

Abstract
Trimethylation of histone H3 lysine 9 and the subsequent binding of heterochromatin protein 1 (HP1) mediate the formation and maintenance of pericentromeric heterochromatin. Trimethylation of H3K9 is governed by the histone methyltransferase SUV39H1. Recent studies of HP1 dynamics revealed that HP1 is not a stable component of heterochromatin but is highly mobile (Cheutin, T., A.J. McNairn, T. Jenuwein, D.M. Gilbert, P.B. Singh, and T. Misteli. 2003. Science. 299:721–725; Festenstein, R., S.N. Pagakis, K. Hiragami, D. Lyon, A. Verreault, B. Sekkali, and D. Kioussis. 2003. Science. 299:719–721). Because the mechanism by which SUV39H1 is recruited to and interacts with heterochromatin is unknown, we studied the dynamic properties of SUV39H1 in living cells by using fluorescence recovery after photobleaching and fluorescence resonance energy transfer. Our results show that a substantial population of SUV39H1 is immobile at pericentromeric heterochromatin, suggesting that, in addition to its catalytic activity, SUV39H1 may also play a structural role at pericentromeric regions. Analysis of SUV39H1 deletion mutants indicated that the SET domain mediates this stable binding. Furthermore, our data suggest that the recruitment of SUV39H1 to heterochromatin is at least partly independent from that of HP1 and that HP1 transiently interacts with SUV39H1 at heterochromatin.
Introduction
Heterochromatin is considered to be the part of the genome that is gene poor, transcriptionally silent, and, furthermore, highly condensed and static in interphase cells. In most eukaryotes, it is characterized by DNA methylation at cytosine guanine dinucleotides and by histone hypoacetylation and methylation. These modifications play a pivotal role in the establishment and function of heterochromatin by creating binding sites for heterochromatin proteins (Jenuwein and Allis, 2001). HP1 (heterochromatin protein 1) is, with a few exceptions, typically associated with heterochromatin and binds to histone H3, which is methylated at lysine 9 (H3K9; Bannister et al., 2001; Lachner et al., 2001). Because HP1 can form homodimers and interacts with several other chromatin proteins, it is thought to nucleate the formation of a higher order structure that is incompatible with transcription. An unexpected feature of HP1 is, however, that it is in continuous flux with chromatin. This suggests that heterochromatin is not a static, inaccessible higher order conformation but is a dynamic structure that has the potential to rapidly adapt to various stimuli that influences gene expression patterns or cell cycle progression (Cheutin et al., 2003; Festenstein et al., 2003).
The histone methyltransferase SUV39H1 has been implicated to play an essential role in the initial steps of heterochromatin formation in mammals by selective methylation of H3K9. In particular, trimethylation of H3K9 is mediated by SUV39H1 and proved to be essential in the establishment of constitutive heterochromatin at pericentromeric and telomeric regions in the genome, both of which consist of tandem repeated DNA sequences (Peters et al., 2003; Garcia-Cao et al., 2004). Mice that were deficient for SUV39H1 were shown to display impaired pericentric H3K9 methylation and chromosomal instability (Peters et al., 2001). Furthermore, loss of methylated H3 tails was shown to be accompanied by delocalization of HP1 from pericentric heterochromatin (Maison et al., 2002). In Drosophila melanogaster, SU(VAR)3-9 is the homologue of human SUV39H1 and of mouse Suv39h1 and Suv39h2. Similar to mouse mutants, D. melanogaster SU(VAR)3-9–null mutants revealed a strong reduction in H3K9 methylation and an almost complete loss of HP1 from chromocenter heterochromatin (Schotta et al., 2002). Together, these findings underscore the essential role of SUV39H1 methyltransferase activity in establishing heterochromatin.
The catalytic methyltransferase activity of SUV39H1 has been mapped to the conserved COOH-terminal SET domain and uses monomethylated H3K9 as a substrate (Peters et al., 2003). The first 44 amino acids at the NH2 terminus of SUV39H1 form an interaction domain for the chromoshadow domain of HP1 and are, together with the adjacent chromodomain, required for binding to heterochromatin (Melcher et al., 2000). Interestingly, in HP1-deficient D. melanogaster, salivary gland nuclei SU(VAR)3-9 was shown to be lost from chromocenter heterochromatin and to spread along euchromatic regions, suggesting that an interaction between SU(VAR)3-9 and HP1 is essential for the association of SU(VAR)3-9 with centromeric heterochromatin (Schotta et al., 2002). However, the exact mechanism by which SUV39H1 interacts with heterochromatin is poorly understood.
Recently, the initial step in centromeric heterochromatin formation was suggested to be mediated by direct or indirect binding of SUV39H1 to components of an RNA interference pathway (Maison et al., 2002), but the mechanism by which SUV39H1 is initially recruited to centromeric repeat units is still enigmatic. Also, it is unclear whether SUV39H1 interacts only temporally with chromatin to methylate histone H3K9 or participates in a more stable multiprotein complex together with HP1 or other chromatin proteins to support a stable heterochromatic, pericentromeric chromatin structure. To gain further insight into the roles that SUV39H1 plays in heterochromatin, we investigated the in vivo kinetics of SUV39H1 in human osteosarcoma U2OS and in mouse NIH3T3 cells. Using FRAP analysis, we showed that SUV39H1 has a significantly slower exchange rate and a larger immobile fraction in heterochromatic regions compared with HP1β. Our results suggest that a substantial fraction of SUV39H1 is immobile at pericentromeric heterochromatin, at least on the time scale of our experiments, and may, therefore, play a structural role. Furthermore, our data indicate that at least a part of the dynamic fraction of SUV39H1 is recruited to heterochromatin independently from HP1 binding. In addition, interactions between various SUV39H1 deletion mutants and HP1β were analyzed in vivo by photobleaching and fluorescence resonance energy transfer (FRET) techniques to characterize the binding of HP1β to SUV39H1 at heterochromatin.
Results
EYFP-SUV39H1 localizes to heterochromatin domains and is catalytically active
To determine the in vivo kinetic properties of SUV39H1, we made a construct coding for the fusion protein EYFP-SUV39H1. This construct was transiently transfected into human U2OS and mouse NIH3T3 cells. Before analyzing the kinetic properties of the fusion protein, we first analyzed its spatial distribution in moderately expressing cells after fixation. In U2OS cells, EYFP-SUV39H1 localized to irregular shaped domains, which were frequently observed to be present at the nuclear periphery around nucleoli but were also dispersed throughout the nucleoplasm (Fig. 1 A, right). In NIH3T3 cells, a more defined distribution pattern of EYFP-SUV39H1 was observed that corresponded to heterochromatic domains, as revealed by DAPI staining as bright fluorescent regions (Fig. 1 A, left). In addition, a diffuse staining throughout euchromatic regions was detected. These staining patterns are in agreement with previous immunocytochemical studies on SUV39H1 localization that used a specific anti-SUV39H1 antibody (Aagaard et al., 1999; Melcher et al., 2000). Notably, these studies show that endogenous Suv39h1 is enriched at heterochromatic foci in mouse interphase cell nuclei. Next, we compared the staining patterns of EYFP-SUV39H1 with the immunocytochemical staining patterns of trimethylated H3K9, HP1α, and HP1β. The latter two are heterochromatin proteins that are predominantly present in constitutive heterochromatin (Wreggett et al., 1994). The results revealed very similar staining patterns in U2OS as well as in NIH3T3 cells (Fig. 1 B), suggesting that EYFP-SUV39H1 localizes mainly to heterochromatic domains. Furthermore, immunocytochemical detection of centromeres in EYFP-SUV39H1–expressing U2OS cells revealed that many, but not all, EYFP-SUV39H1 foci corresponded to centromere localization (Fig. 1 C). Together, these experiments confirm that low to moderate expression levels do not lead to mislocalization of EYFP-SUV39H1. Previously, it was observed that strong overexpression of SUV39H1 fusion proteins led to aberrant localization patterns and to a redistribution of HP1 (Melcher et al., 2000).
Figure 1.

