

Abstract
Life and death decisions are made by integrating a variety of apoptotic and survival signals in mammalian cells. Therefore, there is likely to be a common mechanism that integrates multiple signals adjudicating between the alternatives. In this study, we propose that 14-3-3 represents such an integration point. Several proapoptotic proteins commonly become associated with 14-3-3 upon phosphorylation by survival-mediating kinases such as Akt. We reported previously that cellular stresses induce c-Jun NH2-terminal kinase (JNK)–mediated 14-3-3ζ phosphorylation at Ser184 (Tsuruta, F., J. Sunayama, Y. Mori, S. Hattori, S. Shimizu, Y. Tsujimoto, K. Yoshioka, N. Masuyama, and Y. Gotoh. 2004. EMBO J. 23:1889–1899). Here, we show that phosphorylation of 14-3-3 by JNK releases the proapoptotic proteins Bad and FOXO3a from 14-3-3 and antagonizes the effects of Akt signaling. As a result of dissociation, Bad is dephosphorylated and translocates to the mitochondria, where it associates with Bcl-2/Bcl-xL. Because Bad and FOXO3a share the 14-3-3–binding motif with other proapoptotic proteins, we propose that this JNK-mediated phosphorylation of 14-3-3 regulates these proapoptotic proteins in concert and makes cells more susceptible to apoptotic signals.
Introduction
The balance between signals promoting survival and apoptosis is important for determining cell fate. Survival signals raise the threshold above which apoptotic signals can induce the machinery of cell death, and, accordingly, a reduction in survival signals results in a high sensitivity of the cell to apoptotic signals. A key to understanding cell death regulation, therefore, is to reveal the molecular mechanisms by which survival and apoptotic signals integrate.
The 14-3-3 proteins are a family of phospho-Ser/phospho-Thr–binding molecules that play essential roles in many biological processes, including the regulation of cell death (van Hemert et al., 2001; Masters et al., 2002; Yaffe, 2002). 14-3-3 promotes cell survival by sequestering and inactivating several proapoptotic proteins, including Bad and FOXO3a, after their phosphorylation by survival-inducing kinases such as Akt (Zha et al., 1996; Datta et al., 1997; del Peso et al., 1997; Biggs et al., 1999; Brunet et al., 1999; Kops et al., 1999; Zhang et al., 1999; Masuyama et al., 2001; Masters et al., 2002; Basu et al., 2003). Indeed, the reduction of available 14-3-3 sensitizes cells to apoptotic signals, and overexpression of 14-3-3 renders cells resistant to apoptotic signals (Chan et al., 1999; Zhang et al., 1999; Xing et al., 2000; Masters and Fu, 2001; Samuel et al., 2001; Nomura et al., 2003; Tsuruta et al., 2004). The level of available 14-3-3 may, therefore, be crucial for determining the threshold above which apoptotic signals can initiate the program.
14-3-3 appears to be modulated by phosphorylation. Phosphorylated forms of 14-3-3β and ζ at Ser186 and 184, respectively, are abundantly present in brain tissue (Aitken et al., 1995). We found that 14-3-3ζ and σ isoforms are phosphorylated at their respective phosphorylation sites by c-Jun NH2-terminal kinase (JNK; Tsuruta et al., 2004). This finding prompted us to examine whether phosphorylation of 14-3-3 may have any impact on the regulation of cell death because JNK has been suggested to play a key role in stress-induced apoptosis in a context-dependent manner (Yang et al., 1997; Tournier et al., 2000; Lei et al., 2002; Deng et al., 2003; Kuan et al., 2003). It is important to note that phosphorylation of 14-3-3ζ causes the dissociation of Bax from 14-3-3, leading to Bax translocation to the mitochondria and to apoptosis (Tsuruta et al., 2004). The expression of mutants lacking phosphorylation sites (Ser184A of 14-3-3ζ or Ser186A of 14-3-3σ) effectively attenuates cell death that is induced by stress-activated JNK, suggesting that 14-3-3 is a major target of JNK in the induction of cell death. However, Bax is not a typical 14-3-3 ligand in the sense that this binding does not require the common ligand-binding groove and does not depend on ligand (Bax) phosphorylation (Nomura et al., 2003). Thus, it is unclear whether phosphorylation of 14-3-3 also affects the association of “typical” 14-3-3 ligands, which usually contain the 14-3-3–binding motif RSXpSXP or RXXXpSXP (Muslin et al., 1996; Yaffe et al., 1997). Because Ser184 is located near the ligand-binding groove, the phosphorylation of this residue might regulate the association of many typical 14-3-3 ligands, including the aforementioned proapoptotic proteins. To investigate the hypothesis that the JNK-mediated phosphorylation of 14-3-3 may contribute to cell death by regulating typical 14-3-3 ligands in addition to Bax, we examined the effect of 14-3-3 phosphorylation on the association of Bad, which binds to the common ligand-binding groove of 14-3-3 upon Bad phosphorylation.
Bad is a “BH3-only” member of the Bcl-2 family (Danial and Korsmeyer, 2004). Survival factors suppress the proapoptotic function of Bad by phosphorylation at Ser112, 136, and 155 (Zha et al., 1996; Datta et al., 2000; Lizcano et al., 2000; Zhou et al., 2000). Phosphorylation of Ser112 and 136 creates the 14-3-3–binding motifs RHSpS112YP and RSRpS136AP, respectively, and the consequent interaction with 14-3-3 leads to the cytoplasmic sequestration and inactivation of Bad. Akt, Pak-1, and p70S6 kinase appear to mediate the survival factor–induced phosphorylation of Bad at Ser136, whereas Rsk, mitochondria-associated protein kinase A, Pak-1 and -5, and Pim-1 are responsible for phosphorylation at Ser112 (Datta et al., 1997; del Peso et al., 1997; Blume-Jensen et al., 1998; Bonni et al., 1999; Harada et al., 1999, 2001; Tan et al., 1999; Schurmann et al., 2000; Shimamura et al., 2000; Cotteret et al., 2003; Aho et al., 2004). The dephosphorylated form of Bad is targeted to the mitochondria, where it causes apoptosis by binding and inactivating Bcl-2/Bcl-xL (Wang et al., 1999). Thus, survival factors regulate the interaction between Bad and 14-3-3 by regulating Bad phosphorylation. However, it remains to be determined whether the interaction between Bad and 14-3-3 is also regulated by the phosphorylation state of 14-3-3. Because Bad and JNK are both implicated in stress-induced cell death (Yang et al., 1997; Tournier et al., 2000; Datta et al., 2002; Lei et al., 2002; Deng et al., 2003; Kuan et al., 2003), we focused on Bad as a potential target for JNK-mediated apoptotic signals.
Results
JNK promotes the dissociation of Bad from 14-3-3 proteins in vivo
Ser186 of 14-3-3β and σ (corresponding to Ser184 of 14-3-3ζ) is a surface residue located at the NH2 terminus of helix 8 near the top of the ligand-binding groove (Fig. 1 a; Liu et al., 1995; Xiao et al., 1995). We examined whether phosphorylation of this site would affect the association of Bad, which is a typical 14-3-3 ligand. We first examined whether the activation of JNK affects the interaction between 14-3-3 and Bad. When endogenous Bad was immunoprecipitated from lysates of COS-1 cells, endogenous 14-3-3 was coprecipitated (Fig. 1 b). However, when COS-1 cells were treated with the protein synthesis inhibitor anisomycin, the amount of 14-3-3 that coprecipitated with Bad was markedly reduced (Fig. 1 b). Coprecipitation of Bad with 14-3-3 was also diminished by UV irradiation of HCT116 cells (Fig. 1 c). We chose anisomycin and UV treatment in these experiments because these cellular stresses are known to activate JNK effectively and require JNK activation to induce cell death in mouse embryonic fibroblasts (Tournier et al., 2000) and HCT116 cells (see Fig. 5, a and b). We next examined whether this cellular stress-induced dissociation between 14-3-3 and Bad was mediated by the activation of endogenous JNK. The expression of a dominant negative (DN) JNK or an inhibitory peptide for JNK (called JNK-binding domain [JBD]; Dickens et al., 1997) abrogated the ability of anisomycin or UV treatment to induce the dissociation between 14-3-3 and Bad, demonstrating a prerequisite role of JNK in this event (Fig. 1, b and c). Anisomycin treatment of COS-1 cells or UV irradiation of HCT116 cells resulted in phosphorylation of the JNK substrate c-Jun, and the expression of DN-JNK or JBD blocked these effects (Fig. 1 d). We then asked whether the expression of active JNK is sufficient to induce the dissociation between 14-3-3 and Bad. The expression of a constitutively active (CA) form of JNK (MKK7-JNK wild type [WT]), but not that of a kinase negative (KN) JNK (MKK7-JNK [KN]), resulted in a decrease in the amount of 14-3-3 that coprecipitated with Bad (Fig. 1 e). Although JNK induces activation of the caspase cascade (Lei et al., 2002; Tsuruta et al., 2004), this effect of MKK7-JNK (WT) was not influenced by the coexpression of p35, a pan-caspase inhibitor, suggesting that JNK promotes the dissociation of Bad from 14-3-3 independently of caspase activation (Fig. 1 e).
