

Abstract
c-Jun is induced in many neuronal death paradigms. A critical step in c-Jun regulation involves phosphorylation of Ser63/Ser73 located in the NH2-terminal transactivation domain. To determine the importance of this phosphorylation for neuronal apoptosis, we analyzed the sympathetic neurons of mice carrying a mutant c-Jun gene that lacks Ser63/Ser73 phosphorylation sites (jun aa). Trophic factor–deprivation or DNA damage–induced death was significantly delayed in jun aa/aa neurons. Neuronal c-Jun induction was only partially inhibited, demonstrating that phosphorylation of Ser63/73 is not required for c-Jun activation. The inductions of proapoptotic BH3-only proteins, Bim and PUMA/Bbc3, were delayed during neuronal apoptosis in mutant neurons. These results demonstrate that NH2-terminal c-Jun phosphorylation is important, but not necessary, for the induction of proapoptotic genes and neuronal apoptosis. Thus, additional JNK substrates may be critical for neuronal death. As potential mediators, we identified additional nuclear MLK/JNK substrates, including Nup214 subunit of the nuclear pore complex.
Introduction
One of the hallmarks of neuronal cell death is the activation of Jun–NH2-terminal kinase (JNK) pathway and the rapid induction of its downstream target AP-1 transcription factor c-Jun (for review see Ham et al., 2000; Herdegen et al., 1997). c-Jun induction plays a major role in the transcription of several proapoptotic genes, most notably the BH3-only Bcl-2 family member Bim (Harris and Johnson, 2001; Whitfield et al., 2001). Inhibition of AP-1 activity by dominant-negative c-Jun overexpression, neutralizing antibody injection, or genetic deletion retards sympathetic neuronal apoptosis after NGF deprivation (Estus et al., 1994; Ham et al., 1995; Palmada et al., 2002). Moreover, hippocampal neurons carrying a mutant c-Jun gene (jun aa) that lacks the two critical NH2-terminal phosphorylation sites show increased resistance to kainate-induced excitotoxicity (Behrens et al., 1999).
c-Jun transcriptional activity is regulated by JNK-mediated phosphorylation of the NH2-terminal transactivation domain on Ser63 and Ser73 (Kallunki et al., 1996). JNK is part of a sequential kinase-signaling cascade involving three kinases. MAPK kinase activates JNK by dual Tyr/Thr phosphorylation. MAPK kinase activation is mediated by MAPK kinase kinases, including the mixed lineage kinase (MLK) family in neurons. Selective inhibition of the MLK family in sympathetic neurons by K252a analogue, CEP-1347, or dominant-negative MLKs demonstrates that MLKs mediate JNK activation after trophic factor deprivation, UV irradiation, and oxidative stress (Maroney et al., 1999, 2001; Harris et al., 2002).
Although activation of the JNK pathway is critical for regulating neuronal apoptosis, high levels of activated JNKs occur in neurons under normal conditions, indicating that JNK signaling may also be important for other metabolic processes in neurons (Harris et al., 2002; Besirli and Johnson, 2003). JNK isoforms (JNK1-3) are activated differentially in neurons under normal conditions and after a stress stimulus (Coffey et al., 2002). This isoform-specific activation presumably provides signaling specificity through phosphorylation of distinct substrates. Therefore, identification of proteins regulated by JNK-mediated phosphorylation is critical for understanding the consequences of JNK pathway signaling in neurons.
Despite high levels of activated JNKs, neurons contain low levels of c-Jun under normal conditions. During apoptosis, c-Jun is activated by increased transcription as well as JNK-mediated phosphorylation. Although often assumed to be the basis for the requirement for the JNK activity on neuronal death, the importance of c-Jun phosphorylation alone for neuronal apoptosis is not currently known because preventing JNK signaling by small molecule inhibitors CEP-1347 and SP600125 completely blocks both c-Jun transcription and phosphorylation (Besirli and Johnson, 2003). Similarly, dominant-negative overexpression or targeted deletion of c-Jun cannot separate c-Jun phosphorylation from expression. To determine whether c-Jun phosphorylation is a necessary event for neuronal apoptosis to proceed, we analyzed sympathetic neurons isolated from mice that carry a mutant c-jun allele (jun aa). This c-Jun mutant, JunAA, has alanines in the place of serines 63 and 73 and cannot be phosphorylated at these sites in its NH2-terminal transactivation domain. Neurons from JunAA mutant mice indeed showed resistance to both trophic factor deprivation and DNA damage. This resistance correlated with delayed expression of proapoptotic genes after trophic factor deprivation. The resistance of neurons from the JunAA mice, however, was surprisingly modest. In light of the complete protection provided by the JNK pathway inhibition, these data demonstrate that other critical JNK substrates must exist. We identify at least one component of the nuclear pore complex (NPC) as substrate of the JNK pathway during trophic factor deprivation. Moreover, trophic factor deprivation leads to JNK-mediated phosphorylation of additional nuclear proteins. These nuclear JNK pathway targets, including the Nup214 subunit of the NPC, may play important roles in neuronal death.
Results
More sympathetic neurons are isolated from mutant c-Jun mice
In JunAA mice, endogenous c-jun gene (jun +) is replaced by a mutated copy that encodes alanines in positions 63 and 73 instead of serines (jun aa). These mice are healthy and show no overt abnormalities before or after birth, except for a small difference in body weight (Behrens et al., 1999). Mutant mice had superior cervical ganglion (SCG) that migrated to their normal anatomic position and appeared grossly normal. Upon dissection of SCG, the ganglia from mutant animals appeared consistently larger than the wild type. We isolated sympathetic neurons from the SCGs of littermate mice and maintained them in vitro for 5 d. Neurons with the mutant c-jun alleles (jun aa/aa) were indistinguishable from wild-type neurons (jun +/+) under normal culture conditions (Fig. 1 A). JunAA neurons readily extended processes in culture dishes, made connections with each other and survived up to 15 d (latest time analyzed). Consistent with the enlarged ganglia, the number of neurons obtained from the SCGs of JunAA mice was always more than wild-type mice (Fig. 1 B; P < 0.01, t test). These results indicated that c-Jun phosphorylation is dispensable for the development of sympathetic precursors, but may be important for regulating the naturally occurring cell death of sympathetic neurons during target innervation. Our data, however, does not exclude the possibility that there may be other factors contributing to the increased size of SCG in JunAA mice, such as increased proliferation during development.
Figure 1.

Lack of c-Jun phosphorylation increases the number of sympathetic neurons isolated from the SCG. Sympathetic neurons were isolated from the SCGs of newborn littermate mice and grown in NGF-containing medium. (A) Phase-contrast images of sympathetic neurons from two different genotypes. Bar, 40 μm. (B) Total number of sympathetic neurons was determined by counting viable neurons after 6–12 d in culture. Mean of wild-type neurons was set to 100%. More sympathetic neurons were isolated from the SCGs of mutant mice compared with wild-type littermates (*, P < 0.01, t test). Data are represented as mean ± SE from five independent experiments with 4–10 mice.
JunAA neurons are resistant to trophic factor deprivation
We previously showed that inhibiting neuronal c-Jun activity by neutralizing antibody microinjection retards sympathetic neuronal apoptosis induced by NGF deprivation (Estus et al., 1994). Subsequent studies confirmed the importance of c-Jun for neuronal apoptosis by using other techniques such as dominant-negative c-Jun overexpression and targeted deletion of c-Jun in sympathetic neuronal cultures (Ham et al., 1995; Whitfield et al., 2001; Palmada et al., 2002). However, the importance of c-Jun phosphorylation alone for neuronal apoptosis is not currently known. Dominant-negative overexpression or targeted deletion of c-Jun cannot separate c-Jun phosphorylation from expression. Likewise, pharmacological inhibition of the neuronal JNK signaling by small molecule inhibitors CEP-1347 and SP600125 completely blocks both c-Jun transcription and phosphorylation (Besirli and Johnson, 2003), and the phosphorylation of other JNK substrates (see Fig. 8). To separate the potential prodeath effects of NH2-terminal c-Jun phosphorylation from c-Jun expression, we examined sympathetic neurons from wild-type, heterozygous, and homozygous jun aa knock-in mice. 33 h after the removal of NGF, the majority of wild-type neurons were either dead or had lost their phase-bright appearance and the integrity of their processes, indicating that they were dying (Fig. 2 A). In contrast, jun aa/aa–sympathetic neurons at this time point were largely protected from degeneration induced by NGF withdrawal (Fig. 2 A). To confirm this visual observation, we determined the number of sympathetic neurons that survived after defined periods of trophic factor deprivation. 5-d in vitro (DIV)–sympathetic neurons were subjected to trophic factor deprivation by removing NGF from the culture medium. The deprivation was followed by a 5–7-d rescue period, which included the replacement of NGF for neuronal recovery. This survival assay measured the percentage of sympathetic neurons that made the irreversible decision to die and were no longer rescued by the readdition of NGF. About 30% of wild-type neurons were able to withstand 24 h of NGF deprivation (Fig. 2 B). In contrast, ∼60% of jun +/aa and >90% of jun aa/aa–sympathetic neurons survived NGF deprivation during the same period. After 48 to 72 h of NGF removal, most jun +/+ and jun +/aa neurons were dead, but 20–25% of jun aa/aa neurons still remained alive. This time course analysis demonstrated that trophic factor deprivation-induced neuronal apoptosis was delayed by 22–24 h in sympathetic neurons carrying the mutant c-jun allele. Although JunAA neurons were significantly protected, these results also showed that c-Jun NH2-terminal phosphorylation on serines 63 and 73 was not absolutely required for neuronal cell death.
