

Abstract
Phagocyte recognition and clearance of bacteria play essential roles in the host response to infection. In an on-going forward genetic screen, we identify the Drosophila melanogaster scavenger receptor Croquemort as a receptor for Staphylococcus aureus, implicating for the first time the CD36 family as phagocytic receptors for bacteria. In transfection assays, the mammalian Croquemort paralogue CD36 confers binding and internalization of Gram-positive and, to a lesser extent, Gram-negative bacteria. By mutational analysis, we show that internalization of S. aureus and its component lipoteichoic acid requires the COOH-terminal cytoplasmic portion of CD36, specifically Y463 and C464, which activates Toll-like receptor (TLR) 2/6 signaling. Macrophages lacking CD36 demonstrate reduced internalization of S. aureus and its component lipoteichoic acid, accompanied by a marked defect in tumor necrosis factor-α and IL-12 production. As a result, Cd36 −/− mice fail to efficiently clear S. aureus in vivo resulting in profound bacteraemia. Thus, response to S. aureus requires CD36-mediated phagocytosis triggered by the COOH-terminal cytoplasmic domain, which initiates TLR2/6 signaling.
Introduction
Phagocytosis plays an important role in first line host defense. Phagocytic cells, such as macrophages and dendritic cells, must efficiently differentiate “infectious nonself” from “noninfectious self” and initiate an appropriate inflammatory response (Hoffmann et al., 1999). Central to this process is the expression of pattern recognition receptors, such as the scavenger receptors (SRs), which bind a broad array of modified and foreign ligands (Mukhopadhyay and Gordon, 2004). The B class SR CD36 is an 88-kD glycoprotein expressed by myeloid cells, platelets, endothelial cells, erythroid precursors, and adipocytes (Febbraio et al., 2001). This multifunctional receptor mediates homeostatic functions, including adhesion to thrombospondin and collagen, transport of long-chain fatty acids, and inhibition of angiogenesis (Febbraio et al., 2001). CD36 also shares with its Drosophila melanogaster paralogue, Croquemort, the ability to recognize apoptotic cells (Savill et al., 1992; Franc et al., 1999). In addition to these scavenging functions, CD36 has been implicated in the innate immune response to modified host ligands such as oxidized lipoproteins and fibrillar β-amyloid, as well as foreign antigens such as the Pfemp antigen of Plasmodium falciparum (Ockenhouse and Chulay, 1988; Oquendo et al., 1989; Endemann et al., 1993; Moore et al., 2002; Medeiros et al., 2004). Intriguingly, human CD36 mutations predicted to result in the loss of the COOH-terminal transmembrane domain and cytoplasmic tail (T1264G and G1439C) are relatively common in certain African and Asian populations (occurring at high frequency between 0.5–2%), but are extremely uncommon in Caucasians (Aitman et al., 2000). This suggests that selection pressures exist to maintain or eliminate CD36 mutations within certain populations, and these may include interactions with pathogens. Two such interactions may be CD36-mediated recognition of P. falciparum in malaria and a more recently identified role as a candidate receptor for lipoteichoic acid (LTA), a cell wall component of the Gram-positive bacterium Staphylococcus aureus (Hoebe et al., 2005). However, the mechanism by which CD36 participates in the response to pathogens and their ligands and the consequences of CD36 mutations on the host defense have not been defined.
Mammalian pattern recognition receptors have evolved to identify highly conserved pathogen-associated molecular patterns essential to microbes' existence or pathogenicity, including bacterial lipopolysaccharide (LPS), LTA, peptidoglycan, bacterial CpG-containing DNA, double-stranded RNA, or mannans. A family of receptors related to Toll, a Drosophila receptor involved in dorsal ventral patterning, have emerged as key regulators in the host response to invading pathogens (Akira and Takeda, 2004). The mammalian Toll-like receptor (TLR) family has 10 members, which each recognize a distinct subset of microbial molecules from bacteria, viruses, fungi, and protozoa to activate common signaling pathways that initiate the innate immune response (Akira and Takeda, 2004). The best-characterized TLR is perhaps TLR4, which cooperates with the glycosylphosphatidylinositol-anchored protein CD14 to mediate the signaling response to bacterial LPS (Akira and Takeda, 2004). Although the exact mechanism has yet to be defined, CD14 is believed to cluster LPS at the cell surface where it can deliver this ligand to TLR4. A precedent is emerging for similar cooperation between TLRs and other molecules. As an example, dectin-1, a C-type lectin phagocytic receptor, has been shown to cooperate with TLR2 in mediating response to zymosan (Gantner et al., 2003). Additionally, Rac1, a small Rho-GTPase involved in phagocytosis, has been shown to be required for TLR2-mediated activation of NFκB by S. aureus (Arbibe et al., 2000). Intriguingly, although many TLRs are found on the cell surface, members of this family also localize to endosomes, lysosomes, or the Golgi apparatus (Underhill et al., 1999; Underhill and Gantner, 2004), raising the possibility that these cooperative interactions exist to facilitate ligand delivery to the appropriate compartment after endocytosis or phagocytosis for efficient activation of the immune response.
Here, we show that CD36 and its Drosophila paralogue Croquemort have a similar role in tethering bacteria and derived ligands. This recognition plays an important role in initiating macrophage cytokine production in response to S. aureus, its cell wall component LTA, and, to a lesser extent, Gram-negative E. coli and its derived ligand, LPS. We demonstrate that the COOH-terminal cytoplasmic domain, which is lost in several of the common human mutations of CD36, is required for bacterial internalization and cooperation with TLR2, suggesting that individuals carrying these mutations may be more susceptible to S. aureus infection. Mice deficient in CD36 demonstrate a marked defect in the host response to S. aureus but not E. coli. A combined defect in cytokine response and bacterial phagocytosis in CD36 null mice results in profound bacteraemia and the formation of tissue abscesses. We suggest that like CD14, CD36 functions as an accessory receptor at the cell surface to present bacterial ligands to their cognate TLRs to initiate signaling (Lien et al., 1999; Schwandner et al., 1999; Morr et al., 2002). Furthermore, through its ability to internalize S. aureus and LTA, CD36 also delivers ligands to TLRs recruited to the phagosome/endosome (Underhill et al., 1999).
Results
The Drosophila CD36 paralogue Croquemort is a receptor for S. aureus
In an on-going high-throughput RNA interference (RNAi) screen for Drosophila genes involved in phagocytosis, we identified that the SR Croquemort showed a specific inhibition of phagocytosis of S. aureus but not E. coli (Fig. 1 a). Drosophila S2 cells treated with RNAi to target Croquemort showed a 35% decrease in their ability to internalize S. aureus, whereas targeting of an irrelevant control gene (GFP) showed no effect on bacterial internalization (unpublished data). Mammalian Rac has been shown to have an important role in responses to S. aureus (Arbibe et al., 2000). Therefore, we tested whether Rho-GTPase family members played a role in S. aureus uptake in Drosophila. Silencing of Rac2, but not Rac1, resulted in a specific defect in S. aureus internalization comparable to that seen with Croquemort inhibition, providing circumstantial evidence that Croquemort may activate this Rho-GTPase to mediate downstream phagocytosis. These results suggest that Croquemort and Rac2 play nonredundant roles in the recognition and internalization of S. aureus in Drosophila. However, lethality of Croquemort deficient flies precluded further study of the role of this molecule in innate immune defense in this invertebrate system.
