

Abstract
Viruses have often been observed in association with the dense microvilli of polarized epithelia as well as the filopodia of nonpolarized cells, yet whether interactions with these structures contribute to infection has remained unknown. Here we show that virus binding to filopodia induces a rapid and highly ordered lateral movement, “surfing” toward the cell body before cell entry. Virus cell surfing along filopodia is mediated by the underlying actin cytoskeleton and depends on functional myosin II. Any disruption of virus cell surfing significantly reduces viral infection. Our results reveal another example of viruses hijacking host machineries for efficient infection by using the inherent ability of filopodia to transport ligands to the cell body.
Introduction
Enveloped virus entry is initiated by binding to surface receptors/coreceptors followed by trafficking to specific entry sites where viruses encounter a milieu that provides for activation of their fusion machine, the viral envelope glycoprotein (Env). Fusion induces the mixing of viral and cellular membranes to release viral capsids into the cytoplasm of the host (Hernandez et al., 1996; Young, 2001; Smith and Helenius, 2004). For pH-independent viruses, a critical concentration of receptors/coreceptors may suffice to induce fusion at the plasma membrane, while pH-dependent viruses must reach an endosomal environment for fusion to be triggered by low pH. Little is known about the cell biology of viral trafficking during entry, in particular in the morphological context of the host cell. Viruses have been frequently observed in association with highly dynamic cell surface protrusions such as filopodia, or in the case of mucosal tissues, the dense meshwork of microvilli (Helenius et al., 1980; Duus et al., 2004; Smith and Helenius, 2004). Whether filopodia or microvilli actively contribute to infections is unclear. Here, we demonstrate that upon interaction with filopodia, viruses undergo rapid actin- and myosin II–driven transport, “surfing” to entry sites at the cell body. Our studies suggest that virus cell surfing along filopodia represents a novel mechanism by which viruses use host cell machineries for efficient infection.
Results
Virus cell surfing along filopodia precedes fusion
Murine leukemia virus (MLV), like many other viruses (Helenius et al., 1980), efficiently associates with cellular protrusions such as filopodia and retraction fibers of fibroblast cells (Fig. 1). To gain insights into the dynamics of filopodia-associated MLV, we applied live-cell imaging using fluorescently labeled retroviral particles (McDonald et al., 2002; Sherer et al., 2003). MLV labeled with a fusion-competent envelope–YFP protein (Env–YFP) was added to HEK 293 cells expressing the receptor mCAT-1 fused to CFP (mCAT-1–CFP; Albritton et al., 1989; Masuda et al., 1999). Confocal time-lapse microscopy of HEK 293 cells that exhibited abundant filopodia and retraction fibers revealed that viral particles not only attached to filopodia but also underwent directed rearward movement, surfing toward the cell body of mCAT-1–CFP–expressing cells (Fig. 2, A and B; Video 1 available at http://www.jcb.org/cgi/content/full/jcb.200503059/DC1). After reaching the cell body, viral particles lost the punctate red envelope label, consistent with fusion-mediated diffusion of Env–YFP into the large surface of the plasma membrane (Fig. 2 C; Video 1). To quantitatively analyze particle movement we measured the distance an individual particle traveled within 10 s, the time interval between single frames of the time-lapse video. The resulting speed for each step (in μm/min) was plotted over time, with time “0” representing the moment when an individual particle attached to a filopodium (Fig. 2 D; Fig. S1 A available at http://www.jcb.org/cgi/content/full/jcb.200503059/DC1). The speed of particles moving toward the cell body was defined as positive, whereas particles moving away from the cell body toward the tip of the filopodium generated negative numbers. Such an analysis for 85 MLV particles from six independent experiments revealed that particles initially move randomly along the filopodium before they begin moving toward the cell body (Fig. 2 D). Individual particles progressed at varying speeds. Fast periods of movement reaching maximum speeds of 6–10 μm/min were interrupted by slower intervals, resulting in an average speed of ∼2 μm/min (Fig. 2 D; Fig. S1 A). Particle movement was also very efficient with over 90% of all particles apparently associated with filopodia engaging fast movement toward the cell body. Finally, 2 min after attachment, most particles had disappeared at the base of the filopodium. Over time, Env–YFP accumulated throughout the plasma membrane, inducing neighboring cells to undergo cell–cell fusion (Video 2 available at http://www.jcb.org/cgi/content/full/jcb.200503059/DC1).
Figure 1.

MLV is associated with filopodia. HEK 293 cells expressing receptor mCAT-1 were incubated with infectious MLV for 10 min and then prepared for analysis by SEM. Arrows indicate virions associated with filopodia. Bars, 1 μm.
Figure 2.
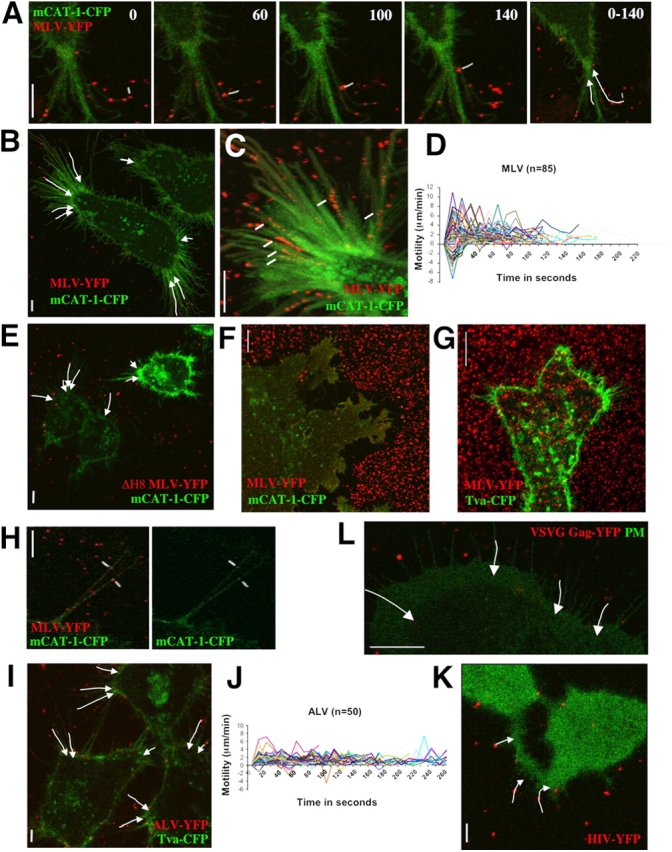
Virus cell surfing along filopodia. (A) An individual MLV particle fluorescently labeled with envelope–YFP (MLV–YFP, pseudocolored in red, marked in white) surfs along the filopodium of a HEK 293 cell transduced with mCAT-1–CFP (pseudocolored in green). The time in seconds is presented in each frame. The overall movements of the two particles are summarized by arrows in the far right panel (0–140 s). (B) The image summarizes the overall movement of selected particles on filopodia of a single HEK 293 cell as observed in Video 1. (C) To visualize the gradual loss of fluorescence of moving particles (marked in white), 31 frames from a region of interest of Video 1 were superimposed. (D) To quantify virus cell surfing, the motility (μm/min) of 85 individual MLV particles was plotted over time (in seconds), with time point 0 representing the moment of virus attachment to a filopodium. Positive values of motility represent particles moving toward the cell body whereas negative numbers represent viruses moving toward the tip of the fiber. (E) MLV bearing a fusion-defective envelope protein also surfs. The image, generated as in B, visually summarizes a time-lapse video not shown. (F) Virus capture by short-lived filopodia and ruffles. HEK 293 cells expressing mCAT-1–CFP were allowed to spread for 30 min on a glass coverslip containing prebound MLV–YFP. Cells formed a large syncytium as a consequence of viral infection. (G) Virus capture is receptor dependent. An experiment as in F was performed with MLV–YFP and HEK 293 cells expressing Tva–CFP, the receptor for the ALV. Note that cells are unable to take up prebound virus from beneath the cell and its surroundings. (H) Receptor mCAT-1–CFP (green) is recruited to MLV particles labeled with Env–YFP (red) when they attach to filopodia of XC cells. (I) ALV surfs on HEK 293 cells expressing Tva–CFP. The image visualizes the movement of individual particles as shown in Video 4. (J) A quantitative analysis as in D was performed for 50 ALV particles. (K) HIV (red) surfs on HEK 293 cells expressing CD4 and CXCR4. To identify receptor-expressing cells, cytoplasmic CFP (green) was coexpressed. The image summarizes the movement of three particles as shown in Video 5. (L) MLV Gag–YFP-labeled virions containing the VSVG protein (red) surf along filopodia of XC cells expressing mCAT-1–CFP (green) as a plasma membrane marker. The entire sequence is shown in Video 6. In all panels, vertical size bars correspond to 5 μm.