Figure 8.
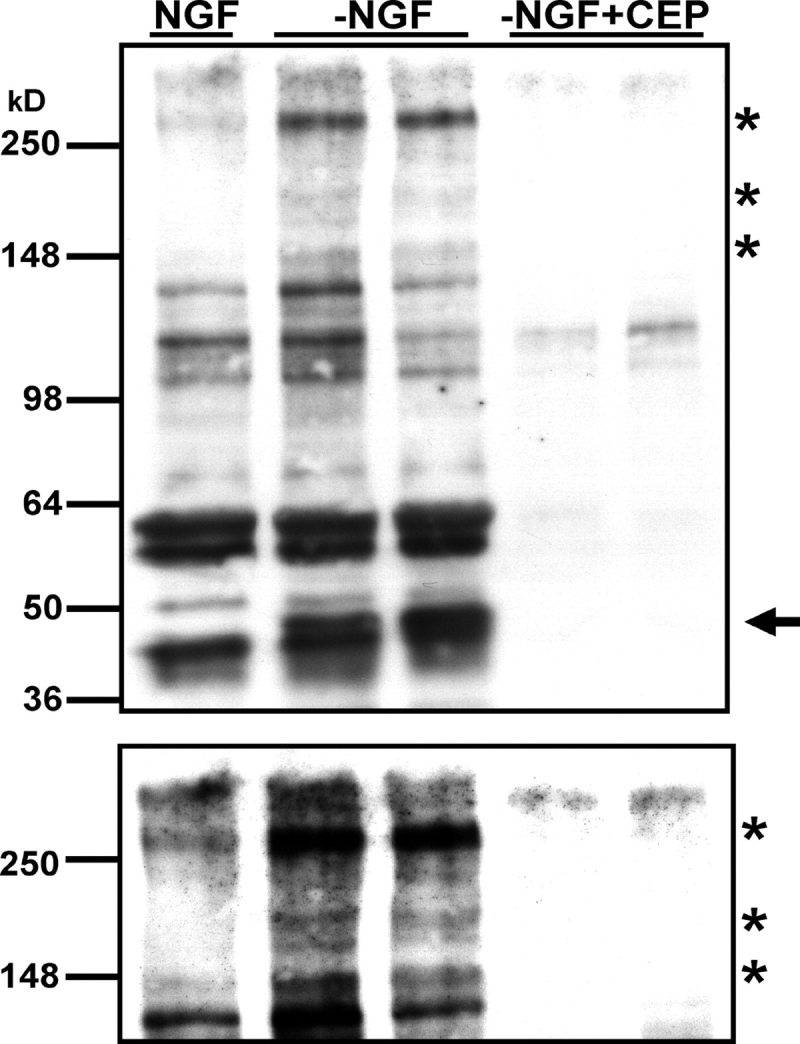
In addition to c-Jun, NGF deprivation induces several other antiphospho-c-Jun Ser73–immunoreactive proteins that are regulated by the JNK pathway. 5-DIV–sympathetic neurons isolated from P0 rats were deprived of NGF (−NGF) or left untreated (NGF) for 24 h in the presence or absence of 1.3 μM CEP-1347 (CEP). Whole cell extracts were subjected to SDS-PAGE, followed by Western analysis with phospho-c-Jun Ser73 antiserum. Arrow indicates the c-Jun protein. Asterisks show other antiphospho-c-Jun immunoreactive proteins. Lower panel is a longer exposure of the same blot. Equal amounts of protein were loaded in each lane (unpublished data).
Figure 2.
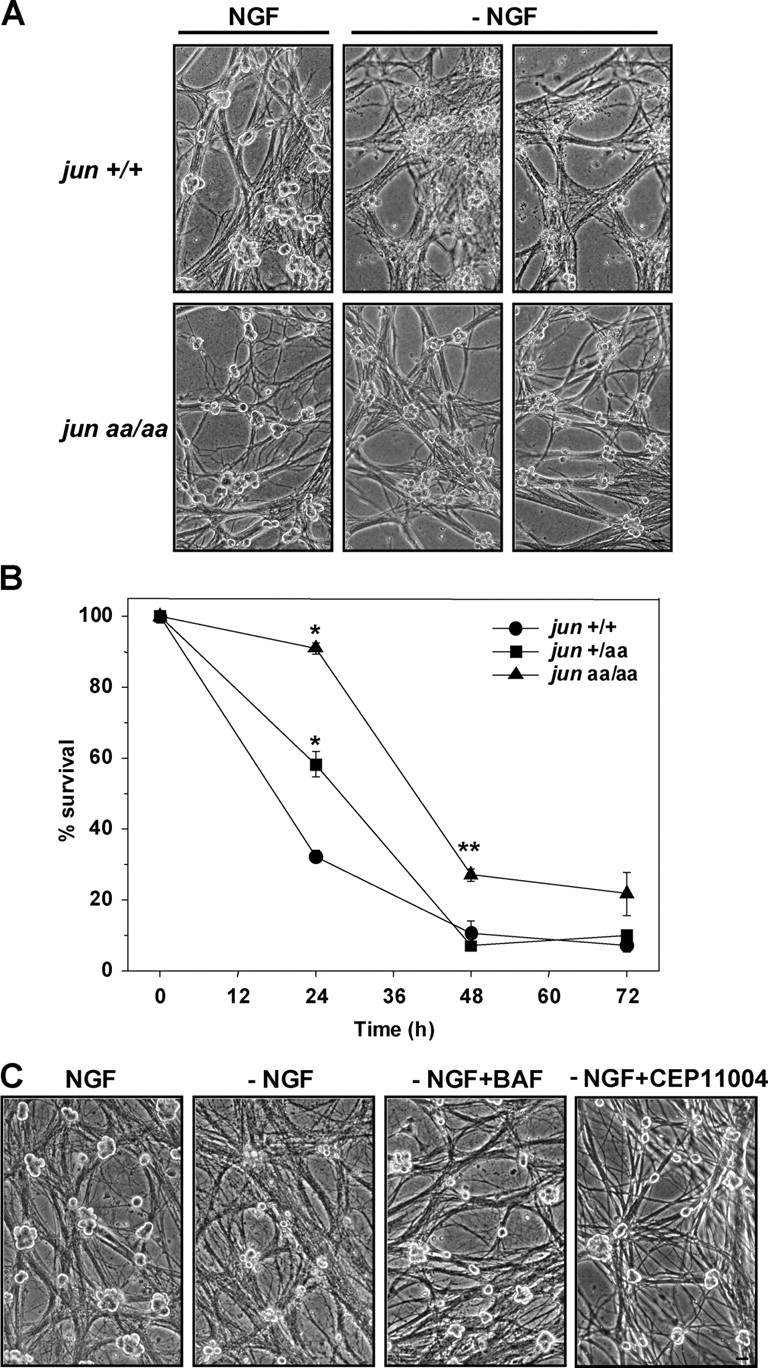
Lack of NH2-terminal c-Jun phosphorylation impairs trophic factor deprivation–induced apoptosis in sympathetic neurons. (A) Phase-contrast images of 5-DIV–sympathetic neurons deprived of NGF (−NGF) for 33 h. Bar, 40 μm. (B) Sympathetic neurons isolated from littermate mice were deprived of NGF, followed by NGF replacement every 24 h for an additional 5–7 d. Cells were fixed after this rescue period and the number of surviving neurons was determined by counting the cells after crystal violet staining. *, P ≤ 0.001, **, P < 0.01, t test, all compared with wild type. Data are represented as mean ± SE from three independent experiments. (C) Sympathetic neurons were isolated from newborn jun aa/aa mice. Medium with no NGF was provided at 5 DIV (−NGF) for 72 h. The medium for sister cultures contained 1.33 μM of selective MLK-inhibitor CEP11004 (−NGF+CEP11004) or 50 μM of pan-caspase inhibitor BAF (−NGF+BAF). Bar, 40 μm.
During NGF deprivation, sympathetic neurons activate the MLK–JNK signaling pathway, which leads to the up-regulation of proapoptotic BH3-only Bcl-2 family members, followed by Bax-mediated cytochrome c release and caspase activation. To determine whether the same cell death pathway is responsible for the death of jun aa/aa neurons, we examined the effect of MLK or caspase inhibition during NGF deprivation. The delayed death seen in jun aa/aa–sympathetic neurons was both MLK and caspase dependent, because neurons deprived of NGF in the presence of selective MLK-inhibitor CEP-11004 or the pan-caspase inhibitor bocaspartyl(OMe)-fluoromethylketone (BAF) did not die (Fig. 2 C). There was no visually detectable death in the presence of CEP-11004 or BAF up to 4 d (latest time point analyzed; unpublished data).