Figure 1.

The Drosophila protein Croquemort and its mammalian paralogue CD36 are receptors for S. aureus. (a) Croquemort and the small GTPase Rac2 are specifically required for S. aureus binding and uptake. RNAi silencing of Croquemort (Crq) and Rac2 in Drosophila S2 cells reduced phagocytosis of S. aureus (left), but not E. coli (right). *, P ≤ 0.05; **, P ≤ 0.001; significantly different from control. (b and c) Expression of CD36 confers cellular binding and internalization of S. aureus and E. coli. (b) HEK293T-CD36 expressing cells (CD36; filled bars) demonstrate a threefold increase in binding of FITC-labeled S. aureus and a twofold increase in binding of FITC-labeled E. coli over vector-transfected cells (Mock; open bars). *, P ≤ 0.05. (c) Expression of CD36 by HEK293 cells confers a threefold increase in internalization of both FITC-labeled S. aureus and E. coli, as compared with vector-transfected cells (Mock; open bars). *, P ≤ 0.05.
CD36 transfection confers both binding and internalization of S. aureus and E. coli
The mammalian SR CD36 is paralogous to Croquemort, and these receptors share the ability to bind apoptotic cells, indicating that they have overlapping ligand specificities (Savill et al., 1992; Franc et al., 1999). Therefore, our observation that Croquemort mediated engulfment of S. aureus raised the possibility that CD36 might also function as a mammalian receptor for Gram-positive bacteria. Thus, to test the ability of CD36 to phagocytose bacteria, we measured binding and internalization of fluorescently labeled S. aureus and E. coli in human embryonic kidney (HEK) 293T cells transfected with a murine CD36 cDNA. Expression of CD36 conferred a threefold increase in binding of S. aureus and a twofold increase with E. coli over mock transfected control cells (Fig. 1 b). Furthermore, expression of CD36 resulted in a threefold increase in cellular internalization of bacteria (Fig. 1 c), indicating that CD36 acts as a phagocytic receptor for both Gram-positive and, to a lesser extent, Gram-negative bacteria.
Tyrosine 463 within the COOH-terminal cytoplasmic tail of CD36 is required to signal engulfment
The COOH-terminal cytoplasmic tail of CD36 is lacking in several common human CD36 mutations (Aitman et al., 2000), and this domain is postulated to interact with signaling molecules (Huang et al., 1991; Bull et al., 1994; Jimenez et al., 2000; Moore et al., 2002). Thus, we addressed the consequences of the lack of the COOH-terminal region on phagocytosis of S. aureus using structure–function analysis. To determine the minimum requirements for CD36 to signal to the actin cytoskeleton and trigger engulfment, HEK293T cells were transfected either with wild-type CD36 (CD36WT) or with a mutant form of CD36 in which the 11 COOH-terminal cytoplasmic amino acid residues had been replaced by alanines (CD36Ala; Fig. 2 a). Equivalent cell surface expression of CD36WT and CD36Ala was confirmed by flow cytometry (Fig. 2 b) and bacterial phagocytosis was measured. Importantly, mutant constructs demonstrated a ≤3% difference in cell surface expression in all experiments, indicating that the tail was not required for cell surface localization. Transfection of CD36WT and CD36Ala resulted in a twofold increase in bacterial binding; however, only CD36WT conferred S. aureus phagocytosis, suggesting that the COOH-terminal tail of CD36 is required to signal the actin cytoskeleton (Fig. 2 c). Included in these COOH-terminal amino acids is a tyrosine residue which, as a potential site of phosphorylation, could provide a docking site for SH2 domain–containing proteins, such as the Src family of tyrosine kinases and the actin regulator p130Cas. Thus, to further define the region responsible for signaling internalization, we mutated this tyrosine at position 463 to phenylalanine (CD36Y463F). Cells transfected with the CD36Y463F mutant bound S. aureus, but, similar to CD36Ala, showed an impairment of phagocytosis, indicating that tyrosine 463 is essential for CD36-mediated bacterial internalization (Fig. 2 c). Together, these data indicate that the COOH-terminal cytoplasmic tail of CD36 regulates receptor-mediated internalization of S. aureus and implicates tyrosine 463 in signaling to the actin cytoskeleton. Thus, we would predict that the reported human CD36 mutations would also be unable to trigger internalization in response to this organism.
Figure 2.

Tyrosine 463 within the COOH-terminal cytoplasmic tail of CD36 is required to signal engulfment of S. aureus . (a) Schematic drawing of CD36 demonstrating the amino acid sequence of the COOH-terminal tail and specific mutations. COOH-terminal mutations of the murine CD36 cDNA were generated by PCR, verified by sequencing, and used to transfect HEK293T cells. (b) Before use in phagocytosis experiments, equivalent cell surface expression of wild-type CD36 (CD36wt) and CD36 mutants (CD36Ala and CD36Y463F) was verified by flow cytometry analysis. (c) Transfection of CD36 COOH-terminal tail mutants fails to confer internalization (open bars), but permits binding (closed bars) of S. aureus. **, P ≤ 0.005, significantly different from wild-type CD36.
CD36-deficient macrophages demonstrate impaired phagocytosis of S. aureus and cytokine production
To define the consequences of lack of functional CD36 signaling, we used CD36 null mice to evaluate phagocytosis and response to S. aureus, E. coli, and their derived ligands. Cd36 −/− macrophages showed a 40–50% reduction in phagocytosis of S. aureus, but comparable levels of internalization of E. coli relative to wild-type macrophages (Fig. 3, a and c). This suggests a relative specificity for Gram-positive organisms conserved between the Drosophila Croquemort protein and mammalian CD36. LTA is an abundant cell wall component of S. aureus that is released after exposure to antibiotics or leukocytic mediators. To better identify the ligand present on S. aureus that is recognized by CD36, we tested whether CD36 could bind and internalize LTA. In concordance with our findings with whole S. aureus, Cd36 −/− macrophages demonstrated a 60% reduction in their ability to internalize BODIPY-LTA (Fig. 3 b), indicating that CD36 mediates phagocytic/endocytic recognition of this important TLR ligand.
Figure 3.