Because the punctate Env–YFP label did not diffuse completely until reaching the base of filopodia, and MLV carrying a fusion defective envelope (Zavorotinskaya et al., 2004) still surfed (Fig. 2 E), we predicted that surfing must precede fusion. To directly examine the membrane topology of viruses associated with filopodia, we performed transmission electron microscopy (TEM) of cells fixed after MLV addition (Fig. 3). TEM micrographs showed individual viruses engaged with filopodia at an average distance of 11.7 ± 0.4 nm (n = 44; P = 0.0001). Spikes, likely representing Env–receptor complexes, were frequently observed between the virus and the cell membrane (Fig. 3, A–D), but fusion intermediates or intracellular capsids were detected only at the widening base of filopodia or at the cell body (Fig. 3, E–H).
Figure 3.
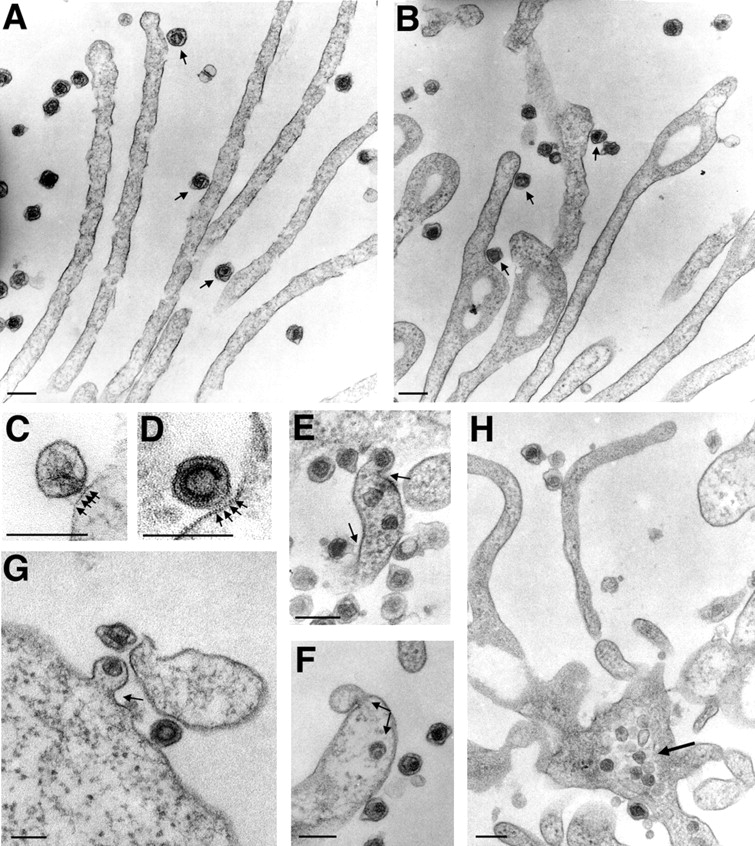
MLV fuses with the plasma membrane at the base of filopodia or at the cell body. (A and B) HEK 293 cells infected with MLV for 30 min were prepared for TEM and cut en face within 100 nm from the glass coverslip to visualize viruses and filopodia. Circular nodules as observed in B are indicative of retraction fibers (Mitchison, 1992). Viruses engaged with filopodia or retraction fibers are marked with arrows. (C and D) Two examples of frequently observed spikes (marked by arrows) between virus and membrane are shown. (E–G) Fusion sites (marked by arrows) were observed at the widening base of fibers or at the cell body. (H) Viral capsids accumulate inside the cytoplasm at the base of fibers. In all panels, size bars correspond to 200 nm.
Virus capture by filopodia and retraction fibers is receptor dependent
Although we had initially studied this process on filopodia and retraction fibers of HEK 293 cells, virus cell surfing was also observed along filopodia of mouse and rat fibroblasts and HEK 293 cells cultured on substrates that induced a spread out and filopodia-abundant morphology (unpublished data). In addition to surfing, ruffles were also active in virus capture and short-lived filopodia were observed to “pick” viral particles from the surrounding areas, efficiently delivering them to the cell body for fusion (Video 3 available at http://www.jcb.org/cgi/content/full/jcb.200503059/DC1). The activity of ruffles and filopodia in virus capture was best illustrated when Env–YFP-labeled MLV was prebound to the glass coverslip before receptor-expressing cells were seeded and allowed to spread. Filopodia captured all of the viral particles surrounding the cells, creating a clear border area lacking fluorescence (Fig. 2 F). Nonviral particle capture and uptake by cells have been frequently observed (Albrecht-Buehler and Lancaster, 1976). However, for viruses this activity was strictly dependent on cognate receptor–envelope interactions (Fig. 2 G). Moreover, the ability of viruses to undergo surfing was not observed for 293 cells lacking mCAT-1 (unpublished data). Consistent with an important role for viral receptors in surfing, mCAT-1–CFP recruitment to attaching MLV particles preceded the onset of surfing. Virus and receptor colocalized shortly after virus attachment and subsequently moved together toward the cell body (Fig. 2 H; unpublished data). Visualization of receptor recruitment was close to the detection limits of the confocal microscope, consistent with very few receptor molecules being concentrated beneath an individual viral particle.
