

Abstract
The two yeast proteins Mlp1p and Mlp2p (homologues of the vertebrate protein Tpr) are filamentous proteins attached to the nuclear face of nuclear pore complexes. Here we perform a proteomic analysis, which reveals that the two Mlps have strikingly different interacting partners, testifying to their different roles within the cell. We find that Mlp2p binds directly to Spc110p, Spc42p, and Spc29p, which are three core components of the spindle pole body (SPB), the nuclear envelope–associated yeast spindle organizer. We further show that SPB function is compromised in mlp2 mutants. Cells lacking Mlp2p form significantly smaller SPBs, accumulate aberrant SPB component-containing structures inside the nucleus, and have stochastic failures of cell division. In addition, depletion of Mlp2p is synthetically lethal with mutants impaired in SPB assembly. Based on these data, we propose that Mlp2p links the SPB to the peripheral Mlp assembly, and that this linkage is required for efficient incorporation of components into the SPB.
Introduction
In eukaryotes a double-membraned coat termed the nuclear envelope (NE) separates the nucleoplasm from the cytoplasm. Exchange between these two compartments is mediated by nuclear pore complexes (NPCs), octagonally symmetric cylindrical structures that span pores in the NE. During mitosis, the spindle microtubules must gain access to the nucleoplasm in order to segregate the daughter genomes. This can be achieved either by breaking down the NE (open mitosis), or by assembling the spindle inside the nucleus (closed mitosis). In the budding yeast Saccharomyces, mitosis is closed and the spindle is assembled by spindle pole bodies (SPBs). SPBs are the sole yeast microtubule-organizing centers (MTOCs), and like NPCs are directly embedded within pores in the NE throughout the cell cycle. The SPB consists of a multilayered disk structure made of hundreds of copies of each of its components (for review see Jaspersen and Winey, 2004; see also Fig. 8). The electron-dense central plaque is assembled around a two-dimensional crystal of Spc42p (Bullitt et al., 1997), which is an anchor for the other core components of the SPB: Spc110p, Cnm67p, Spc29p, and Cmd1p (the yeast homologue of calmodulin; Adams and Kilmartin, 1999). At least 14 other proteins forming the bulk of the cytoplasmic and nuclear plaques and other SPB structures associate with this core. On the cytoplasmic side, Cnm67p connects Spc42p to the nucleation site of the astral microtubules, responsible for nuclear positioning and spindle alignment (Schaerer et al., 2001). The filamentous protein Spc110p acts as a spacer between the central and the inner plaques, from where the nuclear microtubules emanate to form the mitotic spindle responsible for SPB separation and for chromosome segregation (Rout and Kilmartin, 1990; Kilmartin et al., 1993). The spindle also plays a critical role in ensuring that nuclear migration and division occur in concert with DNA replication and bud formation. Despite the observation that the SPB cores are extremely resistant to disruption in vitro, recent results indicate that they are dynamic in vivo. Both the newly synthesized and the old SPB continue to grow in size from G2 to telophase (Bullitt et al., 1997; Yoder et al., 2003). At the same time, the mother SPB is continuously renewed by the exchange of up to 50% of old Spc110p with newly made subunits (Yoder et al., 2003). Several observations also suggest a functional relationship between SPBs and NPCs. For example, some components are shared between the two structures, and proteins known to regulate spindle function are found to reside at the NPC (Chial et al., 1998; Rout et al., 2000; Iouk et al., 2002). Although these results suggest a possible role for the NE in aiding SPB function, they have not provided an explanation for the mechanism underlying this connection.
Figure 8.

Mlp2p links the SPB into the nuclear peripheral Mlp layer and aids the incorporation of new components into its tightly packed core.
The yeast proteins Mlp1p and Mlp2p belong to a well-conserved family of NE proteins, which in vertebrates is represented by the protooncogenic protein Tpr (Kuznetsov et al., 2002). The Mlps are large, coiled-coil filamentous proteins projecting from the nucleoplasmic side of the NPC (Strambio-de-Castillia et al., 1999; Kosova et al., 2000) and they are nonessential in budding yeast. The Mlps have been proposed to form a peripheral nuclear network whose function is still not clear (Strambio-de-Castillia et al., 1999). Consistent with its localization to the NPC, some data indicate a role for the Mlps in nucleocytoplasmic transport (Strambio-de-Castillia et al., 1999; Kosova et al., 2000). Recent observations also indicate that Mlp1p has a role in RNA biogenesis and in particular in nuclear retention of unspliced mRNAs (Galy et al., 2004; Vinciguerra et al., 2005). The Mlps have also been proposed to be telomeric anchoring sites and involved in the establishment of silent chromatin (Galy et al., 2000; Feuerbach et al., 2002; subsequent studies have called these roles into question, although they did indicate that the Mlps might be implicated in telomere maintenance [Andrulis et al., 2002; Hediger et al., 2002a,b]).
In this study we performed a biochemical analysis, which revealed that Mlp2p binds directly to the SPB core. In the absence of Mlp2p, SPB components are not efficiently targeted to the nuclear face of the SPB, leading to smaller SPBs and to delays and errors of cell division. These results show that the NE is an integrated unit whose structures function coordinately to perform essential nuclear functions.
Results
Mlp2p, but not Mlp1p, interacts with key components of the SPB
We set out to identify the interacting partners of Mlp1p and Mlp2p by biochemically purifying these proteins from yeast lysates. Using a variety of extraction conditions, we found striking differences between the proteins copurifying with Mlp1p and Mlp2p (Fig. 1 a). Mlp1p–protein A (PrA) could be reproducibly isolated in a complex exclusively with Mlp2p, indicating that the two proteins are able to bind directly to each other as previously proposed (Strambio-de-Castillia et al., 1999). By contrast, in addition to binding Mlp1p, Mlp2p–PrA formed a tight complex with at least six major proteins, which by mass spectrometry were identified as key SPB components. Five of these represent the components of the SPB core: Spc110p, Cnm67p, Spc42p, Spc29p, and Cmd1p. The sixth, Cdc31p, is a calmodulin-like calcium-binding protein that helps form the bridge structure involved in SPB duplication (Spang et al., 1993). With the exception of Nud1p and the addition of Cdc31p, these proteins match those identified as core components in a previous proteomic study (Adams and Kilmartin, 1999). Lower abundance bands present in the complex were identified to be the inner plaque components Spc98p, Spc97p, and the outer plaque's Spc72p, in addition to Mps2p and Bbp1p, SPB-associated proteins with important roles in SPB duplication. When mild extraction conditions were used we found that both Mlps interact with a number of NPC proteins, as well as components of the mRNA processing machinery and nuclear transport factors, in agreement with previously published functions of the Mlps (Strambio-de-Castillia et al., 1999; Kosova et al., 2000; Green et al., 2003; Galy et al., 2004; and unpublished data).
Figure 1.
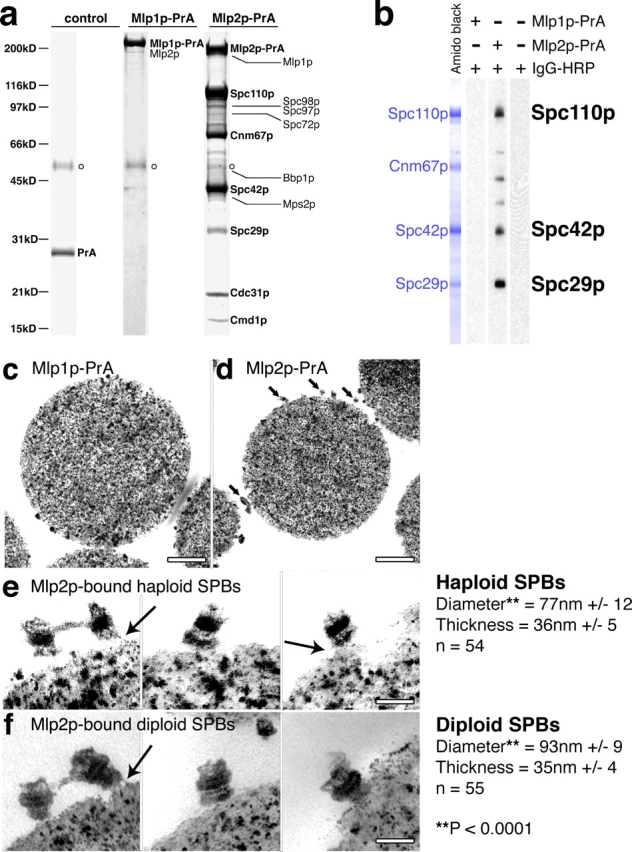
Mlp2p interact with the core of assembled SPBs. (a) Mlp2p, but not Mlp1p, strongly interacts with SPB proteins. PrA-containing complexes were affinity purified from cell lysates expressing Mlp1p–PrA, Mlp2p–PrA, or PrA alone as a control. The complexes were separated by SDS-PAGE, visualized by Coomassie staining, and identified by tandem mass spectrometry. Open circles indicate the position of rabbit IgG heavy chain from the Dynabeads. (b) Mlp2p directly binds Spc110p, Spc42p, and Spc29p, but not Cnm67p. Mlp2p–PrA-associated SPB components were affinity purified as described above and eluted from IgG-Dynabeads using 1 M MgCl2 leaving Mlp2p–PrA attached to the beads. Proteins in the eluate were then resolved by SDS-PAGE and transferred to nitrocellulose. The blot was stained with amido black, cut into strips, and probed with either purified Mlp1p–PrA or Mlp2p–PrA, or with blocking buffer alone. Bound PrA probes were detected with rabbit IgG–HRP conjugate followed by chemiluminescence. The identity of the minor bands has not been determined. (c to f) Mlp2p interacts with assembled SPB cores. Dynabeads carrying Mlp1p–PrA (c) or Mlp2p–PrA (d–f) containing complexes were prepared exactly as described above from either haploid (c–e) or diploid (f) cells and visualized by TEM. Small arrows, haploid SPBs. The electron-dense globular particles visible in panels c and d are also present in empty control beads and are the magnetic material within the Dynabeads. (e) Three representative examples of haploid SPBs bound to Mlp2p–PrA beads at higher magnification. (f) Three examples of diploid SPBs bound to Mlp2p–PrA beads. The diameter and thickness of the central plaque of both haploid and diploid Mlp2p–PrA bound SPBs were measured on calibrated digital images using the ImageJ software and are indicated as the mean value ± 1 SD at the bottom right. Also indicated (**) is the t test P value obtained for the comparison of the distribution of diameters of haploid versus diploid SPBs. Large arrows, filaments attaching the SPBs to the beads. Bars: (c and d) 500 nm; (e and f) 100 nm.
