc., for their generous support.
Footnotes
A Commentary on this article begins on page 11501.
References
- 1. Grimes B F. Ethnologue. 13th Ed. Dallas: Summer Institute of Linguistics; 1996. [Google Scholar]
- 2.Zhao T M, Zhang G, Zhu Y, Zheng S, Liu D, Chen Q, Zhang X. Acta Anthropol Sin. 1986;6:1–8. [Google Scholar]
- 3.Zhao T M, Lee T D. Hum Genet. 1989;83:101–110. doi: 10.1007/BF00286699. [DOI] [PubMed] [Google Scholar]
- 4.Weng Z, Yuan Y, Du R. Acta Anthropol Sin. 1989;8:261–268. [Google Scholar]
- 5.Zhang Z B. Acta Anthropol Sin. 1988;7:314–323. [Google Scholar]
- 6.Zhang H. Acta Anthropol Sin. 1988;7:39–45. [Google Scholar]
- 7.Etler D A. Hum Biol. 1992;64:567–585. [PubMed] [Google Scholar]
- 8.Bowcock A M, Ruiz-Linares A, Tomfohrde J, Minch E, Kidd J R, Cavalli-Sforza L L. Nature (London) 1994;368:455–457. doi: 10.1038/368455a0. [DOI] [PubMed] [Google Scholar]
- 9.Deka R, Jin L, Shriver M D, Yu L M, DeCroo S, Hundrieser J, Bunker C H, Ferrell R E, Chakraborty R. Am J Hum Genet. 1995;56:461–474. [PMC free article] [PubMed] [Google Scholar]
- 10.Jorde L B, Bamshad M J, Watkins W S, Zenger R, Fraley A E, Krakowiak P A, Carpenter K D, Soodyall H, Jenkins T, Rogers A R. Am J Hum Genet. 1995;57:523–538. doi: 10.1002/ajmg.1320570340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang L. Acta Anthropol Sin. 1986;5:243–258. [Google Scholar]
- 12.Brooks A S, Wood B. Nature (London) 1990;344:288–289. doi: 10.1038/344288a0. [DOI] [PubMed] [Google Scholar]
- 13.Li T, Etler D A. Nature (London) 1992;357:404–407. doi: 10.1038/357404a0. [DOI] [PubMed] [Google Scholar]
- 14.Cann R L. In: Prehistoric Mongoloid Dispersals. Akazawa R, Szathmary E J E, editors. Oxford: Oxford Univ. Press; 1996. pp. 41–51. [Google Scholar]
- 15.Nei M, Takezaki N. Mol Biol Evol. 1996;13:170–176. doi: 10.1093/oxfordjournals.molbev.a025553. [DOI] [PubMed] [Google Scholar]
- 16.Cavalli-Sforza L L, Menozzi P, Piazza A. The History and Geography of Human Genes. Princeton, NJ: Princeton Univ. Press; 1994. pp. 280–287. [Google Scholar]
- 17.Ruiz-Linares A. In: The Origin and Past of Modern Humans as Viewed from DNA. Brenner S, Hanihara K, editors. Singapore: World Scientific; 1994. pp. 123–148. [Google Scholar]
- 18.Saitou N, Nei M. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 19.Cavalli-Sforza L L, Edwards A W F. Am J Hum Genet. 1967;19:233–243. [PMC free article] [PubMed] [Google Scholar]
- 20.Shriver M D, Jin L, Boerwinkle E, Deka R, Ferrell R E, Chakraborty R. Mol Biol Evol. 1995;12:914–920. doi: 10.1093/oxfordjournals.molbev.a040268. [DOI] [PubMed] [Google Scholar]
- 21.Goldstein D B, Ruiz-Linares A, Cavalli-Sforza L L, Feldman M W. Genetics. 1995;139:463–471. doi: 10.1093/genetics/139.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Slatkin M. Genetics. 1995;139:457–462. doi: 10.1093/genetics/139.1.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldstein D B, Ruiz-Linares A, Cavalli-Sforza L L, Feldman M W. Proc Natl Acad Sci USA. 1995;92:6723–6727. doi: 10.1073/pnas.92.15.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deka R, Jin L, Shriver M D, Yu L M, Saha N, Barrantes R, Chakraborty R, Ferrell R E. Genome Res. 1996;6:1177–1184. doi: 10.1101/gr.6.12.1177. [DOI] [PubMed] [Google Scholar]
- 25.Underhill P A, Jin L, Lin A A, Mehdi S Q, Jenkins T, Vollrath D, Davis R W, Cavalli-Sforza L L, Oefner P J. Genome Res. 1997;7:996–1005. doi: 10.1101/gr.7.10.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishida H, Dodo Y. In: Prehistoric Mongoloid Dispersals. Akazawa T, Szathmary E J E, editors. Oxford: Oxford Univ. Press; 1996. pp. 113–124. [Google Scholar]
- 27.Konigsberg L W. Hum Biol. 1990;62:49–70. [PubMed] [Google Scholar]
- 28.Ishida H. Z Morphol Anthropol. 1992;79:53–67. [PubMed] [Google Scholar]
- 29.Alekseev V P, Gokhman I I. Izv Sib Otd Akad Nauk SSSR. 1987;3:53–60. [Google Scholar]
- 30.Han K. Acta Anthropol Sin. 1986;5:227–242. [Google Scholar]
- 31.Dodo Y, Ishida H. J Anthropol Soc Nippon. 1987;95:161–177. [Google Scholar]
- 32.Ishida H. Anthropol Sci. 1993;101:47–63. [Google Scholar]
- 33.Ossenberg N S. In: The Evolution and Dispersal of Modern Humans in Asia. Akazawa T, Alki K, Kimura T, editors. Tokyo: Hokusensha; 1992. pp. 493–530. [Google Scholar]
- 34.Alekseev V P, Trubnikova O V. Some Problems of Taxonomy and Genealogy of the Asiatic Mongoloids (Craniometry) Novosibirsk, Russia: Nauka; 1984. [Google Scholar]
- 35.Turner C G, II. Natl Geographic Res. 1986;2:37–46. [Google Scholar]
- 36.Underhill P A, Jin L, Zemans R, Oefner P J, Cavalli-Sforza L L. Proc Natl Acad Sci USA. 1996;93:196–200. doi: 10.1073/pnas.93.1.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schurr T G, Ballinger S W, Gan Y-Y, Hodge J A, Merriwether D A, Lawrence D N, Knowler W C, Weiss K M, Wallace D C. Am J Hum Genet. 1990;46:613–623. [PMC free article] [PubMed] [Google Scholar]
- 38.Torroni A, Schurr T G, Cabell M F, Brown M D, Neel J V, Larsen M, Smith D G, Vullo C M, Wallace D C. Am J Hum Genet. 1993;53:563–590. [PMC free article] [PubMed] [Google Scholar]
- 39.Torroni A, Sukernik R I, Schurr T G, Starkovskays Y B, Cabell M F, Crawford M H, Comuzzie A G, Wallace D C. Am J Hum Genet. 1993;53:591–608. [PMC free article] [PubMed] [Google Scholar]
- 40.Merriwether D A, Hall W W, Vahlne A, Ferrell R E. Am J Hum Genet. 1996;59:204–212. [PMC free article] [PubMed] [Google Scholar]