Cell surfing is shared by other viruses
Surfing was not restricted to MLV. Gag (viral capsid polyprotein precursor)–YFP-labeled MLV capsids containing the envelope glycoprotein of the avian leukosis virus (ALV) surfed along filopodia of HEK 293 cells when they expressed the ALV receptor Tva fused to CFP (Bates et al., 1993; Fig. 2 I; Video 4 available at http://www.jcb.org/cgi/content/full/jcb.200503059/DC1). ALV surfed at a slower speed than MLV, averaging 1 μm/min, and attaching particles lacked the initial random movement seen for MLV (Fig. 2 J). Human immunodeficiency virus (HIV) similarly surfed when cells were expressing the HIV receptors CD4 and CXCR4 (Fig. 2 K; Video 5 available at http://www.jcb.org/cgi/content/full/jcb.200503059/DC1). Surfing was not restricted to retroviruses. Particles bearing the envelope protein of vesicular stomatitis virus (VSV) also surfed along filopodia of rat fibroblasts (Fig. 2 L; Video 6 available at http://www.jcb.org/cgi/content/full/jcb.200503059/DC1). VSVG-containing viruses were found to move along filopodia and then continued to surf along the plasma membrane until they disappeared from the confocal microscope plane, likely due to endocytosis. Parallel immunofluorescence for actin in these cells showed that the areas that support virus movement are rich in actin filaments (Fig. S2, A and B available at http://www.jcb.org/cgi/content/full/jcb.200503059/DC1). Co-expression of clathrin–light chain–YFP confirmed colocalization of viruses with clathrin 15 min after infection (Fig. S2 C). Video microscopy revealed that clathrin-recruitment to surfing viruses was initiated as soon as the virus reached the cell body (Video 7 available at http://www.jcb.org/cgi/content/full/jcb.200503059/DC1). Interestingly, surfing continued during clathrin recruitment (Video 7). These experiments suggest that pH-dependent viruses such as VSV surf along filopodia and actin filaments to reach endocytic hot spots.
Virus cell surfing is actin and myosin dependent
The association of viral particles with actin-rich filaments and the observed highly ordered movement of viral particles implied an actin and motor-driven process. Addition of cytochalasin D, a reagent that blocks the barbed ends of actin filaments, inhibited surfing (Fig. 4, compare B with A; Video 8 available at http://www.jcb.org/cgi/content/full/jcb.200503059/DC1), whereas treatment with nocodazole, which disassembles microtubules, had no effect (Figs. 4 C and 5 C; unpublished data). Addition of the ATPase inhibitor sodium azide immediately blocked particle movement toward the cell body (Fig. 4 D; unpublished data). Quantitative analysis revealed that cytochalasin D or sodium azide treatment led to a diffusive random movement (Fig. 5, compare B and D with A; see Fig. S1 for tracks of individual particles) and a complete loss of directional motility toward the cell body (Fig. 5, H and I). Thus, surfing is an energy-dependent process mediated by the underlying actin cytoskeleton.
Figure 4.
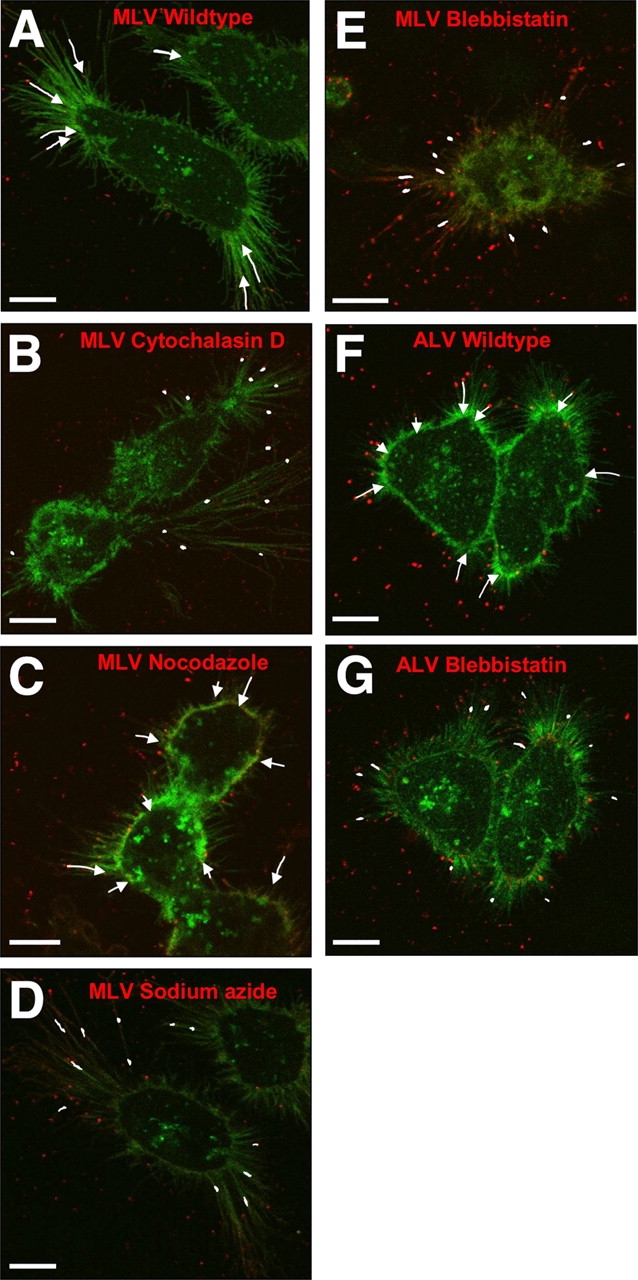
Virus cell surfing is actin and myosin II dependent. (A–G) Images visually summarize the extent of MLV (A–E) and ALV (F and G) surfing in the absence and presence of inhibitors as indicated; cytochalasin D (50 μM), nocodazole (50 μM), sodium azide (10 mM), and blebbistatin (30 μM). The data are based on time-lapse movies, some of them provided as supplemental data (Videos 1, 4, and 9). To compare the surfing activity visually, the motility observed over 120 s (MLV) or 200 s (ALV) is displayed. Bars, 10 μm.
Figure 5.

Quantitative analysis of the effects of inhibitors on virus cell surfing. (A) The motility (μm/min) of 85 individual MLV particles as presented in Fig. 2 D in the absence of any inhibitor (Wildtype) and plotted over time (in seconds). (B-E) An analysis as in (A) was performed in the presence of the inhibitors cytochalasin D (50 μM), nocodazole (50 μM), sodium azide (10 mM), and blebbistatin (30 μM), respectively. The number of particles (n) analyzed in each experiment is indicated. (F and G) Experiments as described in A and E, respectively, were analyzed for ALV movement on 293 cells expressing Tva–CFP. (H) Progressive average movement (μm) was determined using the mean motility for all analyzed particles at each given time point in the absence and presence of inhibitors as indicated. (I) The average directed motility toward the cell body (d, directed) was compared with the overall random motility (r, random) including forward as well as backward movement for all of the above experiments. To exclude the observed initial random motility seen in A, only the period between 30 and 90 s was included in the analysis. Cyto D, Blebb, and Noco are abbreviations for cytochalasin D, blebbistatin, and nocodazole, respectively.