To determine which of the SPB components are capable of directly binding Mlp2p, we performed an in vitro blot binding experiment (Fig. 1 b). The Mlp2p-associated SPB proteins were separated by SDS-PAGE, immobilized on nitrocellulose, and probed either with purified Mlp1p–PrA or Mlp2p–PrA. Whereas Mlp1p–PrA did not bind any SPB components, Mlp2p–PrA bound specifically to three SPB proteins (Spc110p, Spc42p, and Spc29p), but failed to interact with a fourth (Cnm67p). These results are consistent with the immunopurification data above, and indicate that Mlp2p interacts directly with at least three SPB components, all of which are exposed on the nuclear side of the SPB and so accessible to the Mlp2p in vivo.
Mlp2p binds to intact SPB cores
To test if the interaction between Mlp2p and the SPB is limited to unassembled subcomplexes of the SPB or to certain stages during the spindle assembly cycle, we visualized the complex by transmission electron microscopy (TEM). Thin sections of beads from the Mlp1p–PrA immunoprecipitation (Fig. 1 c) and empty control beads (unpublished data) were devoid of any visible protein structures. However, the magnetic beads on which the Mlp2p–PrA complex was immobilized were covered with numerous discrete structures, ∼80 nm in diameter (Fig. 1 d, small arrows), which, at higher magnification, resembled isolated SPB cores (Fig. 1 e).
To confirm that Mlp2p-associated structures were indeed bona fide SPB cores, we also compared the isolated structures from either haploid or diploid cells (Fig. 1 f), as diploid SPBs are characteristically larger than haploid SPBs in diameter but not in thickness, and the layered morphology of diploid SPBs is more easily discernible than that of haploid SPBs (Byers and Goetsch, 1975; Bullitt et al., 1997). The structures on the beads isolated from diploid cells were revealed to be morphologically identical to chemically extracted diploid SPB cores (Adams and Kilmartin, 1999). The layered appearance of SPBs was clearly visible in these structures, with the electron-dense central plaque flanked on either side by filamentous protein layers. Structures resembling the bridge, which connects recently duplicated SPBs during early S phase, were also visible in some examples, accounting for the presence of Cdc31p in the Mlp2p–SPB complex (Fig. 1 a; Spang et al., 1993). Although the average width of the diploid layered structures was significantly larger than that of the haploid structures, their average thicknesses were the same; all measurements are consistent with previously published values for SPBs. Together, these data establish that the structures associated with Mlp2p are SPB-derived cores.
Core components of the SPB coimmunoprecipitate Mlp2p, but not Mlp1p
To test the specificity of the Mlp2p–SPB interaction, we performed reciprocal coimmunoprecipitation experiments followed by immunoblotting. We used Spc42p–PrA or Spc110p–PrA as baits and Mlp1p–Myc or Mlp2p–Myc as targets (Fig. 2, coexpressed). We found that Mlp2p–Myc was indeed coimmunoprecipitated by both Spc42p–PrA and Spc110p–PrA, and Mlp1p–Myc did not interact with either of the baits. This confirms the high specificity of the Mlp2p–SPB interaction, because Mlp1p (a protein similar in location, size and secondary structure to Mlp2p) failed to interact either with Spc42p or Spc110p. We note that Cnm67p-PrA failed to immunoprecipitate either Mlp1p–Myc or Mlp2p–Myc, in agreement with the in vitro binding experiments (unpublished data).
Figure 2.
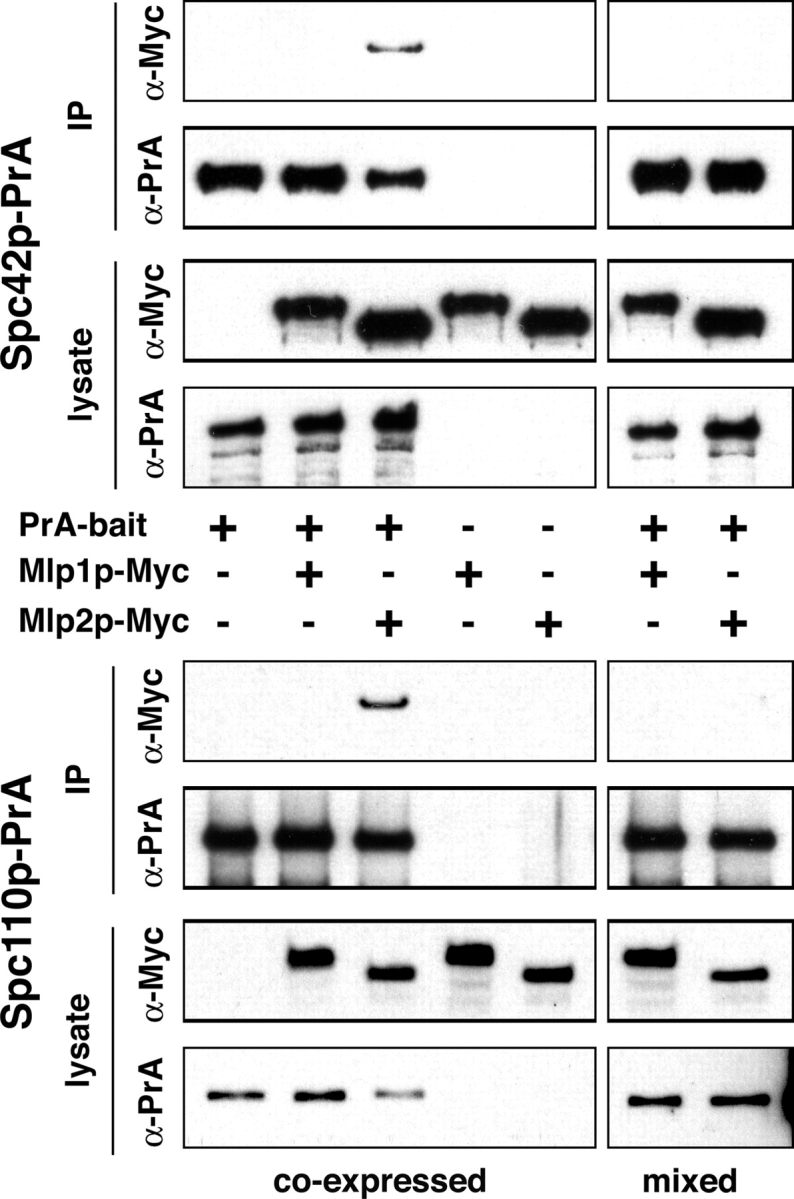
SPB components coimmunoprecipitate Mlp2p, but not Mlp1p. PrA containing affinity-purified complexes (IP) and whole cell lysates (lysate) of strains coexpressing combinations of PrA-tagged SPB components as baits and Myc-tagged Mlp1p or Mlp2p as targets, were probed by immunoblotting for Myc and PrA (coexpressed). To control for interactions occurring after lysis, strains expressing either one PrA-tagged bait or one Myc-tagged target were mixed after cell lysis and analyzed exactly as above (mixed).
Long coiled-coil domains are common to Mlp2p and many SPB components. Because these domains have the potential to artifactually adhere to each other under nonphysiological conditions, we performed the experiment described above under conditions in which the SPB bait and the Mlp targets were expressed independently and mixed only after cell lysis. Under these conditions we detected no interaction between Mlp2p and the tested SPB proteins (Fig. 2, mixed), showing that the Mlp2p–SPB interaction did not occur after lysis in the extracts.
