

Abstract
Serine proteases are proteolytic enzymes that are involved in the regulation of various physiological processes. We generated mice lacking the membrane-anchored channel-activating serine protease (CAP) 1 (also termed protease serine S1 family member 8 [Prss8] and prostasin) in skin, and these mice died within 60 h after birth. They presented a lower body weight and exhibited severe malformation of the stratum corneum (SC). This aberrant skin development was accompanied by an impaired skin barrier function, as evidenced by dehydration and skin permeability assay and transepidermal water loss measurements leading to rapid, fatal dehydration. Analysis of differentiation markers revealed no major alterations in CAP1/Prss8-deficient skin even though the epidermal deficiency of CAP1/Prss8 expression disturbs SC lipid composition, corneocyte morphogenesis, and the processing of profilaggrin. The examination of tight junction proteins revealed an absence of occludin, which did not prevent the diffusion of subcutaneously injected tracer (∼600 D) toward the skin surface. This study shows that CAP1/Prss8 expression in the epidermis is crucial for the epidermal permeability barrier and is, thereby, indispensable for postnatal survival.
Introduction
Serine proteases carry out several physiological and cellular functions ranging from degradation and digestive processes to protein processing and tissue remodelling. Therefore, they are implicated in a wide spectrum of physiological and pathophysiological conditions. They share a high degree of amino acid sequence identity; in particular, they share the histidine-aspartate-serine residues that are necessary for catalytic activity, which are present in highly conserved motifs. Membrane-bound serine proteases are anchored either via a carboxy-terminal transmembrane domain (type I), a glycosyl-phosphatidylinositol (GPI) linkage, or an amino-terminal proximal transmembrane domain (type II). The family of membrane-bound serine proteases is rapidly increasing, which indicates that cell surface proteolysis has emerged as an important mechanism for the generation of biologically active proteins (for review see Netzel-Arnett et al., 2003). Their physiological role remains poorly understood. Members of the relatively small subfamily of GPI-anchored serine proteases are very similar in length, ranging from 310–370 amino acid residues, and contain a typical amino-terminal secretory signal and a hydrophobic domain at their carboxy terminus. These anchoring domains are imbedded in the plasma membrane, whereas their serine protease domains are oriented extracellularly (for review see Netzel-Arnett et al., 2003). GPI-anchored proteins are widely distributed on plasma membranes of eukaryotic cells. They have diverse roles and can function as enzymes, cell adhesion molecules, cell surface antigens, and membrane-bound receptors (Kinoshita et al., 1995).
We recently isolated channel-activating protease (CAP) 1 (also termed protease serine S1 family member 8 [Prss8]), which presents the mouse homologue of the human prostasin. CAP1/Prss8 is found in various mammalian tissues like semen, the prostate gland, and skin (Vuagniaux et al., 2000). The physiological role of this protein is largely unknown, although a deregulation of Prss8 expression is found in high-grade human prostate, breast, and ovarian cancers (Takahashi et al., 2003; Chen et al., 2004). Further roles of this serine protease have been proposed for cellular growth, morphogenesis, and sodium absorption in the kidney and lung (for review see Rossier, 2004).
Skin is the largest organ of the human body, and it functions primarily as a barrier to the physical environment by the skin's relative impermeability to water, water-soluble compounds, and pathogenic micro-organisms. The constant thickness of the epidermis is maintained by a balance between the proliferation of keratinocytes in the basal layer and desquamation from the surface of the stratum corneum (SC). When this process is altered, xerotic and ichthyotic conditions may arise (Pierard et al., 2000). In skin, proteinases and protease inhibitors like gelatinase A, nexin-1, or SC chymotryptic enzyme regulate hair growth and/ or cycling (Ekholm and Egelrud, 1998; Sonoda et al., 1999; Karelina et al., 2000). Mice that are deficient for the papainlike lysosomal cystein protease cathepsin L exhibit structural changes of hair follicles and altered epidermal differentiation (Benavides et al., 2002).
Here, we study the consequences of CAP1/Prss8 deficiency in epidermal function. These mice died early after birth as a result of severe dehydration, thus demonstrating the involvement of CAP1/Prss8 in the epidermal barrier function that is indispensable for postnatal survival.
Results
Generation of mice lacking Prss8 in the epidermis
In the Prss8-modified gene locus, exons 3–5, which contain two essential residues of the catalytic triad (His85 and Asp134), are flanked by loxP sites. In addition, deletion of exons 3–5 results in a frameshift and leads to a premature stop codon in exon 6, thereby generating a truncated protein at the carboxy terminus (Fig. 1 a). When such a truncated construct as cRNA is coinjected with epithelial sodium channel (ENaC) subunits in Xenopus laevis oocytes, activation of the ENaC-mediated sodium transport is completely abolished (4.3-fold vs. 0.88-fold induction of amiloride-sensitive INa in oocytes coinjected with ENaC/CAP1 wild-type and ENaC/CAP1 truncated cRNAs, respectively; n ≥ 21 oocytes, P > 0.001). When ENaC was coinjected with a 1:1 mixture of CAP1/Prss8 wild-type and truncated cRNAs, no difference in ENaC-mediated sodium current was observed, thus rendering unlikely any dominant negative effect of the truncated CAP1/Prss8 cRNA (unpublished data).
Figure 1.

Generation of skin-specific Prss8-deficient mice. (a) Scheme of wild-type, loxP, and Δ allele at the Prss8 gene locus. Coding (gray boxes) and noncoding sequences (white boxes); loxP sites, black triangles. PCR-based genotyping was performed using primers A–C (arrows). For RT-PCR analysis, primers F and G (arrows) were used. (b) PCR analysis with primers A–C distinguishes between wild-type (+; 379 bp), lox (413 bp), and Δ allele (top). Note the shift of the Prss8 lox allele into the Δ allele in epidermis from animals harboring the Cre transgene (lanes 2 and 10). Detection of the Cre transgene using Cre- and myogenin-specific primers (internal control; bottom). (c) RT-PCR analysis using primers F and G demonstrates absence of CAP1/Prss8 mRNA transcripts (309 bp) in the knockout group (lane 1). Wild-type CAP1/Prss8 plasmid (lane 11; positive control). The reaction is controlled by detection of GAPDH transcripts. (d) Immunohistochemistry using the affinity-purified CAP1/Prss8 antibody revealed expression in top layers of the SG and in the transition between SG and SC in the control group (top), but absence of expression in the knockout (KO) group (bottom). BF, bright field; E, epidermis; D, dermis; dotted lines, basal membrane.
To ablate CAP1/Prss8 specifically in the epidermis, we used keratin (K) 14–Cre recombinase transgenic mice with one null (Δ) allele and one conditional allele (lox) at the Prss8 gene locus (Li et al., 2001; Rubera et al., 2002). In the epidermis, this resulted in a majority of cells lacking the Prss8 wild-type allele (Fig. 1 b, lane 2). No recombination was observed in the dermis, brain (Fig. 1 b, lanes 3, 4, 7, 8, 11, and 12), lung, heart (not depicted), or in mice lacking the Cre transgene (Fig. 1 b, lanes 5–8). CAP1/Prss8 mRNA transcripts were detected in the skin and lung but were absent in the brain (Fig. 1 c, lanes 4–9). Knockout mice lacked normal Prss8 mRNA in the epidermis, whereas transcripts were still detected in the lung (Fig. 1 c). In control mice, CAP1/Prss8 expression was detected in the top layers of the granular layer and in the transition between stratum granulosum (SG) and SC. In contrast, CAP1/Prss8 protein was not detected in the skin of knockout mice (Fig. 1 d).
Early postnatal lethality and skin abnormalities in mice lacking CAP1 in the epidermis
Genotype analysis of newborn offspring revealed a ratio that was consistent with the Mendelian pattern of inheritance (Fig. 2 a, left). However, when a total of 119 animals from 10 litters were genotyped at the weaning age, we could not detect Prss8 lox/Δ /K14-Cre animals (Fig. 2 a, right). This was further investigated by monitoring 56 pups (n = 4 litters) during 72 h after birth (Fig. 2 b). None of the 18 genotyped epidermis-specific CAP1/Prss8 knockouts was found alive >60 h after birth, demonstrating that CAP1/Prss8 expression in skin is indispensable for early postnatal survival.