DNA damage–induced neuronal apoptosis is inhibited by c-Jun mutation
To determine whether c-Jun phosphorylation is also important for sympathetic neuronal apoptosis after DNA damage, we exposed jun aa/aa–sympathetic neurons to the DNA-damaging agent Ara-C (Besirli et al., 2003). Wild-type neurons were modestly more susceptible to Ara-C exposure compared with jun +/aa and jun aa/aa neurons (Fig. 3, P < 0.05 for 48 h). Similar results were obtained with another DNA-damaging drug, i.e., topoisomerase II inhibitor etoposide (unpublished data). Similar to sympathetic neurons, DNA damage caused a significant increase in both c-Jun levels and NH2-terminal c-Jun phosphorylation in cerebellar granule neurons (Fig. S1 A available at http://www.jcb.org/cgi/content/full/jcb.200501138/DC1). This induction was comparable to c-Jun activation seen after the removal of trophic factors K+ and serum from cerebellar granule neurons. c-Jun activation during DNA damage–induced sympathetic neuronal death is largely independent of the JNK pathway (Besirli and Johnson, 2003). In contrast, MLK-inhibitor CEP-11004 or JNK-inhibitor SP600125 completely prevented DNA damage–induced c-Jun transcription and phosphorylation in cerebellar granule neurons (Fig. S1 A). In addition, MLK inhibition by CEP-11004 inhibited DNA damage–induced death in cerebellar granule neurons (Fig. S1 B; P < 0.001 at 44 h), in contrast to lack of any prosurvival effect of MLK or JNK inhibitors in DNA-damaged sympathetic neurons. This block was transient, similar to the partial saving effect of MLK inhibition after K+ deprivation of cerebellar granule neurons (Harris and Johnson, 2001). Cerebellar granule neurons isolated from jun aa/aa mice were also more resistant to DNA damage. Treatment with 40 μM etoposide killed 50% of jun +/aa neurons after 44 h, whereas only 35% of jun aa/aa cerebellar granule neurons died at the same time point (Fig. S2; P < 0.01; available at http://www.jcb.org/cgi/content/full/jcb.200501138/DC1). This resistance to DNA damage in mutant neurons was similar to the effect of JNK pathway inhibition by CEP-11004. These results demonstrate that NH2-terminal c-Jun phosphorylation was important, but not absolutely required, for neuronal apoptosis induced by DNA damage.
Figure 3.

DNA damage–induced death is delayed in the absence of NH 2 -terminal c-Jun phosphorylation. 5-DIV–sympathetic neurons from littermate mice were exposed to 100 μM cytosine arabinoside (Ara-C) and cell survival was assessed by counting viable cells after crystal violet staining. Wild-type neurons died faster than the knock-in neurons (*, P < 0.05, t test). Mean ± SE from three independent trials.
NH2-terminal phosphorylation of c-Jun is important in the expression of proapoptotic genes after trophic factor deprivation
Phosphorylation of the c-Jun NH2-terminal transactivation domain greatly enhances AP-1–mediated gene transcription. However, this phosphorylation event is not absolutely required for c-Jun activity, as JunAA protein can induce gene transcription from AP-1 sites in in vitro promoter assays (Behrens et al., 1999). The impaired death of jun aa/aa–sympathetic neurons after NGF deprivation could be explained by a delay in the induction of proapoptotic genes, such as c-Jun itself, as the c-Jun promoter is autoregulated (Angel et al., 1988). Basal expression of c-Jun is low in sympathetic neurons. After a stress stimulus such as trophic factor deprivation, a high level of c-Jun expression is seen in neurons because of an increase in c-jun transcription (Estus et al., 1994; Ham et al., 1995). The basal c-Jun expression was similar in wild-type and mutant neurons (unpublished data). However, when we compared c-Jun expression in jun +/+ and jun aa/aa neurons after NGF removal, we found a consistent delay in c-Jun up-regulation in the mutant-sympathetic neurons (Fig. 4 A).
Figure 4.
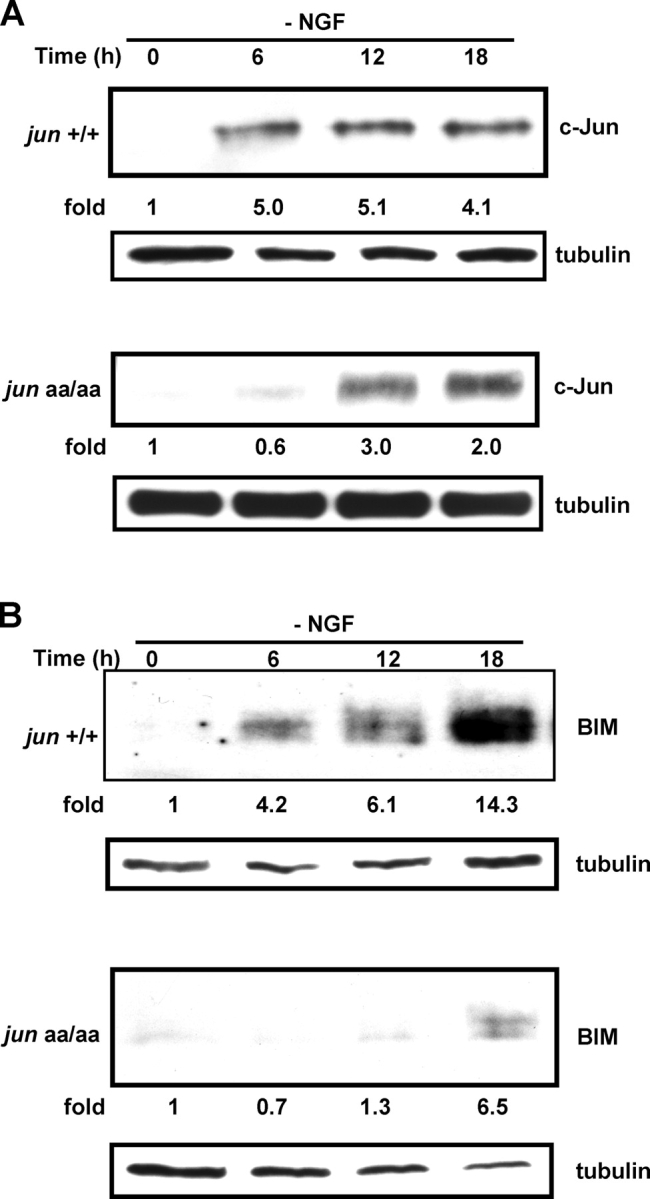
Expression of proapoptotic proteins c-Jun and BIM is inhibited in the absence of NH 2 -terminal c-Jun phosphorylation. Sympathetic neurons from jun +/+ and jun aa/aa mice were deprived of NGF and protein was isolated from the cultures every 6 h for Western blot analysis. Both c-Jun (A) and BIM (B) levels increase more slowly in mutant neurons. The fold induction was calculated by setting time 0 to 1 and normalizing protein levels for loading at each time point by reprobing the blots for tubulin. Similar results were obtained in three independent trials.
Another protein that is regulated by c-Jun is the proapoptotic Bcl-2 family member Bim. Bim is important for sympathetic neuronal apoptosis (Putcha et al., 2001). Pharmacological inhibition of JNK–c-Jun pathway or direct c-Jun inhibition by dominant-negative overexpression significantly reduces Bim induction and retards apoptosis in sympathetic neurons (Harris and Johnson, 2001; Whitfield et al., 2001). Similarly, in the absence of NH2-terminal c-Jun phosphorylation, Bim protein induction was significantly slower in NGF-deprived sympathetic neurons (Fig. 4 B). This result is consistent with incomplete inhibition of Bim expression by JNK pathway inhibitors (Harris and Johnson, 2001).
The BH3-only Bcl-2 family member PUMA is induced in neurons after trophic factor withdrawal and DNA damage
PUMA, first identified as a BH3-only proapoptotic protein from the Bcl-2 family, is activated after cellular stress including DNA damage and serum deprivation (Han et al., 2001; Nakano and Vousden, 2001; Yu et al., 2001). Because two other BH3-only Bcl-2 family members, Bim and DP5, are activated during sympathetic neuronal death, we analyzed protein extracts from NGF-deprived neurons for a potential induction of PUMA. Sympathetic neurons showed a rapid and substantial increase in PUMA expression after NGF deprivation (Fig. 5 A). To determine whether PUMA induction is a generalized neuronal response to trophic factor deprivation, we examined cerebellar granule neurons, which depend on both depolarizing concentrations of potassium and presence of serum for in vitro survival. When potassium was removed from the culture medium of 7-DIV cerebellar granule neurons, PUMA expression was increased by 6 h and remained elevated for 24 h (Fig. 5 B). PUMA is also activated in cells undergoing DNA damage–induced death (Han et al., 2001; Nakano and Vousden, 2001). Exposure to the topoisomerase-II poison etoposide causes c-Jun activation and apoptosis in sympathetic neurons, presumably by creating extensive amounts of double-stranded DNA breaks (Besirli and Johnson, 2003). Similar to trophic factor deprivation, etoposide-induced DNA damage led to rapid PUMA induction in sympathetic neurons (Fig. 5 C). Because the expression of other BH3-only proteins are regulated by c-Jun during neuronal death, we compared PUMA induction in jun +/+ and jun aa/aa–sympathetic neurons. Similar to Bim (Fig. 4 B), PUMA expression was regulated by c-Jun, as lack of c-Jun NH2-terminal phosphorylation delayed PUMA induction after trophic factor deprivation (Fig. 5 D). The delay in PUMA expression was not as significant as that observed for Bim expression, suggesting that other transcription factors also regulate PUMA gene transcription during neuronal apoptosis.