CD36-deficient macrophages demonstrate impaired phagocytosis of S. aureus and its component LTA. Peritoneal macrophages from wild-type and Cd36 −/− mice were incubated with FITC-S. aureus or FITC-E. coli (10:1 bacteria/cell; panel a) or 10 μg/ml BODIPY-LTA (b), and phagocytosis was measured by flow cytometry in the presence of trypan blue. **, P ≤ 0.005, compared with wild-type macrophages. (c) Representative photographs of wild-type and Cd36 −/− macrophages incubated with FITC-S. aureus (green) and stained with DAPI (blue) to show individual nuclei. Macrophages were incubated with 10:1 bacteria/cell and allowed to internalize bacteria for 30 min at 37°C. Cells were washed 3× in PBS to remove extracellular bacteria and fixed with formaldehyde.
Cd36 −/− macrophages treated with S. aureus for 4 h showed a profound reduction (65–75%) in TNFα and IL-12 protein expression as compared with wild-type macrophages (Fig. 4 a). In addition, the absence of CD36 resulted in a modest 25% decrease in the secretion of TNFα and IL-6 (no IL-12 was detected) in response to E. coli (Fig. 4 b). Together, these data indicate that CD36 plays a larger role in the phagocytic and cytokine response to S. aureus than E. coli. As the bacterial ligands LTA and LPS make major contributions to septic shock through their activation of TLR signaling pathways, we investigated whether CD36 was required for their activation of cytokine responses. Similar to our findings with S. aureus, Cd36 −/− macrophages treated with LTA demonstrated a 60–70% defect in TNFα and IL-12 expression (Fig. 5, a and b). Although Cd36 −/− macrophages showed some reduction in TNFα and IL-12 mRNA in response to LPS (Fig. 5 a), TNFα and IL-12 protein expression was not reduced (Fig. 5 b), militating against a general inability of these cells to respond to proinflammatory ligands. Together, these data demonstrate that CD36 mediates not only phagocytic/endocytic recognition of LTA but also is required to initiate the cytokine response to this ligand, suggesting that this receptor is an important adjunct to the TLR signaling pathway.
Figure 4.

Cd36 −/− macrophages demonstrate impaired early responses to whole S. aureus and, to a lesser extent, E. coli. Wild-type and Cd36 −/− macrophages were treated with 10:1 S. aureus (a) or E. coli (b) for 4 h, and TNFα, IL-12, and IL-6 were measured in cell culture supernatants. *, P ≤ 0.05; **, P ≤ 0.005, significantly different from wild-type macrophages.
Figure 5.

CD36 is required for LTA-induced cytokine production. Peritoneal macrophages from wild-type and Cd36 −/− mice were incubated with 100 ng/ml LTA or LPS for 6 h, and cytokine mRNA (a) and protein (b) expression was determined by quantitative RT-PCR and ELISA, respectively. *, P ≤ 0.05; **, P ≤ 0.005, significantly different from wild-type macrophages.
CD36-mediated internalization by the COOH-terminal cytoplasmic domain is required for engagement of TLR2/6 signaling
To define the mechanism of CD36 signaling in response to S. aureus and LTA, and to investigate whether CD36 cooperates with TLRs, we used stably transfected HEK293-TLR2 or -TLR9 expressing cell lines transfected with an NFκB-luciferase reporter system. Transfection of either CD36 or TLR6 alone caused a twofold increase in NFκB activation in TLR2 expressing cells (Fig. 6 a). However, cotransfection of these receptors resulted in a 10-fold increase in NFκB activation, indicating that CD36 cooperates with the TLR2/6 heterodimer to mediate LTA signaling. In response to LTA, CD36 did not cooperate with TLR9, either in the presence or absence of TLR6, demonstrating a specificity for the interaction of CD36 with TLR2/6. CD36 was also found to augment TLR2/6 signaling in response to S. aureus, but to a lesser extent than that observed for LTA (Fig. 6 b; 6- vs. 12-fold increase in NFκB activity).
Figure 6.

CD36-mediated internalization by the COOH-terminal cytoplasmic domain is required for engagement of TLR2/6 signaling. (a) CD36 cooperates with TLR2/TLR6 to initiate NFκB activation in response to LTA. HEK293-TLR2 or -TLR9 cells expressing NFκB reporter constructs and CD36, TLR6, or both were stimulated with 1 μg/ml LTA for 5 h, and cell lysates were analyzed for luciferase activity. (b) CD36 initiates TLR2/6-mediated NFκB activation in response to S. aureus and LTA. HEK293-TLR2 cells expressing NFκB reporter constructs and CD36, TLR6, or both were stimulated with 1 μg/ml LTA or S. aureus (10:1) for 5 h, and cell lysates were analyzed for luciferase activity as described in (a). (c) CD36-mediated TLR2/6-NFκB activation requires internalization of LTA. HEK293-TLR2 cells expressing NFκB-luciferase reporter constructs and CD36, TLR6, or both were pretreated with 1 μg/ml cytochalasin D for 30 min before stimulation with LTA as described in (a). (d and e) Mutations that block CD36-mediated internalization of LTA inhibit TLR2/6 signaling. (d) HEK-293 cells were transfected with wild-type CD36 or CD36 mutant constructs and internalization of BODIPY-LTA (10 μg/ml) was measured by flow cytometry in the presence of trypan blue. (e) HEK293-TLR2 cells expressing NFκB reporter constructs and wild-type or mutant CD36 constructs with TLR6 were stimulated with LTA for 5 h, and NFκB-luciferase expression was measured. (f) Cytochalasin D treatment does not block TLR2/6 signaling by CD36 internalization-defective mutants. HEK293-TLR2 cells expressing NFκB reporter constructs, TLR6, and wild-type or mutant CD36 were pretreated with 1 μg/ml cytochalasin D for 30 min before stimulation with LTA, and cell lysates were analyzed for luciferase activity as described in (a). Data presented are the mean of triplicate samples ± SD. **, P ≤ 0.005.
As CD36 has the additional capacity to internalize ligands, we investigated whether CD36-mediated internalization contributes to activation of TLR2/TLR6 signaling by blocking LTA internalization with cytochalasin D in HEK293-TLR2 cells transfected with CD36, TLR6, or both. Inhibition of LTA internalization abrogated CD36-initiated NFκB activation, indicating a requirement for uptake to initiate TLR2/6 signaling (Fig. 6 c). Similar to our findings with S. aureus, the CD36Ala mutation also blocked internalization of LTA, indicating a requirement for the COOH-terminal tail of the receptor for internalization of not only whole bacteria but also of its cell wall component, LTA (Fig. 6 d). To specifically map the requirement for LTA uptake, we mutated Y463, C464, and C466 and found that only mutation of Y463 and C464, but not the distal cysteine C466, inhibited LTA internalization. Having shown that internalization was necessary for signaling, we hypothesized that these mutations would also disrupt CD36 cooperation with TLR2/6. Similar to the inhibition of LTA internalization, mutation of the entire COOH-terminal tail to alanine resulted in a 70% decrease in NFκB activation compared with wild-type CD36. This deficit could be recapitulated by mutation of either Y463 or C464, but not C466, confirming that mutations that block internalization also block NFκB induction (Fig. 6 e). This is supported by the finding that whereas cytochalasin D inhibited signaling by wild-type CD36, no inhibition of this TLR2/6 signaling was observed with CD36Ala or CD36Y463F (Fig. 6 f). Together, these data indicate that CD36-mediated internalization and cooperation with TLR2/6 is required for effective activation of the inflammatory response to LTA.