Figure 2.
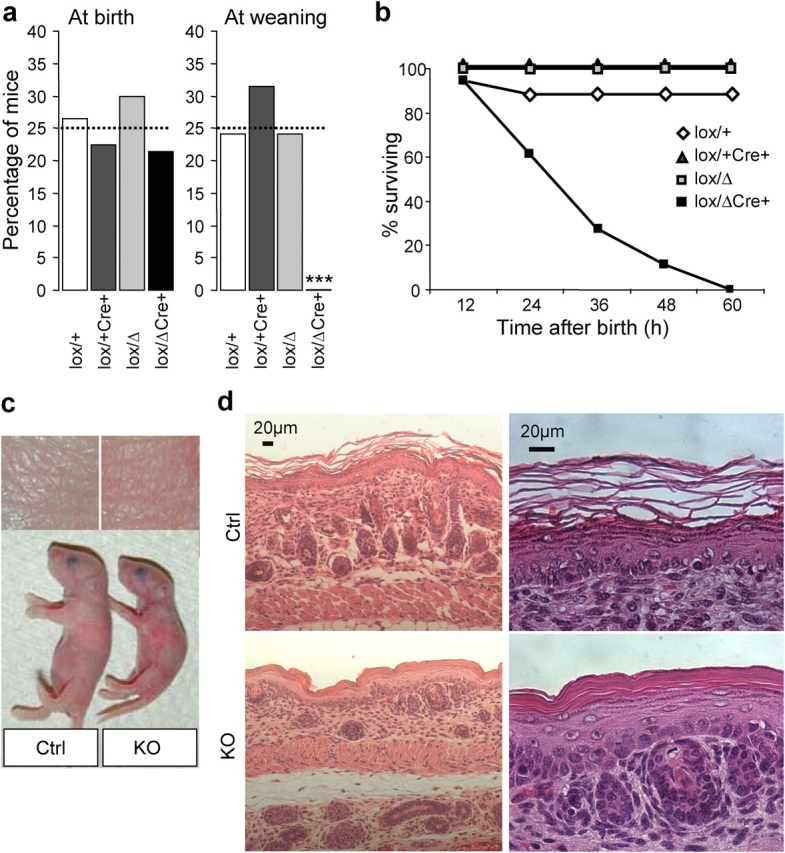
Early postnatal lethality and skin abnormalities in Prss8lox/ Δ /K14-Cre mice. (a) Distribution of genotypes in the knockout (black bar) and control groups (white, light gray, and dark gray bars) at birth indicated in percentages. From a total of 131 pups (n = 14 litters) that were analyzed shortly after birth, 28 were from the knockout group, 32 were from Prss8 lox/+, 40 were from Prss8 lox/Δ, and 31 were from Prss8 lox/+ /K14-Cre (control group; expected distribution in percentage; dotted line). At weaning, 37 Prss8 lox/+, 37 Prss8 lox/Δ, and 44 Prss8 lox/+ /K14-Cre mice were identified. ***, P < 0.001. (b) Survival curves of knockout (black squares) and control mice (white diamonds and gray squares). Note that no knockout mice survived 60 h after birth. (c) Representative example showing different sizes of knockout (right) and control (left) littermates. (d) Representative skin sections from newborn control (top) and knockout (bottom) littermates stained with hematoxylin and eosin. Staining (left) shows abnormal SC formation and a reduced hair follicle number in knockout skin. Higher magnification (right) reveals an orthokeratotic hyperkeratosis and dysmaturation of hair follicles.
Except for the difference in size (nose-caudal extremity), we observed no gross abnormalities at birth (unpublished data). The body weight of newborn knockout mice (1,207 ± 19 mg; n = 4) was lower than that of control mice (1,472 ± 36 mg; n = 3; P < 0.001). After a few hours, the skin appeared more reddish and wrinkled (Fig. 2 c). On the histological level, all skin layers were present, but the SC was disorganized, more compact, and focally detached from the granular layer, thus presenting an orthokeratotic hyperkeratosis phenotype (Fig. 2 d, bottom). The adnexal part of the skin presented a dysmaturation of the hair follicles, which were reduced in number and were incompletely matured. Hair follicles were shorter, and no keratin was visible. Except for medullar hypoplasia in the thymus, whole examination of various organs revealed no further abnormalities (unpublished data).
Impairment of the epidermal barrier function
Next, we tested the functional integrity of the epidermis by measuring the inwards and outwards barrier function. As illustrated in Fig. 3 a, knockout mice severely lost more weight than their littermates (10% in 6 h vs. 2–3% in the control group), which is interpreted as liquid loss by evaporation through the skin. Increased dehydration was further confirmed in these mice by measuring the transepidermal water loss (TEWL), which increased by almost 40% in neonates from the knockout group (Fig. 3 b). These mice also showed increased toluidine blue penetration across the entire epidermal surface and more especially at the ventral side, whereas controls were completely unstained (Fig. 3 c). This severe impairment of both inwards and outwards barrier functions of the epidermis in knockout animals strongly suggests the involvement of CAP1/Prss8 in epidermal barrier function.
Figure 3.

Disruption of epidermal barrier function in Prss8lox/ Δ /K14-Cre mice. (a) Dehydration assay over time. Data are presented as percentages of initial body weight in knockout (black squares; n = 5) and control (Prss8 lox/Δ and Prss8 lox/+ /±K14-Cre; white diamonds and gray squares; n = 5–8) mice. *, P < 0.05; ***, P < 0.001. (b) Transepidermal water loss (TEWL) measured on ventral skin is decreased in Prss8 lox/Δ /K14-Cre (black bar; n = 6) pups compared with control pups (Prss8 lox/Δ, light gray bar; n = 9), Prss8 lox/+ /K14-Cre (dark gray bar; n = 11), and Prss8 lox/+ (white bar; n = 15). **, P < 0.01; ***, P < 0.001. Error bars represent SEM. (c) Barrier-dependent dye exclusion assay in knockout (left; n = 8) and control (right; n = 5) pups. Representative photograph revealing dye penetration of all epidermal surfaces, especially ventrally in knockout, but not in control, pups.
Altered morphogenesis of corneocytes and defective epidermal lipid matrix composition
Components of the SC, corneocytes, and epidermal lipid matrix were further analyzed. The morphology and resistance to mechanical stress of isolated corneocytes was assessed. As in controls, the number of fragilized cells that were resistant to ultrasound treatment after sonication increased but was not significantly different with time. However, cells from the knockout group exhibited an ∼10% larger surface, indicating a perturbation of the morphogenesis of corneocytes (Fig. 4 a; P < 0.001). The surface appearance and shape of these cells were not altered (Fig. 4 b), and the lipid content of isolated SC (dry weight) was slightly reduced (Fig. 4 c; 80 vs. 103 μg/mg in control; n = 5). Densitometric quantification of ceramides and glucosylceramides revealed no changes except for the amount of ceramide (Cer)AS (15 vs. 10 μg/mg dry SC weight in the control group; P< 0.05). The lipid barrier precursor sphingomyelin increased 3.7-fold (Fig. 4 d; P < 0.001). The lipid content of covalent-bound lipids is important for the barrier function (Macheleidt et al., 2002), and, in knockout epidermis, the level of bound ω-hydoxylated fatty acid decreased by 50%, whereas the fatty acid level increased by 56% (Fig. 4 e). Furthermore, the amount of covalently bound Cer(OS) (ceramides with a ω-hydroxy fatty acid and sphingosine groups) significantly decreased by 27% (Fig. 4 e). These data implicated CAP1/Prss8 in the morphogenesis of corneocytes and in the composition of the lipid matrix in the SC.