Figure 5.
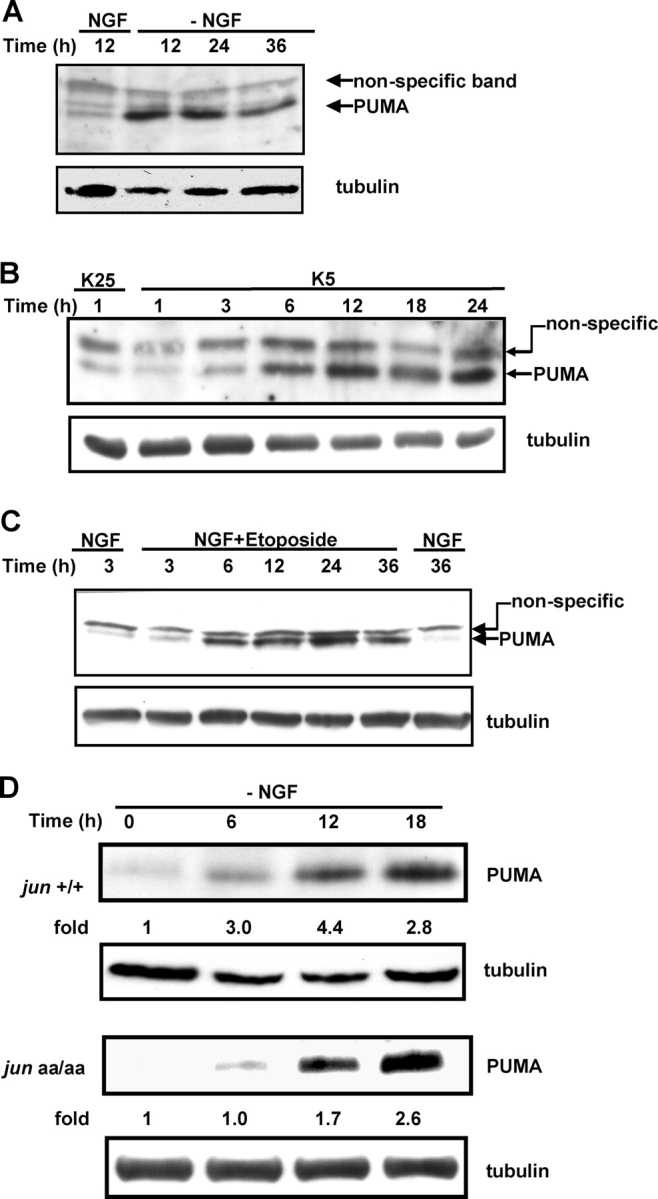
BH3-only Bcl-2 family member PUMA is induced during trophic factor deprivation and DNA damage. (A and C) 5-DIV rat–sympathetic neurons were deprived of NGF (A) or exposed to 10 μM topoisomerase II–inhibitor etoposide (C) and PUMA expression was examined by Western blot analysis. (B) Cerebellar granule neurons were isolated from P7 rats and maintained in 25 mM K+ (K25) and serum for 7 d. PUMA was induced rapidly after reducing K+ concentration to 5 mM (K5), which leads to death of cerebellar neurons. (D) NH2-terminal c-Jun phosphorylation was important for rapid PUMA induction. 5-DIV–sympathetic neurons from jun +/+ and jun aa/aa mice were deprived of NGF and protein lysates were analyzed for PUMA expression. Time 0 PUMA level was set to 1. Protein loading was normalized by reprobing membranes with tubulin antibody.
The rabbit polyclonal antibody against phospho-c-Jun Ser73 recognizes several proteins on Western blots subsequent to NGF deprivation
The delayed death of JunAA-sympathetic neurons after NGF deprivation demonstrated that NH2-terminal c-Jun phosphorylation is important, but not necessary, for neuronal apoptosis. In contrast, inhibition of the JNK signaling pathway prevents sympathetic neuronal apoptosis completely (Harris et al., 2002). Therefore, in addition to c-Jun, sympathetic neurons appear to contain other JNK substrates that are critical for apoptosis. In whole cell extracts obtained from NGF-deprived jun aa/aa neurons, an antibody raised against the phosphorylated Ser73 residue of c-Jun did not detect any phospho-c-Jun (∼45 kD) in Western analysis as expected (Fig. 6 A). However, a band running above the 250-kD marker was seen in phospho-c-Jun Ser73 Westerns specifically after NGF deprivation in both wild-type and mutant neurons (Fig. 6 A). Similar to mouse neurons, NGF-deprived rat-sympathetic neurons showed specific detection of this >250-kD band by the phospho-c-Jun Ser73 antibody on Westerns, but they also contained two additional bands (∼150 and 210 kD) that were also detected during NGF deprivation (see Fig. 8). These bands were not immunoreactive with the phospho-c-Jun Ser63 antibody (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200501138/DC1; and unpublished data). Unlike the phospho-c-Jun Ser63 antibody (Fig. S3), phospho-c-Jun Ser73 antibody stained the nuclei of JunAA-sympathetic neurons after NGF deprivation (Fig. 6 B). The immunocytochemistry results suggested that the phospho-Ser73 antibody-immunoreactive bands detected on Westerns were located in the nucleus. Because other bands were also detected by the phospho-c-Jun Ser73 antibody in whole cell extracts without NGF deprivation (these bands may be “nonspecific” or represent neuronal JNK substrates that are phosphorylated in sympathetic neurons regardless of the trophic status; see Fig. 8), we attempted to increase the sensitivity of the Ser73 antibody by using neuronal lysates after a crude subcellular fractionation to separate the nuclear and postnuclear compartments. More NGF deprivation induced phospho-c-Jun Ser73 antibody-immunoreactive bands (∼40, 65, 150, 210, and >250 kD) were observed in the nuclear fractions (Fig. 7), demonstrating that some of these other phospho-c-Jun Ser73 antibody immunoreactive proteins were masked by the strong background signal coming from the nonnuclear cell compartment in whole cell lysates. These results showed that other neuronal proteins containing sequence motifs possibly similar to the NH2-terminal Ser73 region of c-Jun were recognized by the phospho-c-Jun Ser73 antibody specifically during NGF deprivation. Unlike the phospho-c-Jun Ser73 antibody, phospho-c-Jun Ser63 antibody did not detect any NGF deprivation–regulated proteins other than c-Jun on Western blots (Fig. S3). Immunocytochemistry experiments demonstrated that phospho-c-Jun Ser73 antibody was not specific for c-Jun immunostaining. Therefore, only the phospho-Ser63 antibody should be used for immunostaining of phospho-c-jun because phospho-Ser73 antibody detects other nuclear proteins in a stimulus-dependent manner.
Figure 6.

Phospho-c-Jun Ser73 antibody is immunoreactive against a >250-kD protein located in the nucleus. Sympathetic neurons were isolated from the SCGs of newborn littermate wild-type and jun aa/aa mice and grown in NGF-containing medium. (A) Phosphorylation of c-Jun was detected by antiphospho-c-Jun Ser73 antibody at 48 h after treatment. NGF deprivation (−) induces c-Jun phosphorylation in wild-type, but not in knock-in, neurons (arrow). There is a cross-reactive band in sympathetic neurons appearing only after NGF deprivation in both genotypes (*). 48 h of NGF deprivation induced c-Jun in both genotypes to a similar extent (middle). Tubulin antibody western shows similar protein loading. (B) Phospho-c-Jun Ser73 immunocytochemistry in sympathetic neurons shows nuclear staining after 48 h of NGF deprivation in both jun +/+ and jun aa/aa neurons. Bar, 20 μm.
Figure 7.

Nuclear lysates of NGF-deprived sympathetic neurons show additional antiphospho-c-Jun Ser73–immunoreactive proteins. 5-DIV–sympathetic neurons isolated from P0 rats were deprived of NGF (−NGF) or left untreated (NGF) for 24 h. Nuclear and postnuclear extracts were subjected to SDS-PAGE, followed by Western analysis with phospho-c-Jun Ser73 antiserum. Arrow indicates the c-Jun protein. Stars show the antiphospho-c-Jun–immunoreactive proteins detected in whole cell extracts (see Fig.8). The immunoreactive bands seen only in the nuclear fraction are indicated by arrowheads. Panel on the left is the longer exposure of the nuclear fraction part. Phospho-c-Jun signal (arrow), present mainly in the nuclear fraction, shows that this was a crude subcellular fractionation of the sympathetic neuron nuclei. Equal amounts of protein were loaded in each lane (unpublished data).
