

Abstract
p21-activated kinases (PAKs) regulate many cellular processes, including cytoskeletal rearrangement and cell migration. In this study, we report a direct and specific interaction of PAK1 with a 22-kD Ca2+-binding protein, CIB1, which results in PAK1 activation both in vitro and in vivo. CIB1 binds to PAK1 within discrete regions surrounding the inhibitory switch domain in a calcium-dependent manner, providing a potential mechanism of CIB1-induced PAK1 activation. CIB1 overexpression significantly decreases cell migration on fibronectin as a result of a PAK1-and LIM kinase–dependent increase in cofilin phosphorylation. Conversely, the RNA interference–mediated depletion of CIB1 increases cell migration and reduces normal adhesion-induced PAK1 activation and cofilin phosphorylation. Together, these results demonstrate that endogenous CIB1 is required for regulated adhesion-induced PAK1 activation and preferentially induces a PAK1-dependent pathway that can negatively regulate cell migration. These results point to CIB1 as a key regulator of PAK1 activation and signaling.
Introduction
Upon adhesion to ECM, cytoskeletal rearrangements occur that lead to cell spreading, actin turnover, and cell migration. The p21-activated kinase (PAK) family of serine/threonine kinases plays a significant role in regulating these processes (Kiosses et al., 1999; Sells et al., 1999). The best-described upstream activators of the PAK family are the Rho GTPases Rac and Cdc42. These small GTPases bind within the NH2 terminus of PAK, resulting in PAK autophosphorylation and increased PAK catalytic activity (Leung et al., 1994).
Although it is generally considered that PAK1 activity is primarily regulated via small GTPases, GTPase-independent mechanisms have also been described. Thus, PAK1 activity can be stimulated by sphingosine (Bokoch et al., 1998; Lian et al., 1998), by the actin-binding protein filamin A (Vadlamudi et al., 2002), and by PI3 kinase (Papakonstanti and Stournaras, 2002). Additional PAK1-binding proteins include the family of PAK-interacting exchange factors (Cool/PIX; Bagrodia et al., 1998; Daniels et al., 1999), Etk/Bmx (epithelial and endothelial/bone marrow tyrosine kinase gene in chromosome X; Bagheri-Yarmand et al., 2001), and p35/Cdk5 kinase (Rashid et al., 2001).
Once activated, PAK1 affects multiple pathways to regulate cytoskeletal dynamics and cell migration. However, various studies have described both a positive and negative role for PAK1 in regulating cell migration. For example, overexpression of constitutively active (ca) PAK1 mutants promotes cell migration on collagen (Sells et al., 1997, 1999), possibly via p38-MAPK (Adam et al., 2000; Dechert et al., 2001), whereas in other studies, active PAK1 mutants inhibit cell migration on fibronectin (FN; Kiosses et al., 1999). Furthermore, PAK1 kinase activity is required for directional or haptotactic cell migration (Sells et al., 1999; Adam et al., 2000) but not for random cell movement (Sells et al., 1999). Inhibitory effects of PAK1 on migration appear to involve PAK1 activation of cytoskeletal regulatory proteins such as Lin-11/Isl-1/Mec-3 kinase (LIMK) 1 (Edwards et al., 1999), which, in separate studies, phosphorylates and inactivates the actin depolymerizing factor cofilin (Arber et al., 1998; Yang et al., 1998). Phosphorylation and inactivation of cofilin diminished cell polarity (Dawe et al., 2003; Ghosh et al., 2004) and directed cell movement (Ghosh et al., 2004). However, mechanisms by which PAK1 couples to this negative regulatory pathway are not well understood.
In this study, we report a novel Rac/Cdc42-independent pathway of PAK1 activation by an EF hand–containing regulatory molecule termed CIB1 (also CIB, calmyrin, and KIP [kinase-interacting protein]). CIB1 was originally identified as a 22-kD protein that binds to the platelet integrin αIIb cytoplasmic tail (Naik et al., 1997). However, CIB1 is widely distributed and is likely to have cellular roles that are independent of this platelet-specific integrin. CIB1 contains four EF hand motifs, two of which bind calcium (Gentry et al., 2004; Yamniuk et al., 2004), and is NH2-terminally myristoylated. CIB1 can bind presenilin-2 (Stabler et al., 1999), Rac3 (Haataja et al., 2002), FAK (Naik and Naik, 2003), DNA-dependent protein kinase (Wu and Lieber, 1997), and fibroblast growth factor– and serum-inducible kinases (Kauselmann et al., 1999). However, the functions of endogenous CIB1 and its relationship to relevant intracellular binding partners have not been clearly delineated. We report that CIB1 binds to and specifically activates PAK1 both in vitro and in vivo via a specific CIB1-binding region within PAK1. The CIB1–PAK1 interaction is required for normal adhesion-induced PAK1 activation, which negatively regulates cell migration across FN and appears to involve a PAK1–LIMK–phosphocofilin pathway. Therefore, our results establish CIB1 as a key regulator of PAK1 activation and migration.
Results
CIB1 interacts directly with PAK1
To learn about the in vivo function of CIB1, we sought to identify endogenous CIB1-interacting proteins first by immunoprecipitating CIB1 from platelet lysates. Immunoblotting with an antiphosphoserine antibody revealed a single reactive band of ∼66 kD that specifically coimmunoprecipitated with CIB1 (Fig. 1 A, left). This band was also evident when immunoblotting with an anti-PAK antibody (Fig. 1 A, middle), indicating that CIB1 coimmunoprecipitates with a member of the PAK family. Furthermore, whole platelet lysates were positive for PAK expression (Fig. 1 A, right). Because platelets are primary hematopoietic cells that exist in suspension, we also asked whether endogenous CIB1 and PAK coimmunoprecipitate in an adherent cell line (rat embryo fibroblast [REF] 52). Although CIB1 interacted minimally with PAK in suspended REF52 cells, a dramatic increase in CIB1 binding to PAK was observed within 30 min of cell adhesion to FN. This decreased slightly with extended times of adhesion (Fig. 1 B), suggesting an adhesion-dependent regulation of the endogenous CIB1–PAK interaction.
Figure 1.
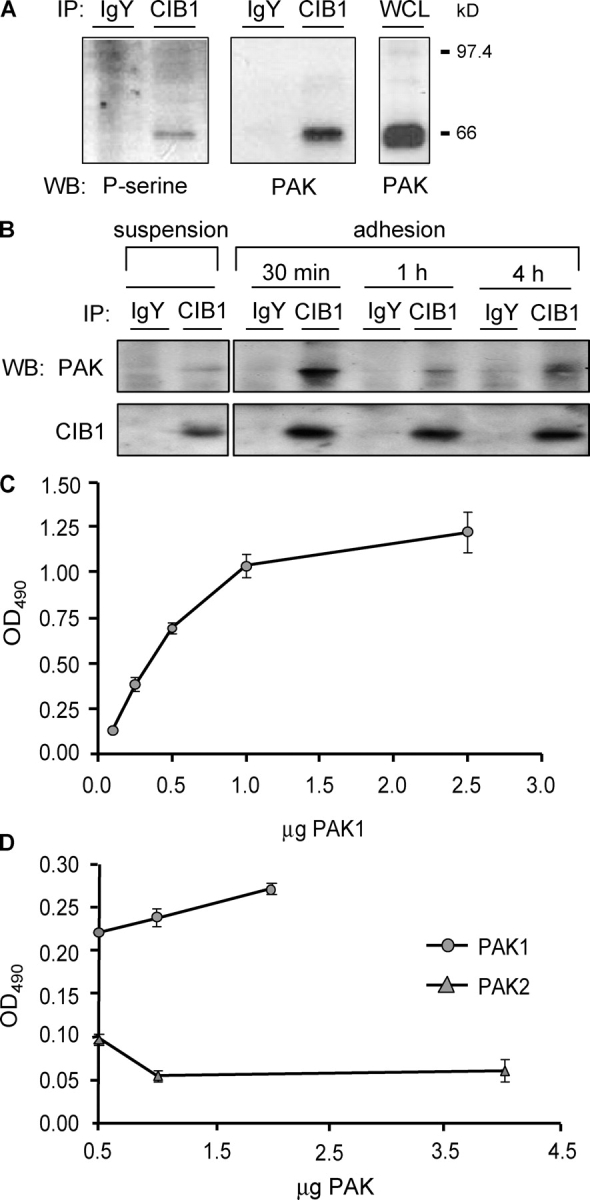
CIB1 binds to PAK1 in vivo and in vitro. (A) Immunoprecipitates from platelet lysates were immunoblotted with an antiphosphoserine (left) or anti-PAK (middle) antibody. Whole cell lysates (WCL) were probed with an anti-PAK antibody (right; n ≥ 3 experiments). (B) CIB1 and control IgY immunoprecipitates from lysates of REF52 cells in suspension or adhered to FN were immunoblotted for PAK (top) and CIB1 (bottom). (C) Solid-phase binding assays using immobilized CIB1 and soluble PAK proteins. Increasing concentrations of His-PAK1 were added to wells coated with and without immobilized CIB1. His-PAK1 binding was detected using an anti-His antibody. (D) PAK isozyme–binding specificity was determined by adding GST-PAK1 or GST-PAK2 to immobilized CIB1. PAK binding was detected with an anti-GST antibody. WB, Western blot; IP, immunoprecipitates. Error bars represent SEM.