Figure 4.
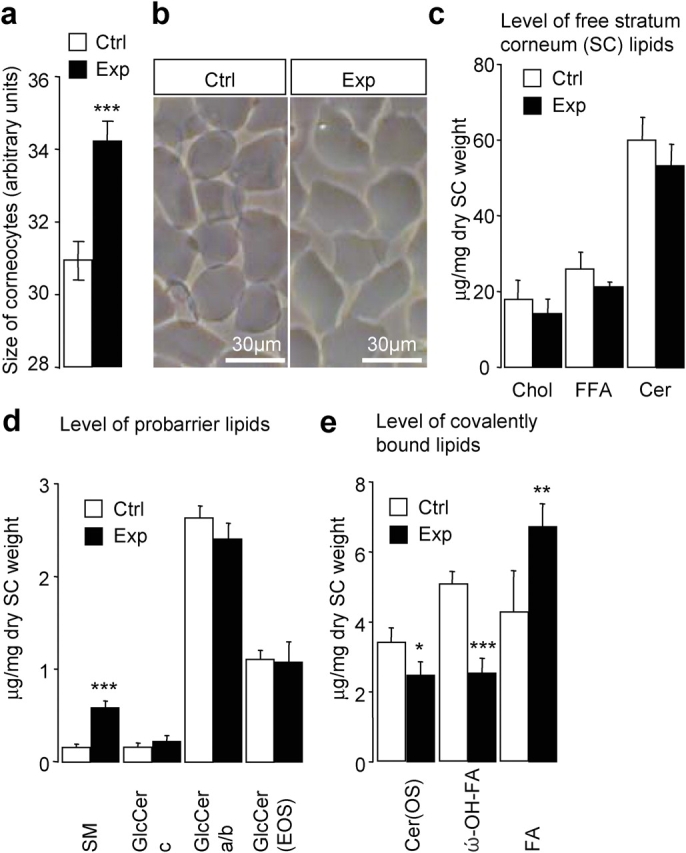
Enlarged corneocytes and altered SC lipid composition in knockout mice. (a) Mean surface area of cells from control (white bar; n = 15–50 corneocytes; n = 6 mice) and knockout (Exp) mice (black bar; n = 15–50 corneocytes; n = 5 mice). ***, P < 0.001. (b) Morphological appearance of purified cells that are isolated from the control (left) and knockout (right) epidermis. (c) Levels of main free SC lipids. Chol, cholesterol; FFA, free fatty acid; Cer, ceramides. (d) Analysis of probarrier lipids. Note the increased amount of sphingomyelin (SM) in the knockout group (***, P < 0.001), whereas levels of different glucosylceramide fractions (GlcCer c, GlcCer a/b, and GlcCer(EOS); N-acyl fatty acid acylated in ω-OH position) were unaltered. (e) Level of covalently bound lipids. Note the 50% reduced amount of ω-hydroxylated fatty acid (ω-OH–FA; ***, P < 0.001), the decrease in ceramides (Cer[OS]); *, P < 0.05), and the significantly increased fatty acid (FA; **, P < 0.01) levels in knockout (Prss8 lox/Δ /K14-Cre; black bar) versus control (Prss8 lox/+ ; white bar) groups. Each value is the mean of five animals ± SEM (error bars).
Defective profilaggrin to filaggrin processing but otherwise normal expression of differentiation markers
To unveil defects in differentiation that are caused by CAP1/Prss8 deficiency, we analyzed the level of expression and distribution of differentiation markers. Western blot analysis of epidermal proteins revealed a normal profilaggrin to filaggrin processing pattern in the control epidermis. Intermediates from profilaggrin (300 kD) to filaggrin monomers (∼25 kD) differed in size (Fig. 5 a). In contrast, the epidermis lacking CAP1 presented an aberrant pattern of profilaggrin-derived proteolytic products, mainly the two-domain intermediate (∼50 kD), which is composed of two monomers of filaggrin. In addition, we found a nearly complete loss of filaggrin monomer (∼25 kD) in Prss8-deficient epidermis (Fig. 5 a). On the other hand, immunofluorescence showed a comparable distribution of the filaggrin expression in both genotypes (Fig. 6). Western blot and immunofluorescence microscopy analysis revealed a normal expression of K14 and no obvious differences in K1, loricrin, and involucrin (Figs. 5 and 6). Both the control and knockout epidermis were clearly negative for K6 (Fig. 6).
Figure 5.

Aberrant profilaggrin to filaggrin processing in Prss8-deficient epidermis. (a) Equal amounts of protein extracts of control (lanes 1 and 3) and knockout (lanes 2 and 4) epidermis were separated through SDS-PAGE under reducing conditions and either were Coomassie blue stained (loading control; lanes 1 and 2) or subjected to Western blot analysis using the anti-filaggrin antibody (lanes 3 and 4). Note the aberrant profilaggrin to filaggrin processing in the knockout epidermis with a nearly complete loss of 25-kD filaggrin monomers (lane 4). The position of profilaggrin (proF) and its proteolytically-derived products (F; e.g., filaggrin three-domain [3F] and two-domain [2F] intermediates) are indicated. The size of the molecular marker (kD) is also indicated. (b) Western blot analysis on epidermal proteins from control (Ctrl) and knockout (KO) mice after SDS-PAGE separation using differentiation markers (loricrin, 57 kD; K1, 67 kD; K14, 55 kD; and involucrin, 56 kD; indicated by arrow). Equal amounts of the proteins were loaded.
Figure 6.
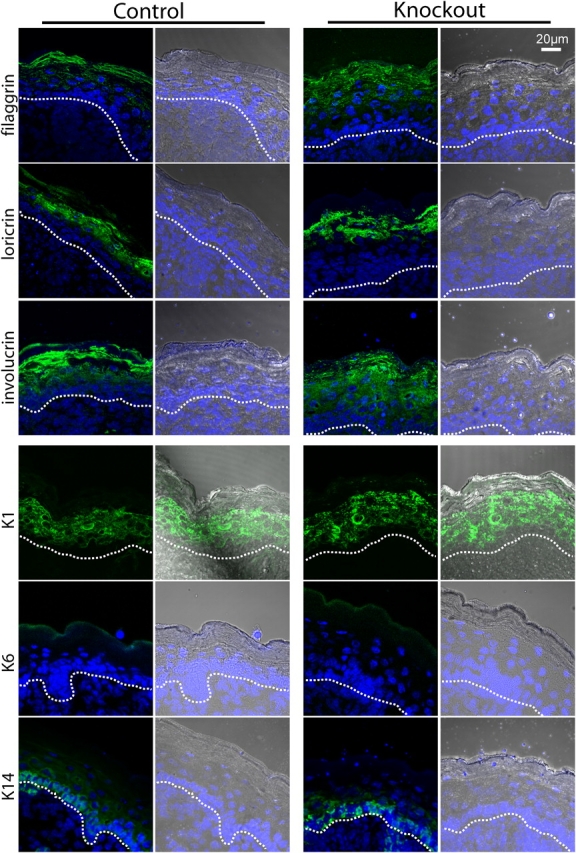
Normal distribution of the differentiation markers in CAP1/Prss8-deficient epidermis. Immunofluorescence LSM for filaggrin, loricrin, involucrin, K1, K6, and K14. Nuclei are counterstained with DAPI. Brightfield/DAPI is equally depicted, with the dotted lines indicating the basal membrane. Filaggrin is mainly localized in the SC but starts in the SG. Loricrin staining is observed in the top SG and continues in the SC. Involucrin staining is strong in the SC but is also expressed in the SG and expressed slightly in the stratum spinosum. K1 is localized from the SP to the SG (intermediate epidermis marker). K6 is not detectable in both control and knockout groups. K14, which is a specific marker for the stratum basale, is localized as expected in the two groups.