Mlp proteins localize to the same nuclear hemisphere as the SPB
With their C-shaped distribution around the rim of the nucleus, the Mlps define a specific hemisphere of the NE, which only partially overlaps with the even distribution of NPCs around the rim of the nucleus and is excluded from the region juxtaposed to the nucleolus (Fig. S1 available at http://www.jcb.org/cgi/content/full/jcb.200504140/DC1; Strambio-de-Castillia et al., 1999; Kosova et al., 2000; Galy et al., 2004). First, to directly compare the localization of the Mlp proteins with respect to each other, we used CFP–Mlp1p and YFP–Mlp2p to visualize their position within the same cell (Fig. 3 a). Though the Mlp proteins occupied the same general area of the nuclear periphery, CFP–Mlp1p was distributed fairly evenly along a C-shaped portion of the nuclear periphery; YFP–Mlp2p was concentrated into fewer foci covering less area.
Figure 3.

The Mlps occupy the same NE hemisphere as the SPB at all stages of the cell cycle. (a) Mlp1p and Mlp2p have different localization patterns. Typical images of homozygous diploid cells expressing both CFP–Mlp1p (red) and YFP–Mlp2p (green). (b) Live fluorescence images of haploid cells expressing either Mlp1p–YFP or YFP–Mlp2p (green) and Spc42-CFP (red). (c) YFP–Mlp2p (green) and Spc42p–CFP (red) double-tagged haploid cells were subjected to differential elutriation to enrich for cells at different stages of the cell cycle. Representative fluorescence images for each cell cycle stage are displayed as indicated. (Merge) Two-dimensional image projections for both YFP and CFP were overlaid over a DIC images of each individual cell. (3D) A three-dimensional rendition of each double-color image was obtained using the Imaris software and overlaid over a digital outline of the whole cell. (a and b) Images represent two-dimensional maximum projections of z-stacks containing 10–15 0.3 μm sections. To visualize the fluorescent signals in the context of the whole cell, a DIC image was merged with both of the CFP and YFP pictures from the same field (Merge). Bars: (a and c) 2 μm; (b) 1 μm.
Next, as a control for our biochemical data, we investigated whether Mlp2p and SPBs colocalize in vivo (Fig. 3 b). When we expressed Spc42p–CFP in combination with Mlp1p–YFP or YFP–Mlp2p, we found that the SPB occupies the Mlps' hemisphere in over 90% of the cases (n = 500). This result is highly significant as testified by the result of the chi-squared test we performed under the test hypothesis that the Mlp proteins are excluded from 20% of the NE surface area (chi-squared value = 2.268 × 10−08). To further investigate the association between the Mlp distribution and the SPBs, we followed the localization of YFP–Mlp2p and Spc42p–CFP at different stages of the cell cycle. We observed that the connection between the Mlp-containing hemisphere and the SPBs was always conserved (Fig. 3 c). In particular, after S phase, the SPBs migrated to opposite ends of the Mlp hemisphere. At anaphase the YFP–Mlp2p signal appeared to trail behind Spc42p–CFP while remaining in contact, as the spindle and the nucleus extended. The close association of Mlp2p with the SPB in vivo is in complete accordance with their direct interaction revealed by our biochemical analyses (Fig. 1). In addition, this result is consistent with published work showing that in yeast the SPB lies opposite the nucleolus (Yang et al., 1989; Bystricky et al., 2005).
mlp2Δ cells display significant delays in proceeding past metaphase
To explore the functional significance of the interaction between the SPB and Mlp2p, we determined the position and morphology of the spindle in asynchronous haploid cells by following GFP-tagged Tub1p. In this test, 52% of cells lacking Mlp2p showed a short spindle, characteristic of cells in late S phase or metaphase, compared with 38% for wild-type cells and 39% for mlp1Δ cells. This result represents a 38% increase of cells in metaphase for mlp2Δ versus wild type (Fig. 4 a), consistent with Mlp2p having a role in the efficient progression of cells past G2 or early M phase. To assess whether Mlp2p could have a role in promoting spindle migration to the bud neck, we measured the spindle migration index in late S to early M cells (Fig. 4 b). We observed no significant difference between wild-type, mlp1Δ, and mlp2Δ cells in this assay. These results suggest that Mlp2p is not involved in the regulation of the activity of cytoplasmic microtubules, consistent with its interaction with SPB components localized only to the nuclear face of the central plaque. To further investigate the function of Mlp2p, we examined wild-type and mlp mutant cells expressing Spc42p–GFP. Cells lacking Mlp2p showed an excess number of cells displaying SPB-“doublets” (i.e., cells with two SPBs per cell body) as compared with wild-type and mlp1Δ cells, consistent with the results we obtained with GFP–Tub1p-expressing cells (Fig. 4 c). To assess whether Mlp2p might have a role in promoting the formation of a fully formed metaphase spindle, we measured the distance between duplicated SPBs in G2 to early M cells (Fig. 4 d). We found that the average SPB-to-SPB distance was significantly reduced in cells lacking Mlp2p with respect to wild type (i.e., 1.58 μm for wild-type cells; 1.10 μm for mlp2Δ cells). Taken together, these results points to a role for Mlp2p in facilitating SPB separation and in promoting the formation of a complete metaphase spindle, but not in astral microtubule-directed nuclear migration to the bud neck.
Figure 4.

mlp2Δ cells display a significant delay in proceeding past metaphase and accumulate with short spindles at the bud neck. All experiments were done on asynchronous cell cultures. (a) Images of haploid wild-type, mlp1Δ, and mlp2Δ cells expressing GFP–Tub1p. Images were obtained as described for Fig. 3. Asynchronous cells (n = 300–400) were scored for short and stubby spindles (i.e., cells in S or early M phase) in two independent experiments, to assess their ability to progress through the cell cycle. The numbers of 38%, 39%, and 52% have SDs of 0.3%, 3.0%, and 4.0%, respectively. (b) Tukey plots displaying the spindle migration index distributions for wild-type and mutant cells lacking either Mlp1p or Mlp2p. Only cells with a metaphase spindle appearance were used for this analysis (n > 60). The top and bottom of each rectangle represent the 75th and 25th percentiles, respectively; the center bar of the rectangle marks the median spindle migration index value. The top and bottom horizontal marks show the 90th and 10th percentiles, respectively. Black dots represent outlying data points. The distributions were not found to be significantly different based on individual pairwise comparisons using the t test. (c) Images of haploid wild-type, mlp1Δ, and mlp2Δ cells expressing Spc42p–GFP. (d) Tukey plots displaying the SPB-to-SPB distance distributions in wild-type and mutant cells lacking either Mlp1p or Mlp2p. Only cells between S and early M were used for this analysis. The median SPB-to-SPB distance was 1.58 μm (n = 60) in wild-type cells, 1.35 μm (n = 54) in mlp1Δ cells and 1.10 μm (n = 120) in mlp2Δ cells. Comparisons of the distributions using the t test showed a high degree of significance in all cases: wild type versus mlp1Δ, P < 0.0003; wild type versus mlp2Δ, P < 0.0001; mlp1Δ versus mlp2Δ, P < 0.0025. Bar, 2 μm.
A significant fraction of cells lacking Mlp2p display aberrant intranuclear microtubule organizers and cytokinesis defects
When looking at cells expressing Spc42p–GFP we noticed that, though we did not find any wild-type and mlp1Δ cells with more than two SPBs, one of every 12 cells lacking MLP2 carried abnormally high numbers of Spc42p–GFP-containing foci (3–6 per cell; Fig. 5 a). Cells that displayed this defect were predominantly found in chains of multiple cell bodies that had failed to complete cytokinesis (hereafter referred to as multicellular chains). Similar results were obtained with cells labeled with Spc110p–GFP or GFP–Tub1p (unpublished data). These results indicated that the aberrant Spc42p–GFP-containing foci found in cells lacking Mlp2p also contain Spc110p and are able to nucleate microtubules. To further study the nature of this defect, we enriched for multicellular chains and large-budded cells by elutriation (Fig. S2 available at http://www.jcb.org/cgi/content/full/jcb.200504140/DC1). When late elutriation fractions were stained with DAPI and examined by fluorescence microscopy, we found that mlp2Δ cells were enriched in multicellular chains, which also displayed abnormal numbers of microtubule organizers (Fig. 5 b). In contrast, equivalent wild-type and mlp1Δ elutriation fractions contained no abnormal cell types. In all cases, the generation of the spurious microtubule organizers appeared to be coupled with the formation of an extra bud before completion of cell division (Fig. 5 b, mlp2Δ, inset).
Figure 5.