Coimmunoprecipitation of endogenous PAK with CIB1 does not indicate whether CIB1 interacts directly with PAK or indirectly via other proteins. To address this, we used purified recombinant CIB1 and PAK1 in solid-phase binding assays and found that PAK1 bound to immobilized CIB1 in a direct, saturable manner (Fig. 1 C), whereas the PAK2 isoform exhibited minimal binding (Fig. 1 D).
Identification of the CIB1-binding site within PAK1
To begin identifying the CIB1-binding site within PAK1, we assessed the CIB1-binding activity of a PAK1 NH2-terminal deletion mutant lacking the first 164 amino acids (N165-PAK1). This mutant failed to bind immobilized CIB1, indicating that the CIB1-binding site is probably within the PAK1 NH2 terminus (Fig. 2 A). Further delineation using peptide SPOT technology (Frank, 2002) demonstrated that CIB1 reacts with peptides corresponding to two distinct sites within PAK1 (Fig. 2 B); the first maps to aa 50–61 (site I) just upstream of the p21-binding domain (PBD; aa 70–113), and the second maps to aa 130–137 (site II) within the NH2-terminal autoinhibitory domain. Sequence homology searches revealed that sites I and II share little sequence identity with other closely related PAK members (30–50% identity to corresponding regions of PAK2 and PAK3) and with other known proteins, which is consistent with a relative lack of CIB1 binding to PAK2.
Figure 2.

Identification of CIB1-binding sites within the PAK1 NH 2 terminus. (A) CIB1 binds to full-length wild-type GST-PAK1 (wtPAK1) and GST-PAK1 K298A (kdPAK1) but not the NH2-terminal deletion GST-N165-PAK1. Solid-phase binding assays were performed as in Fig. 1 C, and soluble GST-PAK1, GST-PAK1 K298A, and GST-N165-PAK1 were added to wells coated with CIB1. (B) Delineation of the CIB1-binding sites in the PAK1 NH2-terminal sequence by SPOT peptide method. Top and middle panels show two sets of CIB1 reactive spots labeled as sites I and II relative to the PBD- or Rac/Cdc42-binding sites (residues 70–113, dotted boxes). The bottom panel indicates the inhibitory switch (IS) and kinase inhibitor (KI) domains. Sequences of corresponding CIB1-binding sites are below. (C) Inhibition of CIB1 binding to PAK1 with site I and II peptides. Ni-NTA agarose beads loaded with purified His-PAK1 were added to recombinant CIB1 that was preincubated with and without PAK1 peptides corresponding to sites I and II and scrambled sites I and II. Peptide sequences of sites I, II, and IIΔ are shown below. The graph represents densitometry of affinity precipitates probed for CIB1 and normalized to His-PAK1 that was immunoblotted from the same membrane. Data represent SEM from two independent experiments (error bars). (D) Loss of CIB1 binding to site I and II PAK1 mutants. Clarified lysates from HEK293 cells cotransfected with CIB1 and myc wild-type (wt)PAK1, myc-PAK1 siteIAAA (myc-AAAPAK1), or myc-PAK1 siteIΔ/siteIIAAA (myc-Δ/AAA PAK1) were immunoprecipitated with a control IgG or anti-CIB1 antibody and with samples immunoblotted for myc (top) and CIB1 (bottom; n ≥ 4). Western blots of the input expression of CIB1 and myc-tagged wtPAK1 (closed arrowheads), AAAPAK (closed arrowheads), or Δ/AAAPAK1 (open arrowheads).
To further confirm these binding sites, we used pull-down assays to test the ability of synthetic peptides (corresponding to sites I and II) to inhibit soluble CIB1 binding to PAK1 that was immobilized on Ni2+–nitrilotriacetic acid (NTA) agarose beads. Preincubation of CIB1 with either site I or II peptide significantly inhibited CIB1 binding to PAK1, although the site II peptide appeared to be slightly more effective. In contrast, corresponding scrambled peptides had a minimal effect (Fig. 2 C). In addition, a peptide lacking the last four amino acids of site II (ΔKTSN and site IIΔ) but possessing additional upstream NH2-terminal residues did not inhibit CIB1 binding to PAK1. This suggests a role of KTSN site II residues in PAK1 recognition of CIB1 (Fig. 2 C).
To further establish the importance of these CIB1-binding sites within the intact PAK1 molecule, the COOH-terminal K135/T136/S137 residues of site II were mutated to alanines (myc-AAAPAK1), and these PAK1 mutants were coexpressed with CIB1 (Fig. 2 D, right). These mutations significantly diminished the ability of PAK1 to coimmunoprecipitate with CIB1. However, overexpression of an AAA site II construct in which the NH2-terminal fragment encompassing site I (aa 8–68) was deleted (myc-Δ/AAAPAK1) did not result in an additional loss of PAK1 binding to CIB1 (Fig. 2 D, left). These results indicate that although both sites in PAK1 may contribute to the CIB1–PAK1 interaction, site II appears to play a more critical role than site I.
Active Cdc42 and Ca2+ affect the interaction between CIB1 and PAK1
Because CIB1 binds near the PBD within PAK1, we asked whether Cdc42 affects the interaction between CIB1 and PAK1. Competition binding assays indicated that PAK1 binding to immobilized CIB1 was largely inhibited by soluble, active Cdc42-GTPγS, but not by inactive Cdc42–GDP (Fig. 3 A). A specific interaction between CIB1 and PAK1 was further confirmed by the ability of soluble CIB1 to inhibit PAK1 binding to immobilized CIB1 (Fig. 3 A).
Figure 3.
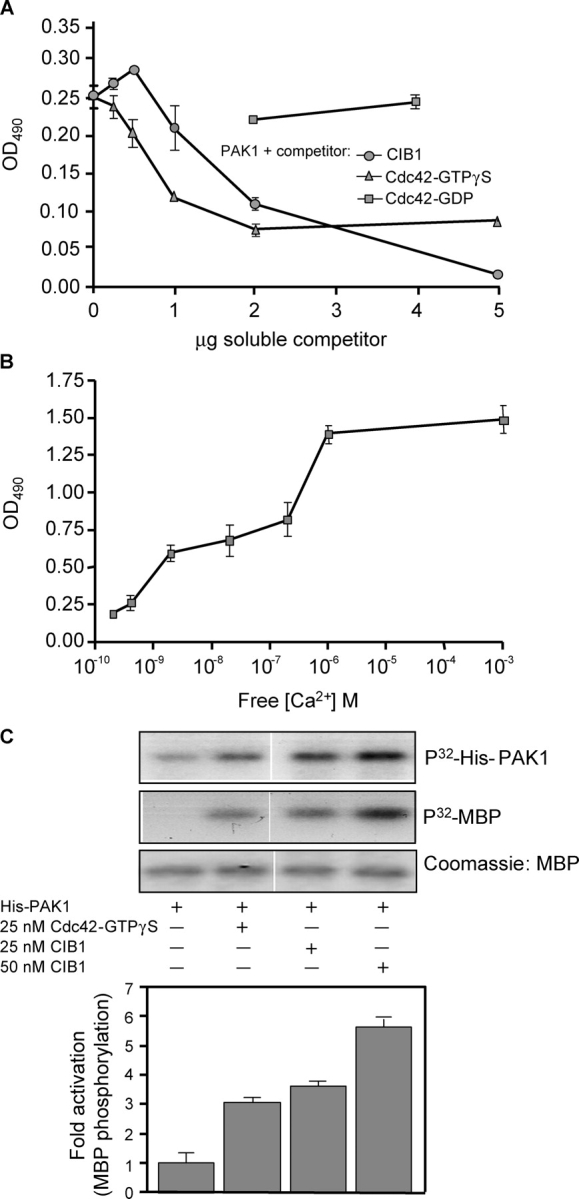
Cdc42 and Ca 2+ affect the CIB1–PAK interaction, and CIB1 stimulates PAK1 activity in vitro. (A) Activated Cdc42-GTPγS or CIB1, but not inactive Cdc42-GDP, competes with immobilized CIB1 for binding to soluble His-PAK1. Soluble His-PAK1 was incubated with increasing concentrations of soluble CIB1 or Cdc42 that was preloaded with GDP or GTPγS before incubation with immobilized CIB1. (B) Determination of Ca2+-dependent binding of His-PAK1 to CIB1. His-PAK1 was diluted in buffer containing 0–5 mM EGTA before addition to immobilized CIB1. Approximate free Ca2+ concentrations were calculated using the MaxChelator program (Bers et al., 1994). (C) Stimulation of recombinant His-PAK1 activity by recombinant CIB1 or Cdc42-GTPγS. His-PAK1 autophosphorylation was assayed in the absence (top) or presence of myelin basic protein (MBP) to detect active kinase (middle). White lines indicate that intervening lanes have been spliced out. Myelin basic protein phosphorylation ([32P]MBP) from densitometry analysis induced by His-PAK1 alone was assigned a value of 1 (bar graph). Data represent means ± SEM (error bars; n = 3).
Because CIB1 contains two Ca2+-binding EF hand domains, we next asked whether PAK1 binding to CIB1 was Ca2+ dependent. In the absence of EGTA, PAK1 binding to immobilized CIB1 was maximized at 1 μM Ca2+ and was not further enhanced by 1 mM Ca2+. However, with decreasing free Ca2+ concentrations, PAK1 binding to immobilized CIB1 diminished significantly at or below 10−8 M free Ca2+ (Fig. 3 B), suggesting that physiologic Ca2+ concentrations modulate the interaction between CIB1 and PAK1.