Impaired barrier function of tight junctions (TJs) in the epidermis
To understand the epidermal barrier defect that is caused by CAP1/Prss8 deficiency, we further analyzed the TJ proteins claudin-1, occludin, and ZO-1 (Zonula occludens 1) in the control and knockout group because an important part of the water barrier is formed by TJs. Localization of claudin-1 (Brandner et al., 2002; Furuse et al., 2002) was not affected and detected in the stratum spinosum or in the lower SG layers (Fig. 7). ZO-1 was localized in the cell periphery of SG cells and seemed to be slightly dispersed in the knockout group (Fig. 7). Occludin appeared as spots in the second layer of SG cells (Fig. 7 a, arrows) as expected from a TJ sealing of the epidermis. However, no expression of occludin was found in the CAP1/Prss8-deficient epidermis (Fig. 7 a). To further examine whether this affects the barrier function of TJs, we performed a tracer experiment as previously described (Chen et al., 1997; Furuse et al., 2002). In control mice, streptavidin labeling of the biotinylation tracer revealed a diffusion through the paracellular spaces from stratum basale to the second layer of SG. This diffusion was abruptly prevented at the “ending” points, where occludin is supposed to be localized (Fig. 7 b, top). In contrast, in CAP1/Prss8-deficient epidermis, the tracer appeared to pass this point and reach the interface between the SG and corneum, where it accumulated (Fig. 7 b, bottom). These findings clearly indicate that TJ function, at least against small molecules of ∼600 D, was severely affected in the knockout epidermis and suggest that the impaired TJ barrier might be causative for rapid and fatal dehydration in these mice.
Figure 7.
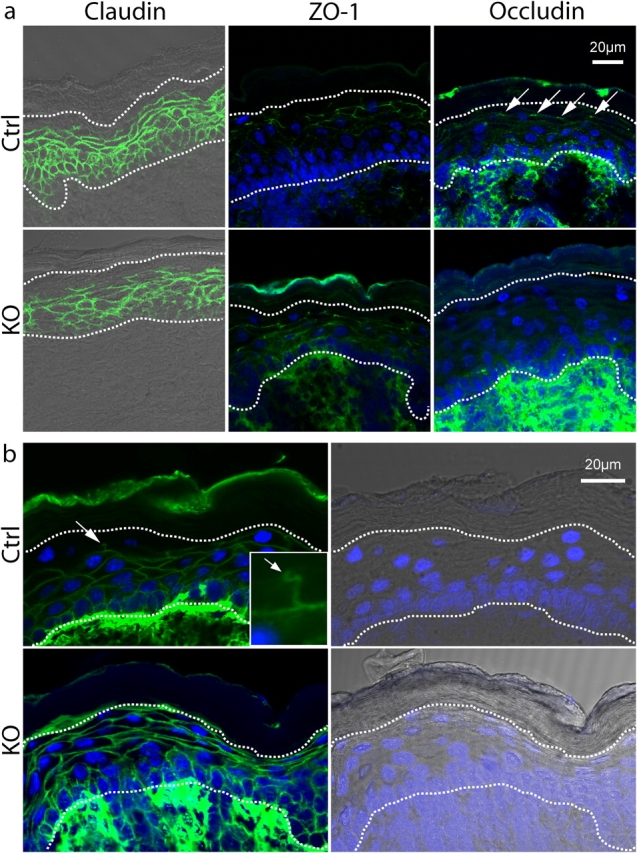
Expression of TJ proteins and TJ permeability assay. (a) Immunofluorescence LSM for claudin-1 (claudin), Zonula occludens 1 (ZO-1), and occludin. For ZO-1 and occludin, the DAPI counterstaining is added. The two dotted lines show the basal membrane (bottom) and the limit SG/SC (top). Claudin staining is localized in the whole epidermis except for the SC and the last layer of SG in both groups. ZO-1 shows a staining in the mid-SG in both groups. In controls, occludin shows a dotted staining, as expected for TJs (arrows), between the second and last layer of the SG. No such staining could be observed in knockouts. (b) TJ permeability assay visualized by LSM. For each genotype, the staining for streptavidin/AlexaFluor488 is shown with DAPI counterstaining, and brightfield/DAPI of the same picture is added for localization. The bottom dotted line indicates the basal membrane, and the top line indicates the limit SG/SC. The top panels show the diffusion of biotin in controls, which is blocked abruptly between the second and last layer of the SG (arrow). The magnification (inset, arrow) indicates the position of TJ. The bottom panel shows the diffusion of biotin in knockout epidermis, which is not blocked by the TJ and, thus, extends up to the SC, where it accumulates.
Discussion
To study the role of GPI-anchored serine protease CAP1/Prss8 in vivo, we used keratinocyte-specific gene targeting of a conditional CAP1/Prss8 allele (Li et al., 2001; Rubera et al., 2002). Disruption of the Prss8 gene locus was confined to the epidermis and to K14-expressing epithelia (e.g., the thymus and tongue; Fig. 1 and not depicted). Histopathological analyses in the skin of CAP1/Prss8-deficient neonates revealed a striking malformation of the SC that was accompanied by defective epidermal barrier function, resulting in severe dehydration and early neonatal lethality (Fig. 2). Interestingly, the observed cutaneous phenotype mostly resembled defects in epidermal barrier function, as seen in mice that were deficient in Klf4 (Krüppel-like factor 4) transcription factor, the transglutaminase I gene, the matriptase/membrane-type serine protease 1 (MT-SP1) gene, or the epidermal-specific GPI anchor (pig-a) gene (for review see Segre, 2003). It is known that proteolytic processing is required for signaling, proprotein activation, structural remodeling, and desquamation. Proteases such as calpain-1 (Yamazaki et al., 1997), furin (Pearton et al., 2001), PEP1 (profilaggrin endopeptidase 1; Resing et al., 1995), and matriptase/MT-SP1 (List et al., 2003) have been identified as being directly implicated in profilaggrin processing.
Identification of the molecular nature of the barrier is still under investigation, with the consensus that a protein lipid layer, which is located in the top layers of the epidermis and TJs, plays an essential role in development of the skin barrier function (Tsuruta et al., 2002). Evidence that cornified envelope assembly is necessary for barrier development in skin is obtained from the study of transglutaminase 1–deficient mice that lack confirmed envelopes. These mice suffer from water loss, resulting in neonatal lethality (Matsuki et al., 1998). Alterations of corneocyte morphology have also been described in various mouse models with epidermal permeability barrier defects ranging from near absence in transglutaminase 1–deficient mice (Matsuki et al., 1998) to irregular shaped corneocytes (e.g., Klf4−/− mice; Segre et al., 1999). Our corneocyte phenotype mostly resembled that of matriptase/MT-SP1–deficient mice, a member of the type II transmembrane serine protease family (List et al., 2003). Equally, mice with a targeted disruption of Fatp4 (fatty acid transport protein 4) showed a disturbed fatty acid composition of epidermal ceramides with a normal distribution of TJ proteins (Herrmann et al., 2003), although a functional TJ assay has not been performed. In the CAP1/Prss8-deficient epidermis, the level of barrier-forming lipids with very long chain fatty acids that were covalently attached to proteins was significantly reduced, suggesting a potential cause for the observed defect in epidermal barrier permeability ( Fig. 4 ). These barrier lipids were formed by mice only a few days before birth (Doering et al., 2002), and their deficiency in prosaposin- and β-glucocerebrosidase knockout mice resulted in disruption of the water permeability barrier and in early death (Doering et al., 1999a,b). Interestingly, the observed imbalance in ceramide composition did not result from an altered keratinocyte differentiation, as epidermal differentiation markers (K14, K1, involucrin, loricrin, and filaggrin) seemed not to be affected (Fig. 6). Defective profilaggrin to filaggrin processing, as seen in the CAP1/Prss8 knockout group, may lead to abnormal SC hydration, which further results in the downstream depletion of humectant amino acids and their deiminated products (Fig. 5; Elias, 2004; for review see Rawlings and Harding, 2004). At the transition between the granular layer and SC, the processing of profilaggrin, which consists of multiple filaggrin repeats joined by linker peptides, involves dephosphorylation and proteolytic steps through three-domain and two-domain intermediates to filaggrin (Fig. 5; Resing et al., 1984). According to the proposed consensus site for proteolytic cleavage by CAP1 (Shipway et al., 2004), two preferred polybasic cleavage sites are situated within filaggrin, thus suggesting a mechanism for CAP1 activation.