mlp2Δ cells accumulate multiple amorphous nuclear Spc42p-containing bodies that nucleate microtubules. (a) Quantitative analysis of the number of Spc42p–GFP containing foci in wild-type, mlp1Δ, and mlp2Δ cells. Asynchronous Spc42p–GFP haploid cells were fixed and stained with DAPI to reveal the position of the nucleus. Two-dimensional image projections of three-dimensional image stacks obtained for both GFP and DAPI were overlaid onto each respective DIC image for scoring. We counted the number of cells with multiple Spc42p–GFP signals (n > 600). (b) Wild-type, mlp1Δ, and mlp2Δ cells expressing Spc42–GFP were separated on the basis of cell sizes by differential elutriation (see Fig. S2). For each sample, cells present in equivalent elutriation fractions were imaged as described for panel a to reveal the presence of Spc42p–GFP (red) and of the DAPI-stained nucleus (blue). Two representative fields of vision are presented for mlp2Δ. Bar, 2 μm. (c) Left panels, thin section, TEM images of diploid mutant cells lacking Mlp2p, after enrichment for large-budded cells and cell chains by elutriation. Arrowheads, amorphous electron-dense material presumably composed of misassembled SPB components. Arrows, spurious microtubule bundles emanating from MTOC positioned perpendicular to the visible SPB. (Inset) Twofold magnification of an area presented in the bottom left panel, showing one of these bundles of microtubules. Right panels, line drawings of the panels shown on the left highlighting the position of microtubule bundles, the NE, SPBs, and amorphous electron-dense SPB-like structures. (d) Postembedding labeling immuno-EM images of diploid mutant cells expressing Spc42p–GFP. Thin sections from embedded samples were labeled with a rabbit anti-GFP polyclonal antibody followed with 10 nm gold-conjugated secondary antibody. Arrowhead, gold particle–decorated fully formed SPB. Arrows, amorphous intranuclear Spc42p–GFP-containing bodies. The bottom right panel has a twofold magnification with respect to the other panels. Bars, 100 nm.
The presence of additional SPB protein containing microtubule organizers could indicate either a defect in the regulation of SPB duplication (Haase et al., 2001) or a defect in SPB assembly and structural integrity (Sundberg et al., 1996). To distinguish between these two possibilities, we performed a detailed ultrastructural analysis on enriched defective cells using thin section TEM (Fig. 5 c). For comparison, an equivalent elutriation fraction from a wild-type culture was also analyzed (unpublished data). In the nuclei of mlp2Δ cells we found numerous examples of aberrant amorphous, electron-dense structures that are absent from the control and are reminiscent of the intranuclear microtubule organizers found in spc110-220 mutant cells (Sundberg et al., 1996). These masses lacked the well-defined architecture characteristic of SPBs and appeared to reside primarily inside the nucleoplasm, although on occasion they were connected to an SPB at the NE. In addition they appeared to nucleate microtubules (Fig. 5 c, top left, arrowheads). We also observed instances in which multiple microtubule bundles appeared to be emanating from MTOCs elsewhere in the nucleus and not forming a normal spindle (Fig. 5 c, bottom left, arrows). The mlp2Δ nuclei often appeared to be fragmented and lobulated and sometimes displayed deep invaginations of the NE carrying what appeared to be fully formed SPBs. These results likely explain the abnormal appearance of the DAPI stained nuclei shown in Fig. 5 b.
To demonstrate that the abnormal electron-dense structures we observed by thin-section TEM are indeed amorphous intranuclear assemblies of un-incorporated SPB components, we performed a post-embedding labeling immuno-EM experiment on enriched defective haploid cells carrying the mlp2Δ allele and expressing Spc42–GFP (Fig. 5 d). Gold particles were specifically found in association either with fully formed SPBs at the NE (Fig. 5 d, top left inset, arrowhead) or with intranuclear amorphous structures resembling the ones shown in Fig. 5 c (Fig. 5 d, arrows). This result underscores the specificity of the labeling and demonstrates that the intranuclear electron-dense bodies found in cells lacking Mlp2p contain Spc42p moieties, which failed to be incorporated in the central plaque of fully formed SPBs. Overall, the ultrastrucutural data are consistent with the results we obtained by fluorescent microscopy and suggest that Mlp2p is involved in the proper incorporation of SPB components into fully formed SPBs.
MLP2 interacts genetically with a mutant affecting SPB assembly
To further investigate the functional interactions between the SPB and Mlp2p, we tested temperature-sensitive mutations of various SPB components for genetic interactions with MLP1 and MLP2. When a strain containing the spc110-220 mutation was mated with the mlp2Δ strain we were unable to recover spores carrying both mutations, although we could generate double mutant strains from all other crosses tested (see Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200504140/DC1). The spc110-220 strain carries a C911R mutation in the Cmd1p-binding domain of Spc110p, which reduces the ability of Spc110p to bind Cmd1p at the restrictive temperature and impairs the incorporation of Spc110p into the central plaque of the SPB, leading to slow growth and the formation of intranuclear mitotic organizers (Sundberg et al., 1996). To study this genetic interaction, we created spc110Δ strains covered by plasmids expressing either SPC110 or spc110-C911R (thereafter referred to as spc110-220). In addition, we introduced a repressible promoter upstream of either a PrA-tagged MLP1 or MLP2 gene. When grown in dextrose this promoter repressed expression to levels below the immunoblotting detection limit (Fig. S3 available at http://www.jcb.org/cgi/content/full/jcb.200504140/DC1).
Although the SPC110 control strains exhibited equal viability at all temperatures tested, the strains carrying spc110-220 had inhibited growth at 37°C (Fig. 6 a). The additional depletion of Mlp2p from spc110-220 cells led to them being inviable at this temperature. Even at permissive temperatures the depletion of Mlp2p decreased the viability of spc110-220 strains significantly. The repression of MLP1 also reduced viability in conjunction with spc110-220, but to a lesser extent than MLP2. We found that the depletion of MLP2 led to a nearly fourfold increase in aberrant nuclear morphology in the spc110-220 mutant at the restrictive temperature (Fig. 6 b). The nuclei appeared enlarged or fragmented with multiple intense DAPI-stained areas distributed throughout the cell, in contrast to the discrete, round wild-type nuclei. This morphology of the nucleus was reminiscent of the one we observed in multicellular chains present in mlp2Δ strains (Fig. 5 b). Other described defects associated with spc110-220 at the restrictive temperature were found to be independent of the expression level of MLP2 (unpublished data). We compared the effects of Mlp depletion in the spc110-220 background with the effect of MLP deletion in other temperature-sensitive SPB protein mutants (Fig. S4 available at http://www.jcb.org/cgi/content/full/jcb.200504140/DC1; Fig. 6 c; Geiser et al., 1991; Davis, 1992; Sundberg and Davis, 1997; Elliott et al., 1999). We find that the other mutants we tested showed either mild or no synthetic lethal interactions with MLP2. These results confirm that a specific functional interaction exists between spc110-220, a mutant in a central plaque component affecting SPB assembly, and the Mlps via Mlp2p.
Figure 6.

Low expression levels of MLP2 in conjunction with spc110-220 cause lethality and loss of nuclear integrity at 37°C. (a) Two individual clones of strains expressing either SPC110 or the spc110-220 allele alone (−) or in combination with either MLP1 or MLP2 under the control of the GAL1-10 promoter (Gal-MLP1 and Gal-MLP2, respectively) were spotted in 10-fold dilution steps on dextrose-containing plates and grown for 2 d at the indicated temperatures. (b) DAPI-stained cells of the indicated genotypes were scored for grossly aberrant nuclear morphology (n > 300). (Inset) Typical appearance of cell with altered nucleus. (c) Semiquantitative relative measure of dilution-adjusted colony density from strains with the indicated genotype and their counterparts carrying the wild-type SPB protein allele grown as described in panel a. This analysis was performed as previously described (Tackett et al., 2005). Data presented in panel a and in Fig. S4 was used for this analysis.
Incorporation of components into the SPB is faulty in mlp2Δ cells
Our data are consistent with a model in which the connection of SPBs to the Mlp assembly via Mlp2p is necessary for the efficient incorporation of SPB components into the SPB core. This model accounts for the binding of Mlp2p to the SPB core, the stochastic failures in cell division in cells lacking Mlp2p, the appearance of intranuclear microtubule organizers in mlp2Δ cells, and the synthetic interaction of Mlps with spc110-220. A prediction of this model is that cells lacking Mlp2p would have a defect in incorporating newly made components into SPBs, resulting in smaller SPBs. We tested this prediction by quantitating the fluorescence signal intensity associated with Spc42p–GFP labeled SPBs in wild-type and mutant cells (Fig. 7 a). We observed a ∼10% decrease in Spc42p–GFP signal intensity in mlp2Δ cells with respect to wild type. A smaller but equally significant reduction in SPB-associated fluorescence was observed in cells lacking Mlp1p (unpublished data). To exclude the possibility that the observed decrease in Spc42p–GFP fluorescent signal is due to a reduction of the Spc42p–GFP expression level in cells lacking Mlp2p, we performed an immunoblot analysis on whole cell lysates from mutant and wild-type strains (Fig. 7 b). We did not observe any significant variation in Spc42p–GFP expression due to the lack of Mlp2p. These results suggest that the anchoring of SPBs to the Mlp assembly via Mlp2p promotes the ability of components to be incorporated into the SPB.