CIB1 directly stimulates PAK1 activity both in vitro and in vivo
Binding of activated Rac and Cdc42 to members of the PAK family results in PAK autophosphorylation and stimulation of kinase activity. Because both Cdc42-GTPγS and CIB1 bind to PAK1 within the NH2-terminal regulatory domain, we explored whether CIB1 also activates PAK1. In vitro kinase assays demonstrated that His-PAK1 alone exhibited little autophosphorylation and kinase activity, but, as expected, 25 nM Cdc42-GTPγS increased His-PAK1 kinase activity threefold (Fig. 3 C). Interestingly, 25 nM CIB1 alone stimulated both PAK1 autophosphorylation and kinase activity to an equal or slightly greater degree than an equimolar amount of Cdc42-GTPγS. Moreover, a twofold higher concentration of CIB1 (50 nM) further increased His-PAK activation to sixfold above basal level (Fig. 3 C), indicating that CIB1 directly activates PAK1 independently of small GTPases.
To examine whether CIB1 can activate PAK1 in vivo, we overexpressed CIB1 in human embryonic kidney (HEK) 293 cells and tested for activation of endogenous PAK1. Consistent with previous reports (Price et al., 1998; Kiosses et al., 1999), vector-transfected cells that were held in suspension exhibited minimal PAK1 activation, whereas cell adhesion to FN up-regulated PAK1 activation (Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200502090/DC1). In contrast, CIB1-overexpressing cells that were held in suspension showed a higher level of PAK1 activation than did vector control cells that were further increased upon cell adhesion relative to the vector (Fig. S1 A). This indicates that CIB1 can stimulate PAK1 activity in vivo.
Because our in vitro binding studies indicate a calcium-dependent interaction between CIB1 and PAK1 (Fig. 3 B), we also asked whether calcium levels affect PAK1 activation in vivo. Pretreatment of control and CIB1-overexpressing cells with the Ca2+ chelator BAPTA-AM resulted in decreased adhesion-dependent PAK activation (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200502090/DC1), suggesting a Ca2+-dependent component to PAK1 signaling.
CIB1 specifically activates PAK1 independently of Rho GTPases and affects cell morphology
To better study the consequences of the CIB1–PAK1 interaction, we chose a nontransformed REF52 cell line, which has been well characterized with respect to cytoskeletal organization, cell spreading, and migration (Pavalko and Burridge, 1991). As predicted by the conserved CIB1-binding sites in rat PAK1 (Fig. S1 B), we found that overexpression of human CIB1 in suspended or FN-adherent REF52 cells activated endogenous rat PAK1 to a similar extent as human PAK1 in HEK293 cells (at least twofold; Fig. 4 A). Although our in vitro binding studies indicated that CIB1 binds to the PAK1, but not to the PAK2, isoform (Fig. 1 D), it is possible that CIB1 may bind to and activate the more closely related PAK3 isoform. However, under the same conditions in which we observed CIB1-induced PAK1 activation, overexpressed CIB1 did not stimulate PAK3 activity in REF52 cells (Fig. 4 A).
Figure 4.

CIB1 specifically stimulates PAK1 activity in vivo independently of small GTPases. (A) CIB1 specifically activates the PAK1 isoform. Endogenous PAK1 (bottom left) and PAK3 (bottom right) were immunoprecipitated from lysates prepared from vector- and CIB1-transfected REF52 cells either held in suspension or replated on FN for 20 min. Immunoprecipitated PAK was subjected to in vitro kinase assays using myelin basic protein (MBP) as substrate. The bar graph (top) depicts [32P]MBP values after normalization for PAK immunoprecipitation. [32P]MBP values from vector control cells were set as 1. Data represent SEM from two independent experiments (error bars). (B) GTPase-independent PAK1 activation. Serum-starved vector and CIB1-transfected REF52 cells held in suspension were treated with or without 100 ng/ml toxin B and either left in suspension or adhered to FN. Cells were lysed at the indicated times, and immunoprecipitated endogenous PAK1 was subjected to in vitro kinase assays using myelin basic protein as substrate. White lines indicate that intervening lanes have been spliced out; n = 3. (C) Efficacy of toxin B treatment was determined by assaying REF52 cell lysates for activated Rac1 and Cdc42 (see PAK1 kinase and Rac/Cdc42 activation assays). Affinity precipitates were analyzed by Western blotting for both Rac1 and Cdc42.
Because PAK1 can also be activated by Rac and Cdc42 (Manser et al., 1994; Bagrodia et al., 1995), we asked whether these GTPases contributed to the CIB1-induced PAK1 activation that was observed in cells adherent to FN. Inactivation of Rho, Rac, and Cdc42 with toxin B from Clostridium difficile (Just et al., 1995; Chaves-Olarte et al., 2003) did not block initial (30 min) adhesion-induced PAK1 activation in control-transfected cells that were replated on FN (Fig. 4 B) but did attenuate PAK1 activation in control cells that were adhered for extended times (180 min). These results suggest a role for Rho GTPases in PAK1 activation at these later time points. Importantly, in cells overexpressing CIB1, toxin B did not inhibit PAK1 activation at each time tested, further demonstrating that CIB1 can stimulate PAK1 activity in the absence of active Rho GTPases. The inhibition of Rho GTPases by toxin B was confirmed with precipitation assays to detect active Cdc42 and Rac under the same conditions tested in Fig. 4 B (Fig. 4 C). In agreement with our toxin B results, the coexpression of both dominant negative (DN) Rac and Cdc42 did not significantly inhibit PAK1 activation upon adhesion to FN (Fig. S3 A, available at http://www.jcb.org/cgi/content/full/jcb.200502090/DC1) but did attenuate PAK1 activation with prolonged adhesion to FN. These results further confirm an initial GTPase-independent component to PAK1 activation upon cellular adhesion to FN.
Because PAK1 activity modulates cytoskeletal architecture (Manser et al., 1997; Sells et al., 1997) and CIB1 stimulates PAK1 activity, we asked whether CIB1 overexpression also modulates cytoskeletal architecture. CIB1-transfected cells exhibited a distinct absence of large, mature focal adhesions that are normally found throughout stably adherent cells (Fig. S4 D, available at http://www.jcb.org/cgi/content/full/jcb.200502090/DC1), which is consistent with the loss of focal adhesions in overexpressed active PAK1 mutants (Manser et al., 1997; Sells et al., 1997). Furthermore, CIB1-overexpressing cells showed extensive membrane ruffling, with CIB1 localizing to these areas of increased actin dynamics (Fig. S4 A, merge). CIB1 overexpression also resulted in decreased cell spreading, and cells became asymmetrical with multiple nonpolarized extensions (Fig. S4, A and B). The exogenous expression of CIB1 with myc-wtPAK1 revealed that both CIB1 and PAK1 are distributed within the cytoplasm and that CIB1 is also expressed within the nucleus (Fig. S4). Importantly, during cell spreading upon readhesion to FN, CIB1 and PAK1 are prominently colocalized within membrane ruffles (Fig. S4 E, left and inset). Furthermore, with extended adhesion to FN (1–2 h), we observed persistent CIB1–PAK1 colocalization within these membrane structures (Fig. S4 E; middle, right, and insets), providing evidence for an interaction between CIB1 and PAK1 in subcellular areas of dynamic actin remodeling.
CIB1 negatively regulates cell migration on FN
Active PAK1 mutants are reported to have both stimulatory and inhibitory effects on cell migration, depending on the type and concentration of ECM substrate (Sells et al., 1997; Kiosses et al., 1999). Therefore, we asked whether overexpression of CIB1 would induce a positive or negative PAK1-dependent effect on cell migration toward FN. Haptotactic migration assays were performed with serum-starved cells that were transfected either with CIB1 or vector control. Visualization of CIB1-transfected cells on the underside of the transwell membrane (Fig. 5 A, middle) indicated a dramatic 2.5-fold decrease in migrating cells relative to vector control or GFP-expressing cells, as quantified in Fig. 5 A (left). To determine whether the inhibitory effect of CIB1 on cell migration was PAK1 dependent, CIB1 was cotransfected with kinase-dead (kd) PAK1-K299R (kdPAK1), which restored cell migration to control levels (Fig. 5 A, left). A role for PAK1 in the CIB1-induced inhibition of migration was further established with mouse embryo fibroblasts (MEFs) that were derived from PAK1-null (PAK−/−) and wild-type (PAK+/+) mice (unpublished data). Consistent with the results in Fig. 5 A, PAK1+/+ cells overexpressing CIB1 exhibited a twofold decrease in haptotactic cell migration toward FN relative to GFP vector control, whereas PAK1−/− MEFs expressing CIB1 migrated normally (Fig. 5 B). This demonstrates a requirement for PAK1 in the CIB-induced inhibition of migration. Interestingly, the lack of PAK1 in PAK1−/− MEFs did not impair cell migration, suggesting that these cells possess compensatory migration pathways and/or that PAK1 is not required for haptotactic migration toward FN under these conditions.
Figure 5.