Moreover, TJs are important for barrier function (Furuse et al., 2002), indicating that two systems for forming a barrier exist in skin. Indeed, CAP1/Prss8-deficient mice exhibited an altered composition of lipids, precursors, and defective filaggrin processing but also exhibited an impaired function of TJ, thus leading to a severely compromised epidermal permeability barrier that likely accounted for early neonatal death. Interestingly, the link between protease action and TJ disruption has been made by the treatment of epithelial cell lines with Derp1, a cysteine protease that reversibly disrupts TJs (Wan et al., 1999). Apparently, Derp1 acts to increase paracellular permeability by direct proteolytic cleavage of occludin and possibly claudin. This proteolysis is assumed to cause the breakdown of the TJ protein complex. In CAP1/Prss8-deficient epidermis, occludin seems to be absent from these TJs despite the normal presence of claudin-1 and ZO-1 ( Fig. 7 a). Interestingly, mice lacking occludin show a complex phenotype, and the TJs themselves are not affected morphologically and functionally (Saitou et al., 2000). By sequence comparison, we found a CAP1-preferred polybasic cleavage site in ZO-1 but not in occludin protein, suggesting that the mechanism of protease action on occludin may be indirect (unpublished data). Nevertheless, the diffusion of injected biotinylation reagent was not prevented, suggesting that the assembly of TJ proteins to functional units is crucial. Thus, the TJ complex may not be that different from wild-type mice, as supported by claudin-1 and occludin-deficient mice (Saitou et al., 2000; Furuse et al., 2002).
The physiological role of most of the membrane-anchored serine proteases is still unclear, and the endogenous targets of the majority of these enzymes have not been identified. Matriptase/MT-SP1 has been proposed to initiate signaling and proteolytic cascades through its ability to activate cell surface–associated proteins like pro-uPA and PAR-2, although an inappropriate processing of these substrates might not contribute to the epidermal phenotype of matriptase/MT-SPI–deficient mice (Takeuchi et al., 2000; List et al., 2002). In vitro and in vivo experiments indicated that membrane-bound serine proteases may be of importance in the activation of the highly amiloride-sensitive sodium channel ENaC (Rossier, 2004). These serine proteases include Prss8 (CAP1 or prostasin), TMPRSS4 (CAP2), matriptase/MT-SP1 (epithin or CAP3), and TMPRSS3 (Vallet et al., 1997; Vuagniaux et al., 2000, 2002; Guipponi et al., 2002). As evidenced by RNA and protein analyses, ENaC subunits are expressed in keratinocytes, whereas the dermis is negative (Brouard et al., 1999). Interestingly, ENaC and CAP1/Prss8 mRNA transcript expression was found in nondifferentiated keratinocytes (Oda et al., 1999) and increased ∼2.2-fold in more differentiated keratinocytes, as evidenced by real-time PCR (unpublished data). In extracellular domains of the α and β subunits of ENaC, CAP1-preferred polybasic cleavage sites were recently found, suggesting a direct interaction with this protease (Shipway et al., 2004). Therefore, ENaC may present a potential substrate for CAP1 (Hughey et al., 2003, 2004), although it is unlikely that ENaC is the only substrate of CAP1. Newborn mice, in which ENaC activity had been deleted, demonstrated epithelial hyperplasia, abnormal nuclei, premature secretion of lipids, and abnormal expression of differentiation markers. This finding suggests that this channel modulates ionic signaling for specific aspects of epidermal differentiation, such as synthesis or processing of differentiation-specific proteins, and lipid secretion (Mauro et al., 2002). It will be interesting to analyze TJs in these knockout mice. Membrane-bound serine proteases might exhibit pleiotropic functions in the activation of growth factors or G protein–coupled receptors and in the activation of proteolytic cascades. A comparison of the skin phenotypes of CAP1/Prss8- with matriptase/MT-SP1–deficient mice indeed suggests that both serine proteases may participate in the same protease-signaling cascade.
In humans, defective epidermal barrier function is seen in a variety of skin disorders that are generally presumed to be ichthyosis. Interestingly, the phenotype of our epidermis CAP1/Prss8-deficient mice strikingly resembles a severe form of congenital ichthyosis, the harlequin disease. Harlequin ichthyosis is an autosomal recessive human disorder affecting skin cornification, and affected infants often die within days to weeks of birth, apparently as a result of massive hyperkeratosis and/ or a severe epidermal permeability barrier defect (for review see Dale and Kam, 1993). A “harlequin-like” mouse showed mutations in cystatin M/E, which is a serine protease inhibitor and substrate for transglutaminase, but no gene mutations have been identified so far in harlequin ichthyosis patients (Zeeuwen et al., 2003). Recently, analysis of a pig-a epidermis-specific knockout mouse suggested that defective GPI-anchored proteins may account for harlequin ichthyosis (types I and II) in humans (Hara-Chikuma et al., 2004). Although the X-linked pig-a gene has been excluded, CAP1/Prss8 may represent a candidate gene for this disease. This study may facilitate further analysis of the role of this membrane-anchored serine protease in normal skin development, the identification of putative target proteins, and its implication in genetic disorders.
Materials and methods
Animals
Mice carrying one knockout allele of Prss8 (Prss8 Δ) were obtained by breeding Prss8 tm1.2Hum (hereafter Prss8 lox) mice to EIIa-Cre (Lakso et al., 1996) mice. To produce Prss8 mutant mice in which CAP1/Prss8 was selectively disrupted in the epidermis, heterozygous mutant Prss8 Δ / + mice were crossed with K14-Cre mice (provided by D. Metzger and P. Chambon, Institut de Genetique et de Biologie Moleculaire et Cellulaire, Illkirch, France; Li et al., 2001). Epidermis-specific CAP1/Prss8-deficient (Prss8 lox/Δ /K14-Cre, knockout group) and heterozygous mutant (Prss8 lox/+ /±K14-Cre and Prss8 Δ /lox, control group) littermates were obtained by interbreeding double transgenic male Prss8 Δ / + /K14-Cre with female Prss8 loxlox mice. All experiments were performed coded. Mice were housed and fed according to federal guidelines, and the local authorities approved all procedures.
Genotyping was performed by using DNA-based PCR analysis from biopsies (tail and organs) and was analyzed by PCR using the following three primers: Prss8 sense, A (5′-GCAGTTGTAAGCTGTCATGTG-3′); Prss8 sense, B (5′-CAGCAGCTGAGGTACCACT-3′); and Prss8 antisense, C (5′-CCAGGAAGCATAGGTAGAAG-3′) to detect Prss8 wild-type– (379 bp), lox- (413 bp), and Δ-specific (473 bp) PCR-amplified products. 36 cycles were run, each consisting of 1 min at 94, 56, and 72°C. The presence of the Cre transgene was detected by PCR using Cre-specific primers (sense, 5′-CCTGGAAAATGCTTCTGTCCG-3′; and antisense, 5′-CAGGGTGTTATAAGCAATCCC-3′) to amplify a 350-bp fragment (36 cycles were run as described above). Myogenin-specific primers (sense, 5′-TTACGTCCATCGTGGACAGC-3′; and antisense, 5′-TGGGCTGGGTGTTAGTCTTA-3′) were used to control the DNA integrity of each sample.