It has been shown that myosin motors are involved in the movement of actin filaments in cell surface protrusions (Ponti et al., 2004; Svitkina et al., 1997), a process known as rearward or retrograde F-actin flow (Forscher and Smith, 1988; Mitchison and Kirschner, 1988; Sheetz et al., 1989; Lin and Forscher, 1993). However, the identity of the myosins involved in this process is unclear. Myosin II is a plus-end motor that localizes to the lamellum and retraction fibers (Miller et al., 1992; Rochlin et al., 1995; Verkhovsky et al., 1995; Fig. S3, A–C). If anchored, this myosin can also mediate minus-end motility by moving entire actin filaments (Mitchison and Kirschner, 1988). We tested the involvement of myosin II in viral surfing using its specific inhibitor, blebbistatin (Straight et al., 2003). Addition of blebbistatin completely blocked MLV, ALV, and VSV surfing along filopodia (Fig. 4, E and G; Videos 9 and 10 available at http://www.jcb.org/cgi/content/full/jcb.200503059/DC1; and unpublished data). Virus movement over the lamellum of rat fibroblast cells was similarly blocked, indicating that this movement over the cell surface is also mediated by the underlying actin–myosin II machinery (Video 10). Parallel immunofluorescence showed that myosin II localized in a patchy distribution to retraction fibers in these cells (Fig. S3, A and B). Myosin II also appeared to localize to the base of filopodia (Fig. S3, A and C). Treatment with blebbistatin resulted in a redistribution of myosin II to a diffuse perinuclear region (Fig. S3, D and E). Thus, in the case of retraction fibers, myosin II is clearly present in the cellular protrusions that promote virus motility. In the case of filopodia, myosin II may affect retrograde F-actin flow from the base of the filament. Quantitative analysis revealed that blebbistatin caused a complete block of directed movement of viruses toward the cell body, but still allowed a diffusive random movement in both directions similar to that observed for azide (Fig. 5, E and G–I). The similar effects of blebbistatin and azide suggest that myosin II is the predominant ATPase involved in virus cell surfing.
Inhibitors that block virus cell surfing interfere with infection of filopodia and microvilli-rich cells
If virus cell surfing represents a physiologically relevant pathway of viral infection, our observed visual block to surfing by blebbistatin and cytochalasin D should result in reduced infectivity. To address this question, we applied an assay that allowed us to test the role of these reversible inhibitors specifically in the earliest stages of infection (Kizhatil and Albritton, 1997). Viruses were added to cells in the presence or absence of drugs for periods of time as short as 5 min before the drugs were washed out and viral particles that had failed to enter cells were inactivated by either a 1-min acid wash or a 10-min treatment with methyl-β-cyclodextrin. Infection was continued in the absence of inhibitors. Applying this assay, we observed significant inhibitory effects of blebbistatin on MLV and VSV infection only when cells were cultured at low confluencies, conditions where cells exhibited abundant filopodia (Table I). At higher confluencies, when cells contacted each other, blebbistatin had reduced effects. Moreover, when cells were rounded completely due to trypsinization (receptor mCAT-1 is trypsin resistant), blebbistatin had no effect on MLV entry as compared with the untreated control. These data indicate that blebbistatin can only affect the earliest steps of MLV entry when cells exhibit abundant filopodia, suggesting a contribution of virus cell surfing to infection.
Table I. The inhibition of virus motility by blebbistatin and cytochalasin D correlates with decreased infectivity.
| Cell type I | Confluency | Incubation | Treatment | Infection in percent
|
|
|---|---|---|---|---|---|
| time | MLV | VSV | |||
| XC | 10% | 5 min | None | 100 ± 5 | 100 ± 8 |
| 5 μM Cyto D | 37 ± 2 | 49 ± 1 | |||
| 50 μM Cyto D | 43 ± 5 | 55 ± 3 | |||
| 50 μM Blebb | 43 ± 4 | 40 ± 3 | |||
| XC | 80–100% | 5 min | None | 100 ± 10 | 100 ± 3 |
| 5 μM Cyto D | 49 ± 5 | 65 ± 6 | |||
| 50 μM Cyto D | 56 ± 9 | 57 ± 3 | |||
| 50 μM Blebb | 85 ± 11 | 74 ± 4 | |||
| XC | Suspension | 5 min | None | 100 ± 3 | 100 ± 16 |
| 5 μM Cyto D | 75 ± 3 | 82 ± 6 | |||
| 50 μM Cyto D | 56 ± 5 | 78 ± 3 | |||
| 50 μM Blebb | 109 ± 4 | 72 ± 7 | |||
| MDCK (apical) | 100% | 30 min | None | 100 ± 4 | 100 ± 5 |
| 50 μM Cyto D | 19 ± 11 | 7 ± 1 | |||
| 30 μM Blebb | 19 ± 6 | 20 ± 1 | |||
| MDCK (apical) | 100% | 120 min | None | 100 ± 3 | 100 ± 4 |
| 50 μM Cyto | 16 ± 3 | 20 ± 4 | |||
| 30 μM Blebb | 31 ± 4 | 33 ± 3 | |||
| MDCK (basolateral) | 100% | 120 min | None | 100 ± 6 | 100 ± 15 |
| 50 μM Cyto D | 137 ± 15 | 93 ± 6 | |||
| 30 μM Blebb | 263 ± 9 | 191 ± 8 | |||
| MDCK (nonpolarized) | Suspension | 30 min | None | 100 ± 4 | 100 ± 1 |
| 5 μM Cyto D | 109 ± 6 | 61 ± 2 | |||
| 50 μM Cyto D | 104 ± 2 | 63 ± 5 | |||
| 30 μM Blebb | 142 ± 5 | 137 ± 5 | |||
| MDCK (nonpolarized) | Suspension | 120 min | None | 100 ± 4 | 100 ± 9 |
| 5 μM Cyto D | 113 ± 7 | 77 ± 3 | |||
| 50 μM Cyto D | 112 ± 3 | 56 ± 2 | |||
| 30 μM Blebb | 148 ± 4 | 130 ± 10 | |||
Cyto D and Blebb are abbreviations for cytochalasin D and blebbistatin, respectively. Basolateral control experiments were performed in duplicate and mean values with simple variations are presented. All other infection experiments were performed three to nine times and standard errors are shown. 100% infection in the absence of inhibitors (none) corresponded to several hundred infected colonies. Infectious titers of MLV and VSV were about 105 in the case of the 5 min infection of XC cells as well as the 30-min infection of nonpolarized MDCK cells. Susceptibility of MDCK cells to MLV and VSV was reduced upon polarization, with both viral preparations reaching titers of about 104.
Like blebbistatin, cytochalasin D had greater inhibitory effects on virus entry when cells exhibited abundant filopodia (Table I). As such, the previously reported requirement for an intact actin cytoskeleton during viral entry may reflect at least in part a need for virus cell surfing (Gottlieb et al., 1993; Kizhatil and Albritton, 1997; Bukrinskaya et al., 1998). However, in contrast to blebbistatin, cytochalasin D showed some pleiotropic effects on confluent and on trypsinized cells, consistent with a more global role for the actin cytoskeleton in early viral entry events (Table I).
Because of the relevance of mucosal epithelia to viral infection, we tested if polarized epithelial cells covered with dense microvilli would require surfing for efficient infection. Indeed, infection of polarized MDCK cells by MLV or VSVG-containing viruses was reduced at least fivefold by blebbistatin and cytochalasin D when virus was added to the microvilli-rich apical side but not to the basolateral side (Table I). As was the case for fibroblasts, no inhibitory effects of blebbistatin were observed for trypsinized MDCK cells that exhibited a highly folded surface but lacked microvilli as visualized by scanning electron microscopy (SEM; Table I; Fig. S4 C). In fact, blebbistatin caused a slight enhancement in infection of trypsinized MDCK cells as well as if the virus was applied to the basolateral side of a monolayer of polarized cells. Although these observations suggest an additional role for myosin II in virus entry, inhibitory effects were strictly associated with the microvilli-rich apical side of polarized epithelial cells.