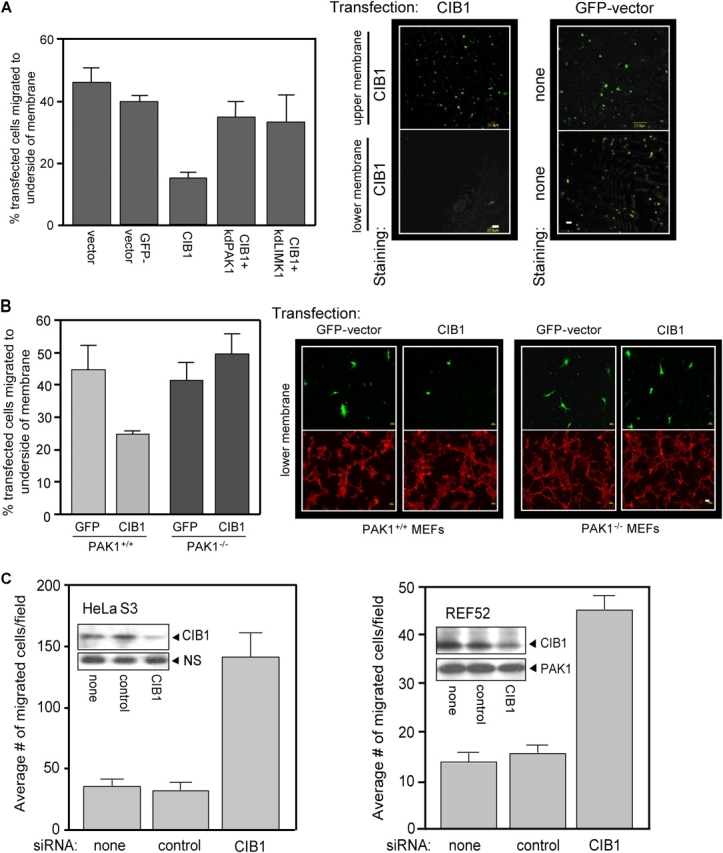
CIB1 overexpression inhibits, and endogenous CIB1 depletion increases, cell migration. (A) Serum-starved REF52 cells transfected with GFP vector, control vector, or CIB1 ± kdPAK1 or kdLIMK1 were subjected to haptotactic transwell migration assays toward FN. Transfected cells on either the top membrane (nonmigrating cells) or bottom membrane (migrating cells) were visualized by staining for CIB1 expression (middle). Control migration was visualized by GFP fluorescence (right). Cells overexpressing vector CIB1 ± kdPAK1 or ± kdLIMK1 on the top and bottom membranes were also stained as described in migration assays and were counted. Migration is represented as the percentage of the total number of transfected cells from the upper and lower membranes (left). Data represent means ± SEM (n = 3). (B) MEFs derived from wild-type (PAK+/+) and PAK1-null (PAK−/−) mice were transfected with GFP vector or CIB1. Serum-starved cells were assayed for haptotactic migration toward 3 μg/ml FN, and migration was determined as in A (left). Data represent means ± SEM (n = 4). Right panels show representative images of migrated transfected cells (green, top) and phalloidin staining from the same field (red, bottom). (A and B) Bars, 20 μm. (C) HeLaS3 (left) and REF52 (right) cells were mock transfected or transfected with control or specific CIB1 siRNA and subjected to haptotactic transwell migration assays toward FN (as in A). Data represent means ± SEM (error bars; n = 4 for each cell type). Inset blots show representative endogenous CIB1 protein expression and nonspecific (NS) band or PAK1 expression from the same blot as the loading control.
Because CIB activates PAK1 and inhibits cell migration, we asked whether ca PAK1 affected migration in a similar manner. REF52 cells overexpressing either ca PAK1-T423E (caPAK1) or CIB1 plus wtPAK1 exhibited a twofold decrease in cell migration toward FN as compared with cells expressing either wtPAK1 or kdPAK1 (Fig. S5 A, available at http://www.jcb.org/cgi/content/full/jcb.200502090/DC1). Because active PAK1 has been shown to stimulate migration of another cell type, NIH3T3, across collagen (Sells et al., 1999), we tested the effects of caPAK1 on NIH3T3 cell migration across FN. Similar to REF52 cells, NIH3T3 cells overexpressing caPAK1 also exhibited an approximately twofold decrease in haptotactic migration compared with cells expressing wtPAK1 (Fig. S5 B). Moreover, CIB1 overexpression in NIH3T3 cells resulted in a similar inhibition of haptotactic migration toward FN (Fig. S5 B). Apparent differences in the effects of caPAK1 are most likely a result of the type and concentration of matrix used in this study (5 μg/ml FN) versus the 50 μg/ml of collagen that were used by Sells et al. (1999). Collectively, our results demonstrate not only an inhibitory role for PAK1 kinase activity in cell migration toward FN but also further implicate PAK1 in mediating CIB1-induced inhibition of cell migration.
A potential downstream effector that may contribute to the CIB1-induced inhibition of cell migration is LIMK1 because PAK1 directly activates LIMK1 (Edwards et al., 1999), which then phosphorylates and inactivates cofilin (Arber et al., 1998; Yang et al., 1998). To test this hypothesis, we cotransfected CIB1 with kd LIMK1-D460N (kdLIMK1) into REF52 cells. Migration was rescued in cells expressing both CIB1 and kdLIMK1 (Fig. 5 A, left), indicating that CIB1–PAK1 signaling to LIMK1 contributes to the inhibitory effects of CIB1 on cell migration. Equivalent CIB1 expression levels were confirmed by immunoblotting (see Fig. 7 C, right).
Figure 7.

CIB1 modulates downstream signaling to cofilin. (A) Lysates were prepared from control or specific CIB1 siRNA-transfected REF52 cells either held in suspension or adhered to FN for the indicated times. Densitometry of p-cofilin levels from lysates that were prepared from control or specific CIB siRNA was normalized to ERK or PAK1 from the same blots. Error bars represent means ± SEM (n = 2). (B) Representative membrane immunoblotted with antibodies against total or phosphorylated cofilin (p-cofilin). The top half of the membrane was also immunoblotted for PAK1 expression (bottom). (C) Lysates prepared from REF52 cells overexpressing empty vector or CIB1 ± kdPAK1 or kdLIMK1 were analyzed for cofilin phosphorylation as in A. The membrane was reprobed for total cofilin (bottom). Lysates from cells expressing control vector, CIB1, or CIB1 coexpressed with kdPAK1 or kdLIMK1 were immunoblotted using anti-CIB1, -PAK1, or -LIMK1 antibodies (middle and right). PAK1 immunoblots show both endogenous PAK1 and overexpressed kdPAK1 (top middle). Immunoblotting for LIMK1 also shows endogenous LIMK1 and overexpressed kdLIMK1 (top right). Middle blots show overexpressed CIB1. Membranes were also probed with an anti-ERK antibody as a loading control (bottom, middle and right). Data represent two separate experiments.
Because overexpressed CIB1 inhibited cell migration, we asked whether decreased endogenous CIB1 expression by RNA interference would have the converse effect on cell migration. Transfection of short inhibitory (si) RNA duplexes that target CIB1 mRNA in two separate cell lines, HeLaS3 and REF52, caused a significant decrease in endogenous CIB1 protein levels without affecting PAK1 expression (Fig. 5 C, insets). Reduced CIB1 expression resulted in a striking five- and threefold increase in haptotactic cell migration toward FN in both HeLaS3 and REF52 cells, respectively, whereas a control siRNA was without effect (Fig. 5 C). Altogether, these results indicate that endogenous CIB1 negatively regulates cell migration across FN.
Endogenous CIB1 affects PAK1 activation
We next asked whether the reduction of endogenous CIB1 affected endogenous PAK1 activation. Upon adhesion to FN, PAK1 activation in CIB1-depleted cells was markedly reduced compared with control cells at all times tested except 2.5 h (Fig. 6 A). At this later time point, when PAK1 activation in control cells had diminished, PAK1 activation in CIB1-depleted cells (REF52 cells, Fig. 6 A; and HeLaS3 cells, not depicted) was moderately elevated, suggesting a disruption of the normal regulation of PAK1 activation. Potential candidates to mediate this later PAK1 activation are the GTPases, Rac, and/or Cdc42. Interestingly, at 3 h of adhesion, increased Cdc42, but not Rac, activity was observed in CIB1-depleted cells compared with control cells (Fig. 6 B). This suggests that in CIB1-depleted cells, Cdc42 is the predominant GTPase mediating the late up-regulation of PAK1 activation. Active Cdc42 promotes the formation of filopodia and microspikes (Nobes and Hall, 1995). Consistent with increased Cdc42 activity at 3 h of adhesion to FN, immunofluorescence analysis of phalloidin-stained cells that adhered for 3 h revealed that CIB1-depleted cells also exhibited extensive filopodia and polarity (Fig. 6 C, right) relative to control cells (Fig. 6 C, left).
Figure 6.

CIB1 is required for adhesion-induced PAK1 activation, and loss of CIB1 disrupts PAK1/GTPase signaling. (A) Mock, control, or specific CIB1 siRNA-transfected REF52 cells were either held in suspension or replated onto FN-coated dishes and were lysed at the indicated times. Immunoprecipitated endogenous PAK1 was subjected to in vitro kinase assays (n = 3). (B) Control or CIB1 siRNA-transfected REF52 cell lysates were assayed for activated and total Rac1 and Cdc42. CIB1 knockdown in REF52 cells was confirmed by immunoblotting cell lysates for endogenous CIB1 expression (right) with ERK as a loading control from the same membrane. (C) REF52 cells transfected with control (left) or CIB1 siRNA (right) were replated on FN for 3 h and were stained with phalloidin. Images are representative of two separate experiments. Bars, 20 μm.