Figure 7.

Cells lacking Mlp2p have a defect in incorporating newly made components into their SPBs with respect to wild type, resulting in significantly smaller SPBs. (a) Haploid wild-type and mutant cells expressing genomically tagged Spc42p–GFP were analyzed by fluorescence microscopy as in Fig. 4. Two-dimensional maximum projections of three-dimensional image stacks were used for quantitative analysis of the GFP signal associated with each individual SPB. In each experiment we scored 100–200 individual SPBs. We repeated the experiment three times with similar results. Data from each experiment were normalized to the average wild-type intensity value and a combination of all data is presented. The 95% confidence interval for the mean of the distributions of relative SPB intensity measurements for each strain is displayed in the graph. The mean SPB intensities ± SD were: wild type 1.00 ± 0.128 and mlp2Δ 0.915 ± 0.114. The significance of the difference between the two normal distributions was evaluated using the t test and the P value is indicated at the bottom of the graph. (b) Deletion of MLP2 does not influence the expression of Spc42p–GFP. Total cell lysates from haploid mutant or wild-type strains used for the experiment in panel a, were run on SDS-PAGE transferred to nitrocellulose. Blots were probed with anti-GFP monoclonal antibody to determine the levels of expression of Spc42p–GFP and with anti-Pgk1p to control for loading levels. The intensity of the Spc42p–GFP signal in each of the indicated strains were quantified using the OpenLab software and normalized against the Pgk1p signals to produce the graph presented (top). The results indicate no significant difference between mutant and wild-type strains.
Discussion
Traditionally, SPBs and NPCs have been considered as functionally separate entities. However, recent observations that NPCs and SPBs share a small number of components point to potential functional links between the two. Our finding that Mlp2p physically interacts with the SPB core is therefore interesting, suggesting that Mlps, SPBs, and NPCs are interconnected as part of a continuous functional unit at the nuclear periphery.
Several lines of evidence suggest that one reason for the connection of SPBs to the peripheral Mlp assembly is to aid the incorporation of new components into the nuclear face of the SPB (Fig. 8). First, new SPBs are inserted normally into the NE in cells lacking Mlp2p, suggesting that the protein has no role in the initial stages of SPB assembly. Second, the nucleus migrates normally, a process associated with the cytoplasmically nucleated SPB microtubules, likely excluding Mlp2p from roles associated with the cytoplasmic face of the SPB. Third, Mlp2p is exclusively nuclear, and attaches to the SPB on its nuclear face exclusively via nucleoplasmically oriented SPB components, suggesting that Mlp2p is intimately involved in the function of the SPB, but only after it has inserted into the NE. Fourth, mlp2Δ cells have an increased failure rate in SPB separation and in progressing past early mitosis, indicating that the nuclear face of SPBs is occasionally compromised in forming proper spindles. Fifth, a significant fraction of the mlp2Δ population accumulates aberrant intranuclear microtubule organizers, indicating a failure in the proper targeting of components to the SPB. This is similar to the phenotype of the spc110-220 mutant, which also fails to properly target SPB components. Here the defect is in the COOH terminus of the Spc110p protein, where it tightly interdigitates with the Spc42p crystalline layer (Kilmartin et al., 1993; Adams and Kilmartin, 1999). The synthetic lethality we observe in cells lacking Mlp2p and carrying the spc110-220 allele suggests that Mlp2p and the COOH terminus of Spc110p act synergistically in the maturation and maintenance of the SPB. Sixth, SPBs in the mlp2Δ mutant are smaller on average in comparison to SPBs of a wild-type strain, as indicated by relative SPB fluorescence intensity. Because the thickness of the individual SPB layers is constant, this reduction in fluorescence intensity represents roughly a 10% decrease in surface area. Even though this appears to be only a minor reduction, the surface area of the SPB in yeast limits the number of microtubules emanating from it. A haploid wild-type SPB can nucleate the 16 required kinetochore microtubules and approximately 2 to 4 pole-to-pole microtubules (O'Toole et al., 1999). Because we neither observe chromosome loss nor increased lethality with spindle checkpoint mutants in the mlp2Δ strain (unpublished data), it appears that capturing of the kinetochores is unimpaired. Thus, the 10% reduction in SPB size might lead to a greater than 50% reduction in the number of pole-to-pole microtubules, consistent with an increase in the number of cells with duplicated but not completely separated SPBs in the mlp2Δ strain. The requirement for Mlp2p in SPB function is not absolute, as mlp2Δ cells are viable. Yet, a significant number of mlp2Δ cells fail to execute mitosis normally. Because loss of Mlp2p can lead to suboptimal SPBs, each new round of SPB duplication might cause the introduction of additional defects due to the dynamic nature of the SPB during S phase. This in turn would lead to stochastic failures of individual SPBs at different stages of the cell cycle. Equally diverse mitotic defects are observed with other (more penetrant) mutants involved in SPB assembly, such as cmd1-1 and SPC110 COOH-terminal mutants (Stirling et al., 1996; Stirling and Stark, 2000).
We hypothesize that Mlp2p helps to incorporate new SPB components into both the mother and daughter SPBs, by recruiting them for exchange or by facilitating their integration into the central plaque. It may be that Mlp2p does this either by providing additional binding sites for SPB components at the SPB; or by anchoring the edges of the SPB to the surrounding Mlp layer, providing the necessary tension to keep the central plaque crystal “open” for the exchange and addition of proteins. This close connection might also reflect a mutual cooperation between these two structures, in which SPBs actively promote segregation of the Mlps to mother and daughter nuclei. Because the Mlp proteins are located asymmetrically along the nuclear periphery, a random bisection of the nucleus does not guarantee their equal distribution at karyokinesis. However, at metaphase the SPBs line up the Mlp layer perpendicular to the axis of nuclear division. This configuration may not only facilitate the segregation of the Mlps; the nucleolar components, which are also closely associated with the NE and excluded from the Mlps, would be aligned perfectly for equal partitioning to mother and daughter nuclei. The equal distribution of the Mlp proteins and other interconnected NE structures to the opposite spindle poles ensure that each cell has a fully functional NE structure immediately upon entering G1. Even though the Mlps are not essential, they do play important roles in a number of nuclear processes; thus their active segregation increases the overall fitness of the new cells.
Our work also shows that Mlp1p and Mlp2p have overlapping but different distributions at the nuclear periphery, and are functionally distinct. So far, yeasts are the only eukaryotes for which two different Mlps have been identified in the same organism (Kuznetsov et al., 2002). Although in Saccharomyces cerevisiae and its close relatives the second copy arose because of a whole-genome duplication event (Kellis et al., 2004), detailed sequence analyses reveal that in fission yeast this second copy of the Mlp protein arose independently (Ding et al., 2000). Alm1p/TC80, the Schizosaccharomyces pombe MLP2 paralog, localizes to the nuclear rim and appears to accumulate at the SPB and at the medial region MTOC (Ding et al., 2000). Because both budding and fission yeast undergo closed mitosis, with the NE remaining intact throughout the cell cycle, it is conceivable that this kind of direct communication between the NE and spindle organizer may facilitate closed mitosis. Alternatively, it may be necessary for any large structure associated with the NE (be it NPC or SPB) to be anchored into the Mlp assembly. Even though many eukaryotes dispense with their NE during mitosis, the distinction between open and closed mitosis appears to be less than absolute. Thus, the MTOC is linked with the NE during nuclear migration and positioning, and varying amounts of NE stay in the vicinity of the MTOCs during spindle assembly, even in cells with an open mitosis (Nadezhdina et al., 1979; Beaudouin et al., 2002; Malone et al., 2003). Although there is no direct evidence that Mlp/Tpr homologues in vertebrates might play a role similar to that of yeast Mlp2p, in Drosophila it appears that the Tpr homologue Megator aids the formation of a spindle matrix, supporting this idea (Qi et al., 2004). Together with our findings, such results suggest that the close relationship between the NE, Mlp/Tpr, and the MTOC might be a universal feature of the cell division process.