Figure 1.

JNK promotes the dissociation of Bad from 14-3-3 in vivo. (a) The putative JNK phosphorylation site of 14-3-3ζ is adjacent to the ligand-binding groove, which is shown in red. The right panel is a close-up view of the region (white box, left) surrounding the putative phosphorylation site (Ser184), which is shown in yellow. (b) COS-1 cells were transfected with the indicated plasmids and were incubated for 3 h in the absence or presence of 10 μg/ml anisomycin. Cell lysates were subjected to immunoprecipitation (IP) with antibodies to Bad or normal rabbit IgG (NRI). (c) HCT116 cells were transfected with the indicated plasmids and were irradiated by 50 J/m2 UV for 6 h. Cell lysates were subjected to immunoprecipitation with antibodies to 14-3-3σ or normal goat IgG (NGI). (d) Cells were transfected and cultured as described in b and c. Cell lysates were subjected to immunoblot analysis with the indicated antibodies. (e) COS-1 cells were transfected for 18 h with expression vectors for MKK7-JNK and p35 as indicated. Cell lysates were subjected to immunoprecipitation with antibodies to Bad. (b–e) The resulting precipitates were subjected to immunoblot analysis (IB) with the indicated antibodies. The same results were obtained in three independent experiments.
Figure 5.

JNK-mediated 14-3-3 phosphorylation regulates Bad-dependent cell death. HCT116 cells were transfected with the indicated plasmids together with a GFP-expressing plasmid for 12 h and were irradiated by 50 J/m2 UV for 6 h (a–d) or for 3 h (e). The cells were stained with Hoechst 33342 for 10 min (a, c, and e). The percentage of GFP-positive cells with pyknotic nuclei was determined. Data shows the means ± SD of values obtained from three fields of 150–200 cells in each of three independent experiments. (a and c) *, P < 0.01; (e) *, P < 0.05. At least 750 cells (a), 600 cells (c), and 500 cells (d) per sample were analyzed. (b and d) Cell lysates were subjected to immunoblot analysis (IB) with the indicated antibodies. p19 and p17 fragments represent cleaved (and activated) caspase-3. SA, Ser186A.
JNK-mediated phosphorylation of 14-3-3 results in the release of Bad from 14-3-3 in vitro
Next, we asked whether the JNK-induced dissociation of Bad from 14-3-3 is dependent on the phosphorylation state of 14-3-3 in an in vitro reconstitution assay. We performed GST pull-down experiments to detect the association between recombinant GST-tagged 14-3-3ζ and His-tagged Bad proteins (Fig. 2). Bad interacts with 14-3-3 proteins only when it is phosphorylated at Ser112 and/or 136 (Zha et al., 1996). To phosphorylate Bad at Ser136, we preincubated recombinant Bad with CA-Akt that was immunoprecipitated from COS-1 cells. We confirmed that Bad coprecipitated with GST–14-3-3ζ, but not with GST alone, only when it was preincubated with CA-Akt in the presence of ATP (Fig. 2 a). We then performed the phosphorylation of GST–14-3-3ζ by MKK7-JNK (WT) before the GST pull-down experiments with Akt-phosphorylated Bad and found that this phosphorylation of 14-3-3ζ reduced the amount of Bad that was coprecipitated with GST–14-3-3ζ. In contrast, when the Ser184A mutant of GST–14-3-3ζ was used, similar amounts of Bad were precipitated with or without MKK7-JNK (WT) preincubation. These results indicate that 14-3-3ζ phosphorylation at Ser184 by JNK reduces its affinity for Bad (Fig. 2 a).
Figure 2.

JNK-mediated phosphorylation of 14-3-3 results in the release of Bad from 14-3-3 in vitro. (a and b) GST-tagged proteins were subjected to an in vitro kinase assay with His-tagged MKK7-JNK and were incubated with His-tagged Bad wild type (WT; a) or Ser128A (b) that was preincubated with or without CA-Akt immune complex. The GST proteins were precipitated using glutathione–Sepharose beads. The amount of Bad protein coprecipitated with either GST or GST–14-3-3ζ was determined by immunoblot analysis (IB) with the indicated antibodies. The same results were obtained in at least two independent experiments.
Recent studies have shown that JNK and Cdc2 phosphorylate Bad at Ser128 in primary cerebellar granule neurons, and phosphorylation of Bad inhibits the interaction of Bad with 14-3-3 (Donovan et al., 2002; Konishi et al., 2002). In addition, JNK phosphorylates mouse Bad at Thr201, although this site is not conserved in human Bad (Yu et al., 2004). Therefore, we investigated whether phosphorylation of 14-3-3ζ at Ser184 can lead to the dissociation of Bad from 14-3-3 regardless of the phosphorylation state of Bad at Ser128 or Thr201. We found that any of the recombinant mutant Bads (Bad Ser128A, Thr201A, or Ser128A-Thr201A) could interact with 14-3-3ζ when phosphorylated by active Akt and that they dissociated from 14-3-3ζ when 14-3-3ζ was phosphorylated by MKK7-JNK (WT; Fig. 2 b and Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200409117.DC1). Therefore, the JNK-mediated phosphorylation of 14-3-3 can induce the dissociation of Bad independently of the Ser128 and Thr201 phosphorylation of Bad.
JNK promotes Bad translocation to mitochondria
Bad is localized mostly in the cytoplasm but redistributes to the mitochondria in response to survival factor deprivation (Li et al., 2004). Because 14-3-3 is regarded as a cytoplasmic anchor for Bad (Muslin and Xing, 2000) and we found that the JNK-mediated phosphorylation of 14-3-3 releases Bad from 14-3-3, we hypothesized that JNK might regulate the localization of Bad. To investigate this, we used Myc-tagged Bad (Myc-Bad). In all of the experiments with Myc-Bad, p35 was cotransfected to prevent caspase activity induced by the overexpression of Myc-Bad. In healthy cells, a large proportion of Myc-Bad localized in the cytoplasm, whereas a small proportion colocalized with MitoTracker CMXRos, as judged by immunocytochemical analysis (Fig. 3 a). We found that the expression of MKK7-JNK (WT), but not MKK7-JNK (KN), resulted in the translocation of Myc-Bad to the mitochondria. To confirm the effect of JNK on promoting Bad translocation, we examined the distribution of endogenous Bad by subcellular fractionation after transfection of COS-1 cells with MKK7-JNK constructs (Fig. 3 b). The amount of endogenous Bad that was detected in the mitochondrial fraction was increased by the expression of MKK7-JNK (WT). In contrast, the expression of MKK7-JNK (KN) did not have these effects. In this assay, the abundance of the mitochondrial marker F1F0-ATPase subunit α and that of the cytosolic marker α-tubulin in these fractions were unaffected by the expression of either construct. Although MKK7-JNK (WT) resulted in the activation of caspase-3 (Lei et al., 2002; Tsuruta et al., 2004), the coexpression of p35 failed to inhibit Bad translocation to the mitochondrial fraction, suggesting that MKK7-JNK (WT) promoted the translocation of endogenous Bad to mitochondria independently of caspase activation.
Figure 3.