For reverse transcription PCR, total RNA was isolated, extracted from newborn mouse tissues (RNeasy Mini Kit; QIAGEN), and reverse transcribed (Superscript II; Invitrogen) as described previously (Olivier et al., 2002). PCR amplification was performed with primers situated in exons 4 and 6 of Prss8 (exon 4 sense, F [5′-CAGCCAATGCCTCCTTTCCC-3′]; and exon 6 antisense, G [5′-TCACCCCAACTCACAATGCC-3′]). 40 cycles (each consisting of 1 min at 95, 57, and 72°C) were run to amplify a Prss8-specific mRNA fragment of 309 bp. GAPDH primers (sense, 5′-CGTCTTCACCACCATGGAGA-3′; and antisense, 5′-CGGCCATCACGCCACAGTTT-3′) were used as loading controls. Amplified PCR products were separated on a 2% agarose gel and were visualized by ethidium bromide staining.
Histopathological analysis and immunohistochemistry
For complete histopathological analysis, whole newborn litters from knockout and control groups were fixed overnight in 4.5% phosphate-buffered formalin, pH 7, and were embedded in paraffin. 4-μm sagittal sections were stained with hematoxylin and eosin and were examined by light microscopy using a photomicroscope (Axioplan; Carl Zeiss MicroImaging, Inc.). Images were acquired with a high sensibility digital color camera (AxionHRc; Carl Zeiss MicroImaging, Inc.).
Immunohistochemistry was performed on newborn skin samples that were frozen in optimum cutting temperature compound (Sakura Finetek). Affinity-purified CAP1/Prss8 rabbit antiserum (Planes et al., 2005) was incubated for 1 h at RT on 10-μm cryosections that were previously fixed for 10 min in 4% PFA. Staining was visualized by laser scanning microscopy (LSM) with a confocal microscope (model LSM510 Meta; Carl Zeiss MicroImaging, Inc.) after 45 min of incubation at RT with CY3 anti–rabbit IgG (Jackson ImmunoResearch Laboratories). Mouse antibodies to K6 (clone Ks6.KA12; Progen Biotechnik GmbH), K14, K1, involucrin, loricrin, and filaggrin were purchased from Covance and were used at a dilution of 1:1,000 except for K14 (1:4,000).
Antibodies against TJ proteins.
Rabbit anti–claudin-1 pAb (Zymed Laboratories) was used (1:100 dilution) in combination with CY3 anti–rabbit IgG (Jackson ImmunoResearch Laboratories). mAb antioccludin (MOC37) and ZO-1 (T8-754) were provided by M. Furuse (Kyoto University, Kyoto, Japan) and were combined with FITC-conjugated anti–mouse antibodies (Calbiochem). After fixation (30 min in 95% ethanol at 4°C and 1 min in acetone at RT) and permeabilization (10 min in 0.2% Triton X-100/PBS), the incubation of cryosections with primary antibody was performed overnight at 4°C after blocking 30 min in PBS/3% BSA, and the reaction was revealed with secondary antibody for 2 h at 4°C. Nuclei were counterstained with 0.2 μg/ml DAPI in mounting medium.
Epidermal protein extraction and Western blot analysis
The epidermis and dermis were separated by heating the skin for 5 min at 54°C in 5 mM EDTA/PBS. Then, the epidermis was homogenized in ice-cold 1 M NaSCN, 50 mM Hepes, 10 mM EDTA, pH 6.8, 0.3 mM orthophenantroline, 20 μg/ml PMSF, and 0.1% isopropanol (Resing et al., 1984), and the lysate was cleared by centrifugation at 12,000 g for 15 min at 0°C after adding 10 vol of ice-cold water. 50 μg of proteins were separated by SDS-PAGE on 4–16% acrylamide gradient gels. Antibodies were detected with donkey anti–rabbit IgG at a dilution 1:10,000 (GE Healthcare), and the signal was developed with the Supersignal West Dura System (Pierce Chemical Co.). Western blot analysis was performed by using rabbit antibodies to filaggrin (1:1,000; Covance), loricrin (1:1,000), K1 (1:1,000), K14 (1:10,000), and involucrin (1:1,000).
Lipid analysis
Whole skin from newborns were removed at autopsy, frozen, and stored at −20°C until further treatment. SC preparation, lipid analysis, and recovery/analysis of covalently bound lipids was performed as described previously (Reichelt et al., 2004).
Functional analyses of the epidermal barrier
Skin permeability assay.
Newborn mice from knockout and control groups were killed and subjected to methanol dehydration and subsequent rehydration as described previously (Koch et al., 2000). They were further washed in PBS, stained overnight at 4°C in 0.1% toluidine blue/PBS (Merck), destained in PBS, and photographed with a digital camera (Coolpix 950; Nikon; List et al., 2002).
Measurement of TEWL.
The rate of TEWL from the ventral skin of newborn mice was determined by using a Tewameter (Courage and Khazaka) as described previously (Barel and Clarys, 1995; Matsuki et al., 1998).
Dehydration assay.
To determine the rate of fluid loss (List et al., 2002), newborns from three independent litters were separated from their mother to prevent fluid intake, and the rate of epithelial water loss was calculated by measuring the reduction of body weight as a function of time.
TJ permeability assay.
A TJ functional test was performed according to methods developed previously (Chen et al., 1997) and was adapted for skin (Furuse et al., 2002). 30 min after injection, the skin was dissected out and frozen in optimum cutting temperature compound (Tissue-Tek). About 10-μm-thick sections were fixed as described previously (Furuse et al., 2002). Staining was visualized by an LSM confocal microscope after the incubation of sections with streptavidin AlexaFluor488 (Invitrogen) overnight at 4°C. Three wild-type and four knockout mice were independently analyzed for this experiment.
Calculations and statistics
All data are expressed as means ± SEM. Values of n refer to the number of mice in each group. Individual groups were compared by using the t test for all pair-wise comparisons. A level of P ≤ 0.05 was accepted as statistically significant for all comparisons.
Acknowledgments
We would like to thank Daniel Metzger and Pierre Chambon for providing the K14-Cre expressing line and Mikio Furuse for the rat anti–mouse occludin mAb (MOC37) and ZO-1 mouse monoclonal antisera (T8-754). We wish to thank the Mouse Pathology platform (Lausanne, Switzerland) for histopathological analysis and Jean-Yves Chatton and Yannis Krempp (Cellular Imaging Facility platform; Lausanne, Switzerland) for help with the cellular imaging. We also thank Anne-Marie Mérillat, Nicole Fowler-Jaeger, and Bettina Kircharz for excellent technical assistance, Hans-Peter Gaeggeler for excellent photographic work, and Friedrich Beermann, Jean-Daniel Horisberger, Miguel van Bemmelen, and Bernard Rossier for helpful suggestions and critical reading of the manuscript. We also thank members of the Hummler lab for discussions.
This work was supported by the Swiss National Science Foundation (grants 31-063801.00 and 3100A0-102125/1 to E. Hummler).
C. Leyvraz and R.-P. Charles contributed equally to this paper.
I. Rubera's present address is Laboratoire de Physiologie Cellulaire et Moléculaire, Université de Nice Sophia-Antipolis, 06108 Nice, Cedex 2, France.
M. Guitard's present address is Cutaneous Biology Research Center, Charlestown, MA 02129.
Abbreviations used in this article: CAP, channel-activating protease; ENaC, epithelial sodium channel; GPI, glycosyl-phosphatidylinositol; K, keratin; LSM, laser scanning microscopy; MT-SP1, membrane-type serine protease 1; Prss8, protease serine S1 family member 8; SC, stratum corneum; SG, stratum granulosum; TEWL, transepidermal water loss; TJ, tight junction.