Phospho-c-Jun Ser73 antibody immunoreactive proteins are downstream targets of the NGF-deprivation-induced MLK–JNK signaling pathway
Similar to c-Jun, expression of, or phosphorylation of, the other proteins recognized by the phospho-c-Jun Ser73 antibody were increased after NGF deprivation. To determine whether these proteins were downstream targets of the JNK pathway or were nonspecific, we examined the phospho-c-Jun Ser73 immunoreactivity in whole cell lysates of sympathetic neurons deprived of NGF in the presence of MLK-inhibitor CEP-1347. As expected, the NH2-terminal phosphorylation of c-Jun was completely blocked in the presence of CEP-1347 (Fig. 8, arrow). Levels of the other phospho-c-Jun Ser73–immunoreactive bands were similarly decreased in the absence of JNK activity (Fig. 8). Therefore, these proteins, both NGF deprivation induced and constitutively phosphorylated, seem to be downstream targets of the JNK signaling pathway in sympathetic neurons. These data indicate that the phospho-c-Jun Ser73 antibody recognized a myriad of JNK substrates in these cells.
The NPC is phosphorylated during NGF deprivation–induced neuronal apoptosis
To identify the nuclear proteins immunoreactive for the phospho-c-Jun Ser73 antibody after NGF deprivation, we performed database searches using the amino acid sequence motif immediately surrounding the NH2-terminal Ser73 of c-Jun. The potential candidates were also evaluated for their subcellular localization, i.e., known nuclear proteins or proteins with a detectable nuclear localization signal. The 358-kD protein nucleoporin 358 (Nup358; Wu et al., 1995), a subunit of NPC, was identified as a good candidate based on sequence homology and molecular weight (Fig. 9 A). To examine Nup expression in sympathetic neurons, we used the monoclonal antibody, Mab414, which recognizes the Nups that carry the conserved phenylalanine-glycine rich (FG) region (Davis and Blobel, 1987). Mab414 detected several Nups in whole cell extracts of sympathetic neurons, including Nup358, Nup214, Nup153, and Nup62 (Fig. 9 B). Consistent with Nup358 and Nup214 as JNK substrates, both Nup358 and Nup214 comigrated with the phospho-c-Jun Ser73–immunoreactive bands in the same samples (unpublished data). To determine more definitely if Nup358 and Nup214 were recognized by the phospho-c-Jun Ser73 antibody, we immunoprecipitated Nups with the Mab414 antibody and immunoblotted with the phospho-c-Jun Ser73 antibody. Nup214 was specifically detected with the phospho-c-Jun Ser73 antibody only in samples coming from the NGF-deprived neurons (Fig. 9 C). We were unable to detect Nup358 with the phospho-c-Jun Ser73 antibody in the same samples, though Nup358 was immunoprecipitated by Mab414 antibody (Fig. 9 C). Based on sequence homology, we had expected that phospho-c-Jun Ser73 antibody would be more likely to recognize Nup358 than Nup214. Nup214 also contains many potential JNK phosphorylation sites (Ser-Pro motifs), but sequence similarities between these sites and Ser73 motif of c-Jun are lower (unpublished data). This may explain the improved detection of this 214-kD band after immunoprecipitation compared with whole cell lysates (Fig. 8 vs. Fig. 9 C). Similar to Mab414, phospho-c-Jun Ser73 antibody specifically immunoprecipitated Nup214, but not Nup358, after NGF deprivation (unpublished data). Although we could not confirm with the available tools that Nup358 was phosphorylated by the JNK pathway, NGF deprivation–specific recognition of the other NPC subunit, Nup214, by the phospho-c-Jun Ser73 antibody strongly indicates that NPC is a downstream target of the JNK pathway during neuronal apoptosis.
Figure 9.

Nup214 is phosphorylated after NGF deprivation. (A) Alignment of the human and mouse Nup358 sequences with the amino acid sequence surrounding Ser73 of human c-Jun protein. (B) Whole cell extract from 5-DIV–sympathetic neurons were subjected to Western analysis with monoclonal Nup antibody MAb414. Both lanes contain extracts of NGF-maintained neurons. (C) Sympathetic neurons were deprived of NGF (−NGF) or left untreated (NGF) for 24 h. Whole cell lysates were subjected to MAb414 (414) or IgG immunoprecipitation. Immunoprecipitated proteins were analyzed for phospho-c-Jun Ser73 (left) or MAb414 (right) immunoreactivity.
Discussion
In this study, we analyzed the role of NH2-terminal c-Jun phosphorylation during neuronal apoptosis induced by trophic factor deprivation and DNA damage. Neurons with one or two mutated c-jun genes were resistant to stress-induced cell death in a gene dose-dependent manner. Our results demonstrate that c-Jun phosphorylation is important for the increased expression of downstream proapoptotic genes. In the absence of this phosphorylation, expression of c-Jun and Bim were impaired, consistent with the slower death of neurons after NGF deprivation. We also identified PUMA as another BH3-only proapoptotic Bcl-2 family member induced by trophic factor deprivation and DNA damage. Similar to c-Jun and Bim, expression of PUMA was delayed in neurons expressing the mutant c-Jun protein. These results indicate that NH2-terminal c-Jun phosphorylation on Ser 63 and 73 by JNKs was important in the induction of proapoptotic genes leading to sympathetic neuronal apoptosis. In addition to c-Jun, MLK–JNK pathway regulated other nuclear targets during NGF deprivation. One of these targets was identified as the Nup214 subunit of the NPC.
c-Jun phosphorylation on Ser63 and 73 is important, but not necessary, for neuronal apoptosis
The importance of c-Jun activity for neuronal death has been implicated in several in vitro systems (Estus et al., 1994; Ham et al., 1995; Behrens et al., 1999; Palmada et al., 2002). However, other studies in nonneuronal systems suggest that c-Jun expression can be a protective measure against cellular injury and may actually prevent death (Potapova et al., 2001). Data presented in this paper further demonstrate that c-Jun is a proapoptotic factor in sympathetic and cerebellar granule neurons. Importantly, our results also show that phosphorylation of c-Jun promoted neuronal apoptosis, but it was not absolutely necessary for death. Because the JunAA protein is still functional, whether in vivo deletion of c-Jun from sympathetic and cerebellar granule neurons would completely prevent apoptosis after trophic factor deprivation or DNA damage remains to be determined.
Ser63/73 phosphorylation is not required for neuronal c-Jun induction during apoptosis
Mutation of c-Jun NH2-terminal phosphorylation sites significantly inhibited neuronal apoptosis. The underlying cause for this is most likely due to the impaired induction of proapoptotic c-Jun transcriptional targets. One of these targets is c-jun, the expression of which is autoregulated (Angel et al., 1988). Under normal conditions, c-Jun exists in neurons at low levels with no detectable NH2-terminal phosphorylation despite high levels of constitutive JNK activity. One suggested model is that during stress, isoform-specific JNK activation or altered JNK localization to specific cell compartments causes NH2-terminal phosphorylation of c-Jun on Ser63 and Ser73, increasing transcriptional activity (Coffey et al., 2002). As a result, NH2-terminal phosphorylation activates the c-Jun autoregulatory cycle, leading to more c-jun transcription and NH2-terminal phosphorylation. Our results show that this is not the sole mechanism of c-Jun activation during neuronal apoptosis, because c-Jun expression was impaired, but not completely prevented in JunAA neurons. Several explanations may exist for this induction in the absence of NH2-terminal phosphorylation at Ser 63 and 73. First, JunAA protein is still functional at some low level and can induce c-Jun transcription, though inefficiently. Second, two other known phosphorylation sites in the NH2-terminal region, Thr91 and Thr93, exist. Phosphorylation of these residues by JNKs or other kinases may be sufficient to activate c-Jun, and this may explain the residual activity of the JunAA mutant. Finally, besides c-Jun, other JNK substrates may regulate c-Jun expression, such as other Jun, Fos, or ATF family members. Trophic factor deprivation induces the expression of other AP-1 transcription factors, including JunB, c-Fos, and ATF-2 (Estus et al., 1994; Eilers et al., 2001). ATF-2 is phosphorylated by JNKs, leading to its transcriptional activation. ATF-2 regulates c-jun transcription in sympathetic neurons after trophic factor deprivation (Eilers et al., 2001). The ability of the JNK pathway to activate c-Jun in jun aa/aa neurons strongly suggests that JNKs may be modulating the activity of several transcription factors that concomitantly regulate the c-jun promoter.
Expressions of BH3-only Bcl-2 family members Bim and PUMA are regulated by c-Jun during neuronal death
Bcl-2 family members are critical for making life-or-death decisions in neurons. The proapoptotic Bcl-2 family protein Bax is necessary for neuronal death in both sympathetic and cerebellar granule neurons (Deckwerth et al., 1996; Miller et al., 1997). Bax activity is regulated by the BH3-only members of the Bcl-2 family. Two BH3-only proteins, Bim and DP5, are increased in sympathetic and cerebellar granule neurons during cell death (Harris and Johnson, 2001; Putcha et al., 2001). Expression of both Bim and DP5 is regulated by MLK–JNK signaling and blocked by pharmacological inhibitors of this pathway (Harris and Johnson, 2001). In addition, c-Jun is important for Bim induction in vitro because dominant-negative c-Jun inhibits Bim expression (Whitfield et al., 2001). Our results confirm these findings, showing that Bim expression is attenuated in neurons carrying a hypomorphic c-Jun protein. We also identified another BH3-only protein that is up-regulated during neuronal death. PUMA/Bbc3, first identified as a proapoptotic protein in thymocytes and tumor cells, is induced by DNA damage, dexamethasone treatment, or serum deprivation (Han et al., 2001; Nakano and Vousden, 2001). In primary murine thymocytes, PUMA expression is dependent on p53 activity after DNA damage, but not after dexamethasone treatment or serum deprivation (Han et al., 2001). In the work reported here, both trophic factor deprivation and DNA damage induced PUMA expression in sympathetic and cerebellar granule neurons. PUMA expression occurred well before the terminal stages of death, similar to Bim and DP5. The delay of PUMA expression in JunAA neurons suggests that c-Jun activity contributes to PUMA induction during neuronal death. Identification of yet another BH3-only proapoptotic protein (in addition to Bim and DP5) induced during neuronal death demonstrates the redundancy built into the apoptotic machinery in neurons and probably accounts for the modest phenotype associated with the deletion of Bim or DP5 (Putcha et al., 2001; Imaizumi et al., 2004).