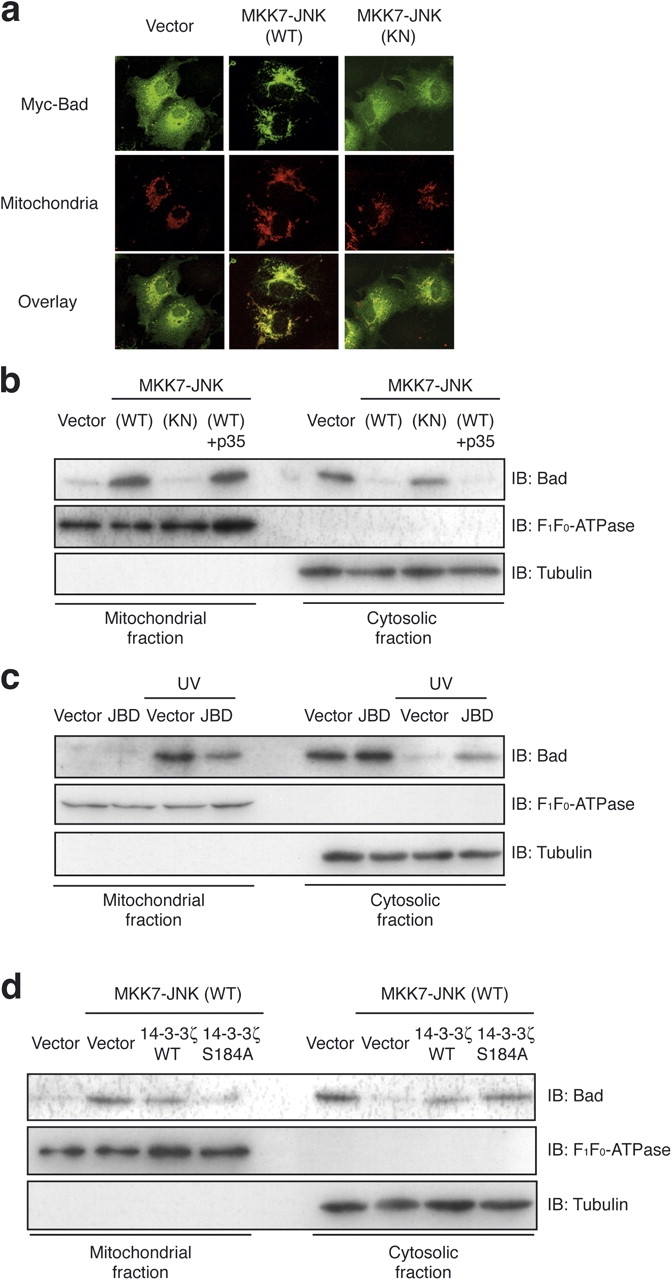
Active JNK promotes Bad translocation to the mitochondria. (a) COS-1 cells were transfected for 18 h with expression vectors for Myc-Bad and p35 together with a vector for MKK7-JNK as indicated. They were examined for the distribution of Myc-Bad (anti-Myc staining, green) and mitochondria (CMXRos, red) by fluorescence microscopy; the two separate images for each representative cell are shown superimposed (overlay). (b) COS-1 cells were transfected for 18 h with expression vectors for GFP and MKK7-JNK in the absence or presence of the vector for p35 as indicated. The transfection efficiency was ∼75% as determined by monitoring the expression of GFP. (c) HCT116 cells were transfected with expression vectors for indicated plasmids and were irradiated by 50 J/m2 UV for 6 h. (d) COS-1 cells were transfected for 18 h with expression vectors for GFP, MKK7-JNK (WT), and either 14-3-3ζ (WT) or Ser184A as indicated. (b–d) Then, the cells were subjected to subcellular fractionation, and the amount of endogenous Bad in the mitochondrial and cytosolic fractions was assessed by immunoblot analysis (IB) with antibodies to Bad. The mitochondrial marker F1F0-ATPase and the cytosolic marker α-tubulin were used as internal standards. The same results were obtained in three independent experiments.
We also determined whether UV induces Bad translocation to the mitochondria through the activation of endogenous JNK. The UV irradiation of HCT116 cells resulted in an increase in endogenous Bad in the mitochondrial fraction (Fig. 3 c). Moreover, the expression of JBD inhibited this translocation of Bad from the cytosol to the mitochondria (Fig. 3 c), supporting the idea that JNK mediates cellular UV-induced Bad translocation.
If JNK promotes Bad translocation to the mitochondria through phosphorylation of 14-3-3 proteins, the expression of a 14-3-3 mutant lacking the phosphorylation site should block JNK-induced Bad translocation because the unphosphorylated form of 14-3-3 should retain Bad in the cytoplasm. As expected, the coexpression of 14-3-3ζ Ser184A, and to a lesser extent of 14-3-3ζ (WT), inhibited MKK7-JNK (WT)–induced Bad translocation to the mitochondrial fraction (Fig. 3 d). These results strongly suggest that 14-3-3 is an essential target for JNK in promoting Bad translocation to the mitochondria.
JNK promotes the association of Bad with Bcl-2/Bcl-xL
The BH3-only subfamily of Bcl-2 family proteins, including Bad, has been postulated to promote apoptosis by antagonizing the antiapoptotic functions of Bcl-2/Bcl-xL (Yang et al., 1995; Danial and Korsmeyer, 2004). Because we found that active JNK can induce Bad translocation to the mitochondria through phosphorylation of 14-3-3, we next asked whether JNK also induces Bad association with Bcl-2/Bcl-xL. When endogenous Bad in healthy cells was immunoprecipitated with anti-Bad antibody, small amounts of Bcl-2/Bcl-xL were coimmunoprecipitated (Fig. 4 a). In contrast, when MKK7-JNK (WT), but not MKK7-JNK (KN), was expressed in these cells, the amounts of endogenous Bcl-2/Bcl-xL that was associated with endogenous Bad greatly increased, corresponding to the reduction of 14-3-3 associated with Bad (Fig. 4 a). If JNK promotes the association of Bad with Bcl-2/Bcl-xL through phosphorylation of 14-3-3, the expression of 14-3-3ζ Ser184A should block the JNK-induced interaction between Bad and Bcl-2/Bcl-xL. Indeed, the expression of 14-3-3ζ Ser184A inhibited the increase of Bad protein that interacted with Bcl-2/Bcl-xL (Fig. 4 b). Altogether, these results suggest that the activation of JNK induces the activation of Bad at the mitochondria through phosphorylation of 14-3-3.
Figure 4.

Expression of active JNK promotes association of Bad with Bcl-2/Bcl-xL and regulates the phosphorylation state of Bad. (a and b) COS-1 cells were transfected for 18 h with expression vectors for MKK7-JNK and p35 (a) or for Myc–14-3-3ζ (WT or Ser184A) and MKK7-JNK (b) as indicated and were subjected either to immunoblot analysis (IB) with indicated antibodies or to immunoprecipitation (IP) with antibodies to Bad; the resulting precipitates were subjected to immunoblot analysis with indicated antibodies. (b) The asterisk indicates the position of Myc-tagged 14-3-3ζ. (c) COS-1 cells were transfected for the indicated times with expression vectors for MKK7-JNK. (d) COS-1 cells were transfected for 12 h with the indicated plasmids and were incubated for 6 h in the absence or presence of 500 nM okadaic acid (OA). (c and d) Cell lysates were subjected to immunoprecipitation with antibodies to Bad, and the resulting precipitates were subjected to immunoblot analysis with indicated antibodies. (e) COS-1 cells were transfected for 18 h with expression vectors for MKK7-JNK (WT) and 14-3-3ζ Ser184A. Cell lysates were subjected to immunoblot analysis as indicated. The same results were obtained in two independent experiments.