CD36 null mice fail to clear S. aureus in vivo
To establish the role of CD36 in the innate immune response to bacteria, Cd36 −/− and wild-type mice were infected intravenously with S. aureus and E. coli (Fig. 7 a). When infected with encapsulated S. aureus (Reynolds strain), Cd36 −/− failed to clear the initial bacteraemia, resulting in a mean bacterial load of 3.8 × 109 S. aureus colony forming units per milliliter of blood on day 4 (Fig. 7 b). In contrast, wild-type mice had efficiently cleared the S. aureus and no circulating bacteria were detectable. Furthermore, 20% of Cd36 −/− mice died 6–8 d after inoculation, whereas all wild-type mice survived the infection (Fig. 7 a). Necropsy examination of these mice on day 22 demonstrated that the prolonged early bacteraemia in Cd36 −/− mice resulted in abscess formation in 50% of the animals (Fig. 7 c). In contrast, only 1 of 10 wild-type mice had an apparent abscess of the kidney, indicating that these mice had successfully cleared the bacteria. Thus, loss of CD36-mediated clearance and cytokine signaling appeared to have profound biological consequences on the host response to S. aureus infection. In unencapsulated S. aureus strains, LTA is exposed on the bacterial cell surface. As CD36 directly recognizes LTA, we hypothesized that it may make a greater contribution to host defense against unencapsulated S. aureus. When injected with the unencapsulated S. aureus strain 3825-4 (CP5−), 50% of Cd36 −/− mice succumbed to the infection, whereas all wild-type mice survived (Fig. 7 a). Furthermore, 100% of the surviving Cd36 −/− mice developed perinephric and/or pericardial abscesses. Similar experiments using E. coli failed to demonstrate any significant difference in the survival of wild-type and CD36 null mice, indicating that this was not a nonspecific immune paresis, but rather that CD36 has an important and nonredundant role in combating S. aureus infection, particularly those unencapsulated strains in which LTA is exposed.
Figure 7.
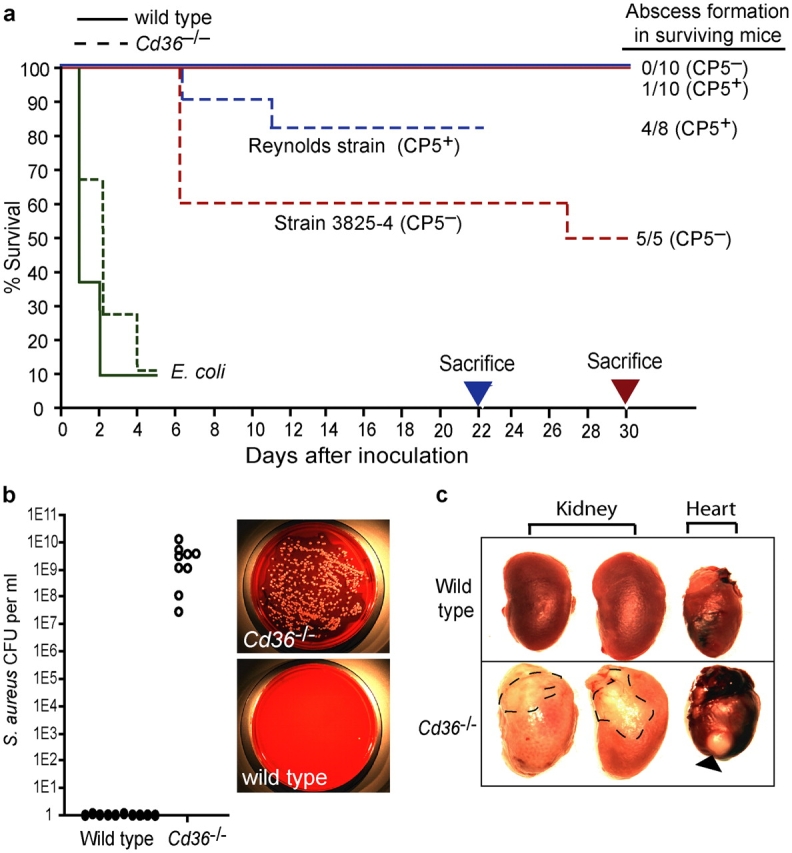
CD36 null mice are more susceptible to S. aureus and fail to clear circulating bacteria. (a) Wild-type and Cd36 −/− mice were inoculated i.v. with S. aureus (capsular strain Reynolds or the unencapsulated 3825-4 strain) or E. coli (018K) and monitored for survival. (b) Cd36−/− mice fail to effectively clear S. aureus. Blood collected from the tail vein after 4 d was cultured on tryptic soy agar plates supplemented with 5% sheep's blood and the mean colony forming units were counted from each mouse. Representative bacterial plates are presented. (c) Cd36−/− mice develop perinephric and pericardial abscesses. Necropsies performed on surviving mice showed abundant abscess formation in the hearts and kidneys of Cd36 −/− mice (absolute numbers reported in panel a). Arrowhead indicates large abscess in heart.
Discussion
In a forward genetic screen, we identify that Croquemort, a Drosophila SR, plays a specific role in the phagocytosis of S. aureus, demonstrating for the first time that the members of this family of CD36-related molecules act as phagocytic receptors for bacteria. We extend this initial observation to confirm that the mammalian paralogue of Croquemort, CD36, also directly phagocytoses bacteria and plays a role in the host immune response to S. aureus. Transfection of cells with CD36 confers the ability to bind and internalize both S. aureus and E. coli; however, macrophages deficient in CD36 are impaired only in their ability to phagocytose S. aureus. Through structure–function analysis, we show that CD36-mediated internalization of S. aureus and LTA is dependent on the COOH-terminal cytoplasmic portion of this receptor proposed to interact with signaling molecules. Mutations of the cytoplasmic residues, specifically tyrosine 463, abolish CD36-mediated phagocytosis, but not binding, indicating that this domain is essential to signal to the actin cytoskeleton and trigger engulfment. Furthermore, in the absence of CD36, macrophages show a profound defect in cytokine production in response to both S. aureus and its cell wall component (LTA). In vivo, CD36 null mice fail to efficiently clear S. aureus, resulting in profound bacteraemia and abscess formation. Recent data has identified a role for CD36 in the recognition of diacylglycerides and in the activation of signaling via TLR2 and TLR6 (Hoebe et al., 2005). However, the mechanism by which CD36 augments this response is unknown. We now show that CD36-mediated internalization of LTA, mediated by its cytoplasmic tail, is required to fully activate TLR2/6 signaling. Our data suggest that CD36 functions to both clear circulating bacteria and to bind bacterial ligands where they may be presented to TLR signaling pathways either at the cell surface or the phagosome.