References
- Barel, A.O., and P. Clarys. 1995. Study of the stratum corneum barrier function by transepidermal water loss measurements: comparison between two commercial instruments: Evaporimeter and Tewameter. Skin Pharmacol. 8:186–195. [DOI] [PubMed] [Google Scholar]
- Benavides, F., M.F. Starost, M. Flores, I.B. Gimenez-Conti, J.-L. Guénet, and C.J. Conti. 2002. Impaired hair follicle morphogenesis and cycling with abnormal epidermal differentiation in nackt mice, a cathepsin L-deficient mutation. Am. J. Pathol. 161:693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandner, J.M., S. Kief, C. Grund, M. Rendl, P. Houdek, C. Kuhn, E. Tschachler, W.W. Franke, and I. Moll. 2002. Organization and formation of the tight junction system in human epidermis and cultured keratinocytes. Eur. J. Cell Biol. 81:253–263. [DOI] [PubMed] [Google Scholar]
- Brouard, M., M. Casado, S. Djelidi, Y. Barrandon, and N. Farman. 1999. Epithelial sodium channel in human epidermal keratinocytes: expression of its subunits and relation to sodium transport and differentiation. J. Cell Sci. 112:3343–3352. [DOI] [PubMed] [Google Scholar]
- Chen, L.M., X. Zhang, and K.X. Chai. 2004. Regulation of prostasin expression and function in the prostate. Prostate. 59:1–12. [DOI] [PubMed] [Google Scholar]
- Chen, Y.-H., C. Merzdorf, D.L. Paul, and D.A. Goodenough. 1997. COOH terminus of occludin is required for tight junction barrier function in early Xenopus embryos. J. Cell Biol. 138:891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doering, T., W.M. Holleran, A. Potratz, G. Vielhaber, P.M. Elias, K. Suzuki, and K. Sandhoff. 1999. a. Sphingolipid activator proteins are required for epidermal permeability barrier function. J. Biol. Chem. 274:11038–11045. [DOI] [PubMed] [Google Scholar]
- Doering, T., R.L. Proia, and K. Sandhoff. 1999. b. Accumulation of protein-bound epidermal glucosylceramides in beta-glucocerebrosidase-deficient type 2 Gaucher mice. FEBS Lett. 447:167–170. [DOI] [PubMed] [Google Scholar]
- Doering, T., H. Brade, and K. Sandhoff. 2002. Sphingolipid metabolism during epidermal barrier development in mice. J. Lipid Res. 43:1727–1733. [DOI] [PubMed] [Google Scholar]
- Dale, B.A., and E. Kam. 1993. Harlequin ichthyosis. Variability in expression and hypothesis for disease mechanism. Arch. Dermatol. 129:1471–1477. [DOI] [PubMed] [Google Scholar]
- Ekholm, E., and T. Egelrud. 1998. The expression of stratum corneum chymotrypsic enzyme in human anagen hair follicles: further evidence for its involvement in desquamation-like processes. Br. J. Dermatol. 139:585–590. [DOI] [PubMed] [Google Scholar]
- Elias, P.M. 2004. The epidermal permeability barrier: from the early days at Harvard to emerging concepts. J. Invest. Dermatol. 122:xxxvi–xxxix. [DOI] [PubMed] [Google Scholar]
- Furuse, M., M. Hata, K. Furuse, Y. Yoshida, A. Haratake, Y. Sugitani, T. Noda, A. Kubo, and S. Tsukita. 2002. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1–deficient mice. J. Cell Biol. 156:1099–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guipponi, M., G. Vuagniaux, M. Wattenhofer, K. Shibuya, M. Vazquez, L. Dougherty, N. Scamuffa, E. Guida, M. Okui, and C. Rossier, et al. 2002. The transmembrane serine protease (TMPRSS3) mutated in deafness DFNB8/10 activates the epithelial sodium channel (ENaC) in vitro. Hum. Mol. Genet. 11:2829–2836. [DOI] [PubMed] [Google Scholar]
- Hara-Chikuma, M., J. Takeda, M. Tarutani, Y. Uschida, W.M. Holleran, Y. Endo, P.M. Elias, and S. Inoue. 2004. Epidermal-specific defect of GPI anchor in pig-a null mice results in harlequin ichthyosis-like features. J. Invest. Dermatol. 123:464–469. [DOI] [PubMed] [Google Scholar]
- Herrmann, T., F. Van der Hoeven, H.-J. Gröne, A.F. Stewart, L. Langbein, I. Kaiser, G. Liebisch, I. Gosch, F. Buchkremer, and W. Drobnik, et al. 2003. Mice with targeted disruption of the fatty acid transport protein 4 (Fatp 4, Slc27a4) gene show features of lethal restrictive dermopathy. J. Cell Biol. 161:1105–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughey, R.P., G.M. Mueller, J.B. Bruns, C.L. Kinlough, P.A. Poland, K.L. Harkleroad, M.D. Carattino, and T.R. Kleyman. 2003. Maturation of the epithelial Na+ channel involves proteolytic processing of the alpha- and gamma-subunits. J. Biol. Chem. 278:37073–37082. [DOI] [PubMed] [Google Scholar]
- Hughey, R.P., J.B. Bruns, C.L. Kinlough, K.L. Harkleroad, Q. Tong, M.D. Carattino, J.P. Johnson, J.D. Stockand, and T.R. Kleyman. 2004. Epithelial sodium channels are activated by furin-dependent proteolysis. J. Biol. Chem. 279:18111–18114. [DOI] [PubMed] [Google Scholar]
- Karelina, T.V., G.A. Bannikov, and A.Z. Eisen. 2000. Basement membrane zone remodeling during appendageal development in human fetal skin. The absence of type VII collagen is associated with gelatinase-A (MMP2) activity. J. Invest. Dermatol. 114:371–375. [DOI] [PubMed] [Google Scholar]
- Kinoshita, T., N. Inoue, and J. Takeda. 1995. Defective glycosyl phosphatidylinositol anchor synthesis and paroxysmal nocturnal hemoglobinuria. Adv. Immunol. 60:57–103. [DOI] [PubMed] [Google Scholar]
- Koch, P.J., P.A. de Viragh, E. Scharer, D. Bundman, M.A. Longley, J. Bickenbach, Y. Kawachi, Y. Suga, Z. Zhou, and M. Huber, et al. 2000. Lessons from loricrin-deficient mice: compensatory mechanisms maintaining skin barrier function in the absence of a major cornified envelope protein. J. Cell Biol. 151:389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakso, M., J.G. Pichel, J.R. Gorman, B. Sauer, Y. Okamoto, E. Lee, F.W. Alt, and H. Westphal. 1996. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc. Natl. Acad. Sci. USA. 93:5860–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M., H. Chiba, X. Warot, N. Messaddeq, C. Gerard, P. Chambon, and D. Metzger. 2001. RXT-alpha ablation in skin keratinocytes results in alopecia and epidermal alterations. Development. 128:675–688. [DOI] [PubMed] [Google Scholar]
- List, K., C.C. Haudenschild, R. Szabo, W. Chen, S.M. Wahl, W. Swaim, L.H. Engelholm, N. Behrendt, and T.H. Bugge. 2002. Matriptase/MT-SP1 is required for postnatal survival, epidermal barrier function, hair follicle development, and thymic homeostasis. Oncogene. 21:3765–3779. [DOI] [PubMed] [Google Scholar]
- List, K., R. Szabo, P.W. Wertz, J. Segre, C.C. Haudenschild, S.-Y. Kim, and T.H. Bugge. 2003. Loss of proteolytically processed filaggrin caused by epidermal deletion of matriptase/MT-SP1. J. Cell Biol. 163:901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macheleidt, O., H.W. Kaiser, and K. Sandhoff. 2002. Deficiency of epidermal protein-bound omega-hydroxyceramides in atopic dermatitis. J. Invest. Dermatol. 119:166–173. [DOI] [PubMed] [Google Scholar]