JNK regulates the phosphorylation state of Bad through 14-3-3
Although Bad is released from 14-3-3 by JNK-mediated 14-3-3 phosphorylation, this release by itself should not be sufficient for its association in a complex of Bcl-2/Bcl-xL because phosphorylation of Bad at Ser112, 136, or 155 has been shown to hamper its association with Bcl-2/Bcl-xL (Zha et al., 1996; Datta et al., 2000; Zhou et al., 2000). The finding that activation of JNK results in the association of Bad with Bcl-2/Bcl-xL prompted us to examine the phosphorylation state of Bad in cells expressing active JNK. Our most important finding was that the expression of MKK7-JNK (WT) resulted in a gradual reduction in the level of Bad phosphorylation at Ser112 and 136 (Fig. 4 c). The dissociation between 14-3-3 and Bad started to take place 16 h after the transfection of MKK7-JNK (WT), whereas the reduction of Bad phosphorylation was evident at ∼18 h after transfection (Fig. 4 c). Phosphorylation of Bad at Ser155, which is a key site inhibiting Bcl-2/Bcl-xL binding, was also reduced by the expression of MKK7-JNK (WT; Fig. S2 a, available at http://www.jcb.org/cgi/content/full/jcb.200409117.DC1). We do not believe that JNK causes dephosphorylation of Bad by inactivating kinases that are responsible for Bad phosphorylation, because we observed that the expression of MKK7-JNK (WT) did not reduce the activity of Akt or Rsk, two major kinases responsible for Bad phosphorylation, under conditions where it reduced Bad phosphorylation (Fig. S2 b).
Therefore, JNK appears to induce both the release of Bad from 14-3-3 and the dephosphorylation of Bad. Is there any causal relationship between these two events? To examine this, we treated COS-1 cells with the phosphatase inhibitor okadaic acid (OA) to prevent dephosphorylation of Bad at Ser112 and 136 (Fig. 4 d). We confirmed that OA maintained the level of Bad phosphorylation, yet we observed that the expression of MKK7-JNK (WT) still reduced the amount of 14-3-3 that is associated with Bad (Fig. 4 d). This suggests that dephosphorylation of Bad is not a prerequisite for the JNK-mediated dissociation of Bad from 14-3-3. On the other hand, the expression of 14-3-3ζ Ser184A suppressed Bad dephosphorylation at Ser112 and 136 (Fig. 4 e). This strongly supports the notion that Bad release from 14-3-3 is necessary for Bad dephosphorylation after JNK activation. Because Chiang et al. (2003) has shown that 14-3-3 and protein phosphatase 2A compete for Bad and that 14-3-3 maintains the phosphorylated state of Bad, it is conceivable that the JNK-mediated release from 14-3-3 makes Bad accessible to phosphatases and that it is dephosphorylated as a result.
JNK-mediated 14-3-3 phosphorylation regulates Bad-dependent cell death
We next examined the relative contribution of the JNK-induced phosphorylation of 14-3-3 to cell death regulation in a cellular context in which cell death is dependent on Bad and is counteracted by Akt. In HCT116 cells, UV-induced apoptosis, which is dependent on JNK (Fig. 5, a and b), and MKK7-JNK (WT)–induced apoptosis were suppressed by small interference RNA (siRNA) against Bad (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200409117.DC1), suggesting that they are dependent on endogenous Bad, at least in part. It is important to note that the expression of 14-3-3σ Ser186A, and to a lesser extent the expression of 14-3-3σ (WT), suppressed both UV-induced apoptosis (Fig. 5, c and d) and MKK7-JNK (WT)–induced apoptosis (Tsuruta et al., 2004). The ectopic expression of a low level of Bad caused enhanced UV-induced apoptosis; and, again, this enhancement was suppressed, in part, by the expression of 14-3-3σ Ser186A and, to a lesser extent, by the expression of 14-3-3σ (WT; Fig. 5 e). These results clearly indicate that 14-3-3 suppresses Bad-mediated apoptosis in a phosphorylation-dependent manner.
In HCT116 cells, the survival-promoting effects of serum appear to be mediated by the Akt pathway because the expression of DN-Akt abrogated these effects (Fig. 6 a). The survival-promoting effects of serum and CA-Akt were suppressed by MKK7-JNK (WT), whereas the apoptosis-promoting effects of MKK7-JNK (WT) were suppressed by serum and CA-Akt (Fig. 6, a and b), indicating antagonistic interactions between JNK and Akt in this cell line. The enhanced apoptosis caused by serum deprivation or the expression of DN-Akt was in part suppressed by siRNA against Bad (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200409117.DC1). Importantly, the expression of 14-3-3σ Ser186A also suppressed apoptosis that was induced by serum deprivation and DN-Akt and did so more effectively than 14-3-3σ (WT; Fig. 6, c and d). These results support the notion that 14-3-3 suppresses Bad-mediated apoptosis in a phosphorylation-dependent manner in a context in which serum promotes cell survival via the Akt pathway.
Figure 6.

Antagonistic interaction between Akt and JNK in the regulation of cell death. (a) HCT116 cells were transfected with expression vectors for GFP, MKK7-JNK (WT), and Akt mutants and were incubated with or without 10% serum for 6 h. Then, they were stained with Hoechst 33342 and were subjected to cell death assay by the detection of chromatin condensation. The right panel shows that endogenous Akt was dephosphorylated 6 h after serum deprivation. At least 540 cells (a) and 480 cells (b) per sample were analyzed (*, P < 0.01 and **, P < 0.04). (b) HCT116 cells were transfected with expression vectors for GFP, CA-Akt, and the indicated amounts of MKK7-JNK (WT) and were incubated with or without 10% serum for 6 h. (c and d) HCT116 cells were transfected with the indicated plasmids together with a GFP-expressing plasmid for 20 h. Then, they were incubated with or without 10% serum for 8 h (d) when necessary and were subjected to cell death assay by the detection of chromatin condensation. (c) *, P < 0.05; (d) *, P < 0.001; and **, P < 0.04. At least 460 cells (c) and 640 cells (d) per sample were analyzed. (a–d) Data are means ± SD (error bars) of values obtained from three fields of 150–200 cells in each of three independent experiments.
JNK-mediated 14-3-3 phosphorylation affects another 14-3-3 ligand: FOXO3a
As mentioned previously, many 14-3-3 ligands bind to the common ligand-binding groove of 14-3-3 and share 14-3-3–binding motifs. It is likely, therefore, that JNK-mediated phosphorylation results in the dissociation of 14-3-3 ligands other than just Bad. To test this idea, we examined whether the JNK-mediated phosphorylation of 14-3-3 releases FOXO3a from 14-3-3 (Fig. S5, available at http://www.jcb.org/cgi/content/full/jcb.200409117.DC1), as Akt inactivates the proapoptotic protein FOXO3a by direct phosphorylation, inducing its association with 14-3-3 (Brunet et al., 1999). In an in vitro reconstitution assay, Akt-mediated phosphorylation increased the amount of GST-FOXO3a that was coprecipitated with His–14-3-3ζ. However, phosphorylation of 14-3-3ζ by MKK7-JNK (WT) reduced the amount of FOXO3a that was coprecipitated with 14-3-3ζ even when FOXO3a was phosphorylated by Akt. On the other hand, when the Ser184A mutant of His–14-3-3ζ was used, similar amounts of FOXO3a were precipitated with or without preincubation with MKK7-JNK (WT), suggesting that direct 14-3-3ζ phosphorylation at Ser184 by JNK reduces its affinity for FOXO3a. Although we found that the expression of FOXO3a promoted apoptosis, which was suppressed by the expression of 14-3-3 in a Ser184-dependent manner, the down-regulation of endogenous FOXO3a expression by RNA interference (RNAi) did not affect apoptosis that was induced by UV treatment in our preliminary experiments (unpublished data). Therefore, we think that FOXO3a may not be a key mediator in UV-induced apoptosis in this cell line, although this result does not rule out the possible existence of proapoptotic factors that function redundantly with FOXO3a in this system. In fact, several 14-3-3–binding proteins in extracts that were prepared from [35S]methionine-labeled COS-1 cells were dissociated from GST–14-3-3ζ when subjected to the JNK-mediated phosphorylation of GST–14-3-3ζ at Ser184 in an in vitro GST pull-down assay (Fig. 7). This suggests that JNK regulates these 14-3-3–binding proteins in concert.
Figure 7.