Microbial internalization is accompanied by inflammatory responses that are driven by TLR signaling pathways. Particles are internalized into a membrane-limited organelle, the phagosome, which matures into a highly hydrolytic environment facilitating degradation of internalized bacteria into their component parts. Although TLRs are not believed to function directly as phagocytic receptors, many TLRs are recruited to the newly formed phagosome after particle ingestion, where they are thought to “sample” the phagosome for microbial ligands (Underhill et al., 1999; Underhill and Gantner, 2004). Thus, CD36-mediated delivery of S. aureus to this organelle might facilitate the release of ligands such as LTA from the cell wall, thereby delivering these particles to TLRs to initiate inflammatory signaling and cytokine production. On the cell surface, CD36 would be able to cluster LTA released from S. aureus after exposure to antibiotics or leukocytic mediators, coordinating engagement of TLRs at the cell surface in a manner similar to CD14. An alternative hypothesis is that CD36 may mediate its effects independent of TLR signaling pathways. However, CD36 transfection alone was insufficient to stimulate NFκB activation in response to S. aureus or LTA, suggesting that CD36 cooperates with TLRs to initiate NFκB signaling. The majority of the CD36 protein forms an extracellular loop containing distinct ligand binding domains, and its short NH2- and COOH-terminal cytoplasmic tails are palmitoylated, localizing this receptor to lipid rafts (Tao et al., 1996). These domains are rich in signaling molecules, and CD36 has been shown to interact with Src kinase family members that reside in lipid rafts to engage downstream signaling cascades, including p38 and p44/42 MAPK pathways (Huang et al., 1991; Jimenez et al., 2000; McGilvray et al., 2000; Moore et al., 2002; Bamberger et al., 2003; Medeiros et al., 2004). Recent work has highlighted the importance of MAPK pathways in regulating endocytosis, phagocytosis, and phagosome maturation in both macrophages and dendritic cells (Blander and Medzhitov, 2004; West et al., 2004). Thus, it is also possible that CD36 regulation of MAPK pathways contributes to augment S. aureus– and LTA-induced cytokine production. However, the CD36 residues vital for augmentation of TLR2/6 signaling in response to LTA (Y463 and C464) are not required for CD36-mediated activation of MAPK by its other ligands, β-amyloid and oxidized low-density lipoprotein (unpublished data). Together, these data provide compelling evidence that CD36-mediated internalization is an important mechanism by which it augments TLR2/6 signaling.
Phagocyte recognition of foreign or modified ligands, such as bacteria and apoptotic cells, is an essential component of both the innate and adaptive immune responses. Particle recognition and internalization is mediated by a variety of phagocytic receptors including the Fcγ receptor, CR3, the mannose receptor, and members of the SR family, including SR-A, LOX-1, and MARCO (Mukhopadhyay and Gordon, 2004; Underhill and Gantner, 2004). Engagement of these receptors ultimately results in activation of RhoGTPases, including Rac, to induce cytoskeletal reorganization, actin polymerization, and particle internalization (Cox et al., 1997). Although, CD36 and its Drosophila homologue Croquemort have been shown to mediate binding of apoptotic cells (Savill et al., 1992; Ren et al., 1995; Franc et al., 1999), it was unclear whether CD36 alone was sufficient to trigger engulfment. We have recently found that β-amyloid engagement of CD36 leads to assembly of focal adhesion molecules that act upstream of Rac1, including p130cas, Pyk2, and paxillin (unpublished data). These observations lead us to hypothesize that CD36, either alone or in cooperation with a coreceptor, can regulate downstream targets such as Rac and the actin cytoskeleton to mediate internalization. Our finding that expression of CD36 confers both binding and internalization of bacteria supports this role of CD36 as a phagocytic receptor. Furthermore, in Drosophila S2 cells, the absence of Croquemort and Rac2 causes a similar degree of inhibition of S. aureus, but not E. coli phagocytosis, suggesting that these molecules may function in the same pathway to signal engulfment. Importantly, we show that the COOH-terminal cytoplasmic residues of CD36 are essential to trigger phagocytic engulfment, indicating that several of the human CD36 mutations known to lack this region are unlikely to support internalization or activation in response to S. aureus or LTA. However, whether this domain of CD36 interacts directly with the actin cytoskeleton through adaptor molecules such as p130Cas, or whether it recruits coreceptors such as the integrins to perform this function, remains to be determined.
To investigate the in vivo consequences of lack of CD36-mediated phagocytosis and signaling, we used mice with a targeted deletion of CD36. We demonstrate that loss of CD36 has profound biological consequences on the early response to S. aureus infection, particularly in the ability to mount a response to an unencapsulated strain (3825-4) of this pathogen. Cd36 −/− mice exhibit 5–8 log order greater bacterial loads, which ultimately lead to perinephric and pericardial abscess formation. However, although CD36 is essential for early responses to S. aureus and LTA, including macrophage phagocytosis and cytokine responses, alternative pathways appear to compensate to protect the host against S. aureus–induced lethality, indicating that the innate immune system has evolved overlapping mechanisms to combat this important pathogen. Several other molecules have been implicated in the immune response to S. aureus, including the SRs AI and II and soluble opsonins such as mannose-binding lectin, thrombospondin, and LPS-binding protein (Dunne et al., 1994; Haworth et al., 1997; Thomas et al., 2000; Schroder et al., 2003; Mukhopadhyay and Gordon, 2004; Shi et al., 2004), emphasizing the complexity of this in vivo model system.
Interestingly, CD36 did appear to play a small role in response to E. coli, as TNFα and IL-6 secretion by macrophages was decreased in its absence. However, macrophage TNFα production in response to isolated LPS did not require CD36. Importantly, neither E. coli engulfment by macrophages nor in vivo host response was compromised by the absence of CD36, indicating a specific failure to respond to S. aureus rather than a general immune depression. It is likely that in these complex and partially redundant systems, other receptors such as the LPS receptor CD14 and CD11b/CD18 are more important to mediate host cytokine responses to E. coli. Nonetheless, that CD36 binds both Gram-negative and -positive bacteria is entirely consistent with its function as a multi-ligand SR.
The data presented demonstrate that CD36 acts as a phagocytic receptor able to recognize S. aureus and its cell wall component LTA. These data demonstrate that CD36 functions in a capacity analogous to the LPS receptor CD14, clustering LTA at the cell surface where it may engage TLR2 and TLR6. Moreover, CD36 has the additional capacity to internalize whole S. aureus and LTA, concentrating them in the phagosome or endosome where they can also engage TLR signaling pathways. This internalization is vital for full activation of TLR2/6 signaling in response to LTA. Loss of this ability to internalize ligands in individuals with CD36 mutations altering the COOH-terminal cytoplasmic domain would be predicted to impair the host response, rendering these individuals more susceptible to S. aureus infection. These findings provide a novel mechanism likely to be common to many SRs that links host detection of pathogens to engagement of TLR signaling pathways, both at the cell surface and intracellularly, to activate host proinflammatory responses.