Materials and methods
Plasmids and strains
Strains are isogenic to W303 unless otherwise specified. All yeast strains were constructed using standard genetic techniques (Table S1 available at http://www.jcb.org/cgi/content/full/jcb.200504140/DC1). COOH-terminal genomically tagged strains were generated using the PCR method previously described (Rout et al., 2000; Reid et al., 2002). For the NH2-terminal YFP or CFP tagging of Mlp1p and Mlp2p we used the “pop-in/pop-out” method previously described (Reid et al., 2002). In all cases correct integration was verified by PCR analysis, immunoblotting, and, in the case of fluorescent tags, by direct in vivo fluorescence microscopy. The GAL1-10 promoter was inserted upstream of MLP2-PrA in strain yCS115 using the vector pFA6a-kanMX6-PGAL1 (Longtine et al., 1998) to create yMN437. Wild-type SPC110 (pHS29) and spc110-220 (C911R; pHS38) expressing plasmids (Sundberg et al., 1996) were transformed into yMN290 to create yMN443 and yMN444. yMN443 and yMN444 were crossed with yMN437 and sporulated to create strains yMN439-442. A similar strategy was followed to construct the corresponding MLP1 strains.
pRS314-DsRed-Nop1 (Gadal et al., 2001) and pXYNup49-CFP were used in strains expressing either Mlp1p–YFP or YFP–Mlp2p. For pXYNup49-CFP, the enhanced-CFP (eCFP) open reading frame was PCR amplified from pECFP-N1 (BD Biosciences) and inserted into the HindIII and SalI sites of pYX242 (Novagen) to produce pYX242-CFP. The NUP49 open reading frame was PCR amplified from the pET28b-NUP49 plasmid (gift from S. Dokudovskaya, The Rockefeller University, New York, NY) and inserted into the unique BamHI and HindIII sites of pXY242-CFP to produce pXYNup49-CFP. In all cases the plasmid sequence was verified by DNA sequencing.
Affinity PrA purification
The protocol for the purification of PrA-containing complexes was modified from published methods (Aitchison et al., 1996; Schultz et al., 1997). In brief, frozen cells were ground with a motorized grinder (Retsch) and 1 g (PrA control, Mlp1p–PrA) or 2 g (Mlp2p–PrA) were thawed into 9 ml of extraction buffer 1 (EB1; 20 mM Na-Hepes, pH 7.4, 0.5% Triton X-100, 1 mM DTT, 4 μg/ml pepstatin, 0.2 mg/ml PMSF) supplemented with 300 mM NaCl. Different amounts of frozen cell powder had to be used in order to recover comparable amounts of Mlp-PrA. Cell lysates were homogenized with a Polytron for 25 s (PT 10/35; Brinkman Instruments) and cleared by centrifugation at 2,000 g av for 10 min. 7.5 mg of epoxy-activated Dynabeads (Dynal) cross-linked to rabbit IgG (ICN Biomedicals) were added to each lysate and rotated for 2 h at 4°C. The IgG-Dynabeads were collected with a magnet, washed five times with 1 ml of EB1 and once with 1 ml of 100 mM ammonium acetate, pH 7.4, 0.1 mM MgCl2. The PrA-containing complexes were eluted off the beads in 1 ml of 0.5 M NH4OH, 0.5 mM EDTA at 25°C for 20 min and lyophilized in a SpeedVac (Thermo Savant). Protein samples were resolved by SDS-PAGE with Novex 4–20% Tris-glycine polyacrylamide gels (Invitrogen) and visualized by Coomassie blue staining.
For the preparation of purified Mlp1p- and Mlp2p–PrA for use as probes in the in vitro blot binding assay, the procedure described above was modified as follows. The ground cell powder (1.2 g for Mlp1p–PrA and 4 g for Mlp2p–PrA) was resuspended in 10 ml of extraction buffer 2 (EB2; 20 mM Hepes/KOH, pH 7.4, 1% Triton X-100, 0.5% Na-deoxycholate, 0.3% sodium N-lauroyl-sarcosine, 0.1 mM MgCl2, 1 mM DTT, 4 μg/ml pepstatin, 0.2 mg/ml PMSF) per gram. After homogenization and clarification, the lysate was incubated over night at 4°C with IgG-Sepharose resin (10 μl of bed volume per gram of cell powder). After extensive washing in EB2 without DTT, bound material was eluted off the resin using a biotinylated derivative of a previously published PrA-mimicking peptide (Biotin-DCAWHLGELVWCT; DeLano et al., 2000). The eluting peptide was removed on a G25 sizing column (Amersham Biosciences) and the probe was quantified by running an aliquot on SDS-PAGE alongside BSA standards.
Identification of proteins by mass spectrometry
Protein bands were excised and tryptic digestions were prepared according to standard protocols (Krutchinsky et al., 2001). Extracted peptides were analyzed by a modified matrix-assisted laser desorption/ionization ion trap mass spectrometer (Krutchinsky et al., 2001) based on a LCQ Deca XP ion trap mass spectrometer (Thermo Finnigan). An automated protocol was used to perform tandem mass spectrometry and proteins were identified searching the National Center for Biotechnology Information nonredundant protein database with the program Sonar (Genomic Solutions) to identify proteins from the tryptic peptide fragmentation masses (Field et al., 2002).
In vitro blot binding assay
The Mlp2p complex was immobilized on IgG-Dynabeads as described for affinity PrA purification. After binding, Dynabeads were incubated using 1 M MgCl2, 20 mM Hepes/KOH, pH 7.4, 0.5% Tween 20 in order to preserve the binding of Mlp2p–PrA to IgG and selectively elute the Mlp2p–PrA bound proteins off the beads. The eluate was subsequently incubated with fresh IgG-Dynabeads to remove contaminating Mlp2p–PrA and with PrA-Sepharose beads to remove IgG that might have bled through from the IgG-Dynabeads. Proteins were then recovered by TCA precipitation, separated on SDS-PAGE, and transferred to nitrocellulose. After amido black staining the blot was cut into three 0.2-cm wide identical strips. The strips were blocked for 1 h at 25°C in 5% milk, 2% BSA, 20 mM Hepes/KOH, 110 mM KOAc, 2 mM MgCl2, 0.1% Tween 20, 1 mM DTT, 4 μg/ml pepstatin, 0.2 mg/ml PMSF, before incubation with 1.2 μg of purified Mlp probes in 500 μl of the same buffer at 4°C overnight. After four brief washes with the same buffer at 25°C, the strips were incubated with 1:2,000 rabbit IgG-HRP conjugate (Jackson ImmunoResearch Laboratories, Inc.) in binding buffer for 1 h at 25°C. The presence of bound HRP was detected by chemiluminescence.
Coimmunoprecipitation immuoblot experiments
For each strain 0.5 g of cell powder was used. To adjust for total protein amount, 0.5 g of untagged cell powder was added to strains marked “coexpressed.” The powder was thawed into 5 ml of EB1 supplemented with 300 mM NaCl, 1 mg/ml Heparin for Spc42p–PrA, and 150 mM NaCl, 1 mg/ml Heparin for Spc110p–PrA, cleared by centrifugation and incubated with 7.5 mg of rabbit IgG–conjugated Dynabeads. In all cases the lysates were rotated with the beads for 2 h at 4°C and subsequently treated as described for affinity PrA purification. Samples of each cell lysate and each isolated complex were resolved in duplicate by SDS PAGE and transferred onto nitrocellulose. The presence of PrA and Myc in the samples was detected by immunoblotting using a 1:1,000 dilution of a rabbit IgG (ICN Biomedicals) and a mouse monoclonal anti–Myc antibody (Santa Cruz Biotechnology, Inc.).
Microscopy
For in vivo fluorescence experiments cells expressing DsRed-, GFP-, CFP-, or YFP-tagged proteins were grown overnight in YPD or the appropriate selective medium containing 200 μg/ml adenine to reduce autofluorescence. The next morning cells were diluted in the same medium and grown to mid-log phase for 4–5 h. When ready, cells were harvested and concentrated in SC medium with 200 μg/ml adenine. A small drop (1–2 μl) of the concentrated cell suspension was spotted on a poly-L-lysine–coated slides before immobilization using a coverslip. Cells were observed immediately after immobilization at 25°C. Cell were visualized with a 100× 1.4 numerical aperture Planapochromat objective using an inverted Carl Zeiss Axiovert 200 wide-field confocal microscope fitted with a Perkin-Elmer Ultra-View spinning disk confocal head on side-port optimized for real-time confocal imaging. The microscope was equipped with a Hamamatsu Orca ER-cooled CCD camera. Image analysis was performed using the MetaMorph software provided by Universal Imaging Corp. For three-dimensional volume and surface reconstruction we used the Imaris and Imaris-Surpass (Bitplane AG) software.
For DAPI staining, cells were grown to mid-log phase in YPD containing adenine as above. Cells were fixed in 4% parafomaldehyde, 3.4% sucrose, and 0.1 M KPO4 for 5–15 min at 25°C. Cells were washed with 1.2 M sorbitol, 0.1 M KPO4, pH 7.5, sonicated, and subsequently permeabilized with 0.1% Triton X-100 in the same buffer for 1 min at 25°C. Cells were stained with 0.06 μg/ml DAPI in sorbitol/KPO4 for 10 min at 25°C, washed in the same buffer, and immobilized on slides. For image acquisition we used an Axioplan 2 microscope (Carl Zeiss MicroImaging Corp.), with a 100× 1.4 numerical aperture Planapochromat objective, fitted with a Hamamatsu C4742-95 cooled charge-coupled device camera (Sciscope Instrument) interfaced with the OpenLab software (Improvision).
Samples were prepared for thin-section TEM essentially as described (Rout and Blobel, 1993). For postembedding labeling immuno-EM samples were prepared essentially as reported (Wente et al., 1992). The primary antibody was a 1:2,000 dilution of a polyclonal rabbit anti-GFP raised against the whole GFP molecule. The secondary antibody was 10 nm gold particles conjugated goat anti–rabbit IgG.