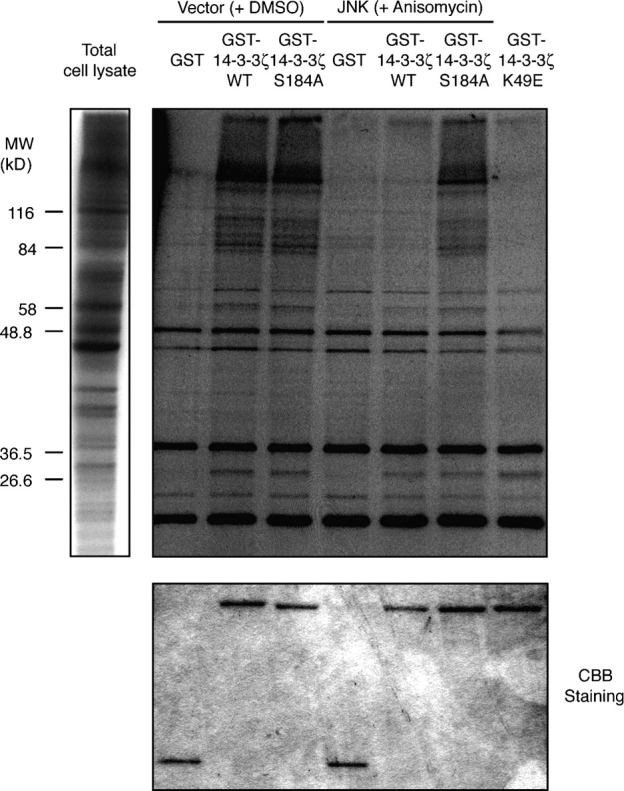
Phosphorylation of 14-3-3ζ at Ser184 by JNK releases a subset of 14-3-3ζ–binding proteins. COS-1 cells were transfected for 12 h with either an expression vector for Flag-tagged MKK7-JNK (WT) or control vector and were incubated for 1 h in the presence of either 10 μg/ml anisomycin or DMSO. Flag-tagged MKK7-JNK (WT) was immunoprecipitated from cell lysates with antibodies to Flag and were incubated with recombinant GST, GST–14-3-3ζ (WT), or Ser184A in the presence of ATP for 45 min. These GST proteins were incubated for 2 h with [35S]methionine-labeled COS-1 cell lysates. The GST proteins were precipitated using glutathione–Sepharose beads, and the bound proteins were resolved by 10% SDS-PAGE and were detected by autoradiography and by staining with Coomassie brilliant blue (CBB). Essentially the same results were obtained in at least two independent experiments. In control experiments (vector + DMSO), many proteins were found to be associated with GST–14-3-3ζ (WT) and GST–14-3-3ζ Ser184A, but not with GST alone. A subset of these proteins did not associate with JNK-phosphorylated GST–14-3-3ζ (WT). This dissociation appears to be dependent on the phosphorylation of 14-3-3ζ at Ser184 because GST–14-3-3ζ Ser184A associated with almost the same set of proteins regardless of whether it was phosphorylated by JNK or not. The proteins that dissociated from GST–14-3-3ζ (WT) upon JNK phosphorylation also did not bind to the GST–14-3-3ζ mutant (K49E), which harbors a point mutation within the common ligand-binding groove. This strongly supports the notion that JNK phosphorylation of 14-3-3ζ at Ser184 releases typical 14-3-3 ligands.
Discussion
14-3-3: an integration point of survival and apoptotic signals
In this study, we described a novel mechanism by which the JNK signaling pathway regulates cell death. We found that the JNK-mediated phosphorylation of 14-3-3 releases Bad from 14-3-3 and induces its translocation to mitochondria and its association with Bcl-2/Bcl-xL, which presumably results in their inactivation. It is important to note that this phosphorylation also leads to the release of several 14-3-3 ligands from 14-3-3, including the proapoptotic protein FOXO3a. Because many proapoptotic proteins share a common 14-3-3–binding motif (RSXpSXP or RXXXpSXP) and the JNK phosphorylation site in 14-3-3 resides in the vicinity of its ligand-binding groove, it is likely that JNK phosphorylation affects this ligand-binding site, leading to the release and consequent activation of these proapoptotic proteins through a common mechanism. Our results support the notion that the JNK-mediated phosphorylation of 14-3-3 affects the activities of a broad range of 14-3-3 ligands, ultimately leading to the onset of the cell death program.
Recent studies have shown that JNK directly phosphorylates Bad at Ser128 and Thr201 (Donovan et al., 2002; Konishi et al., 2002; Yu et al., 2004). Phosphorylation of the former residue reduces the affinity of Bad for 14-3-3 and activates Bad, whereas phosphorylation of the latter reduces the affinity of Bad for Bcl-2/Bcl-xL and inactivates Bad. We found that the JNK-mediated phosphorylation of 14-3-3 reduced its interaction with WT and Ser128A Bad proteins to a similar extent. This suggests that JNK can cause the release of Bad from 14-3-3 independently of phosphorylation at Bad Ser128. However, it is possible that these two JNK-mediated phosphorylations (on Bad and 14-3-3) act in parallel or in a concerted fashion to effectively activate Bad. Thr201, on the other hand, exists in mouse Bad but not in human Bad and may not be generally involved in Bad regulation.
There appears to be some threshold below which apoptotic signaling can occur but cannot lead to cell death. In this context, 14-3-3 can function to raise this hypothetical threshold by acting as a “buffer” that sequesters several proapoptotic proteins in response to survival signals, including those mediated by Akt. This is also consistent with the observation that increasing or decreasing the amount of available 14-3-3 can shift the balance between survival and apoptosis, in effect raising or lowering the threshold for apoptotic signaling. There would be at least three ways to override the threshold set by 14-3-3. The first and probably the simplest way is to increase the total amount of proapoptotic proteins so that they exceed the threshold level. The second way is to reduce the amount of survival signals, which would result in dephosphorylation and release of proapoptotic proteins from 14-3-3 (Fig. 8 b). The third way is to reduce the level of available 14-3-3 by the JNK-mediated phosphorylation of 14-3-3, resulting in the release of proapoptotic proteins (Fig. 8 c). In this scenario, JNK functions to lower this hypothetical threshold, thus rendering cells more susceptible to apoptotic signals.
Figure 8.

A model for antagonism between Akt (survival) and JNK (apoptotic) signals via 14-3-3 proteins. (a) Exposure of a cell to survival signals results in inhibiting apoptosis by the 14-3-3–mediated sequestration of Akt-phosphorylated proapoptotic proteins such as Bad and FOXO3a. (b) Reduced survival signals promote dephosphorylation of proapoptotic proteins, resulting in the release of these proteins from 14-3-3 and, thus, leading to cell death. (c) Potent (sustained) apoptotic signals promote JNK-mediated phosphorylation of 14-3-3, resulting in the release of proapoptotic proteins from 14-3-3 and cell death. P shown in pink and blue circles represents Akt- and JNK-mediated phosphorylation, respectively.
As mentioned previously, the balance between apoptosis and survival signals is essential for understanding cell death regulation. We propose that 14-3-3 may serve as a key integration point of both JNK-mediated stress signals and Akt-mediated survival signals, and thus contributes to the decision whether a cell should live or die.
Materials and methods
Materials
Anisomycin was obtained from Sigma-Aldrich, OA was obtained from Calbiochem, and CMXRos was obtained from Molecular Probes.
Cell culture and transfection
COS-1 cells were maintained in DME (Sigma-Aldrich) supplemented with 10% FBS and 1% penicillin/streptomycin. HCT116 cells were maintained in McCoy's 5A Medium (Sigma-Aldrich) with 10% FBS, 1% penicillin/streptomycin, and 1.5 mM l-glutamine. COS-1 cells were transfected with plasmids by the use of Fugene6 (Roche Diagnostics), and HCT116 cells were transfected with LipofectAMINE 2000 (Invitrogen).