Materials and methods
Mice
The Cd36 −/− mice (backcrossed nine generations to C57BL/6J) were generated in our laboratory as described, and Cd36 +/+ mice were generated from Cd36 +/− intercrosses (Moore et al., 2002). All mice were maintained in a pathogen-free facility with free access to rodent chow and water. Experimental procedures were approved by the Massachusetts General Hospital's Subcommittee on Research Animal Care and were conducted in accordance with the US Department of Agriculture Animal Welfare Act and the Public Health Service Policy for the Humane Care and Use of Laboratory Animals.
Peritoneal macrophage culture
Elicited peritoneal macrophages were collected from mice by peritoneal lavage 4 d after i.p. injection with 3% thioglycollate as described previously (Moore et al., 2002) and cultured in DME containing 2% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin overnight at 37°C before use.
Transfection assays
COOH-terminal mutations of the murine CD36 cDNA (Fig. 2 a) were generated by PCR, verified by sequencing, and cloned into the pcDNA3.1 vector containing a CMV promoter. HEK293 EBNA T cells were maintained in DME containing 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin and were cultured in medium lacking penicillin and streptomycin for 24 h before transfection. Cells (6 × 105 cells/well; 6-well plate) were transfected with pcDNA 3.1 plasmids containing wild-type or mutant CD36 cDNAs, or no insert (mock), in OptiMEM containing Lipofectamine 2000 as previously described (Kim et al., 2004). Equivalent expression of wild-type and mutant CD36 was confirmed 48 h after transfection by flow cytometry using a monoclonal anti-murine CD36 antibody (1:650 dilution; Cascade Biologics, Inc.) and an anti–mouse IgA-FITC secondary antibody (1:200 dilution; BD Biosciences) as previously described (Medeiros et al., 2004). The anti-CD36 antibody recognizes an epitope in the extracellular portion of CD36 that is not affected by the mutations generated.
Fluorescent labeling of LTA
LTA preparations from S. aureus (Sigma-Aldrich) were labeled with 4,4-difluoro-1,3-dimethyl-5-(4-methoxyphenyl)-4-bora-3a,4a-diaza-S-indacene-2-propionoyl-tetramethyl-rhodamine (BODIPY-TMR; Molecular Probes Inc.) using the manufacturer's protocol for oligosaccharide labeling as previously described (Levels et al., 2003).
RNAi treatment
Double stranded RNA interference was used to specifically silence genes in Drosophila S2 cells as described previously (Ramet et al., 2002). Drosophila S2 cells were cultured in Schneider's Drosophila media (GIBCO BRL) at 26°C. S2 cells (106) were treated with 15 μg dsRNA for 60 h and viability was confirmed by trypan blue exclusion. The ability of RNAi-treated cells to phagocytose heat-killed FITC-labeled S. aureus and E. coli (Molecular Probes) was measured using flow cytometry as described in the following section and compared with cells treated with irrelevant GFP control dsRNA. dsRNA was generated by in vitro transcription from a PCR amplicon either from cDNA, template plasmids, or PCR products using gene-specific primers flanked by a T7 polymerase site. Primer sequences were as follows: GFP forward T7, CCTACATACCTCGCTCTGC; GFP reverse T7, GCGCTTTCTCATAGCTCAC; Crq forward T7, TTGTACGTAACCAAGCCG; Crq reverse T7, CTTCATTCCACTGACAGCA; Rac1 forward T7, TCAGCTACACGACCAATGCCTTTC; Rac1 reverse T7, CAGATACTTGACCGCTCCGATTTC; Rac2 forward T7, GGCCATCAAGTGTGTGGTTGTGGG; Rac2 reverse T7, CATCGCCAGTCCTTGGGGATAGGT.
Phagocytosis assays
Quantification of binding and uptake of heat-killed FITC-labeled S. aureus and E. coli (both from Molecular Probes) was performed as described previously (Ramet et al., 2002; Shi et al., 2004). In brief, 2 × 105 cells in 48-well plates were cooled to 4°C for 20 min, 2 × 106 bacteria were added for 30 min at 4°C, and then cells were incubated at 37°C (26°C for S2 cells) for 30 min to allow internalization. Cells were placed on ice to stop internalization and were maintained on ice for the remainder of the assay. Total cell-associated fluorescence (binding and uptake) was measured by flow cytometry (10,000 events) using the CellQuest program (Becton Dickinson). Bacterial internalization was measured by quenching extracellular fluorescence with 0.2% trypan blue shortly before analysis. Binding was calculated as the difference between total cell-associated fluorescence and intracellular fluorescence. The amount of phagocytosis (phagocytic index) was expressed as the percentage of fluorescence-positive cells multiplied by the mean fluorescence of these cells. All treatments were performed in triplicate and data are presented as the mean ± SD.
Microscopy
Peritoneal macrophages were allowed to adhere to glass coverslips for 1 h before the addition of bacteria at a ratio of 10:1. FITC-bacteria were spun onto the cells, and phagocytosis was synchronized by first binding on ice for 30 min. Cells were then warmed to 37°C for 30 min. Internalization was stopped by vigorously washing, and cells were fixed using formaldehyde before mounting with Vectashield fluorescent mounting medium containing DAPI stain (Vector Laboratories). Slides were imaged on an inverted fluorescent microscope (model TE 2000-U; Nikon) equipped with a camera (Nikon) using Openlab software (version 4.0.2), original magnification ×40, NA 0.60, at RT. After acquisition, manipulation of brightness, color balance, and contrast was applied uniformly across images using Adobe Photoshop software.
Measurement of cytokine expression
TNFα, IL-12, and IL-6 mRNA levels were measured by real-time quantitative RT-PCR using an iCycler (Bio-Rad Laboratories) as described previously (Medeiros et al., 2004). Total RNA was extracted using Trizol reagent and reverse transcribed using Moloney Murine Leukemia Virus reverse transcriptase. Each reaction contained 3 μl cDNA, 0.3 μM TNFα, IL-12, IL-6, or GAPDH primers, SYBR green (QIAGEN), and HotStarTaq polymerase. PCR conditions were 15 min at 95°C, followed by 30 cycles of 30 s at 95°C, 30 s at 55°C, and 30 s at 72°C. Each sample was analyzed in triplicate and the amount of mRNA in each sample was calculated from a standard curve of known template and normalized to GAPDH. The amount of TNFα, IL-12, and IL-6 protein in cell culture supernatants was measured by ELISA (R&D Systems) as described previously (Shi et al., 2004). Data are presented as the mean of triplicate samples ± SD.