CIB1 affects downstream signaling to cofilin
The previous experiments demonstrated that endogenous CIB1 regulates endogenous PAK1 activation and that CIB1-induced inhibition of cell migration requires PAK1 and LIMK1 activity. Presently, the only known substrate of LIMK1 is the actin depolymerizing and severing protein cofilin (Arber et al., 1998). Nonphosphorylated active cofilin (Yang et al., 1998) is required for rapid actin filament turnover and cell motility (Chen et al., 2001; Dawe et al., 2003). Therefore, we assessed cofilin phosphorylation in cells under- and overexpressing CIB1. CIB1-depleted REF52 cells exhibited decreased phosphorylation of endogenous cofilin upon adhesion to FN, which persisted up to 3 h (Fig. 7 A); conversely, CIB1-overexpressing cells showed increased cofilin phosphorylation (Fig. 7 C, left). Decreased cofilin phosphorylation in CIB1-depleted cells further links CIB1 to a PAK1–LIMK–cofilin pathway.
To determine whether PAK1 signaling to LIMK1 was required for the CIB1-induced increase in cofilin phosphorylation, we coexpressed CIB1 with kdPAK1 or kdLIMK1. Coexpression of CIB1 with either kdPAK1 or kdLIMK1 blocked the CIB1-induced increase in cofilin phosphorylation (Fig. 7 C, left). To confirm that the decreased cofilin phosphorylation observed in cells coexpressing CIB1 with kdPAK1 or kdLIMK1 was not caused by decreased CIB1 expression, lysates were analyzed for CIB1, PAK1, and LIMK1 expression (Fig. 7 C, right), which showed no difference in CIB1 levels among transfected cells. Therefore, these results demonstrate that CIB1-mediated inhibition of cell migration most likely occurs via PAK1–LIMK-dependent phosphorylation and inactivation of cofilin.
Discussion
In this study, we show that CIB1 binds directly to PAK1 to regulate PAK1 activation, cell spreading, and cell migration. CIB1 binding to PAK1 is calcium dependent and is inhibited by active GTP-Cdc42. The CIB1-binding sites within PAK1 map to the NH2-terminal residues 130–137 (site II) within the inhibitory switch (IS) domain, thereby providing a mechanism by which CIB1 directly activates PAK1 both in vitro and in vivo. In addition, CIB1 overexpression and depletion experiments indicate an important role for endogenous CIB1 in activating endogenous PAK1 upon cell adhesion and in suppressing cell migration toward FN. CIB1 appears to suppress cell migration by signaling to PAK1, thereby promoting a LIMK-dependent phosphorylation and inactivation of the actin regulatory protein cofilin.
Our results show that purified CIB1 activates PAK1 in vitro independently of small GTPases. However, because the Rho GTPases Rac and Cdc42 are the best-characterized PAK activators and because CIB1 reportedly binds directly to Rac3 (Haataja et al., 2002), it was necessary to determine whether CIB1 also activates PAK1 in vivo independently of small GTPases. In support of this possibility, CIB1 and Cdc42 compete for binding to PAK1 in vitro. We extended this observation in vivo by assessing PAK1 activation in cells in which Rac and Cdc42 were inhibited by toxin B or DN mutants. Because neither toxin B nor DN Rac and Cdc42 inhibited the peak of PAK1 activation, which was induced upon cell adhesion in vector control cells, it appears that molecules other than Rac and Cdc42 (for example CIB1) might contribute to the initial adhesion-induced PAK1 activation. This is also consistent with the observation that CIB1-depleted cells exhibit impaired PAK1 activation immediately upon cell adhesion. However, with extended adhesion to FN, PAK1 activation in control cells was sensitive to both toxin B and DN Rac/Cdc42, implicating a role for Rac- and Cdc42-mediated PAK1 activation at these later time points. Consistent with this observation, prolonged adhesion resulted in an apparent CIB1-independent up-regulation of PAK1 activation in CIB1-depleted cells. In contrast, PAK1 activation in CIB1-overexpressing cells was insensitive to toxin B at all times tested, demonstrating the ability of CIB1 to override any GTPase component of PAK1 activation. These results further demonstrate that CIB1 can activate PAK1 in vivo independently of small GTPases.
CIB1 has two putative binding sites within PAK1 NH2-terminal residues (site I, aa 50–60; and site II, aa 130–137) that flank the Rac/Cdc42 PBD (aa 70–113). Substitution of site II residues 135KTS137 with alanines results in a significant loss of CIB1 binding to PAK1, further emphasizing the direct interaction between CIB1 and PAK1. Interestingly, these site II residues lie within a COOH-terminal extension of the IS domain (aa 136–149) that is referred to as the kinase inhibitor (KI) domain (aa 136–149), which is a region that is critical to regulating PAK1 activation (Lei et al., 2000). The PAK1 crystal structure indicates that PAK1 exists as a homodimer and that in the autoinhibited conformation, the KI domain, which contains site I, is positioned within and directly contacts the catalytic cleft of the kinase domain (Lei et al., 2000). Structural information on site I within the context of PAK1 is unknown at this time because this region was not included in the PAK1 crystal structure. However, nearby NH2-terminal PBD residues are hypothesized to form initial contacts with Cdc42 as a result of their relative exposure within the structure (Lei et al., 2000). Therefore, it is possible that CIB1 binding to PAK1 occurs as a two-stage event in which CIB1 may initially interact with the more accessible site I residues and alter the PAK1 conformation, which would then allow binding within site II to fully activate PAK1.
Because CIB1 is widely expressed and interacts with a variety of proteins, it is possible that CIB1 may have different functions in different cell types. It was recently reported that overexpression of human CIB1 in CHO cells also overexpressing two additional CIB1 binding partners, Rac3 and integrin αIIbβ3, resulted in αIIbβ3-dependent cell spreading on fibrinogen (Haataja et al., 2002). Because the presence of additional CIB1-binding partners may induce a very different distribution of CIB1 relative to that found in our cells, which lack endogenous αIIbβ3 and also may not express endogenous Rac3, it is difficult to directly compare these results with our study. Also, a separate report indicated that stably overexpressed human CIB1 in CHO cells induced cell migration on FN (Naik and Naik, 2003). However, the phenotype of the clone that is characterized in this study may represent a clonal variant, or these cells may express different ratios or combinations of CIB1-binding proteins. In this study, we determined that transient overexpression of human CIB1 effectively activated both human and rat PAK1 but not the closely related PAK3. Although overexpression studies indicate potential functions of CIB1, depletion experiments reveal the role of endogenous CIB1. In agreement with overexpression studies, depletion of CIB1 from both human (HeLaS3) and rat (REF52) cells resulted in a dramatic increase in cell migration, further supporting an inhibitory effect of CIB1 on cell migration.
A fundamental question is how CIB1, via PAK1, regulates actin cytoskeletal dynamics that ultimately impact cell migration. A potential mechanism is through the activation of a LIMK- and cofilin-dependent pathway. PAK1 directly activates LIMK1 via phosphorylation of threonine 508 (Edwards et al., 1999). Presently, the only known substrate of active LIMK1 is the actin depolymerizing and severing protein cofilin (Arber et al., 1998; Yang et al., 1998), which is important for rapid actin filament turnover and cell motility (Chen et al., 2001; Dawe et al., 2003). Cofilin activity is negatively regulated by phosphorylation on serine 3, causing accumulation of F-actin (Moriyama et al., 1996). Recently, active, nonphosphorylated cofilin was shown to set the direction and increase the rate of cell migration (Ghosh et al., 2004). In agreement with these studies, we found that depletion of CIB1 results in decreased cofilin phosphorylation and increased cell migration, whereas CIB1 overexpression had the opposite effects. CIB1-overexpressing cells also typically displayed decreased spreading with nonpolarized lamellipodia and altered actin polymerization, which may have contributed to the observed decrease in cell migration. Conversely, CIB1-depleted cells showed increased cell polarity and loss of an organized actin cytoskeleton (in agreement with their increased migratory phenotype), suggesting an important role for CIB1 in modulating cytoskeletal dynamics (Manser et al., 1998). Moreover, coexpression of either DN PAK1 or LIMK1 with CIB1 blocked CIB1-induced cofilin phosphorylation and rescued cell migration, indicating a role for PAK1 and LIMK1 in mediating the inhibitory effects of CIB1 on cell migration. However, unlike LIMK overexpression, which can promote focal adhesion formation (Edwards et al., 1999; Sumi et al., 1999), CIB1 overexpression did not increase the number and size of focal adhesions, suggesting that LIMK1 mediates a subset of the effects of CIB1.