Plasmid construction
The constructs encoding mice Bad (full length) and FOXO3a (aa 1–525) were provided by M.E. Greenberg (Harvard Medical School, Boston, MA). We obtained the constructs encoding 14-3-3ζ from H. Fu (Emory University School of Medicine, Atlanta, GA) and obtained 14-3-3σ from B. Vogelstein (The Johns Hopkins University School of Medicine, Baltimore, MD). For the Myc-tagged Bad expression vector, the Bad cDNA fragment was inserted into the BglII site of pCS4-Myc. Site-directed mutagenesis was performed with the QuikChange kit (Stratagene) to generate the Ser128 and Thr201 into Ala change in Bad and to generate the Lys49 into Glu change in 14-3-3ζ. WT, Ser128A, Thr201A, and Ser128A-Thr201A Bad cDNA were cloned into the BamHI sites of pET28a (Novagen), and K49E 14-3-3ζ cDNA was cloned into the BglII site of pCS4-Myc. pcDNA3-MKK7-FLAG-JNK (WT and KN), pET28a-MKK7-FLAG-JNK (WT and KN), pGEX6P-1–14-3-3ζ (WT and Ser184A), pCS4-Myc–14-3-3ζ (WT and Ser184A), pCS4-Myc–14-3-3σ (WT and Ser186A), the constructs encoding p35 (a gift from M. Miura, University of Tokyo, Tokyo, Japan), CA-Akt, DN-Akt, WT-Akt, DN-JNK, and DN-JBD (a gift from R. Davis, University of Massachusetts Medical School, Worcester, MA) have been described previously (Masuyama et al., 2001; Tsuruta et al., 2002, 2004).
Antibodies
Antibodies to 14-3-3 (K-19; Santa Cruz Biotechnology, Inc.), 14-3-3σ (C-18; Santa Cruz Biotechnology, Inc.), Bad (C-20; Santa Cruz Biotechnology, Inc.), phospho-Bad (Ser112; Cell Signaling), phospho-Bad (Ser136; Cell Signaling), Bcl-2 (N-19; Santa Cruz Biotechnology, Inc.), Bcl-xL (S-18; Santa Cruz Biotechnology, Inc.), cleaved caspase-3 (Cell Signaling), F1F0-ATPase subunit α (7H10; Molecular Probes), α-tubulin (DM1A; Sigma-Aldrich), GAPDH (MAB374; Chemicon), GST (B-14; Santa Cruz Biotechnology, Inc.), c-Jun (Cell Signaling), phospho–c-Jun (Cell Signaling), Akt (Cell Signaling), phospho-Akt (Ser473; 58F11; Cell Signaling), Rsk (Upstate Biotechnology), or phospho-p90Rsk (Thr359-Ser363; Cell Signaling) were used for immunoblot analysis. An antibody to phospho-Bad (Ser155) was provided by M.E. Greenberg (Datta et al., 2000). Antibodies to Bad (C-20; Santa Cruz Biotechnology, Inc.), 14-3-3σ (C-18; Santa Cruz Biotechnology, Inc.), HA (Y-11; Santa Cruz Biotechnology, Inc.), Flag (M2; Sigma-Aldrich), normal rabbit IgG (Santa Cruz Biotechnology, Inc.), or normal goat IgG (Santa Cruz Biotechnology, Inc.) were used for immunoprecipitation.
Immunoblot analysis
Cells were washed with PBS and were lysed in an extraction buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10 mM β-glycerophosphate, 5 mM EGTA, 1 mM sodium pyrophoshate, 5 mM NaF, 1 mM Na3VO4, 0.5% Triton X-100, and 1 mM DTT) supplemented with protease inhibitors (1 mM PMSF, 5 μg/ml leupeptin, 5 μg/ml pepstatin A, and 5 μg/ml aprotinin). Proteins were separated by SDS-PAGE and were electrically transferred to a polyvinylidene difluoride membrane. The membrane was probed with the appropriate primary antibody and with an HRP-conjugated secondary antibody. Blots were visualized by Western Lightning (PerkinElmer).
Protein purification
His6-tagged Bad (WT, Ser128A, Thr201A, or Ser128A-Thr201A) was expressed in Escherichia coli BL21 by pET28a-based vectors. After IPTG was added, induction occurred; and His-tagged proteins were purified with Ni2+-bound column (ProBond resin; Invitrogen). The purification of GST–14-3-3ζ, GST-FOXO3a (aa 1–525), and His6-tagged MKK7-JNK was performed as described previously (Brunet et al., 1999; Tsuruta et al., 2004).
Subcellular fractionation
Cells were washed with PBS, suspended in 300 μl of ice-cold isotonic buffer (200 mM mannitol, 70 mM sucrose, 1 mM EDTA, 10 mM Hepes-NaOH, pH 7.4, and 1 mM DTT) supplemented with the protease inhibitors described above, and homogenized with a Potter-Elvehjem homogenizer (model Mazela Z; Eyela). Nuclei and unbroken cells were removed by centrifugation at 500 g for 10 min, and the supernatant was further centrifuged at 100,000 g for 60 min. The resulting supernatant was saved as the cytosolic fraction, and the pellet was washed with isotonic buffer, resuspended in extraction buffer supplemented with protease inhibitors, and centrifuged at 20,000 g for 5 min to remove debris. The resulting supernatant was saved as the mitochondrial fraction.
Coimmunoprecipitation assay
COS-1 cells were lysed in lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10 mM β-glycerophosphate, 5 mM EGTA, 1 mM sodium pyrophoshate, 5 mM NaF, 1 mM Na3VO4, 0.05% Triton X-100, and 1 mM DTT). The lysates were then incubated with antibodies to Bad (C-20) for 1 h and were subsequently incubated with protein A–Sepharose beads (GE Healthcare) for 1 h. HCT116 cells were lysed in the lysis buffer described above, and the lysates were precleared with protein G–Sepharose beads (GE Healthcare). The lysates were incubated with antibodies to 14-3-3σ (Santa Cruz Biotechnology, Inc.) and with protein G–Sepharose beads for 2 h. The protein–antibody complexes that were recovered on beads was subjected to immunoblot analysis after separation by SDS-PAGE.
GST pull-down assay
Recombinant GST–14-3-3ζ (WT or Ser184A) was incubated with or without purified MKK7-JNK (WT or KN) in the presence of a kinase reaction buffer (100 μM ATP, 20 mM Tris-HCl, pH 7.5, and 15 mM MgCl2) for 30 min at 30°C. His6-Bad (WT, Ser128A, Thr201A, or Ser128A-Thr201A) protein was preincubated with or without active Akt immunoprecipitate in a kinase reaction buffer. Subsequently, Bad protein was mixed with GST–14-3-3ζ and glutathione–Sepharose 4B beads for 1 h at 4°C, and the bead-bound proteins were subjected to immunoblot analysis with antibodies to Bad and GST.
Immunofluorescence analysis
COS-1 cells were grown on poly-d-lysine–coated coverslips and were transfected with a plasmid expressing Myc-tagged Bad and p35 and/or expression plasmids encoding MKK7-JNK proteins. Cells were fixed in PBS containing 4% PFA for 10 min at RT. The fixed coverslips were permeabilized in PBS containing 0.5% Triton X-100 for 10 min, washed twice in PBS, and incubated in a blocking solution (PBS containing 2% BSA) for 30 min. The cells were then incubated in the blocking solution with anti-Myc antibody (9E10; Santa Cruz Biotechnology, Inc.) for 1 h and with AlexaFluor488 anti–mouse IgG antibody (Molecular Probes) for 1 h in the blocking solution. Where indicated, cells were stained with CMXRos before fixation to visualize mitochondria. Fluorescence images were recorded by a confocal laser-scanning microscope (model LSM-510; Carl Zeiss MicroImaging, Inc.). All images were captured at 40× by using objective lenses (1.4 NA; C-Apochromat). Pictures were analyzed by using LSM5 Image Browser (Carl Zeiss MicroImaging, Inc.).
RNAi experiment
The siRNA for human Bad mRNA was obtained from Japan Bio Services Co., Ltd. The siRNA sequences used in this study for Bad were sense (5′-UGAGUGACGAGUUUGUGGAdTdT-3′) and antisense (5′-UCCACAAACUCGUCACUCAdTdT-3′). The 5′ terminus of the sense sequence was labeled with Texas red, and the sequences were randomized for the control siRNA (sense, 5′-AGUUCGAUUUUAGGGGGGAdTdT-3′; antisense, 5′-UCCCCCCUAAAAUCGAACUdTdT-3′). Transfection to HCT116 cells was performed using Oligofectamine reagent (Invitrogen) according to the manufacturer's instructions except that the incubation time with transfection reagents was 120 min. The transfection efficiency was >95%, as assessed by Texas red.