Cytochalasin D behaved identically to blebbistatin in its ability to inhibit the entry of MLV via microvilli-rich surfaces suggesting that the role of actin is limited to the movement along microvilli (Table I). In contrast, infection of MDCK cells by VSV, which uses a pH-dependent endocytic entry route, remained moderately sensitive to cytochalasin D even on trypsinized MDCK cells.
In contrast to the filopodia of fibroblasts, microvilli are too small to be imaged by live microscopy. To test if the observed inhibitory effects of blebbistatin were due to a block in virus cell surfing, we applied SEM to visualize microvilli-rich surfaces and viruses at magnifications of ∼20,000 and 40,000×. To this end, viruses were bound on ice to polarized MDCK cells. After 30 min, the samples were washed with cold media to remove unbound virus. One sample was immediately prepared for SEM while the other two were incubated for an additional 30 min at 37°C in the absence or presence of blebbistatin. Scanning electron micrographs of all samples revealed efficient binding of viruses to the microvilli of MDCK cells. A clustering and bundling of tips of microvilli and viruses, both of similar size, was observed (Fig. 6 A, upper panel). This effect was not due to aggregation of virions, rather, as revealed by parallel TEM, was caused by the multivalency of virus/microvilli interactions (Fig. 6 B). Incubation at 37°C in the absence of blebbistatin resulted in a complete loss of microvilli-associated viruses (Fig. 6 A, middle panel). Parallel TEM demonstrated that under these conditions viruses had entered cells (Fig. 6 C). In contrast, the presence of blebbistatin completely blocked virus movement along microvilli, arresting the process at the step of virus binding (Fig. 6 A, lower panel). These data demonstrate that viruses efficiently bind to the microvilli of polarized epithelial cells, suggest that they use surfing to enter cells at the cell body and indicate that surfing is controlled by myosin II.
Figure 6.

MLV enters polarized epithelial cells via myosin II–dependent surfing along microvilli. (A) Individual steps of MLV infection of polarized MDCK cells were visualized by SEM in the presence and absence of blebbistatin as indicated. Arrows indicate virions associated with microvilli. Bars, 500 nm. (B) To analyze the nature of clusters seen in A TEM was performed revealing multivalent interactions between viruses and microvilli. Viruses similar in size to microvilli are recognized by their electron dense capsid core. (C) Parallel TEM of infected MDCK cells shows that at 30 min viruses are internalized. Bars: (B and C) 200 nm.
Discussion
We have identified a novel process by which viruses induce rapid and directed trafficking along filopodia to reach the cell body for entry into cells. The frequently observed association of viruses with filopodia is not coincidental, but represents an efficient infectious pathway that in the case of endocytic viruses such as Semliki Forest virus and VSV can result in a direct transport of viruses into clathrin-coated pits (Helenius et al., 1980; and this paper). Interference with surfing results in a modest reduction in infection of cultured fibroblast cells where viruses have relatively easy access to the cell body. In contrast, a block to surfing leads to a significant reduction in infection of polarized epithelial cells that are covered with dense microvilli. We hypothesize that in vivo, virus cell surfing enables the penetration of the microvilli-rich mucosal surfaces to efficiently infect the host. Inhibitors such as blebbistatin should be tested in vivo for their potential to reduce mucosal transmission, which accounts for ∼80% of all HIV infections (Bomsel and Alfsen, 2003).
With virus cell surfing, our understanding of viral entry is now modified by an early stage involving virus attachment, receptor recruitment, the establishment of a link to the underlying actin cytoskeleton and myosin II–mediated transport of the virus to the cell body. This process lies upstream of cell entry regardless of whether the virus enters cells by pH-independent (MLV, HIV) or pH-dependent (ALV, VSV) mechanisms (Stein et al., 1987; Maddon et al., 1988; Mothes et al., 2000; Kolokoltsov and Davey, 2004; Matsuyama et al., 2004). Surfing may have evolved to avoid having to penetrate the dense cortical actin cytoskeleton by moving along the plasma membrane to reach endocytic hot spots, which are areas of active actin remodeling. MLV and HIV, which do not require endocytosis for fusion, also surf to these areas before fusion, thereby avoiding the delivery of capsid into actin filament-dense areas.
Virus cell surfing depends on cognate Env–receptor interactions. Visually, receptor recruitment precedes the onset of surfing. The initial random movements of MLV observed within the first 10 s of attachment to filopodia likely reflects a period of mCAT-1 receptor recruitment as well as the establishment of a link between oligomerized receptor and actin filaments.
Transport along filopodia is mediated by the underlying actin cytoskeleton and is controlled by myosin II. Because myosin II is a plus end motor that apparently mediates minus end motility toward the cell body, it must regulate the movement of entire actin filaments, a process called retrograde F-actin flow (Forscher and Smith, 1988; Mitchison and Kirschner, 1988; Sheetz et al., 1989; Lin and Forscher, 1993). Thus, our data suggest a role for myosin II in retrograde flow. How myosin II controls retrograde flow remains to be determined. Although myosin II clearly localizes to retraction fibers, it is absent from filopodia. In the latter case, myosin II may control actin filament movement at the base of filopodia by regulating actin filament disassembly. Although we would anticipate the involvement of other myosins in retrograde flow, azide does not block virus cell surfing beyond the level observed for blebbistatin, suggesting that myosin II plays a predominant role.
How viruses engage retrograde F-actin flow remains to be determined. Signaling from cytoplasmic tails of oligomerized receptor appears to be a prerequisite for the establishment of a link to the actin cytoskeleton (Felsenfeld et al., 1996; Suter et al., 1998). Tva receptors carrying a GPI anchor instead of a transmembrane domain do not support efficient surfing of ALV (unpublished data), consistent with the observed slow kinetics of ALV infection mediated by these receptors (Narayan et al., 2003). The link to the actin cytoskeleton may only briefly be tight when viruses travel at maximum speed. In general, however, surfing viruses appear to engage a molecular clutch resulting in a fast-and-slow or slippage mode of movement rather than a rigid link to actin filaments. The ability of different viruses and their receptors to engage retrograde flow varies. MLV and ALV move at different average speeds along filopodia of the same cell type. In addition, movement of individual viral particles along the same filopodium occurs independently with no apparent coordination (unpublished data).
Virus cell surfing was observed on a number of different actin-rich protrusions such as filopodia, retraction fibers, and microvilli. In each case, surfing was mediated by the underlying actin cytoskeleton and was dependent on myosin II. Even in the case of short-lived filopodia that were observed to occasionally capture viral particles, particles were brought to the cell body by retrograde flow of actin filaments. These observations suggest that all instances of virus cell surfing are based on a related basic mechanism despite being morphologically different.