Our finding that CIB1 inhibits cell migration in a PAK1-dependent manner may seem paradoxical relative to some other studies in which active PAK1 or Rac and Cdc42 were reported to induce cell migration (Sells et al., 1997, 1999; Ridley et al., 1999). However, these results are most likely explained by differences in experimental conditions, because the type and concentration of ECM can have distinct effects on the cellular migratory response. Thus, our results are consistent with Kiosses et al. (1999), who showed that active PAK1 inhibits endothelial cell migration, and with Sander et al. (1999), who found that active Rac inhibits MDCK cell migration on relatively low concentrations of FN (2–10 μg/ml). Specifically, we found that overexpression of either PAK1 or CIB1 inhibited both REF52 and NIH3T3 cell migration toward low FN concentrations (Fig. S3, A and B). Importantly, overexpression of CIB1 in PAK1-null MEFs, unlike wild-type MEFs, did not suppress cell migration on low concentrations of FN, further demonstrating the requirement of PAK1 in CIB1-induced inhibition of cell migration (Fig. 5 B). In contrast, in studies in which PAK1 or Rac/Cdc42 induced cell migration, either relatively high FN concentrations (e.g., 50 μg/ml) or different substrates (e.g., collagen) were used and, therefore, may reflect differences in integrin engagement and signaling. Additional studies are underway to determine how CIB1 affects cell migration across different ECM substrates.
Several lines of evidence indicate that the interaction of PAK1 with CIB1 versus Rac/Cdc42 may be temporally regulated and, thus, differentially impact downstream signaling pathways. First, with extended times of adhesion to FN, control cells continued to exhibit PAK1-dependent cofilin phosphorylation (Fig. 7, A–C). Second, this phosphorylation also appeared to involve a CIB1-dependent component because CIB1 depletion significantly decreased cofilin phosphorylation at all times examined (Fig. 7, A and B). Third, adhesion-induced PAK1 and cofilin activation were not inhibited by the coexpression of DN Rac and Cdc42. However, with extended adhesion to FN, PAK1 (Fig. S3 A) and cofilin (Fig. S3 C) activity decreased in cells coexpressing DN Rac and Cdc42. These findings are of interest because Rac and Cdc42 are currently the only known activators of PAK1 upon cell adhesion (Price et al., 1998; del Pozo et al., 2000). However, our results suggest a model in which the activation of PAK1 upon cell adhesion to FN transitions from a CIB1-dependent to a more Cdc42/Rac-dependent process. Furthermore, CIB1 depletion resulted in increased Cdc42 but not in Rac activity, and this increased Cdc42 activity correlated with extensive filopodia formation and increased cell polarity (Fig. 7 C), suggesting a disregulation of GTPase signaling. Altogether, these results suggest that Rac/Cdc42 may mediate PAK1-dependent signaling in a manner temporally distinct from that of CIB1 and that this temporal and spatial regulation of the interaction of PAK1 with different upstream activators can ultimately affect the coupling of PAK1 to specific downstream signaling components. Thus, the differential regulation of PAK1 by CIB1 relative to Cdc42/Rac appears to be important in modulating cytoskeletal dynamics and cell migration.
In conclusion, we found that CIB1 binds to and activates PAK1 both in vitro and in vivo and that this interaction is required for the normal temporal sequence of adhesion-induced PAK1 activation. Although Cdc42 and Rac bind multiple PAK isoforms, CIB1 shows preferential binding to PAK1. The specificity of CIB1 for PAK1 may define a unique Ca2+-dependent PAK1 signaling pathway that leads to the phosphorylation and deactivation of cofilin, resulting in the inhibition of cell migration. Further study of CIB1 and its relationship to PAK1, as well as other binding partners, will provide a greater understanding of cytoskeletal regulation and migration in both normal and pathological conditions such as metastases and invasion.
Materials and methods
Platelet preparation and immunoprecipitation
Washed platelets were prepared as previously described (Shock et al., 1999), were resuspended to 4–8 × 108 platelets/ml in Tyrode's buffer without inhibitors or BSA, and were lysed with CHAPS lysis buffer (20 mM Hepes, pH 7.4, 0.15 M NaCl, 10 mM CHAPS, 0.5% deoxycholate, 50 mM NaF, 1 mM NaVO3, 10 mM sodium pyrophosphate, Protease Inhibitor Cocktail Set III [Calbiochem-Novabiochem], and 1 mM each of CaCl2 and MgCl2). Clarified lysates were incubated with control IgY or with chicken polyclonal anti-CIB1 antibodies, and immune complexes were isolated with anti–chicken IgY–conjugated agarose (Promega). Samples were immunoblotted with either a polyclonal antiphosphoserine (Zymed Laboratories) or with an anti-αPAK antibody (Santa Cruz Biotechnology, Inc.).
Recombinant proteins and in vitro kinase assays
Recombinant GST-CIB1, -PAK1, and -PAK2 were purified, and the GST tag was removed and separated from CIB1 as described previously (Shock et al., 1999). GST-PAK1 mutants were provided by M. Cobb (University of Texas Southwestern Medical Center, Dallas, TX) and were purified as described previously (Frost et al., 1997). The His-tagged wild-type PAK1 construct was provided by G. Bokoch (Scripps Research Institute, La Jolla, CA) and was purified under native conditions as described previously (Zenke et al., 1999). Kinase reactions with recombinant His-PAK1, CIB1, and GTP/GDP-loaded Cdc42 (provided by J. Sondek, University of North Carolina [UNC], Chapel Hill, NC) were performed as described previously (Zenke et al., 1999), stopped with 6× sample buffer, and separated by SDS-PAGE. Coomassie blue–stained gels were dried, and samples were visualized by autoradiography.
In vitro binding assays
Microtiter wells were coated with and without 5 μg/ml CIB1 overnight at RT and were blocked with 3% BSA. 50 μl/well PAK proteins were added for 1 h at 37°C. For inhibition studies, 7 μg/ml His-PAK1 was incubated for 30 min with soluble CIB1 or Cdc42 that was preloaded with GDP or GTPγS before addition to wells coated with and without CIB1. For Ca2+-dependent binding studies, His-PAK1 was diluted in buffer containing 1 μM Ca2+ with increasing concentrations of EGTA (0–5 μM) before addition to CIB1-coated wells. In all cases, wells were washed with TBS containing 0.05% Tween 20, and PAK1 binding was detected with an anti-His or anti-GST antibody (Santa Cruz Biotechnology, Inc.) followed by HRP-conjugated sheep anti–mouse IgG (GE Healthcare). The reactions were developed by using o-phenylenediamine as a substrate, and absorbance was measured at 490 nm (OD490). To assess the effects of PAK1 peptides on CIB1–PAK1 binding, 0.5 μg/ml CIB1 was preincubated for 30 min at 22°C with and without 5 μM PAK1 peptides. His-PAK1 coupled to Ni-NTA agarose (QIAGEN) was then added to the CIB1/peptide samples and were mixed for 1 h at 22°C. His-PAK1 beads were washed with 20 mM Hepes/saline, pH 7.4, with 0.05% Tween 20, prepared for SDS-PAGE, and immunoblotted for CIB1. The top half of the membrane was also blotted for His-PAK1 as a loading control.
CIB1-binding site characterization
The CIB1-binding site on PAK1 was identified by using SPOT peptide technology (Frank, 2002). Peptides of the first 164 amino acids of the PAK1 NH2 terminus were synthesized as 12mers that overlapped by seven amino acids, were spotted onto nitrocellulose (gift from F. Gertler, Massachusetts Institute of Technology, Cambridge, MA), and were incubated with 2 μg/ml CIB1 diluted in 20 mM Hepes, pH 7.4, 150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 0.5% BSA, and 5% glycerol. After three washes, CIB1 binding was detected with a chicken polyclonal anti-CIB1 IgY followed by HRP-conjugated donkey anti-IgY (Jackson ImmunoResearch Laboratories).
Generation and immunoprecipitation of Pak1 mutants
The QuikChange Site-Directed Mutagenesis Kit (Stratagene) was used to generate the myc-PAK1 siteIIAAA (AAA myc-PAK1, aa 135–137/AAA). Primers for the triple mutations are listed as follows (mutated sites are in bold face): forward primer, 5′-GTTTTACAACTCGAAGGCGGCAGCCAACAGCCAGAAATAC-3′; and reverse primer, 5′-GTATTTCTGGCTGTTGGCTGCCGCCTTCGAGTTGTAAAAC-3′. Myc-PAK1 siteIΔ/siteIIAAA (Δ/AAA myc-PAK1) mutant with deletion of aa 8–68 was generated by PCR using pfu Turbo (Stratagene) with an ExSite PCR-based site-directed mutagenesis strategy (Stratagene). Primers for generating Pak1Δ8–68 are listed as follows: forward primer, 5′-GTCTAGGCCGTTATTTGA-3′; and reverse primer, 5′-GAGAAAGAGCGGCCAGAG-3′. For coimmunoprecipitation experiments, HEK293 cells were cotransfected with CIB1 and myc-tagged wild-type or mutant PAK1 using LipofectAMINE 2000 (Invitrogen). CIB1 was immunoprecipitated from clarified lysates by using a mAb anti-CIB1 (UN2), and the immunoprecipitates were immunoblotted with anti-myc (Santa Cruz Biotechnology, Inc.) and anti-CIB1 antibodies.