- Matsuki, M., F. Ymashita, A. Ishida-Yamamoto, K. Yamada, C. Kinoshita, S. Fushiki, E. Ueda, Y. Morishima, K. Tabata, and H. Yasuno, et al. 1998. Defective stratum corneum and early neonatal death in mice lacking the gene for transglutaminase 1(keratinocyte transglutaminase). Proc. Natl. Acad. Sci. USA. 95:1044–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauro, T., M. Guitard, Y. Oda, D. Crumrine, L. Komuves, M. Behne, U. Rassner, P.M. Elias, and E. Hummler. 2002. The ENaC channel is required for normal epidermal differentiation. J. Invest. Dermatol. 118:589–594. [DOI] [PubMed] [Google Scholar]
- Netzel-Arnett, S., J.D. Hooper, R. Szabo, E.L. Madison, J.P. Quigley, T.H. Bugge, and T.M. Antalis. 2003. Membrane anchored serine proteases: a rapidly expanding group of cell surface proteolytic enzymes with potential roles in cancer. Cancer Metastasis Rev. 22:237–258. [DOI] [PubMed] [Google Scholar]
- Oda, Y., A. Imanzahrei, A. Kwong, L. Komuves, P.M. Elias, C. Largman, and T. Mauro. 1999. Epithelial sodium channel are upregulated during epidermal differentiation. J. Invest. Dermatol. 113:796–801. [DOI] [PubMed] [Google Scholar]
- Olivier, R., U. Scherrer, J.-D. Horisberger, B.C. Rossier, and E. Hummler. 2002. Selected contribution: limiting Na(+) transport rate in airway epithelia from alpha-ENaC transgenic mice: a model for pulmonary edema. J. Appl. Physiol. 93:1881–1887. [DOI] [PubMed] [Google Scholar]
- Pearton, D.J., W. Nirunsuksiri, A. Rehemtulla, W.P. Lewis, R.B. Presland, and B.A. Dale. 2001. Proprotein convertase expression and localization in epidermis: evidence for multiple roles and substrates. Exp. Dermatol. 10:193–203. [DOI] [PubMed] [Google Scholar]
- Pierard, G.E., V. Goffin, T. Hermanns-Le, and C. Pierard-Franchimont. 2000. Corneocyte desquamation. Int. J. Mol. Med. 6:217–221. [DOI] [PubMed] [Google Scholar]
- Planes, C., C. Leyvraz, T. Uchida, M.A. Angelova, G. Vuagniaux, E. Hummler, M. Matthay, C. Clerici, and B.C. Rossier. 2005. In vitro and in vivo regulation of transepithelial lung alveolar sodium transport by serine proteases. Am. J. Physiol. Lung Cell. Mol. Physiol. 288:L1099–L1109. [DOI] [PubMed] [Google Scholar]
- Rawlings, A.V., and C.R. Harding. 2004. Moisturization and skin barrier function. Dermatol. Ther. 17:43–48. [DOI] [PubMed] [Google Scholar]
- Reichelt, J., B. Breiden, K. Sandhoff, and T.M. Magin. 2004. Loss of keratin 10 is accompanied by increased sebocyte proliferation and differentiation. Eur. J. Cell Biol. 83:747–759. [DOI] [PubMed] [Google Scholar]
- Resing, K.A., K.A. Walsh, and B.A. Dale. 1984. Identification of two intermediates during processing of profilaggrin to filaggrin in neonatal mouse epidermis. J. Cell Biol. 99:1372–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resing, K.A., C. Thulin, K. Whiting, N. Al-Alawi, and S. Mostad. 1995. Characterization of profilaggrin endoproteinase 1. J. Biol. Chem. 270:28193–28198. [DOI] [PubMed] [Google Scholar]
- Rossier, B.C. 2004. The epithelial sodium channel: activation by membrane-bound serine proteases. Proc. Am. Thorac. Soc. 1:4–9. [DOI] [PubMed] [Google Scholar]
- Rubera, I., E. Meier, G. Vuagniaux, A.-M. Mérillat, F. Beermann, B.C. Rossier, and E. Hummler. 2002. A conditional allele at the mouse channel activating protease 1 (Prss8) gene locus. Genesis. 32:173–176. [DOI] [PubMed] [Google Scholar]
- Saitou, M., M. Furuse, H. Sasaki, J.-D. Schulzke, M. Fromm, H. Takano, T. Noda, and S. Tsukita. 2000. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol. Biol. Cell. 11:4131–4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segre, J. 2003. Complex redundancy to build a simple epidermal permeability barrier. Curr. Opin. Cell Biol. 15:776–782. [DOI] [PubMed] [Google Scholar]
- Segre, J.A., C. Bauer, and E. Fuchs. 1999. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat. Genet. 22:356–360. [DOI] [PubMed] [Google Scholar]
- Shipway, A. H. Danahay, J.A. Williams, D.C. Tully, B.J. Backes, and J.L. Harris. 2004. Biochemical characterization of prostasin, a channel activating protease. Biochem. Biophys. Comm. 324:953–963. [DOI] [PubMed] [Google Scholar]
- Sonoda, T., Y. Asada, S. Kurata, and S. Takayasu. 1999. The mRNA for protease nexin-1 is expressed in human dermal papilla cells and its level is affected by androgen. J. Invest. Dermatol. 113:308–313. [DOI] [PubMed] [Google Scholar]
- Takahashi, S., S. Suzuki, S. Inaguma, Y. Ikeda, Y.-M. Cho, N. Hayashi, T. Inoue, Y. Sugimura, N. Nishiyama, and T. Fujita, et al. 2003. Down-regulated expression of prostasin in high-grade or hormone-refractory human prostate cancers. Prostate. 54:187–193. [DOI] [PubMed] [Google Scholar]
- Takeuchi, T., J.L. Harris, W. Huang, K.W. Yan, S.R. Coughlin, and C.S. Craik. 2000. Cellular localization of membrane-type serine protease I and identification of protease-activated receptor-2 and single-chain urokinase-type plasminogen activator as substrates. J. Biol. Chem. 275:26333–26342. [DOI] [PubMed] [Google Scholar]
- Tsuruta, D., K.J. Green, S. Getsios, and C.R. Jones. 2002. The barrier function of skin: how to keep a tight lid on water loss. Trends Cell Biol. 12:355–357. [DOI] [PubMed] [Google Scholar]
- Vallet, V., A. Chraibi, H.-P. Gaeggeler, J.-D. Horisberger, and B.C. Rossier. 1997. An epithelial serine protease activates the amiloride-sensitive sodium channel. Nature. 389:607–610. [DOI] [PubMed] [Google Scholar]
- Vuagniaux, G., V. Vallet, N. Jaeger-Fowler, M. Bens, N. Farman, N. Courtois-Coutry, A. Vandewalle, B.C. Rossier, and E. Hummler. 2000. Activation of the amiloride-sensitive epithelial sodium channel by the serine protease mCAP1 expressed in a mouse cortical collecting duct cell line. J. Am. Soc. Nephrol. 11:828–834. [DOI] [PubMed] [Google Scholar]
- Vuagniaux, G., V. Vallet, N.F. Jaeger, E. Hummler, and B.C. Rossier. 2002. Synergistic activation of ENaC by three membrane-bound channel activating serine proteases (mCAP1, mCAP2 and mCAP3) and serum- and glucocorticoid-regulated kinase (Sgk1) in Xenopus oocytes. J. Gen. Physiol. 120:191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan, H., H.L. Winton, C. Soeller, E.R. Tovey, D.C. Gruenert, P.J. Thompson, G.A. Stewart, G.A. Taylor, D.R. Garrod, M.B. Cannell, and C. Robinson. 1999. Der p 1 facilitates transepithelial allergen delivery by disruption of tight junctions. J. Clin. Invest. 104:123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki, M., K. Ishidoh, Y. Suga, T.C. Saido, S. Kawashima, K. Suzuki, E. Kominami, and H. Ogawa. 1997. Cytoplasmic processing of human profilaggrin by active mu-calpain. Biochem. Biophys. Res. Commun. 235:652–656. [DOI] [PubMed] [Google Scholar]
- Zeeuwen, P.L.J.M., B.A. Dale, G.J. de Jongh, I.M.J.J. Van Vlijmen-Willems, P. Fleckman, J.R. Kimball, K. Stephens, and J. Schalkwijk. 2003. The human cystatin M/E gene (CST6): exclusion candidate gene for harlequin ichthyosis. J. Invest. Dermatol. 121:65–68. [DOI] [PubMed] [Google Scholar]