In both sympathetic and cerebellar granule neurons, the lack of NH2-terminal c-Jun phosphorylation on Ser63 and 73 retarded, but did not prevent, cell death. Therefore, NH2-terminal c-Jun phosphorylation by JNKs is important, but not absolutely necessary, for neuronal apoptosis. Because JunAA protein is a c-Jun hypomorph, but not a complete functional null, c-Jun activity may still be critical for promoting neuronal death in jun aa/aa mice. This is supported by the delayed expression of proapoptotic c-Jun transcription targets. In the absence of any c-Jun activity, these genes may never get transcribed and apoptosis may be completely suppressed without these important cell death mediators. If c-Jun is necessary for neuronal apoptosis, the phenotype of c-Jun–null neurons should mirror that of Bax-deficient neurons. Bax expression is absolutely required for trophic factor deprivation-induced death and sympathetic ganglia of bax-null mice contain twice as many neurons (Deckwerth et al., 1996). Although not as impressive as the bax-null mice, at birth JunAA mice also appear to have more neurons in their sympathetic ganglia (Fig. 1 B). The difference between bax deletion and c-jun mutation on the number of neurons in a SCG is not surprising because JunAA neurons eventually die after NGF deprivation. The role of c-Jun during naturally occurring or stress-induced cell death may also be different in distinct neuronal populations. In adult mice, conditional deletion of the neuronal c-jun gene causes a significant decrease in axotomy-induced death of facial nucleus motor neurons, but the total number of motor neurons is increased by only ∼20% (Raivich et al., 2004), which is significantly less than the 51% increase seen in Bax-null mice (Deckwerth et al., 1996). In addition, conditional c-jun knockout mice had no significant increase in the number of dorsal root ganglion sensory neurons. Analysis of SCGs isolated from conditional c-Jun knockout mice may allow a determination of whether c-Jun is necessary for the developmental programmed cell death of sympathetic neurons, resembling the cell death phenotype of Bax-null SCG.
The NPC subunit Nup214 is recognized by the phospho-c-Jun Ser73 antibody in a JNK-dependent manner specifically after NGF deprivation of sympathetic neurons
Inhibition of the MLK–JNK signaling completely prevents NGF deprivation–induced death of sympathetic neurons. In contrast, lack of NH2-terminal c-Jun phosphorylation delays death only transiently. These findings indicate that MLK–JNK signaling regulates other proteins critical for neuronal programmed cell death after trophic factor deprivation. One of these proteins is Bim. Ser65 phosphorylation of Bim potentiates its proapoptotic function during NGF deprivation–induced death of sympathetic neurons (Putcha et al., 2003). To identify additional JNK pathway targets, we used the rabbit polyclonal antibody against Ser73-phosphorylated c-Jun. This antibody proved to be a useful tool in identifying downstream targets of the JNK pathway. The loss of detectable bands on Westerns upon treatment with CEP-1347 indicates that this antibody acted as a general JNK substrate antibody, showing immunoreactivity against other neuronal proteins that were either constitutively phosphorylated in sympathetic neurons or were specifically phosphorylated after NGF deprivation. Increased phosphorylation or expression of these proteins after NGF deprivation was dependent on the JNK signaling and failed to occur in the presence of selective MLK-inhibitor CEP-1347. Database searches using the Ser73 motif identified Nup358, a 270-kD protein on SDS-PAGE, as a potential JNK substrate. Although Nup358 comigrated with the >250-kD phospho-c-Jun Ser73 immunoreactive band in whole cell lysates (resolution of the gel is poor above the 250-kD marker), immunoprecipitated Nup358 was not detectable with phospho-c-Jun Ser73 antibody. This may indicate that Nup358 is not phosphorylated after NGF deprivation. Alternatively, Mab414 antibody could have immunoprecipitated only the nonphosphorylated form of Nup358 if the phosphorylation changes the conformation of Nup358 or otherwise inhibits Mab414 recognition (e.g., by facilitating the association of Nup358 with another protein that blocks antibody binding). Unlike Nup358, the same immunoprecipitation experiments definitively showed that the related Nup214 became immunoreactive for the phospho-c-Jun antibody only after NGF deprivation. This result indicates that NPC was specifically phosphorylated by JNK during neuronal apoptosis.
NPC is a large protein assembly that penetrates the nuclear membrane and is the only known channel between the cytoplasm and the nucleus (Suntharalingam and Wente, 2003). NPC mediates the bidirectional permeability of the nucleus and facilitates nucleocytoplasmic exchange. Small molecules can diffuse through NPC in and out of the nucleus, but proteins that are larger than 40 kD require active transport. Although the primary function of NPC is to regulate the nucleocytoplasmic exchange, recent studies demonstrate that NPC function is also important for other physiological process such as mitosis, gene expression, and cell death (Talcott and Moore, 1999). Deletion of Nup358 leads to defects in chromosome segregation and kinetochore structure, indicating that some components of NPC are involved in mitosis (Salina et al., 2003). During apoptosis, Nup153, Nup358, and Nup214 are cleaved by caspases. Other non-Nup components of NPC, such as RanGTP and karyopherins, redistribute across the nuclear envelope during cell death before caspase activation and limit the permeability of NPC (Ferrando-May et al., 2001).
Both Nup358 and Nup214 are located in the cytoplasmic fibril compartment of NPC, which also contains two other domains, i.e., central core and the nuclear basket. Karyopherins recognize nuclear import and export signals in their specific protein cargo (Weis, 2003). During nuclear import karyopherin–cargo complex interacts with the FG repeat domains of the cytoplasmic fibril Nups such as Nup358 (Weis, 2003). JNK phosphorylation of Ser or Thr residues in this region may alter the kinetics of karyopherin–cargo binding to cytoplasmic Nups. Alternatively, JNK-dependent phosphorylation may change the tertiary structure of the Nups and modify the nucleocytoplasmic permeability.
One of the immediate effects of the reduced nucleocytoplasmic transport is the accumulation of mRNA in the nucleus. A similar process may be occurring in neuronal cells undergoing trophic factor deprivation-induced apoptosis. Sympathetic neurons show reduced metabolic activity after trophic factor deprivation (Martin et al., 1988). This loss of metabolism is largely prevented by the inhibition of the JNK pathway by CEP-1347 (Harris et al., 2002), which maintains protein and mRNA synthesis rates in NGF-deprived sympathetic neurons. The loss of nuclear permeability due to JNK-mediated NPC phosphorylation may be one of the underlying causes of the reduced metabolic activity and trophic status of sympathetic neurons after trophic factor deprivation. Phosphorylation of NPC may be causing the buildup of the neuronal mRNA in the nucleus. This hypothesis is consistent with our inability to demonstrate detectable changes in the activity of protein translation machinery in sympathetic neurons after NGF deprivation (unpublished data), suggesting that at least part of the underlying defect may not be inherent to the protein translation machinery but may be a direct result of impaired nuclear mRNA export. Therefore, inhibition of the JNK pathway may be beneficial for sympathetic neurons by inhibiting the induction of proapoptotic genes such as c-jun and bim and protecting the integrity of NPC and nucleocytoplasmic exchange to maintain neuronal metabolism.
In summary, we demonstrated that NH2-terminal c-Jun phosphorylation by the JNK pathway was important, but not necessary, for neuronal death after trophic factor deprivation or DNA damage. The resistance of mutant sympathetic neurons to NGF deprivation in the absence of c-Jun phosphorylation was consistent with delayed expression of proapoptotic genes c-Jun, Bim, and PUMA. The complete suppression of sympathetic neuronal death by JNK pathway inhibition, but not lack of NH2-terminal c-Jun phosphorylation, indicated that other JNK substrates were required for neuronal apoptosis. It was found that phospho-c-Jun Ser73 antibody was an effective experimental tool in identifying downstream substrates of the JNK pathway. Using this antibody, we identified Nup214 subunit of the NPC as a target of the JNK pathway during trophic factor deprivation. In addition to Nup214, the phospho-c-Jun Ser73 antibody detected other nuclear JNK pathway targets that remain to be identified. Similar to c-Jun and Bim, JNK-mediated regulation of NPC and possibly other nuclear proteins may be important for neuronal programmed cell death.