Cell culture and immunohistochemistry
Cells were maintained in DME (REF52 and HEK293) and Ham's F-12 (HeLaS3) media supplemented with 10% FBS. MEFs were isolated from day 13.5 wild-type or Pak1−/− embryos. Wild-type and Pak1−/− MEFs were immortalized with SV40 T antigen and were maintained in DME supplemented with 12% FBS. Plasmids encoding V5 epitope–tagged kinase-inactive K299R and ca T423E PAK1(gift of R. Juliano, UNC, Chapel Hill, NC), kinase-inactive LIMK1 D460N (gift of G. Bokoch), and wild-type CIB1 in pcDNA3.1 were transiently transfected into REF52 cells with LipofectAMINE Plus (Invitrogen). Cells were serum starved 30 h posttransfection, harvested by trypsinization, and added to DME containing 2 mg/ml of soybean trypsin inhibitor and 0.2% delipidated BSA. After washing, cells were resuspended in DME/0.2% BSA and were incubated for 1 h in suspension before assays. For immunostaining, REF52 cells were seeded onto coverslips coated with 10 μg/ml FN and were allowed to adhere for various times at 37°C. Cells were permeabilized with 0.2% Triton X-100 in PBS, blocked with normal serum, and stained with antibodies raised against CIB1 (chicken pAb anti-CIB1 or UN2) and vinculin (Sigma-Aldrich). Polymerized actin was visualized with AlexaFluor594 phalloidin (Molecular Probes). Goat anti–mouse AlexaFluor594, goat anti–chicken AlexaFluor488, and goat anti–mouse AlexaFluor488 (Molecular Probes) were used as secondary antibodies. Images were collected using a laser scanning confocal imaging system (Fluoview 300; Olympus) that was configured with a fluorescence microscope (model IX70; Olympus) fitted with a 60× oil objective (PlanApo; Olympus). Confocal images were processed using Adobe Photoshop.
Migration assays
Haptotactic migration was performed using 8-μm pore polycarbonate transwell membranes (Costar) coated on the underside with 5 (REF52) or 20 μg/ml FN (HeLaS3), and 5–7.5 × 104 serum-starved cells were added to the top chamber. For siRNA depletion studies, nonmigrating cells were removed from the top membrane surface, and migrated cells were fixed and stained with the Diff-Quik Stain Kit (Dade Behring). Cells from 10 fields per membrane were counted and averaged. For overexpression studies, transfected cells on the top (nonmigrating cells) or bottom (migrating cells) membrane were fixed and visualized by GFP expression, by immunostaining with antibodies directed against CIB1 alone, by CIB1 with PAK1 (anti-V5 epitope; Bethyl Labs), or by LIMK1 (Santa Cruz Biotechnology, Inc.). Cells were also stained with phalloidin to detect the total cell number. Images were collected using laser scanning confocal imaging as described above with a 10× objective (PlanApo; Olympus). Cells were counted under 20×, and transfected cells migrating to the underside of the membrane were represented as the percentage of the total number of transfected cells.
PAK1 kinase and Rac/Cdc42 activation assays
Serum-starved transfected cells were held in suspension or were seeded onto tissue culture plates coated with 10 μg/ml FN. Lysates were prepared by the addition of ice-cold 1% NP-40 lysis buffer as described in Platelet preparation and immunoprecipitation, but with the addition of 10% glycerol and 0.2 mM pervanadate. Equal amounts of lysate protein on Western blots were probed with total and phosphospecific anticofilin antibodies (Cytoskeleton, Inc. and Santa Cruz Biotechnology, Inc., respectively). Lysates were also subjected to immunoprecipitation with either an NH2-terminal–specific anti-PAK1 or -PAK3 antibody (Santa Cruz Biotechnology, Inc.). Immunoprecipitated PAK kinase activity was assayed using myelin basic protein as the substrate (Zenke et al., 1999). Activated Cdc42/Rac1 was assayed using the GST-fusion protein of PAK PBD (GST-PBD; aa 70–132), which binds specifically to active GTP-bound Rac and Cdc42 (del Pozo et al., 2000). Immobilized GST-PBD was provided by K. Burridge (UNC, Chapel Hill, NC). Affinity-precipitated and total Rac1 and Cdc42 were detected by Western blotting with antibodies against Rac1 and Cdc42 (Santa Cruz Biotechnology, Inc.).
siRNA
RNA interference for human CIB1 was performed as described previously (Elbashir et al., 2001). CIB1 siRNA oligonucleotides 5′-AAGCAGGAGATCCTCCTAGCC(TT)-3′ (corresponding to bases 136–156) or 5′-AAGAGTCACTGCATACCCGAG(TT)-3′ (bases 177–197) within the human or rat cDNA ORFs, respectively (Dharmacon Research), were transfected at a final concentration of 80 (REF52) and 60 nM (HeLaS3) by using a TransIT-TKO or SiQuest reagent (Mirus). Assays were performed 48–72 h posttransfection.
Online supplemental material
Fig. S1 shows in vivo activation of PAK1 by CIB1 and a comparison of the CIB1-binding sites in human and rat PAK1 versus PAK3. Fig. S2 provides evidence for Ca2+-dependent PAK1 activation in adherent cells. Fig. S3 shows the effects of DN GTPases on signaling to PAK1 and cofilin. Fig. S4 shows the effects of CIB1 overexpression on cell spreading, actin dynamics, and colocalization with PAK1. Fig. S5 provides additional evidence for PAK1 activity in inhibiting REF52 and NIH3T3 cell migration. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200502090/DC1.
Acknowledgments
We thank G. Bokoch, M. Cobb, and R. Juliano for providing the various LIMK1, PAK, and Rac/Cdc42 constructs; J. Sondek for purified Cdc42; K. Burridge for GST-PBD; and F. Gertler for the PAK1 1–165 peptide SPOT membrane. We also thank A. Howe for helpful discussions.
This work was supported by grants to L.V. Parise (2PO1 HL 45100) and J. Chernoff (RO1 GM 54168) from the National Institutes of Health (NIH). T.M. Leisner was supported by an NIH postdoctoral fellowship (CA09156) to the Lineberger Comprehensive Cancer Center.
Abbreviations used in this paper: ca, constitutively active; DN, dominant negative; FN, fibronectin; HEK, human embryonic kidney; IS, inhibitory switch; kd, kinase dead; KI, kinase inhibitor; LIMK, Lin-11/Isl-1/Mec-3 kinase; MEF, mouse embryo fibroblast; NTA, nitrilotriacetic acid; PAK, p21-activated kinase; PBD, p21-binding domain; REF, rat embryo fibroblast; si, short inhibitory.
References
- Adam, L., R. Vadlamudi, M. Mandal, J. Chernoff, and R. Kumar. 2000. Regulation of microfilament reorganization and invasiveness of breast cancer cells by kinase dead p21-activated kinase-1. J. Biol. Chem. 275:12041–12050. [DOI] [PubMed] [Google Scholar]
- Arber, S., F.A. Barbayannis, H. Hanser, C. Schneider, C.A. Stanyon, O. Bernard, and P. Caroni. 1998. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 393:805–809. [DOI] [PubMed] [Google Scholar]
- Bagheri-Yarmand, R., M. Mandal, A.H. Taludker, R.A. Wang, R.K. Vadlamudi, H.J. Kung, and R. Kumar. 2001. Etk/Bmx tyrosine kinase activates Pak1 and regulates tumorigenicity of breast cancer cells. J. Biol. Chem. 276:29403–29409. [DOI] [PubMed] [Google Scholar]
- Bagrodia, S., S.J. Taylor, C.L. Creasy, J. Chernoff, and R.A. Cerione. 1995. Identification of a mouse p21Cdc42/Rac activated kinase. J. Biol. Chem. 270:22731–22737. [DOI] [PubMed] [Google Scholar]
- Bagrodia, S., S.J. Taylor, K.A. Jordon, L. Van Aelst, and R.A. Cerione. 1998. A novel regulator of p21-activated kinases. J. Biol. Chem. 273:23633–23636. [DOI] [PubMed] [Google Scholar]
- Bers, D.M., C.W. Patton, and R. Nuccitelli. 1994. A practical guide to the preparation of Ca2+ buffers. Methods Cell Biol. 40:3–29. [DOI] [PubMed] [Google Scholar]
- Bokoch, G.M., A.M. Reilly, R.H. Daniels, C.C. King, A. Olivera, S. Spiegel, and U.G. Knaus. 1998. A GTPase-independent mechanism of p21-activated kinase activation. Regulation by sphingosine and other biologically active lipids. J. Biol. Chem. 273:8137–8144. [DOI] [PubMed] [Google Scholar]
- Chaves-Olarte, E., E. Freer, A. Parra, C. Guzman-Verri, E. Moreno, and M. Thelestam. 2003. R-Ras glucosylation and transient RhoA activation determine the cytopathic effect produced by toxin B variants from toxin A-negative strains of Clostridium difficile. J. Biol. Chem. 278:7956–7963. [DOI] [PubMed] [Google Scholar]
- Chen, J., D. Godt, K. Gunsalus, I. Kiss, M. Goldberg, and F.A. Laski. 2001. Cofilin/ADF is required for cell motility during Drosophila ovary development and oogenesis. Nat. Cell Biol. 3:204–209. [DOI] [PubMed] [Google Scholar]
- Daniels, R.H., F.T. Zenke, and G.M. Bokoch. 1999. alphaPix stimulates p21-activated kinase activity through exchange factor-dependent and -independent mechanisms. J. Biol. Chem. 274:6047–6050. [DOI] [PubMed] [Google Scholar]