It is the nature of host–pathogen interactions that pathogens exploit existing cellular pathways. In this case, viruses likely engage retrograde F-actin flow, a process that was discovered and has been studied extensively using artificial beads (Abercrombie et al., 1970; Harris and Dunn, 1972; Albrecht-Buehler and Lancaster, 1976; Forscher and Smith, 1988; Mitchison and Kirschner, 1988; Sheetz et al., 1989). The ability of beads to establish a link to the actin cytoskeleton had been used to study how the forces of the actin cytoskeleton can translate into cell motility if beads were immobilized. As such, the field has focused on adhesion molecules such as integrins and members of the immunoglobulin family of CAM proteins (Felsenfeld et al., 1996; Suter et al., 1998). Our work with viruses now reemphasizes the role of the actin cytoskeleton in ligand uptake, transport, and endocytosis and offers viruses as physiological “beads” to study this process. In addition to adhesion molecules we extend the list of receptors capable of promoting rearward movement toward the cell body to also include transporters such as mCAT-1 for MLV (Albritton et al., 1989), receptors of the LDL receptor-like family Tva for ALV (Bates et al., 1993) as well as CD4/chemokine receptors in the case of HIV (Littman, 1998). As such, virus cell surfing likely reflects the fundamental ability of receptors to transport ligands along filopodia. Movement of EGF quantum dots along filopodia has recently been reported (Lidke et al., 2004). Ferritin and toxins have also been observed to travel along microvilli in an actin-dependent fashion (Gottlieb et al., 1993; Shurety et al., 1996). Thus, viral cell surfing appears to represent the exploitation of retrograde F-actin flow to efficiently transport signaling molecules from the periphery toward endocytic zones at the cell body. One prediction of this hypothesis is that signaling cascades are spatially separated into peripheral events that induce cell surfing and downstream events in the cell body that trigger signaling toward the nucleus. As such, cell surfing, powered by the underlying F-actin flow, and endocytosis in areas of actin remodeling, likely represent two steps of a coordinated process of ligand signaling and uptake.
Materials and methods
Reagents and cell lines
Cytochalasin D, nocodazole, and sodium azide were obtained from Sigma-Aldrich. Blebbistatin was purchased from Toronto Research Chemicals. Reagents and cell lines used for the generation of labeled virions were as described (Sherer et al., 2003). Plasmids encoding CD4 and CXCR4 were a gift from Heinrich Gottlinger (University of Massachusetts, Worcester, MA). 293 cells were obtained from American Type Culture Collection, a small number of which exhibited extensive filopodia under normal culture conditions. Extensive filopodia were also evident when 293 cells were grown on alcian blue–treated glass coverslips. For this purpose glass coverslips were rinsed in 70% ethanol, heated to boiling in 1% alcian blue (Sigma-Aldrich), incubated for 10 min, rinsed three times in water, and allowed to dry (Hammond et al., 1998). 293 cells were bound to treated glass coverslips in the absence of serum. DFJ8 cells represent DF-1 chicken cells stably expressing mCAT-1 (Sherer et al., 2003). Mouse Swiss fibroblasts were a gift from Philip Allen (Harvard Medical School, Boston, MA) and Rat XC sarcoma cells (Klement et al., 1969) stably expressing low amounts of mCAT-1–CFP, actin–YFP (a gift from Melissa Rolls, Pascal Stein, and Tom Rapoport, Harvard Medical School, Boston, MA) or clathrin light chain–YFP (Gaidarov et al., 1999) were generated by selection using G418 (Invitrogen). 293 and DFJ8 cells were cultured in DME high glucose (Invitrogen) containing 10% FBS plus Pen/Strep/Glutamine. XC and MDCK cells (strain II; gift of Ira Mellman, Yale University, New Haven, CT) were grown in αMEM (Invitrogen) with 10% FBS. MDCK cells stably expressing mCAT-1–CFP were selected using G418 (Invitrogen).
Generation of fluorescently labeled viruses
Most reagents for the generation of fluorescent viruses have previously been described (Sherer et al., 2003). Env–YFP-labeled MLV is a replication competent virus produced in large quantities from infected DFJ8 cells cultured in roller bottles. Viruses obtained from filtered supernatants were spun through a 15% sucrose cushion, resuspended in DME containing 10% FBS, filtered through 0.2 μm filters (Nalgene) and stored at −80°C. Gag–YFP-labeled MLV cores (Sherer et al., 2003) were used for the generation of viruses bearing fusion-defective MLV Env (delta H8; James Cunningham, Harvard Medical School, Boston, MA), ALV-A Env, and VSVG (Mothes et al., 2000). Fluorescent HIV particles were generated by cotransfection of a 10 cm plate of 293 cells with plasmids encoding HIVBRU3, −Env (5 μg; McDonald et al., 2002), HIV-Env (5 μg), and HIV-Gag–YFP (1 μg; Sherer et al., 2003). Medium was replaced 36 h after transfection. Virus-containing supernatants were collected at 40 and 44 h and directly used for imaging experiments.
Imaging
Transfection and live cell imaging of 293, DFJ8, and XC cells were as previously described (Sherer et al., 2003) using the 100× oil objective (numerical aperture 1.4) of an LSM 510 confocal microscope equipped with a Zeiss axiovert 100 M base (Zeiss MicroImaging, Inc.). For live cell imaging, cells were grown on poly-L-lysine–coated 35-mm glass-bottom plates (MatTek). Before imaging at 37°C, media was replaced with DME/10% FBS supplemented with 10 mM Hepes, pH 7.4. In most imaging experiments, a high viral multiplicity of infection of 100–1,000 was used in order to capture a suitable number of particles in an individual confocal plane within a short period of time. In time-lapse videos, CFP and YFP channels were imaged every 10 s and pseudocolored in green and red, respectively. All videos were exported from the LSM 510 as QuickTime videos, edited using Openlab (Improvision) or Photoshop software (Adobe) and compressed using Apple video (Videos 1–6 and 8–10) or the Apple animation mode (Video 7). For quantitative analysis, the distance each individual particle traveled within 10 s was determined using LSM 510 software.
In some experiments MatTek glass-bottom plates were pretreated with 1 mg/ml fibronectin (Invitrogen) for 10 min at room temperature and spun at 1,000 rpm to generate an evenly coated surface. Fluorescent viruses were allowed to bind for 30 min at room temperature. After removal of excess virus, HEK 293 cells expressing viral receptors were seeded for 30 min at 37°C before fixation and imaging.
For myosin II–indirect fluorescence, XC cells stably expressing actin–YFP were incubated with 0.02% saponin–PBS for 30 s before 10 min fixation in 4% paraformaldehyde/0.25% Triton X-100. Cells were washed with PBS, quenched with 50 mM NH4Cl, and washed with 0.05% Triton X-100–PBS before incubation for 1 h with rabbit antimyosin II antisera (Biomedical Technologies Inc.). After washing, cells were subsequently incubated for 40 min with Alexa Fluor 568–conjugated anti–rabbit secondary antisera (Invitrogen), washed, and mounted on glass slides with Gelmount (Biomeda). For three-colored imaging of virus, clathrin, and actin, VSV-G–carrying MLV Gag–CFP virions were added to XC cells stably transfected with clathrin light chain–YFP and fixed at various times after infection using 4% paraformaldehyde. After permeabilization using −20°C methanol immunostaining was as described above using rabbit antiactin antisera (Sigma-Aldrich) and Alexa Fluor 568–conjugated anti–rabbit secondary antisera (Invitrogen). All immunofluorescence was imaged using the 60× oil objective (numerical aperture 1.4) of the Nikon TE2000 microscope and Openlab acquisition software (Improvision).