Quantitative image analyses
All the quantitative analyses were performed on digital images. Scoring of SPB numbers on DAPI-stained nuclei was performed on two-dimensional projections of three-dimensional image stacks containing 8 0.35 μm optical sections after overlaying them onto differential interference contrast images for clearer positioning of SPB signals and nuclei. Scoring of the colocalization between Spc42p–CFP signal versus YFP–Mlp1p was performed on two-dimensional maximum projections of three-dimensional image stacks containing 10–15 0.27 μm optical sections. Only nuclei that presented with a typical C-shaped Mlp distribution (i.e., nuclei in which the nucleolus was positioned around the mid-plane of the stack) were scored for the localization of the Spc42p–CFP signal. All other analyses were performed directly using Z-calibrated three-dimensional digital image stacks, which contained 15–20 0.27 μm optical sections. Scoring of spindle morphology frequencies in live cells expressing GFP–Tub1p was performed using the Manually Count Objects module included in the MetaMorph software. All length measurements were obtained using the Measure XYZ Distance module of the same software. The spindle migration index of a cell is proportional to the proximity of its spindle to the bud neck and is calculated as the minimal distance between the neck and the nearest edge of spindle, divided by the maximal mother cell axis (i.e., distance between the bud neck and the most distal edge of the mother cell; Jacobs et al., 1988).
Differential elutriation
Haploid strains yMN291 and yMN293 and diploid strains yCS101 and yCS251 were subjected to differential elutriation as previously described (Miller and Cross, 2001). 12 fractions were collected from each experiment. Cells were harvested by filtration and prepared for either DAPI staining and fluorescence microscopy (yMN291 and yMN293), for thin section EM analysis (yCS101 and yCS251) or for postembedding-labeling EM (yMN291 and yMN293). Progression of cells through the cell cycle was monitored by budding index and by FACS (Becton Dickinson) as described previously (Epstein and Cross, 1992).
Online supplemental material
Fig. S1 shows the distribution of YFP–Mlp2p in comparison to the nucleolus and the NPCs by fluorescence microscopy. Fig. S2 shows the budding state and the DNA content of the elutriation fractions used in Fig. 5. Fig. S3 shows the repression of PrA-tagged Mlp2p in dextrose containing media to levels below the detection limit by immunoblotting. Fig. S4 shows the growth effects of depleting or deleting the Mlp proteins in a series of SPB temperature sensitive mutant backgrounds. Table S1 shows the S. cerevisiae strains used in this study. Online supplemental materials are available at http://www.jcb.org/cgi/content/full/jcb.200504140/DC1.
Acknowledgments
We are deeply indebted to Helen Shio for performing the EM studies. We are very grateful to Wenzhu Zhang and Andrew Krutchinsky in the Chait laboratory for essential technical guidance in the mass spectrometric analyses. Furthermore, we would like to thank Alison North, Director, and Dan Elreda of the Bio-Imaging Resource Center (http://www.rockefeller.edu/bioimaging/) at The Rockefeller University for their continual help and assistance with the collection and analysis of all light fluorescence images. We wish to thank Rosemary Williams for her skilled technical assistance throughout the course of this study. We would like to thank Xiaolan Zhao for helpful discussion and for providing us with reagents without which this work would not have been possible. Thanks to all members of the Rout laboratory, past and present, for their continual help and unwavering support. Thanks to Fred Cross for critically reviewing the manuscript and to all members of his laboratory for help in yeast manipulations. Thanks to Michael Eisenstein for critically reviewing the manuscript.
This work was supported by a grant from the American Cancer Society (RSG0404251), an Irma T. Hirschl Career Scientist Award, a Sinsheimer Scholar Award, and a grant from the Rita Allen Foundation to M.P. Rout; by grants from the National Institutes of Health to M.P. Rout (GM062427), B.T. Chait (RR00862), and B.T. Chait and M.P. Rout (CA89810); and by a fellowship from the Boehringer Ingelheim Fonds to M. Niepel.
M. Niepel and C. Strambio-de-Castillia contributed equally to this paper.
J. Fasolo's present address is Yale University, New Haven, CT 06520.
Abbreviations used: IMO, intranuclear microtubule organizer; MTOC, microtubule organizer center; NE, nuclear envelope; NPC, nuclear pore complex; PrA, protein A; SPB, spindle pole body; TEM, transmission electron microscopy.
References
- Adams, I.R., and J.V. Kilmartin. 1999. Localization of core spindle pole body (SPB) components during SPB duplication in Saccharomyces cerevisiae. J. Cell Biol. 145:809–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitchison, J.D., G. Blobel, and M.P. Rout. 1996. Kap104p: a karyopherin involved in the nuclear transport of messenger RNA binding proteins. Science. 274:624–627. [DOI] [PubMed] [Google Scholar]
- Andrulis, E.D., D.C. Zappulla, A. Ansari, S. Perrod, C.V. Laiosa, M.R. Gartenberg, and R. Sternglanz. 2002. Esc1, a nuclear periphery protein required for Sir4-based plasmid anchoring and partitioning. Mol. Cell. Biol. 22:8292–8301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudouin, J., D. Gerlich, N. Daigle, R. Eils, and J. Ellenberg. 2002. Nuclear envelope breakdown proceeds by microtubule-induced tearing of the lamina. Cell. 108:83–96. [DOI] [PubMed] [Google Scholar]
- Bullitt, E., M.P. Rout, J.V. Kilmartin, and C.W. Akey. 1997. The yeast spindle pole body is assembled around a central crystal of Spc42p. Cell. 89:1077–1086. [DOI] [PubMed] [Google Scholar]
- Byers, B., and L. Goetsch. 1975. Electron microscopic observations on the meiotic karyotype of diploid and tetraploid Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 72:5056–5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bystricky, K., T. Laroche, G. van Houwe, M. Blaszczyk, and S.M. Gasser. 2005. Chromosome looping in yeast: telomere pairing and coordinated movement reflect anchoring efficiency and territorial organization. J. Cell Biol. 168:375–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chial, H.J., M.P. Rout, T.H. Giddings, and M. Winey. 1998. Saccharomyces cerevisiae Ndc1p is a shared component of nuclear pore complexes and spindle pole bodies. J. Cell Biol. 143:1789–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, T.N. 1992. A temperature-sensitive calmodulin mutant loses viability during mitosis. J. Cell Biol. 118:607–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano, W.L., M.H. Ultsch, A.M. de Vos, and J.A. Wells. 2000. Convergent solutions to binding at a protein-protein interface. Science. 287:1279–1283. [DOI] [PubMed] [Google Scholar]
- Ding, D.Q., Y. Tomita, A. Yamamoto, Y. Chikashige, T. Haraguchi, and Y. Hiraoka. 2000. Large-scale screening of intracellular protein localization in living fission yeast cells by the use of a GFP-fusion genomic DNA library. Genes Cells. 5:169–190. [DOI] [PubMed] [Google Scholar]
- Elliott, S., M. Knop, G. Schlenstedt, and E. Schiebel. 1999. Spc29p is a component of the Spc110p subcomplex and is essential for spindle pole body duplication. Proc. Natl. Acad. Sci. USA. 96:6205–6210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein, C.B., and F.R. Cross. 1992. CLB5: a novel B cyclin from budding yeast with a role in S phase. Genes Dev. 6:1695–1706. [DOI] [PubMed] [Google Scholar]
- Feuerbach, F., V. Galy, E. Trelles-Sticken, M. Fromont-Racine, A. Jacquier, E. Gilson, J.C. Olivo-Marin, H. Scherthan, and U. Nehrbass. 2002. Nuclear architecture and spatial positioning help establish transcriptional states of telomeres in yeast. Nat. Cell Biol. 4:214–221. [DOI] [PubMed] [Google Scholar]
- Field, H.I., D. Fenyo, and R.C. Beavis. 2002. RADARS, a bioinformatics solution that automates proteome mass spectral analysis, optimises protein identification, and archives data in a relational database. Proteomics. 2:36–47. [PubMed] [Google Scholar]
- Gadal, O., D. Strauss, J. Kessl, B. Trumpower, D. Tollervey, and E. Hurt. 2001. Nuclear export of 60s ribosomal subunits depends on Xpo1p and requires a nuclear export sequence-containing factor, Nmd3p, that associates with the large subunit protein Rpl10p. Mol. Cell. Biol. 21:3405–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galy, V., J.C. Olivo-Marin, H. Scherthan, V. Doye, N. Rascalou, and U. Nehrbass. 2000. Nuclear pore complexes in the organization of silent telomeric chromatin. Nature. 403:108–112. [DOI] [PubMed] [Google Scholar]