- Dawe, H.R., L.S. Minamide, J.R. Bamburg, and L.P. Cramer. 2003. ADF/cofilin controls cell polarity during fibroblast migration. Curr. Biol. 13:252–257. [DOI] [PubMed] [Google Scholar]
- Dechert, M.A., J.M. Holder, and W.T. Gerthoffer. 2001. p21-activated kinase 1 participates in tracheal smooth muscle cell migration by signaling to p38 Mapk. Am. J. Physiol. Cell Physiol. 281:C123–C132. [DOI] [PubMed] [Google Scholar]
- del Pozo, M.A., L.S. Price, N.B. Alderson, X.D. Ren, and M.A. Schwartz. 2000. Adhesion to the extracellular matrix regulates the coupling of the small GTPase Rac to its effector PAK. EMBO J. 19:2008–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards, D.C., L.C. Sanders, G.M. Bokoch, and G.N. Gill. 1999. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat. Cell Biol. 1:253–259. [DOI] [PubMed] [Google Scholar]
- Elbashir, S.M., W. Lendeckel, and T. Tuschl. 2001. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 15:188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank, R. 2002. The SPOT-synthesis technique. Synthetic peptide arrays on membrane supports-principles and applications. J. Immunol. Methods. 267:13–26. [DOI] [PubMed] [Google Scholar]
- Frost, J.A., H. Steen, P. Shapiro, T. Lewis, N. Ahn, P.E. Shaw, and M.H. Cobb. 1997. Cross-cascade activation of ERKs and ternary complex factors by Rho family proteins. EMBO J. 16:6426–6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry, H.R., A.U. Singer, L. Betts, C. Yang, J.D. Ferrara, J. Sondek, and L.V. Parise. 2004. Structural and biochemical characterization of CIB1 delineates a new family of EF-hand containing proteins. J. Biol. Chem. 280:8407–8415. [DOI] [PubMed] [Google Scholar]
- Ghosh, M., X. Song, G. Mouneimne, M. Sidani, D.S. Lawrence, and J.S. Condeelis. 2004. Cofilin promotes actin polymerization and defines the direction of cell motility. Science. 304:743–746. [DOI] [PubMed] [Google Scholar]
- Haataja, L., V. Kaartinen, J. Groffen, and N. Heisterkamp. 2002. The small GTPase Rac3 interacts with the integrin-binding protein CIB and promotes integrin alpha(IIb)beta(3)-mediated adhesion and spreading. J. Biol. Chem. 277:8321–8328. [DOI] [PubMed] [Google Scholar]
- Just, I., J. Selzer, M. Wilm, C. Eichel-Streiber, M. Mann, and K. Aktories. 1995. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature. 375:500–503. [DOI] [PubMed] [Google Scholar]
- Kauselmann, G., M. Weiler, P. Wulff, S. Jessberger, U. Konietzko, J. Scafidi, U. Staubli, J. Bereiter-Hahn, K. Strebhardt, and D. Kuhl. 1999. The polo-like protein kinases Fnk and Snk associate with a Ca(2+)- and integrin-binding protein and are regulated dynamically with synaptic plasticity. EMBO J. 18:5528–5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiosses, W.B., R.H. Daniels, C. Otey, G.M. Bokoch, and M.A. Schwartz. 1999. A role for p21-activated kinase in endothelial cell migration. J. Cell Biol. 147:831–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei, M., W. Lu, W. Meng, M.C. Parrini, M.J. Eck, B.J. Mayer, and S.C. Harrison. 2000. Structure of PAK1 in an autoinhibited conformation reveals a multistage activation switch. Cell. 102:387–397. [DOI] [PubMed] [Google Scholar]
- Leung, T., B.E. How, E. Manser, and L. Lim. 1994. Cerebellar beta 2-chimaerin, a GTPase-activating protein for p21 ras- related rac is specifically expressed in granule cells and has a unique N-terminal SH2 domain. J. Biol. Chem. 269:12888–12892. [PubMed] [Google Scholar]
- Lian, J.P., R. Huang, D. Robinson, and J.A. Badwey. 1998. Products of sphingolipid catabolism block activation of the p21- activated protein kinases in neutrophils. J. Immunol. 161:4375–4381. [PubMed] [Google Scholar]
- Manser, E., T. Leung, H. Salihuddin, Z.S. Zhao, and L. Lim. 1994. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature. 367:40–46. [DOI] [PubMed] [Google Scholar]
- Manser, E., H.Y. Huang, T.H. Loo, X.Q. Chen, J.M. Dong, T. Leung, and L. Lim. 1997. Expression of constitutively active alpha-PAK reveals effects of the kinase on actin and focal complexes. Mol. Cell. Biol. 17:1129–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manser, E., T. Leung, and L. Lim. 1998. Identification and characterization of small GTPase-associated kinases. Methods Mol. Biol. 84:295–305. [DOI] [PubMed] [Google Scholar]
- Moriyama, K., K. Iida, and I. Yahara. 1996. Phosphorylation of Ser-3 of cofilin regulates its essential function on actin. Genes Cells. 1:73–86. [DOI] [PubMed] [Google Scholar]
- Naik, M.U., and U.P. Naik. 2003. Calcium- and integrin-binding protein regulates focal adhesion kinase activity during platelet spreading on immobilized fibrinogen. Blood. 102:3629–3636 [DOI] [PubMed] [Google Scholar]
- Naik, U.P., P.M. Patel, and L.V. Parise. 1997. Identification of a novel calcium-binding protein that interacts with the integrin alphaIIb cytoplasmic domain. J. Biol. Chem. 272:4651–4654. [DOI] [PubMed] [Google Scholar]
- Nobes, C.D., and A. Hall. 1995. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 81:53–62. [DOI] [PubMed] [Google Scholar]
- Papakonstanti, E.A., and C. Stournaras. 2002. Association of PI-3 kinase with PAK1 leads to actin phosphorylation and cytoskeletal reorganization. Mol. Biol. Cell. 13:2946–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavalko, F.M., and K. Burridge. 1991. Disruption of the actin cytoskeleton after microinjection of proteolytic fragments of α-actinin. J. Cell Biol. 114:481–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, L.S., J. Leng, M.A. Schwartz, and G.M. Bokoch. 1998. Activation of Rac and Cdc42 by integrins mediates cell spreading. Mol. Biol. Cell. 9:1863–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid, T., M. Banerjee, and M. Nikolic. 2001. Phosphorylation of Pak1 by the p35/Cdk5 kinase affects neuronal morphology. J. Biol. Chem. 276:49043–49052. [DOI] [PubMed] [Google Scholar]
- Ridley, A.J., W.E. Allen, M. Peppelenbosch, and G.E. Jones. 1999. Rho family proteins and cell migration. Biochem. Soc. Symp. 65:111–123. [PubMed] [Google Scholar]
- Sander, E.E., J.P. ten Klooster, S. van Delft, R.A. van der Kammen, and J.G. Collard. 1999. Rac downregulates Rho activity: reciprocal balance between both GTPases determines cellular morphology and migratory behavior. J. Cell Biol. 147:1009–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sells, M.A., U.G. Knaus, S. Bagrodia, D.M. Ambrose, G.M. Bokoch, and J. Chernoff. 1997. Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr. Biol. 7:202–210. [DOI] [PubMed] [Google Scholar]
- Sells, M.A., J.T. Boyd, and J. Chernoff. 1999. p21-activated kinase 1 (Pak1) regulates cell motility in mammalian fibroblasts. J. Cell Biol. 145:837–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shock, D.D., U.P. Naik, J.E. Brittain, S.K. Alahari, J. Sondek, and L.V. Parise. 1999. Calcium-dependent properties of CIB binding to the integrin alphaIIb cytoplasmic domain and translocation to the platelet cytoskeleton. Biochem. J. 342:729–735. [PMC free article] [PubMed] [Google Scholar]
- Stabler, S.M., L.L. Ostrowski, S.M. Janicki, and M.J. Monteiro. 1999. A myristoylated calcium-binding protein that preferentially interacts with the Alzheimer's disease presenilin 2 protein. J. Cell Biol. 145:1277–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumi, T., K. Matsumoto, Y. Takai, and T. Nakamura. 1999. Cofilin phosphorylation and actin cytoskeletal dynamics regulated by rho- and Cdc42-activated LIM-kinase 2. J. Cell Biol. 147:1519–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadlamudi, R.K., F. Li, L. Adam, D. Nguyen, Y. Ohta, T.P. Stossel, and R. Kumar. 2002. Filamin is essential in actin cytoskeletal assembly mediated by p21-activated kinase 1. Nat. Cell Biol. 4:681–690. [DOI] [PubMed] [Google Scholar]
- Wu, X., and M.R. Lieber. 1997. Interaction between DNA-dependent protein kinase and a novel protein, KIP. Mutat. Res. 385:13–20. [DOI] [PubMed] [Google Scholar]
- Yamniuk, A.P., L.T. Nguyen, T.T. Hoang, and H.J. Vogel. 2004. Metal ion binding properties and conformational states of calcium- and integrin-binding protein. Biochemistry. 43:2558–2568. [DOI] [PubMed] [Google Scholar]
- Yang, N., O. Higuchi, K. Ohashi, K. Nagata, A. Wada, K. Kangawa, E. Nishida, and K. Mizuno. 1998. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature. 393:809–812. [DOI] [PubMed] [Google Scholar]
- Zenke, F.T., C.C. King, B.P. Bohl, and G.M. Bokoch. 1999. Identification of a central phosphorylation site in p21-activated kinase regulating autoinhibition and kinase activity. J. Biol. Chem. 274:32565–32573. [DOI] [PubMed] [Google Scholar]