Apoptosis assay
HCT116 cells were seeded into 60-mm culture dishes and were transfected with a plasmid expressing GFP together with expression plasmids encoding JBD, Bad, MKK7-JNK (WT), and various Akt or 14-3-3 proteins. After 24 h, cells were irradiated by UV for 6 h when necessary in the presence of 10% serum for 6 h. After staining with Hoechst 33342 for 10 min, the percentage of GFP-positive cells with a pyknotic nucleus was determined. For the RNAi experiments, 2.5 × 105 cells were seeded into 35-mm dishes and were transfected with 100 pmol siRNA. After 36 h, cells were transfected with 0.625 μg of expression vectors for GFP, MKK7-JNK (WT), or various Akt proteins or, after 48 h, cells were irradiated by UV. Then, they were stained with Hoechst 33342.
Statistical analysis
Results were expressed as the mean ± SD and were analyzed by using the unpaired t test.
Online supplemental material
Fig. S1 shows that the JNK-mediated phosphorylation of 14-3-3 can induce the dissociation of Bad regardless of the Ser128 and Thr201 phosphorylation of Bad in vitro. Fig. S2 shows that the expression of active JNK results in the reduction of Bad phosphorylation at Ser112, 136, and 155 without inhibiting phosphorylation of Akt and Rsk. Fig. S3 shows that UV- and active JNK-induced cell death is partially dependent on Bad. Fig. S4 shows that the introduction of siRNA against Bad suppressed cell death, which is induced by serum deprivation or DN-Akt. Fig. S5 shows that the JNK-mediated phosphorylation of 14-3-3ζ results in the release of FOXO3a from 14-3-3ζ in vitro. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200409117.DC1.
Acknowledgments
We thank Drs. M. Miura, H. Fu, B. Vogelstein, R. Davis, and M.E. Greenberg for providing reagents. We also thank Dr. Marc Lamphier for reading the manuscript and members of the Gotoh laboratory for helpful discussions.
This work was supported by Grants-in-Aid from the Ministry of Education, Science, Sports and Culture of Japan and by Precursory Research for Embryonic Science and Technology 21 of the Japan Science and Technology Corporation.
Abbreviations used in this paper: CA, constitutively active; DN, dominant negative; JBD, JNK-binding domain; JNK, c-Jun NH2-terminal kinase; KN, kinase negative; OA, okadaic acid, siRNA, small interference RNA; RNAi, RNA interference; WT, wild type.
References
- Aho, T.L., J. Sandholm, K.J. Peltola, H.P. Mankonen, M. Lilly, and P.J. Koskinen. 2004. Pim-1 kinase promotes inactivation of the pro-apoptotic Bad protein by phosphorylating it on the Ser112 gatekeeper site. FEBS Lett. 571:43–49. [DOI] [PubMed] [Google Scholar]
- Aitken, A., S. Howell, D. Jones, J. Madrazo, and Y. Patel. 1995. 14-3-3 alpha and delta are the phosphorylated forms of raf-activating 14-3-3 beta and zeta. In vivo stoichiometric phosphorylation in brain at a Ser-Pro-Glu-Lys MOTIF. J. Biol. Chem. 270:5706–5709. [DOI] [PubMed] [Google Scholar]
- Basu, S., N.F. Totty, M.S. Irwin, M. Sudol, and J. Downward. 2003. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol. Cell. 11:11–23. [DOI] [PubMed] [Google Scholar]
- Biggs, W.H., III, J. Meisenhelder, T. Hunter, W.K. Cavenee, and K.C. Arden. 1999. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl. Acad. Sci. USA. 96:7421–7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume-Jensen, P., R. Janknecht, and T. Hunter. 1998. The kit receptor promotes cell survival via activation of PI 3-kinase and subsequent Akt-mediated phosphorylation of Bad on Ser136. Curr. Biol. 8:779–782. [DOI] [PubMed] [Google Scholar]
- Bonni, A., A. Brunet, A.E. West, S.R. Datta, M.A. Takasu, and M.E. Greenberg. 1999. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 286:1358–1362. [DOI] [PubMed] [Google Scholar]
- Brunet, A., A. Bonni, M.J. Zigmond, M.Z. Lin, P. Juo, L.S. Hu, M.J. Anderson, K.C. Arden, J. Blenis, and M.E. Greenberg. 1999. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 96:857–868. [DOI] [PubMed] [Google Scholar]
- Chan, T.A., H. Hermeking, C. Lengauer, K.W. Kinzler, and B. Vogelstein. 1999. 14-3-3Sigma is required to prevent mitotic catastrophe after DNA damage. Nature. 401:616–620. [DOI] [PubMed] [Google Scholar]
- Chiang, C.W., C. Kanies, K.W. Kim, W.B. Fang, C. Parkhurst, M. Xie, T. Henry, and E. Yang. 2003. Protein phosphatase 2A dephosphorylation of phosphoserine 112 plays the gatekeeper role for BAD-mediated apoptosis. Mol. Cell. Biol. 23:6350–6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotteret, S., Z.M. Jaffer, A. Beeser, and J. Chernoff. 2003. p21-Activated kinase 5 (Pak5) localizes to mitochondria and inhibits apoptosis by phosphorylating BAD. Mol. Cell. Biol. 23:5526–5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial, N.N., and S.J. Korsmeyer. 2004. Cell death: critical control points. Cell. 116:205–219. [DOI] [PubMed] [Google Scholar]
- Datta, S.R., H. Dudek, X. Tao, S. Masters, H. Fu, Y. Gotoh, and M.E. Greenberg. 1997. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 91:231–241. [DOI] [PubMed] [Google Scholar]
- Datta, S.R., A. Katsov, L. Hu, A. Petros, S.W. Fesik, M.B. Yaffe, and M.E. Greenberg. 2000. 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol. Cell. 6:41–51. [PubMed] [Google Scholar]
- Datta, S.R., A.M. Ranger, M.Z. Lin, J.F. Sturgill, Y.C. Ma, C.W. Cowan, P. Dikkes, S.J. Korsmeyer, and M.E. Greenberg. 2002. Survival factor-mediated BAD phosphorylation raises the mitochondrial threshold for apoptosis. Dev. Cell. 3:631–643. [DOI] [PubMed] [Google Scholar]
- del Peso, L., M. Gonzalez-Garcia, C. Page, R. Herrera, and G. Nunez. 1997. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 278:687–689. [DOI] [PubMed] [Google Scholar]
- Deng, Y., X. Ren, L. Yang, Y. Lin, and X. Wu. 2003. A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell. 115:61–70. [DOI] [PubMed] [Google Scholar]
- Dickens, M., J.S. Rogers, J. Cavanagh, A. Raitano, Z. Xia, J.R. Halpern, M.E. Greenberg, C.L. Sawyers, and R.J. Davis. 1997. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science. 277:693–696. [DOI] [PubMed] [Google Scholar]
- Donovan, N., E.B. Becker, Y. Konishi, and A. Bonni. 2002. JNK phosphorylation and activation of BAD couples the stress-activated signaling pathway to the cell death machinery. J. Biol. Chem. 277:40944–40949. [DOI] [PubMed] [Google Scholar]
- Harada, H., B. Becknell, M. Wilm, M. Mann, L.J. Huang, S.S. Taylor, J.D. Scott, and S.J. Korsmeyer. 1999. Phosphorylation and inactivation of BAD by mitochondria-anchored protein kinase A. Mol. Cell. 3:413–422. [DOI] [PubMed] [Google Scholar]
- Harada, H., J.S. Andersen, M. Mann, N. Terada, and S.J. Korsmeyer. 2001. p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc. Natl. Acad. Sci. USA. 98:9666–9670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi, Y., M. Lehtinen, N. Donovan, and A. Bonni. 2002. Cdc2 phosphorylation of BAD links the cell cycle to the cell death machinery. Mol. Cell. 9:1005–1016. [DOI] [PubMed] [Google Scholar]