NFκB-luciferase reporter assays
Dual luciferase reporter assays for NFκB activation were performed in HEK293 cells that stably express TLR2 or TLR9 (provided by D. Golenbock, University of Massachusetts Medical School, Worcester, MA) as previously described (Latz et al., 2002). Cells were transfected with a NFκB-luciferase reporter construct and/or pcDNA3.1-CD36 and/or TLR6 cDNAs using lipofectamine 2000 as per the manufacturer's instructions. Before assays, equivalent expression of CD36 and TLR6 was confirmed in all samples. The cells were stimulated with 1 μg/ml LTA or S. aureus (10:1) for 5 h and lysed, and reporter gene activity was measured using the Dual Luciferase Assay Reporter System (Promega) using a plate-reader luminometer. Data were normalized to cellular protein. In some assays, cells were treated with 1 μg/ml cytochalasin D (Sigma-Aldrich) for the duration of the assay. The data shown represent one of at least four separate experiments and are presented as the mean values ± SD of triplicate samples.
Bacterial infection in vivo
S. aureus was grown overnight in Columbia medium with 2% NaCl and resuspended in saline. Wild-type and Cd36 −/− mice (n = 10; 6–8 wk old) were inoculated with S. aureus Reynolds capsular serotype 5 (107), unencapsulated S. aureus 3825-4 strain (2 × 107), or E. coli 018K (5 × 106) by tail vein injection and monitored for survival and complications from infection as described previously (Shi et al., 2004). Moribund animals were killed by CO2 inhalation. In mice injected with S. aureus, blood was collected from the tail vein after 4 and 15 d and mixed with heparin, and serial dilutions of the blood were cultured on tryptic soy agar plates supplemented with 5% sheep's blood to determine the number of S. aureus colony forming units. Necropsies were performed on surviving mice at the end of the study and abscess formation was quantified.
Statistical analysis
Quantitative data are expressed as the mean ± SD of triplicate samples and are representative of at least three separate experiments. The difference between two groups was statistically analyzed by t test or an analysis of variance (one-way ANOVA). A p-value of <0.05 was considered significant. Survival data was analyzed using Log-Rank and Wilcoxon tests using JMP5 software (SAS Institute, Inc.).
Acknowledgments
We would like to thank Dr. Douglas Golenbock for the HEK293-TLR2 and TLR9 expressing cell lines and NFκB reporter system.
This work was supported by the National Institutes of Health (grant R01AG20255 to K.J. Moore), the Ellison Medical Foundation (K.J. Moore), and the Wellcome Trust (Clinician Scientist grant R37631 to L.M. Stuart).
Abbreviations used in this paper: HEK, human embryonic kidney; LPS, lipopolysaccharide; LTA, lipoteichoic acid; RNAi, RNA interference; SR, scavenger receptor; TLR, Toll-like receptor.
References
- Aitman, T.J., L.D. Cooper, P.J. Norsworthy, F.N. Wahid, J.K. Gray, B.R. Curtis, P.M. McKeigue, D. Kwiatkowski, B.M. Greenwood, R.W. Snow, et al. 2000. Malaria susceptibility and CD36 mutation. Nature. 405:1015–1016. [DOI] [PubMed] [Google Scholar]
- Akira, S., and K. Takeda. 2004. Toll-like receptor signalling. Nat. Rev. Immunol. 4:499–511. [DOI] [PubMed] [Google Scholar]
- Arbibe, L., J.P. Mira, N. Teusch, L. Kline, M. Guha, N. Mackman, P.J. Godowski, R.J. Ulevitch, and U.G. Knaus. 2000. Toll-like receptor 2-mediated NF-kappa B activation requires a Rac1-dependent pathway. Nat. Immunol. 1:533–540. [DOI] [PubMed] [Google Scholar]
- Bamberger, M.E., M.E. Harris, D.R. McDonald, J. Husemann, and G.E. Landreth. 2003. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J. Neurosci. 23:2665–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blander, J.M., and R. Medzhitov. 2004. Regulation of phagosome maturation by signals from toll-like receptors. Science. 304:1014–1018. [DOI] [PubMed] [Google Scholar]
- Bull, H.A., P.M. Brickell, and P.M. Dowd. 1994. Src-related protein tyrosine kinases are physically associated with the surface antigen CD36 in human dermal microvascular endothelial cells. FEBS Lett. 351:41–44. [DOI] [PubMed] [Google Scholar]
- Cox, D., P. Chang, Q. Zhang, P.G. Reddy, G.M. Bokoch, and S. Greenberg. 1997. Requirements for both Rac1 and Cdc42 in membrane ruffling and phagocytosis in leukocytes. J. Exp. Med. 186:1487–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunne, D.W., D. Resnick, J. Greenberg, M. Krieger, and K.A. Joiner. 1994. The type I macrophage scavenger receptor binds to gram-positive bacteria and recognizes lipoteichoic acid. Proc. Natl. Acad. Sci. USA. 91:1863–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endemann, G., L.W. Stanton, K.S. Madden, C.M. Bryant, R.T. White, and A.A. Protter. 1993. CD36 is a receptor for oxidized low density lipoprotein. J. Biol. Chem. 268:11811–11816. [PubMed] [Google Scholar]
- Febbraio, M., D.P. Hajjar, and R.L. Silverstein. 2001. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J. Clin. Invest. 108:785–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franc, N.C., P. Heitzler, R.A. Ezekowitz, and K. White. 1999. Requirement for croquemort in phagocytosis of apoptotic cells in Drosophila. Science. 284:1991–1994. [DOI] [PubMed] [Google Scholar]
- Gantner, B.N., R.M. Simmons, S.J. Canavera, S. Akira, and D.M. Underhill. 2003. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J. Exp. Med. 197:1107–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haworth, R., N. Platt, S. Keshav, D. Hughes, E. Darley, H. Suzuki, Y. Kurihara, T. Kodama, and S. Gordon. 1997. The macrophage scavenger receptor type A is expressed by activated macrophages and protects the host against lethal endotoxic shock. J. Exp. Med. 186:1431–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoebe, K., P. Georgel, S. Rutschmann, X. Du, S. Mudd, K. Crozat, S. Sovath, L. Shamel, T. Hartung, U. Zahringer, and B. Beutler. 2005. CD36 is a sensor of diacylglycerides. Nature. 433:523–527. [DOI] [PubMed] [Google Scholar]
- Hoffmann, J.A., F.C. Kafatos, C.A. Janeway, and R.A. Ezekowitz. 1999. Phylogenetic perspectives in innate immunity. Science. 284:1313–1318. [DOI] [PubMed] [Google Scholar]