Infection assays
To gauge the effects of various drugs on viral infection, MLV particles carrying a viral genome for the expression of β-galactosidase (pMMP-LTR-LacZ; Richard Mulligan, Harvard Medical School, Boston, MA) were generated as previously described (Mothes et al., 2000; Sherer et al., 2003). Viruses were added in the presence of polybrene to cells either untreated or pretreated for 5 min with 5–50 μM cytochalasin D or 30–50 μM blebbistatin. Infection was performed for 5, 30, or 120 min before nonfused virus was inactivated using either a 1-min acid wash (Kizhatil and Albritton, 1997) or a 15-min incubation with 10 mM methyl-β-cyclodextrin (MCD). MCD-treated MLV was five orders of magnitude less infectious. Cells were cultured for 2 d before infectivity was scored by X-gal staining.
Polarized MDCK cells were prepared for infection experiments by growing a confluent cell layer for at least 12 d on transwell filters (pore size 0.4 μm; Corning) until the transepithelial electrical resistance (TER) plateaued (Fig. S4 A). Addition of inhibitors for up to 2 h did not affect the TER. TEM of fully polarized MDCK cells revealed the apical formation of a typical dense meshwork of microvilli (Fig. S4 B). Infection was performed as described above except that virus was added to either the apical or basolateral side of the transwell. Cells were transferred into six well dishes after MCD treatment and infection was scored as above.
Electron microscopy
Cells were rinsed with serum-free buffer and then quick-fixed with 1% osmium tetroxide for 10 s, immediately followed by aldehyde fixation for 1 h (2.5% glutaraldehyde, 2% paraformaldehyde in 100 mM cacodylate buffer, pH 7.4). Cells were rinsed three times for 5 min with 100 mM cacodylate buffer, postfixed for 1 h in 1% osmium tetroxide, rinsed three times with HPLC water, en bloc uranyl acetate stained, dehydrated through a graded ethanol series, and finally embedded using EMBed 812 (EMS).
293 cells were cut en face and 70–90 nm sections were collected within the first 200 nm of the coverglass surface (Fig. 3). Scraped transwell plate samples of MDCK cells were cut perpendicular to the substrate, at the same section thickness (Fig. S4 B). In both cases, sections were counterstained with either 2% (wt/vol) or 4% (wt/vol) uranyl acetate followed by lead citrate. All samples were imaged on an FEI Tecnai 12 (Philips).
For analysis by SEM, cells were fixed for 30 min with 2.5% glutaraldehyde/2% paraformaldehyde in 100 mM cacodylate buffer (pH 7.4), rinsed three times with 100 mM cacodylate buffer, and dehydrated through a graded ethanol series. After washing three times with hexamethyldisilazane (EMS), cells were dried for 5 min at 60°C, coated with platinum, and analyzed on a FEI ESEM scanning electron microscope (Philips).
Online supplemental material
Descriptions of the data presented in supplemental figures and videos are introduced upon citation in the text. Online supplemental materials are available at http://www.jcb.org/cgi/content/full/jcb.200503059/DC1.
Acknowledgments
We thank Luisa Jimenez-Soto for the construction of mCAT-1–CFP and Tva–CFP, Ella Hinson for initial infection experiments, as well as Amelie Peryea and Brian Quinlan for assistance. We are grateful to Jorge Galan for his support, Mark Mooseker and Paul Forscher for advice, Tom Rapoport, Pradeep Uchil, and Wai-Tsing Chan for critical reading of the manuscript and the 2004 virology class of the Albert Einstein College of Medicine for discussions.
This work was supported by National Institutes of Health grant R01CA098727 and the Searle Scholars Program to W. Mothes, as well as an Anna Fuller Fund Fellowship and a Leopoldina Fellowship BMBF-LPD 9901/8-75 to M. Lehmann.
C.B. Marks' present address is University of Richmond, Richmond, VA 23173.
Abbreviations used: ALV, avian leukosis virus; Env, viral envelope glycoprotein; HIV, human immunodeficiency virus; MLV, murine leukemia virus; SEM, scanning electron microscopy; TEM, transmission electron microscopy; VSV, vesicular stomatitis virus.
References
- Abercrombie, M., J.E. Heaysman, and S.M. Pegrum. 1970. The locomotion of fibroblasts in culture. 3. Movements of particles on the dorsal surface of the leading lamella. Exp. Cell Res. 62:389–398. [DOI] [PubMed] [Google Scholar]
- Albrecht-Buehler, G., and R.M. Lancaster. 1976. A quantitative description of the extension and retraction of surface protrusions in spreading 3T3 mouse fibroblasts. J. Cell Biol. 71:370–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albritton, L.M., L. Tseng, D. Scadden, and J.M. Cunningham. 1989. A putative murine ecotropic retrovirus receptor gene encodes a multiple membrane-spanning protein and confers susceptibility to virus infection. Cell. 57:659–666. [DOI] [PubMed] [Google Scholar]
- Bates, P., J.A. Young, and H.E. Varmus. 1993. A receptor for subgroup A Rous sarcoma virus is related to the low density lipoprotein receptor. Cell. 74:1043–1051. [DOI] [PubMed] [Google Scholar]
- Bomsel, M., and A. Alfsen. 2003. Entry of viruses through the epithelial barrier: pathogenic trickery. Nat. Rev. Mol. Cell Biol. 4:57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukrinskaya, A., B. Brichacek, A. Mann, and M. Stevenson. 1998. Establishment of a functional human immunodeficiency virus type 1 (HIV-1) reverse transcription complex involves the cytoskeleton. J. Exp. Med. 188:2113–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duus, K.M., V. Lentchitsky, T. Wagenaar, C. Grose, and J. Webster-Cyriaque. 2004. Wild-type Kaposi's sarcoma-associated herpesvirus isolated from the oropharynx of immune-competent individuals has tropism for cultured oral epithelial cells. J. Virol. 78:4074–4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenfeld, D.P., D. Choquet, and M.P. Sheetz. 1996. Ligand binding regulates the directed movement of beta1 integrins on fibroblasts. Nature. 383:438–440. [DOI] [PubMed] [Google Scholar]
- Forscher, P., and S.J. Smith. 1988. Actions of cytochalasins on the organization of actin filaments and microtubules in a neuronal growth cone. J. Cell Biol. 107:1505–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidarov, I., F. Santini, R.A. Warren, and J.H. Keen. 1999. Spatial control of coated-pit dynamics in living cells. Nat. Cell Biol. 1:1–7. [DOI] [PubMed] [Google Scholar]
- Gottlieb, T.A., I.E. Ivanov, M. Adesnik, and D.D. Sabatini. 1993. Actin microfilaments play a critical role in endocytosis at the apical but not the basolateral surface of polarized epithelial cells. J. Cell Biol. 120:695–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond, C., L.K. Denzin, M. Pan, J.M. Griffith, H.J. Geuze, and P. Cresswell. 1998. The tetraspan protein CD82 is a resident of MHC class II compartments where it associates with HLA-DR, -DM, and -DO molecules. J. Immunol. 161:3282–3291. [PubMed] [Google Scholar]