Materials and methods
Animals and materials
All reagents were purchased from Sigma-Aldrich unless otherwise stated. Timed-pregnant Sprague Dawley rats were obtained from Harlan Bioproducts. Generation and initial characterization of JunAA mice has been described previously (Behrens et al., 1999). Animal use and treatment complied fully with the Animal Studies Committee of Washington University and the U.S. Animal Welfare Act (1985). Collagenase and trypsin were purchased from Worthington Biochemical Corporation. Mouse 2.5S NGF was from Harlan Bioproducts. The goat anti–mouse 2.5S NGF-neutralizing antiserum has been characterized previously (Ruit et al., 1992). CEP-11004 and CEP-1347 were gifts of Cephalon. SP600125 was purchased from BioMol.
Neuronal cultures
Primary sympathetic neuronal cultures were prepared from postnatal day (P) 0–P1 rat or mouse SCG by using previously described methods (Johnson and Argiro, 1983; Deckwerth and Johnson, 1993). Cultures were maintained in AM50 medium (90% minimum essential medium [Invitrogen], 2 mM glutamine, 10% FBS [Hyclone], 50 ng/ml 2.5S NGF, 20 μM fluorodeoxyuridine, 20 μM uridine, 100 U/ml penicillin, and 100 U/ml streptomycin), supplemented with 3.3 μg/ml aphidicolin (A.G. Scientific) to reduce the number of nonneuronal cells. Total number of neurons from each animal was determined by counting all viable neurons in three to four wells containing 1/5–1/7 of a SCG at 6–12 DIV. Because SCGs of JunAA mice had significantly more sympathetic neurons in initial experiments, higher dilution of SCG per well was used in later experiments. To deprive neurons of NGF, 5-DIV neurons were washed three times with AM0 (AM50 medium lacking NGF) and fed with fresh AM0 containing 0.01% anti-NGF antiserum. Anti-NGF treatment was stopped by rinsing the cultures three times with AM0 and adding back fresh AM50 for 5–7 d. To induce neuronal DNA damage, the culture medium was replaced with fresh AM50 containing 10 μM of topoisomerase-II–inhibitor etoposide. In neuronal cultures used to harvest protein or measure somal diameter, 50 μM of broad-spectrum caspase inhibitor, BAF (Enzyme Systems), was included in all treatment conditions to inhibit neuronal death.
Cerebellar granule cell culture protocol was described in detail previously (Miller et al., 1997). In brief, P6–P8 mouse or rat cerebellum was dissected, dissociated by 1 mg/ml trypsin for 15 min, followed by mechanical trituration. Cells were plated onto poly-L-lysine–coated dishes (Nunc) and were maintained in K25+S medium (Eagle's basal medium, 25 mM potassium chloride, 10% dialyzed FBS, 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin). 3.3 μg/ml aphidicolin was added 1 or 2 d later to reduce the number of nonneuronal cells. At 7 DIV, the cells were washed twice with K5−S medium (K25+S medium without the serum and with only 5 mM potassium) and treated with K5−S or fresh K25+S.
Neuronal survival
The number of viable sympathetic neurons was assessed after fixing the cultures with 4% PFA (Fischer Scientific) in PBS and staining with crystal violet. Neurons were scored as viable by a naive observer if the crystal violet-positive cells had large, well-defined cellular outlines. Dead neurons and debris stain faintly or show no staining with crystal violet. The percentage of viable neurons was calculated by dividing the number of crystal violet-positive neurons at each time point by the total number of neurons in NGF-maintained, untreated sister cultures.
Immunocytochemistry
Cultures were fixed with fresh 4% PFA in PBS, washed with Tris-buffered saline (TBS: 0.1 M Tris-HCl, pH 7.6, 0.9% NaCl), and incubated in blocking solution (5% normal goat serum in TBS, containing 0.3% Triton X-100) for 1 h at RT. The cultures were then incubated with phospho-c-Jun (Ser73; 1:1000; Cell Signaling) antibody in antibody solution (1% normal goat serum in TBS, containing 0.3% Triton X-100) overnight at 4°C. The cultures were next washed three times with TBS and incubated in antibody solution containing Cy-3–labeled secondary antibody (1:400; Jackson ImmunoResearch Laboratories) for 4 h at 4°C and counterstained with 1 μg/ml bisbenzimide (Hoechst 33258; Molecular Probes). After four washes with TBS, the cultures were mounted for fluorescence microscopy. Sympathetic neurons were visualized with a Zeiss Axiophot microscope.
Western blot analysis
Neuronal cultures were rinsed twice with cold PBS, lysed in reducing sample buffer (125 mM Tris-HCl, pH 6.8, 10% 2-mercaptoethanol, 4% SDS, 0.1% bromophenol blue, and 20% glycerol), boiled for 5 min, and stored at −20°C until use. Proteins were separated by SDS-PAGE on Tris-Glycine mini-gels (Invitrogen) and transferred to Immobilon-P PVDF membranes (Millipore). Blots were blocked for 1 h at RT with TBST (10 mM Tris-HCl, pH 7.5, 100 mM NaCl, and 0.1% Tween 20) containing 5% nonfat dry milk or BSA and incubated overnight at 4°C with primary antibody diluted in blocking solution recommended by manufacturer. The following primary antibodies were used: c-Jun (0.25 μg/ml; BD Transduction Labs), phospho-c-Jun (Ser73; 1:1,000; Cell Signaling), Bim (1:1,000; Stressgen), PUMA (1:250; Axxora), Mab414 (1 μg/ml; Covance), and tubulin (1:50,000; Sigma-Aldrich). After washing, blots were incubated for 1 h at RT with HRP-linked secondary antibodies (Cell Signaling) diluted 1:2,500–1:10,000 in blocking solution. The blots were washed three times with TBST and developed with a chemiluminescent substrate (Supersignal; Pierce Chemical Co.). To strip and reprobe blots, the membranes were incubated in 100 mM glycine (pH 2.5) twice for 25 min and then washed with TBST; Western analysis was repeated. A Bio-Rad Laboratories ChemiDoc system with QuantiOne Software was used to quantify the immunoblots. All values were normalized against the values obtained for tubulin-loading controls.
Immunoprecipitation
Neuronal cultures were rinsed twice with cold PBS and lysed in NP-40 immunoprecipitation buffer (Tris-buffered saline, pH 7.4, 1% Nonidet P-40, 10% glycerol, protease inhibitors, and 1 mM sodium orthovanadate, 1 mM sodium fluoride, 50 mM β-glycerophosphate, 1 mM DTT, and 50 mM NaF) with gentle rocking at 4°C. The detergent extracts were cleared of insoluble debris and nuclei by centrifugation at 13,000 g in a refrigerated microcentrifuge for 10 min. The cleared lysates were immunoprecipitated with 1 μg of Mab414 antibody or normal mouse IgG.
Subcellular fractionation
Sympathetic neurons were harvested into isotonic fractionation buffer (250 mM sucrose, 0.5 mM EDTA, 20 mM Hepes, 500 μM sodium orthovanadate, pH 7.2) supplemented with protease inhibitors (inhibitor cocktail complete; Roche Molecular Biochemicals) and centrifuged at 900 g for 5 min. The pellet was resuspended into 500 μl of fractionation buffer, homogenized with a ball-bearing homogenizer, and centrifuged at 900 g for 5 min to remove nuclei. The nuclear pellet and the postnuclear supernatant were resuspended to equivalent volumes with reducing sample buffer and evaluated by Western blotting.
Online supplemental material
DNA damage–induced c-Jun activation is MLK and JNK dependent in cerebellar granule neurons (Fig.S1). DNA damage–induced death of cerebellar granule neurons is delayed in the absence of NH2-terminal c-Jun phosphorylation (Fig. S2). Phosphorylation of c-Jun detected by antiphospho-c-Jun Ser63 antibody in wild-type and jun aa/aa neurons (Fig. S3). Online supplemental materials are available at http://www.jcb.org/cgi/content/full/jcb.200501138/DC1.
Acknowledgments
We thank P.A. Osborne for expert technical assistance; M. Bloomgren for secretarial assistance; and, members of the Johnson lab for their critical review of this manuscript. C.G. Besirli is a member of the Medical Scientist Training Program at Washington University School of Medicine.
This work was supported by National Institutes of Health grants R37AG-12947 and RO1NS38651.
Abbreviations used: DIV, days in vitro; JNK, Jun–NH2-terminal kinase; MLK, mixed lineage kinase; NPC, nuclear pore complex; Nup, nucleoporin; P, postnatal day; SCG, superior cervical ganglion.