- Kops, G.J., N.D. de Ruiter, A.M. De Vries-Smits, D.R. Powell, J.L. Bos, and B.M. Burgering. 1999. Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature. 398:630–634. [DOI] [PubMed] [Google Scholar]
- Kuan, C.Y., A.J. Whitmarsh, D.D. Yang, G. Liao, A.J. Schloemer, C. Dong, J. Bao, K.J. Banasiak, G.G. Haddad, R.A. Flavell, et al. 2003. A critical role of neural-specific JNK3 for ischemic apoptosis. Proc. Natl. Acad. Sci. USA. 100:15184–15189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei, K., A. Nimnual, W.X. Zong, N.J. Kennedy, R.A. Flavell, C.B. Thompson, D. Bar-Sagi, and R.J. Davis. 2002. The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH(2)-terminal kinase. Mol. Cell. Biol. 22:4929–4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W.Q., Q. Jiang, A.R. Khaled, J.R. Keller, and S.K. Durum. 2004. Interleukin-7 inactivates the pro-apoptotic protein Bad promoting T cell survival. J. Biol. Chem. 279:29160–29166. [DOI] [PubMed] [Google Scholar]
- Liu, D., J. Bienkowska, C. Petosa, R.J. Collier, H. Fu, and R. Liddington. 1995. Crystal structure of the zeta isoform of the 14-3-3 protein. Nature. 376:191–194. [DOI] [PubMed] [Google Scholar]
- Lizcano, J.M., N. Morrice, and P. Cohen. 2000. Regulation of BAD by cAMP-dependent protein kinase is mediated via phosphorylation of a novel site, Ser155. Biochem. J. 349:547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters, S.C., and H. Fu. 2001. 14-3-3 proteins mediate an essential anti-apoptotic signal. J. Biol. Chem. 276:45193–45200. [DOI] [PubMed] [Google Scholar]
- Masters, S.C., R.R. Subramanian, A. Truong, H. Yang, K. Fujii, H. Zhang, and H. Fu. 2002. Survival-promoting functions of 14-3-3 proteins. Biochem. Soc. Trans. 30:360–365. [DOI] [PubMed] [Google Scholar]
- Masuyama, N., K. Oishi, Y. Mori, T. Ueno, Y. Takahama, and Y. Gotoh. 2001. Akt inhibits the orphan nuclear receptor Nur77 and T-cell apoptosis. J. Biol. Chem. 276:32799–32805. [DOI] [PubMed] [Google Scholar]
- Muslin, A.J., and H. Xing. 2000. 14-3-3 proteins: regulation of subcellular localization by molecular interference. Cell. Signal. 12:703–709. [DOI] [PubMed] [Google Scholar]
- Muslin, A.J., J.W. Tanner, P.M. Allen, and A.S. Shaw. 1996. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell. 84:889–897. [DOI] [PubMed] [Google Scholar]
- Nomura, M., S. Shimizu, T. Sugiyama, M. Narita, T. Ito, H. Matsuda, and Y. Tsujimoto. 2003. 14-3-3 interacts directly with and negatively regulates pro-apoptotic Bax. J. Biol. Chem. 278:2058–2065. [DOI] [PubMed] [Google Scholar]
- Samuel, T., H.O. Weber, P. Rauch, B. Verdoodt, J.T. Eppel, A. McShea, H. Hermeking, and J.O. Funk. 2001. The G2/M regulator 14-3-3sigma prevents apoptosis through sequestration of Bax. J. Biol. Chem. 276:45201–45206. [DOI] [PubMed] [Google Scholar]
- Schurmann, A., A.F. Mooney, L.C. Sanders, M.A. Sells, H.G. Wang, J.C. Reed, and G.M. Bokoch. 2000. p21-activated kinase 1 phosphorylates the death agonist bad and protects cells from apoptosis. Mol. Cell. Biol. 20:453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura, A., B.A. Ballif, S.A. Richards, and J. Blenis. 2000. Rsk1 mediates a MEK-MAP kinase cell survival signal. Curr. Biol. 10:127–135. [DOI] [PubMed] [Google Scholar]
- Tan, Y., H. Ruan, M.R. Demeter, and M.J. Comb. 1999. p90(RSK) blocks bad-mediated cell death via a protein kinase C-dependent pathway. J. Biol. Chem. 274:34859–34867. [DOI] [PubMed] [Google Scholar]
- Tournier, C., P. Hess, D.D. Yang, J. Xu, T.K. Turner, A. Nimnual, D. Bar-Sagi, S.N. Jones, R.A. Flavell, and R.J. Davis. 2000. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 288:870–874. [DOI] [PubMed] [Google Scholar]
- Tsuruta, F., N. Masuyama, and Y. Gotoh. 2002. The phosphatidylinositol 3-kinase (PI3K)-Akt pathway suppresses Bax translocation to mitochondria. J. Biol. Chem. 277:14040–14047. [DOI] [PubMed] [Google Scholar]
- Tsuruta, F., J. Sunayama, Y. Mori, S. Hattori, S. Shimizu, Y. Tsujimoto, K. Yoshioka, N. Masuyama, and Y. Gotoh. 2004. JNK promotes Bax translocation to mitochondria through phosphorylation of 14-3-3 proteins. EMBO J. 23:1889–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hemert, M.J., H.Y. Steensma, and G.P. van Heusden. 2001. 14-3-3 proteins: key regulators of cell division, signalling and apoptosis. Bioessays. 23:936–946. [DOI] [PubMed] [Google Scholar]
- Wang, H.G., N. Pathan, I.M. Ethell, S. Krajewski, Y. Yamaguchi, F. Shibasaki, F. McKeon, T. Bobo, T.F. Franke, and J.C. Reed. 1999. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 284:339–343. [DOI] [PubMed] [Google Scholar]
- Xiao, B., S.J. Smerdon, D.H. Jones, G.G. Dodson, Y. Soneji, A. Aitken, and S.J. Gamblin. 1995. Structure of a 14-3-3 protein and implications for coordination of multiple signalling pathways. Nature. 376:188–191. [DOI] [PubMed] [Google Scholar]
- Xing, H., S. Zhang, C. Weinheimer, A. Kovacs, and A.J. Muslin. 2000. 14-3-3 proteins block apoptosis and differentially regulate MAPK cascades. EMBO J. 19:349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe, M.B. 2002. How do 14-3-3 proteins work?–Gatekeeper phosphorylation and the molecular anvil hypothesis. FEBS Lett. 513:53–57. [DOI] [PubMed] [Google Scholar]
- Yaffe, M.B., K. Rittinger, S. Volinia, P.R. Caron, A. Aitken, H. Leffers, S.J. Gamblin, S.J. Smerdon, and L.C. Cantley. 1997. The structural basis for 14-3-3:phosphopeptide binding specificity. Cell. 91:961–971. [DOI] [PubMed] [Google Scholar]
- Yang, D.D., C.Y. Kuan, A.J. Whitmarsh, M. Rincon, T.S. Zheng, R.J. Davis, P. Rakic, and R.A. Flavell. 1997. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 389:865–870. [DOI] [PubMed] [Google Scholar]
- Yang, E., J. Zha, J. Jockel, L.H. Boise, C.B. Thompson, and S.J. Korsmeyer. 1995. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 80:285–291. [DOI] [PubMed] [Google Scholar]
- Yu, C., Y. Minemoto, J. Zhang, J. Liu, F. Tang, T.N. Bui, J. Xiang, and A. Lin. 2004. JNK suppresses apoptosis via phosphorylation of the proapoptotic Bcl-2 family protein BAD. Mol. Cell. 13:329–340. [DOI] [PubMed] [Google Scholar]
- Zha, J., H. Harada, E. Yang, J. Jockel, and S.J. Korsmeyer. 1996. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell. 87:619–628. [DOI] [PubMed] [Google Scholar]
- Zhang, L., J. Chen, and H. Fu. 1999. Suppression of apoptosis signal-regulating kinase 1-induced cell death by 14-3-3 proteins. Proc. Natl. Acad. Sci. USA. 96:8511–8515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, X.M., Y. Liu, G. Payne, R.J. Lutz, and T. Chittenden. 2000. Growth factors inactivate the cell death promoter BAD by phosphorylation of its BH3 domain on Ser155. J. Biol. Chem. 275:25046–25051. [DOI] [PubMed] [Google Scholar]