- Galy, V., O. Gadal, M. Fromont-Racine, A. Romano, A. Jacquier, and U. Nehrbass. 2004. Nuclear retention of unspliced mRNAs in yeast is mediated by perinuclear Mlp1. Cell. 116:63–73. [DOI] [PubMed] [Google Scholar]
- Geiser, J.R., D. van Tuinen, S.E. Brockerhoff, M.M. Neff, and T.N. Davis. 1991. Can calmodulin function without binding calcium? Cell. 65:949–959. [DOI] [PubMed] [Google Scholar]
- Green, D.M., C.P. Johnson, H. Hagan, and A.H. Corbett. 2003. The C-terminal domain of myosin-like protein 1 (Mlp1p) is a docking site for heterogeneous nuclear ribonucleoproteins that are required for mRNA export. Proc. Natl. Acad. Sci. USA. 100:1010–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase, S.B., M. Winey, and S.I. Reed. 2001. Multi-step control of spindle pole body duplication by cyclin-dependent kinase. Nat. Cell Biol. 3:38–42. [DOI] [PubMed] [Google Scholar]
- Hediger, F., K. Dubrana, and S.M. Gasser. 2002. a. Myosin-like proteins 1 and 2 are not required for silencing or telomere anchoring, but act in the Tel1 pathway of telomere length control. J. Struct. Biol. 140:79–91. [DOI] [PubMed] [Google Scholar]
- Hediger, F., F.R. Neumann, G. Van Houwe, K. Dubrana, and S.M. Gasser. 2002. b. Live imaging of telomeres: yKu and Sir proteins define redundant telomere-anchoring pathways in yeast. Curr. Biol. 12:2076–2089. [DOI] [PubMed] [Google Scholar]
- Iouk, T., O. Kerscher, R.J. Scott, M.A. Basrai, and R.W. Wozniak. 2002. The yeast nuclear pore complex functionally interacts with components of the spindle assembly checkpoint. J. Cell Biol. 159:807–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs, C.W., A.E. Adams, P.J. Szaniszlo, and J.R. Pringle. 1988. Functions of microtubules in the Saccharomyces cerevisiae cell cycle. J. Cell Biol. 107:1409–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspersen, S.L., and M. Winey. 2004. The budding yeast spindle pole body: structure, duplication, and function. Annu. Rev. Cell Dev. Biol. 20:1–28. [DOI] [PubMed] [Google Scholar]
- Kellis, M., B.W. Birren, and E.S. Lander. 2004. Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature. 428:617–624. [DOI] [PubMed] [Google Scholar]
- Kilmartin, J.V., S.L. Dyos, D. Kershaw, and J.T. Finch. 1993. A spacer protein in the Saccharomyces cerevisiae spindle pole body whose transcript is cell cycle-regulated. J. Cell Biol. 123:1175–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosova, B., N. Pante, C. Rollenhagen, A. Podtelejnikov, M. Mann, U. Aebi, and E. Hurt. 2000. Mlp2p, a component of nuclear pore attached intranuclear filaments, associates with Nic96p. J. Biol. Chem. 275:343–350. [DOI] [PubMed] [Google Scholar]
- Krutchinsky, A.N., M. Kalkum, and B.T. Chait. 2001. Automatic identification of proteins with a MALDI-quadrupole ion trap mass spectrometer. Anal. Chem. 73:5066–5077. [DOI] [PubMed] [Google Scholar]
- Kuznetsov, N.V., L. Sandblad, M.E. Hase, A. Hunziker, M. Hergt, and V.C. Cordes. 2002. The evolutionarily conserved single-copy gene for murine Tpr encodes one prevalent isoform in somatic cells and lacks paralogs in higher eukaryotes. Chromosoma. 111:236–255. [DOI] [PubMed] [Google Scholar]
- Longtine, M.S., A. McKenzie III, D.J. Demarini, N.G. Shah, A. Wach, A. Brachat, P. Philippsen, and J.R. Pringle. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 14:953–961. [DOI] [PubMed] [Google Scholar]
- Malone, C.J., L. Misner, N. Le Bot, M.C. Tsai, J.M. Campbell, J. Ahringer, and J.G. White. 2003. The C. elegans hook protein, ZYG-12, mediates the essential attachment between the centrosome and nucleus. Cell. 115:825–836. [DOI] [PubMed] [Google Scholar]
- Miller, M.E., and F.R. Cross. 2001. Mechanisms controlling subcellular localization of the G(1) cyclins Cln2p and Cln3p in budding yeast. Mol. Cell. Biol. 21:6292–6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadezhdina, E.S., D. Fais, and Y.S. Chentsov. 1979. On the association of centrioles with the interphase nucleus. Eur. J. Cell Biol. 19:109–115. [PubMed] [Google Scholar]
- O'Toole, E.T., M. Winey, and J.R. McIntosh. 1999. High-voltage electron tomography of spindle pole bodies and early mitotic spindles in the yeast Saccharomyces cerevisiae. Mol. Biol. Cell. 10:2017–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi, H., U. Rath, D. Wang, Y.Z. Xu, Y. Ding, W. Zhang, M.J. Blacketer, M.R. Paddy, J. Girton, J. Johansen, and K.M. Johansen. 2004. Megator, an essential coiled-coil protein that localizes to the putative spindle matrix during mitosis in Drosophila. Mol. Biol. Cell. 15:4854–4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid, R.J., M. Lisby, and R. Rothstein. 2002. Cloning-free genome alterations in Saccharomyces cerevisiae using adaptamer-mediated PCR. Methods Enzymol. 350:258–277. [DOI] [PubMed] [Google Scholar]
- Rout, M.P., and G. Blobel. 1993. Isolation of the yeast nuclear pore complex. J. Cell Biol. 123:771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout, M.P., and J.V. Kilmartin. 1990. Components of the yeast spindle and spindle pole body. J. Cell Biol. 111:1913–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout, M.P., J.D. Aitchison, A. Suprapto, K. Hjertaas, Y. Zhao, and B.T. Chait. 2000. The yeast nuclear pore complex: composition, architecture, and transport mechanism. J. Cell Biol. 148:635–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaerer, F., G. Morgan, M. Winey, and P. Philippsen. 2001. Cnm67p is a spacer protein of the Saccharomyces cerevisiae spindle pole body outer plaque. Mol. Biol. Cell. 12:2519–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz, M.C., D.J. Hockman, T.A. Harkness, W.I. Garinther, and B.A. Altheim. 1997. Chromatin assembly in a yeast whole-cell extract. Proc. Natl. Acad. Sci. USA. 94:9034–9039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spang, A., I. Courtney, U. Fackler, M. Matzner, and E. Schiebel. 1993. The calcium-binding protein cell division cycle 31 of Saccharomyces cerevisiae is a component of the half bridge of the spindle pole body. J. Cell Biol. 123:405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling, D.A., and M.J. Stark. 2000. Mutations in SPC110, encoding the yeast spindle pole body calmodulin-binding protein, cause defects in cell integrity as well as spindle formation. Biochim. Biophys. Acta. 1499:85–100. [DOI] [PubMed] [Google Scholar]
- Stirling, D.A., T.F. Rayner, A.R. Prescott, and M.J. Stark. 1996. Mutations which block the binding of calmodulin to Spc110p cause multiple mitotic defects. J. Cell Sci. 109:1297–1310. [DOI] [PubMed] [Google Scholar]
- Strambio-de-Castillia, C., G. Blobel, and M.P. Rout. 1999. Proteins connecting the nuclear pore complex with the nuclear interior. J. Cell Biol. 144:839–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundberg, H.A., and T.N. Davis. 1997. A mutational analysis identifies three functional regions of the spindle pole component Spc110p in Saccharomyces cerevisiae. Mol. Biol. Cell. 8:2575–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundberg, H.A., L. Goetsch, B. Byers, and T.N. Davis. 1996. Role of calmodulin and Spc110p interaction in the proper assembly of spindle pole body compenents. J. Cell Biol. 133:111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tackett, A.J., D.J. Dilworth, M.J. Davey, M. O'Donnell, J.D. Aitchison, M.P. Rout, and B.T. Chait. 2005. Proteomic and genomic characterization of chromatin complexes at a boundary. J. Cell Biol. 169:35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinciguerra, P., N. Iglesias, J. Camblong, D. Zenklusen, and F. Stutz. 2005. Perinuclear Mlp proteins downregulate gene expression in response to a defect in mRNA export. EMBO J. 24:813–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wente, S.R., M.P. Rout, and G. Blobel. 1992. A new family of yeast nuclear pore complex proteins. J. Cell Biol. 119:705–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, C.H., E.J. Lambie, J. Hardin, J. Craft, and M. Snyder. 1989. Higher order structure is present in the yeast nucleus: autoantibody probes demonstrate that the nucleolus lies opposite the spindle pole body. Chromosoma. 98:123–128. [DOI] [PubMed] [Google Scholar]
- Yoder, T.J., C.G. Pearson, K. Bloom, and T.N. Davis. 2003. The Saccharomyces cerevisiae spindle pole body is a dynamic structure. Mol. Biol. Cell. 14:3494–3505. [DOI] [PMC free article] [PubMed] [Google Scholar]