- Huang, M.M., J.B. Bolen, J.W. Barnwell, S.J. Shattil, and J.S. Brugge. 1991. Membrane glycoprotein IV (CD36) is physically associated with the Fyn, Lyn, and Yes protein-tyrosine kinases in human platelets. Proc. Natl. Acad. Sci. USA. 88:7844–7848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez, B., O.V. Volpert, S.E. Crawford, M. Febbraio, R.L. Silverstein, and N. Bouck. 2000. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat. Med. 6:41–48. [DOI] [PubMed] [Google Scholar]
- Kim, W.S., M.L. Fitzgerald, K. Kang, K.I. Okuhira, S.A. Bell, J.J. Manning, S.L. Koehn, N. Lu, K.J. Moore, and M.W. Freeman. 2004. Abca7 null mice retain normal macrophage phosphatidylcholine and cholesterol efflux activity despite alterations in adipose mass and serum cholesterol levels. J. Biol. Chem. 280:3989–3995. [DOI] [PubMed] [Google Scholar]
- Latz, E., A. Visintin, E. Lien, K.A. Fitzgerald, B.G. Monks, E.A. Kurt-Jones, D.T. Golenbock, and T. Espevik. 2002. Lipopolysaccharide rapidly traffics to and from the Golgi apparatus with the toll-like receptor 4-MD-2-CD14 complex in a process that is distinct from the initiation of signal transduction. J. Biol. Chem. 277:47834–47843. [DOI] [PubMed] [Google Scholar]
- Levels, J.H., P.R. Abraham, E.P. van Barreveld, J.C. Meijers, and S.J. van Deventer. 2003. Distribution and kinetics of lipoprotein-bound lipoteichoic acid. Infect. Immun. 71:3280–3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien, E., T.J. Sellati, A. Yoshimura, T.H. Flo, G. Rawadi, R.W. Finberg, J.D. Carroll, T. Espevik, R.R. Ingalls, J.D. Radolf, and D.T. Golenbock. 1999. Toll-like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. J. Biol. Chem. 274:33419–33425. [DOI] [PubMed] [Google Scholar]
- McGilvray, I.D., L. Serghides, A. Kapus, O.D. Rotstein, and K.C. Kain. 2000. Nonopsonic monocyte/macrophage phagocytosis of Plasmodium falciparum-parasitized erythrocytes: a role for CD36 in malarial clearance. Blood. 96:3231–3240. [PubMed] [Google Scholar]
- Medeiros, L.A., T. Khan, J.B. El Khoury, C.L. Pham, D.M. Hatters, G.J. Howlett, R. Lopez, K.D. O'Brien, and K.J. Moore. 2004. Fibrillar amyloid protein present in atheroma activates CD36 signal transduction. J. Biol. Chem. 279:10643–10648. [DOI] [PubMed] [Google Scholar]
- Moore, K.J., J. El Khoury, L.A. Medeiros, K. Terada, C. Geula, A.D. Luster, and M.W. Freeman. 2002. A CD36-initiated signaling cascade mediates inflammatory effects of beta-amyloid. J. Biol. Chem. 277:47373–47379. [DOI] [PubMed] [Google Scholar]
- Morr, M., O. Takeuchi, S. Akira, M.M. Simon, and P.F. Muhlradt. 2002. Differential recognition of structural details of bacterial lipopeptides by toll-like receptors. Eur. J. Immunol. 32:3337–3347. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay, S., and S. Gordon. 2004. The role of scavenger receptors in pathogen recognition and innate immunity. Immunobiology. 209:39–49. [DOI] [PubMed] [Google Scholar]
- Ockenhouse, C.F., and J.D. Chulay. 1988. Plasmodium falciparum sequestration: OKM5 antigen (CD36) mediates cytoadherence of parasitized erythrocytes to a myelomonocytic cell line. J. Infect. Dis. 157:584–588. [DOI] [PubMed] [Google Scholar]
- Oquendo, P., E. Hundt, J. Lawler, and B. Seed. 1989. CD36 directly mediates cytoadherence of Plasmodium falciparum parasitized erythrocytes. Cell. 58:95–101. [DOI] [PubMed] [Google Scholar]
- Ramet, M., P. Manfruelli, A. Pearson, B. Mathey-Prevot, and R.A. Ezekowitz. 2002. Functional genomic analysis of phagocytosis and identification of a Drosophila receptor for E. coli. Nature. 416:644–648. [DOI] [PubMed] [Google Scholar]
- Ren, Y., R.L. Silverstein, J. Allen, and J. Savill. 1995. CD36 gene transfer confers capacity for phagocytosis of cells undergoing apoptosis. J. Exp. Med. 181:1857–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savill, J., N. Hogg, Y. Ren, and C. Haslett. 1992. Thrombospondin cooperates with CD36 and the vitronectin receptor in macrophage recognition of neutrophils undergoing apoptosis. J. Clin. Invest. 90:1513–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder, N.W., S. Morath, C. Alexander, L. Hamann, T. Hartung, U. Zahringer, U.B. Gobel, J.R. Weber, and R.R. Schumann. 2003. Lipoteichoic acid (LTA) of Streptococcus pneumoniae and Staphylococcus aureus activates immune cells via Toll-like receptor (TLR)-2, lipopolysaccharide-binding protein (LBP), and CD14, whereas TLR-4 and MD-2 are not involved. J. Biol. Chem. 278:15587–15594. [DOI] [PubMed] [Google Scholar]
- Schwandner, R., R. Dziarski, H. Wesche, M. Rothe, and C.J. Kirschning. 1999. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J. Biol. Chem. 274:17406–17409. [DOI] [PubMed] [Google Scholar]
- Shi, L., K. Takahashi, J. Dundee, S. Shahroor-Karni, S. Thiel, J.C. Jensenius, F. Gad, M.R. Hamblin, K.N. Sastry, and R.A. Ezekowitz. 2004. Mannose-binding lectin-deficient mice are susceptible to infection with Staphylococcus aureus. J. Exp. Med. 199:1379–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao, N., S.J. Wagner, and D.M. Lublin. 1996. CD36 is palmitoylated on both N- and C-terminal cytoplasmic tails. J. Biol. Chem. 271:22315–22320. [DOI] [PubMed] [Google Scholar]
- Thomas, C.A., Y. Li, T. Kodama, H. Suzuki, S.C. Silverstein, and J. El Khoury. 2000. Protection from lethal gram-positive infection by macrophage scavenger receptor-dependent phagocytosis. J. Exp. Med. 191:147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underhill, D.M., and B. Gantner. 2004. Integration of Toll-like receptor and phagocytic signaling for tailored immunity. Microbes Infect. 6:1368–1373. [DOI] [PubMed] [Google Scholar]
- Underhill, D.M., A. Ozinsky, A.M. Hajjar, A. Stevens, C.B. Wilson, M. Bassetti, and A. Aderem. 1999. The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature. 401:811–815. [DOI] [PubMed] [Google Scholar]
- West, M.A., R.P. Wallin, S.P. Matthews, H.G. Svensson, R. Zaru, H.G. Ljunggren, A.R. Prescott, and C. Watts. 2004. Enhanced dendritic cell antigen capture via toll-like receptor-induced actin remodeling. Science. 305:1153–1157. [DOI] [PubMed] [Google Scholar]