References
- Angel, P., K. Hattori, T. Smeal, and M. Karin. 1988. The jun proto-oncogene is positively autoregulated by its product, Jun/AP-1. Cell. 55:875–885. [DOI] [PubMed] [Google Scholar]
- Behrens, A., M. Sibilia, and E.F. Wagner. 1999. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat. Genet. 21:326–329. [DOI] [PubMed] [Google Scholar]
- Besirli, C.G., and E.M. Johnson Jr. 2003. JNK-independent activation of c-Jun during neuronal apoptosis induced by multiple DNA-damaging agents. J. Biol. Chem. 278:22357–22366. [DOI] [PubMed] [Google Scholar]
- Besirli, C.G., T.L. Deckwerth, R.J. Crowder, R.S. Freeman, and E.M. Johnson Jr. 2003. Cytosine arabinoside rapidly activates Bax-dependent apoptosis and a delayed Bax-independent death pathway in sympathetic neurons. Cell Death Differ. 10:1045–1058. [DOI] [PubMed] [Google Scholar]
- Coffey, E.T., G. Smiciene, V. Hongisto, J. Cao, S. Brecht, T. Herdegen, and M.J. Courtney. 2002. c-Jun N-terminal protein kinase (JNK) 2/3 is specifically activated by stress, mediating c-Jun activation, in the presence of constitutive JNK1 activity in cerebellar neurons. J. Neurosci. 22:4335–4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, L.I., and G. Blobel. 1987. Nuclear pore complex contains a family of glycoproteins that includes p62: glycosylation through a previously unidentified cellular pathway. Proc. Natl. Acad. Sci. USA. 84:7552–7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckwerth, T.L., and E.M. Johnson Jr. 1993. Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J. Cell Biol. 123:1207–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckwerth, T.L., J.L. Elliott, C.M. Knudson, E.M. Johnson Jr., W.D. Snider, and S.J. Korsmeyer. 1996. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron. 17:401–411. [DOI] [PubMed] [Google Scholar]
- Eilers, A., J. Whitfield, B. Shah, C. Spadoni, H. Desmond, and J. Ham. 2001. Direct inhibition of c-Jun N-terminal kinase in sympathetic neurones prevents c-jun promoter activation and NGF withdrawal-induced death. J. Neurochem. 76:1439–1454. [DOI] [PubMed] [Google Scholar]
- Estus, S., W.J. Zaks, R.S. Freeman, M. Gruda, R. Bravo, and E.M. Johnson Jr. 1994. Altered gene expression in neurons during programmed cell death: identification of c-jun as necessary for neuronal apoptosis. J. Cell Biol. 127:1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrando-May, E., V. Cordes, I. Biller-Ckovric, J. Mirkovic, D. Gorlich, and P. Nicotera. 2001. Caspases mediate nucleoporin cleavage, but not early redistribution of nuclear transport factors and modulation of nuclear permeability in apoptosis. Cell Death Differ. 8:495–505. [DOI] [PubMed] [Google Scholar]
- Ham, J., C. Babij, J. Whitfield, C.M. Pfarr, D. Lallemand, M. Yaniv, and L.L. Rubin. 1995. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 14:927–939. [DOI] [PubMed] [Google Scholar]
- Ham, J., A. Eilers, J. Whitfield, S.J. Neame, and B. Shah. 2000. c-Jun and the transcriptional control of neuronal apoptosis. Biochem. Pharmacol. 60:1015–1021. [DOI] [PubMed] [Google Scholar]
- Han, J., C. Flemington, A.B. Houghton, Z. Gu, G.P. Zambetti, R.J. Lutz, L. Zhu, and T. Chittenden. 2001. Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc. Natl. Acad. Sci. USA. 98:11318–11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, C.A., and E.M. Johnson Jr. 2001. BH3-only Bcl-2 family members are coordinately regulated by the JNK pathway and require Bax to induce apoptosis in neurons. J. Biol. Chem. 276:37754–37760. [DOI] [PubMed] [Google Scholar]
- Harris, C.A., M. Deshmukh, B. Tsui-Pierchala, A.C. Maroney, and E.M. Johnson Jr. 2002. Inhibition of the c-Jun N-terminal kinase signaling pathway by the mixed lineage kinase inhibitor CEP-1347 (KT7515) preserves metabolism and growth of trophic factor-deprived neurons. J. Neurosci. 22:103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herdegen, T., P. Skene, and M. Bahr. 1997. The c-Jun transcription factor–bipotential mediator of neuronal death, survival and regeneration. Trends Neurosci. 20:227–231. [DOI] [PubMed] [Google Scholar]
- Imaizumi, K., A. Benito, S. Kiryu-Seo, V. Gonzalez, N. Inohara, A.P. Leiberman, H. Kiyama, and G. Nunez. 2004. Critical role for DP5/Harakiri, a Bcl-2 homology domain 3-only Bcl-2 family member, in axotomy-induced neuronal cell death. J. Neurosci. 24:3721–3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, M.I., and V. Argiro. 1983. Techniques in the tissue culture of rat sympathetic neurons. Methods Enzymol. 103:334–347. [DOI] [PubMed] [Google Scholar]
- Kallunki, T., T. Deng, M. Hibi, and M. Karin. 1996. c-Jun can recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell. 87:929–939. [DOI] [PubMed] [Google Scholar]
- Maroney, A.C., J.P. Finn, D. Bozyczko-Coyne, T.M. O'Kane, N.T. Neff, A.M. Tolkovsky, D.S. Park, C.Y. Yan, C.M. Troy, and L.A. Greene. 1999. CEP-1347 (KT7515), an inhibitor of JNK activation, rescues sympathetic neurons and neuronally differentiated PC12 cells from death evoked by three distinct insults. J. Neurochem. 73:1901–1912. [PubMed] [Google Scholar]
- Maroney, A.C., J.P. Finn, T.J. Connors, J.T. Durkin, T. Angeles, G. Gessner, Z. Xu, S.L. Meyer, M.J. Savage, L.A. Greene, et al. 2001. Cep-1347 (KT7515), a semisynthetic inhibitor of the mixed lineage kinase family. J. Biol. Chem. 276:25302–25308. [DOI] [PubMed] [Google Scholar]
- Martin, D.P., R.E. Schmidt, P.S. DiStefano, O.H. Lowry, J.G. Carter, and E.M. Johnson Jr. 1988. Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J. Cell Biol. 106:829–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, T.M., K.L. Moulder, C.M. Knudson, D.J. Creedon, M. Deshmukh, S.J. Korsmeyer, and E.M. Johnson Jr. 1997. Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J. Cell Biol. 139:205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano, K., and K.H. Vousden. 2001. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell. 7:683–694. [DOI] [PubMed] [Google Scholar]
- Palmada, M., S. Kanwal, N.J. Rutkoski, C. Gufstafson-Brown, R.S. Johnson, R. Wisdom, and B.D. Carter. 2002. c-jun is essential for sympathetic neuronal death induced by NGF withdrawal but not by p75 activation. J. Cell Biol. 158:453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potapova, O., S. Basu, D. Mercola, and N.J. Holbrook. 2001. Protective role for c-Jun in the cellular response to DNA damage. J. Biol. Chem. 276:28546–28553. [DOI] [PubMed] [Google Scholar]
- Putcha, G.V., K.L. Moulder, J.P. Golden, P. Bouillet, J.A. Adams, A. Strasser, and E.M. Johnson. 2001. Induction of BIM, a proapoptotic BH3-only BCL-2 family member, is critical for neuronal apoptosis. Neuron. 29:615–628. [DOI] [PubMed] [Google Scholar]
- Putcha, G.V., S. Le, S. Frank, C.G. Besirli, K. Clark, B. Chu, S. Alix, R.J. Youle, A. LaMarche, A.C. Maroney, and E.M. Johnson Jr. 2003. JNK-mediated BIM phosphorylation potentiates BAX-dependent apoptosis. Neuron. 38:899–914. [DOI] [PubMed] [Google Scholar]
- Raivich, G., M. Bohatschek, C. Da Costa, O. Iwata, M. Galiano, M. Hristova, A.S. Nateri, M. Makwana, L. Riera-Sans, D.P. Wolfer, et al. 2004. The AP-1 transcription factor c-Jun is required for efficient axonal regeneration. Neuron. 43:57–67. [DOI] [PubMed] [Google Scholar]
- Ruit, K.G., J.L. Elliott, P.A. Osborne, Q. Yan, and W.D. Snider. 1992. Selective dependence of mammalian dorsal root ganglion neurons on nerve growth factor during embryonic development. Neuron. 8:573–587. [DOI] [PubMed] [Google Scholar]
- Salina, D., P. Enarson, J.B. Rattner, and B. Burke. 2003. Nup358 integrates nuclear envelope breakdown with kinetochore assembly. J. Cell Biol. 162:991–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suntharalingam, M., and S.R. Wente. 2003. Peering through the pore: nuclear pore complex structure, assembly, and function. Dev. Cell. 4:775–789. [DOI] [PubMed] [Google Scholar]
- Talcott, B., and M.S. Moore. 1999. Getting across the nuclear pore complex. Trends Cell Biol. 9:312–318. [DOI] [PubMed] [Google Scholar]
- Weis, K. 2003. Regulating access to the genome: nucleocytoplasmic transport throughout the cell cycle. Cell. 112:441–451. [DOI] [PubMed] [Google Scholar]
- Whitfield, J., S.J. Neame, L. Paquet, O. Bernard, and J. Ham. 2001. Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron. 29:629–643. [DOI] [PubMed] [Google Scholar]
- Wu, J., M.J. Matunis, D. Kraemer, G. Blobel, and E. Coutavas. 1995. Nup358, a cytoplasmically exposed nucleoporin with peptide repeats, Ran-GTP binding sites, zinc fingers, a cyclophilin A homologous domain, and a leucine-rich region. J. Biol. Chem. 270:14209–14213. [DOI] [PubMed] [Google Scholar]
- Yu, J., L. Zhang, P.M. Hwang, K.W. Kinzler, and B. Vogelstein. 2001. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol. Cell. 7:673–682. [DOI] [PubMed] [Google Scholar]