- Harris, A., and G. Dunn. 1972. Centripetal transport of attached particles on both surfaces of moving fibroblasts. Exp. Cell Res. 73:519–523. [DOI] [PubMed] [Google Scholar]
- Helenius, A., J. Kartenbeck, K. Simons, and E. Fries. 1980. On the entry of Semliki forest virus into BHK-21 cells. J. Cell Biol. 84:404–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez, L.D., L.R. Hoffman, T.G. Wolfsberg, and J.M. White. 1996. Virus-cell and cell-cell fusion. Annu. Rev. Cell Dev. Biol. 12:627–661. [DOI] [PubMed] [Google Scholar]
- Kizhatil, K., and L.M. Albritton. 1997. Requirements for different components of the host cell cytoskeleton distinguish ecotropic murine leukemia virus entry via endocytosis from entry via surface fusion. J. Virol. 71:7145–7156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klement, V., W.P. Rowe, J.W. Hartley, and W.E. Pugh. 1969. Mixed culture cytopathogenicity: a new test for growth of murine leukemia viruses in tissue culture. Proc. Natl. Acad. Sci. USA. 63:753–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolokoltsov, A.A., and R.A. Davey. 2004. Rapid and sensitive detection of retrovirus entry by using a novel luciferase-based content-mixing assay. J. Virol. 78:5124–5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidke, D.S., P. Nagy, R. Heintzmann, D.J. Arndt-Jovin, J.N. Post, H.E. Grecco, E.A. Jares-Erijman, and T.M. Jovin. 2004. Quantum dot ligands provide new insights into erbB/HER receptor-mediated signal transduction. Nat. Biotechnol. 22:198–203. [DOI] [PubMed] [Google Scholar]
- Lin, C.H., and P. Forscher. 1993. Cytoskeletal remodeling during growth cone-target interactions. J. Cell Biol. 121:1369–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littman, D.R. 1998. Chemokine receptors: keys to AIDS pathogenesis? Cell. 93:677–680. [DOI] [PubMed] [Google Scholar]
- Maddon, P.J., J.S. McDougal, P.R. Clapham, A.G. Dalgleish, S. Jamal, R.A. Weiss, and R. Axel. 1988. HIV infection does not require endocytosis of its receptor, CD4. Cell. 54:865–874. [DOI] [PubMed] [Google Scholar]
- Masuda, M., N. Kakushima, S.G. Wilt, S.K. Ruscetti, P.M. Hoffman, and A. Iwamoto. 1999. Analysis of receptor usage by ecotropic murine retroviruses, using green fluorescent protein-tagged cationic amino acid transporters. J. Virol. 73:8623–8629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama, S., S.E. Delos, and J.M. White. 2004. Sequential roles of receptor binding and low pH in forming prehairpin and hairpin conformations of a retroviral envelope glycoprotein. J. Virol. 78:8201–8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald, D., M.A. Vodicka, G. Lucero, T.M. Svitkina, G.G. Borisy, M. Emerman, and T.J. Hope. 2002. Visualization of the intracellular behavior of HIV in living cells. J. Cell Biol. 159:441–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, M., E. Bower, P. Levitt, D. Li, and P.D. Chantler. 1992. Myosin II distribution in neurons is consistent with a role in growth cone motility but not synaptic vesicle mobilization. Neuron. 8:25–44. [DOI] [PubMed] [Google Scholar]
- Mitchison, T., and M. Kirschner. 1988. Cytoskeletal dynamics and nerve growth. Neuron. 1:761–772. [DOI] [PubMed] [Google Scholar]
- Mitchison, T.J. 1992. Actin based motility on retraction fibers in mitotic PtK2 cells. Cell Motil. Cytoskeleton. 22:135–151. [DOI] [PubMed] [Google Scholar]
- Mothes, W., A.L. Boerger, S. Narayan, J.M. Cunningham, and J.A. Young. 2000. Retroviral entry mediated by receptor priming and low pH triggering of an envelope glycoprotein. Cell. 103:679–689. [DOI] [PubMed] [Google Scholar]
- Narayan, S., R.J. Barnard, and J.A. Young. 2003. Two retroviral entry pathways distinguished by lipid raft association of the viral receptor and differences in viral infectivity. J. Virol. 77:1977–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponti, A., M. Machacek, S.L. Gupton, C.M. Waterman-Storer, and G. Danuser. 2004. Two distinct actin networks drive the protrusion of migrating cells. Science. 305:1782–1786. [DOI] [PubMed] [Google Scholar]
- Rochlin, M.W., K. Itoh, R.S. Adelstein, and P.C. Bridgman. 1995. Localization of myosin II A and B isoforms in cultured neurons. J. Cell Sci. 108:3661–3670. [DOI] [PubMed] [Google Scholar]
- Sheetz, M.P., S. Turney, H. Qian, and E.L. Elson. 1989. Nanometre-level analysis demonstrates that lipid flow does not drive membrane glycoprotein movements. Nature. 340:284–288. [DOI] [PubMed] [Google Scholar]
- Sherer, N.M., M.J. Lehmann, L.F. Jimenez-Soto, A. Ingmundson, S.M. Horner, G. Cicchetti, P.G. Allen, M. Pypaert, J.M. Cunningham, and W. Mothes. 2003. Visualization of retroviral replication in living cells reveals budding into multivesicular bodies. Traffic. 4:785–801. [DOI] [PubMed] [Google Scholar]
- Shurety, W., N.A. Bright, and J.P. Luzio. 1996. The effects of cytochalasin D and phorbol myristate acetate on the apical endocytosis of ricin in polarised Caco-2 cells. J. Cell Sci. 109:2927–2935. [DOI] [PubMed] [Google Scholar]
- Smith, A.E., and A. Helenius. 2004. How viruses enter animal cells. Science. 304:237–242. [DOI] [PubMed] [Google Scholar]
- Stein, B.S., S.D. Gowda, J.D. Lifson, R.C. Penhallow, K.G. Bensch, and E.G. Engleman. 1987. pH-independent HIV entry into CD4-positive T cells via virus envelope fusion to the plasma membrane. Cell. 49:659–668. [DOI] [PubMed] [Google Scholar]
- Straight, A.F., A. Cheung, J. Limouze, I. Chen, N.J. Westwood, J.R. Sellers, and T.J. Mitchison. 2003. Dissecting temporal and spatial control of cytokinesis with a myosin II inhibitor. Science. 299:1743–1747. [DOI] [PubMed] [Google Scholar]
- Suter, D.M., L.D. Errante, V. Belotserkovsky, and P. Forscher. 1998. The Ig superfamily cell adhesion molecule, apCAM, mediates growth cone steering by substrate-cytoskeletal coupling. J. Cell Biol. 141:227–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina, T.M., A.B. Verkhovsky, K.M. McQuade, and G.G. Borisy. 1997. Analysis of the actin-myosin II system in fish epidermal keratocytes: mechanism of cell body translocation. J. Cell Biol. 139:397–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhovsky, A.B., T.M. Svitkina, and G.G. Borisy. 1995. Myosin II filament assemblies in the active lamella of fibroblasts: their morphogenesis and role in the formation of actin filament bundles. J. Cell Biol. 131:989–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, J.A. 2001. Virus entry and uncoating. Fields Virology. Vol. 1. D.M. Knipe, editor. Lippincott Williams & Wilkins, Philadelphia. 87–103.
- Zavorotinskaya, T., Z. Qian, J. Franks, and L.M. Albritton. 2004. A point mutation in the binding subunit of a retroviral envelope protein arrests virus entry at hemifusion. J. Virol. 78:473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]