Localization of EYFP-SUV39H1 in mouse NIH3T3 as well as in human U2OS cells. (A) After transfection, EYFP-SUV39H1 localizes in NIH3T3 cells to distinct nuclear regions that are also visible by DAPI staining. In U2OS cells, EYFP-SUV39H1 also localizes at sites that are stained by DAPI, but these areas are less well defined. (B) NIH3T3 cells transfected with EYFP-SUV39H1 were labeled with antibodies specific for trimethylated H3K9 and HP1α. (C) Human U2OS cells transfected with EYFP-SUV39H1 were labeled with antibodies against centromeres. Single optical sections show the YFP-tagged protein, centromere labeling, and an overlay. Line scans (diagonal lines through the images) show the local intensity distributions of the EYFP fusion protein in yellow and of the centromere labeling in red. Bars, 10 μm.
To confirm that EYFP-SUV39H1 is catalytically active in vivo, Suv39h double-null (dn) primary mouse embryonic fibroblasts (PMEFs; Bannister et al., 2001) were transfected with EYFP-SUV39H1 and were stained for trimethylated H3K9 and HP1β by using specific antibodies. In nontransfected cells, there appeared to be little trimethylated H3K9 present (Fig. 2 A, bottom; cells are indicated by arrows), and HP1β displayed a diffuse nuclear staining (Fig. 2 B, bottom). In EYFP-SUV39H1–expressing dn PMEFs, however, trimethylated H3K9 was found to be enriched in foci (Fig. 2 A, middle and bottom), which are similar to the trimethylated H3K9–stained foci that were observed in wild-type PMEFs (Fig. 2 A, top). Also, HP1β relocated to heterochromatic foci in dn PMEFs expressing EYFP-SUV39H1 (Fig. 2 B, middle), which resembled the HP1β-stained foci in wild-type PMEFs expressing EYFP-SUV39H1 (Fig. 2 B, top). These results strongly indicate that the Suv39h dn phenotype can be rescued by EYFP-SUV39H1 and that this fusion protein is functional.
Figure 2.

EYFP-SUV39H1 expression rescues the phenotype of Suv39h dn PMEFs. Suv39h dn and wild-type PMEFs transfected with EYFP-SUV39H1 were labeled with antibodies against trimethylated H3K9 (A) or HP1β (B). In nontransfected Suv39h dn PMEFs, a little trimethylated H3K9 is present throughout the nucleus (A, arrows). Transfected cells show an increase in trimethylated H3K9 that is localized to heterochromatic areas (A, middle and bottom). This localization is comparable to wild-type cells (A, top). Bars, 5 μm.
SUV39H1 is a more stable component of heterochromatin than HP1β
To gain insight into the dynamic properties of SUV39H1 and its recruitment to heterochromatin, FRAP experiments were performed using NIH3T3 cells. Cells were transiently transfected with EYFP-SUV39H1, and defined areas of ∼1 μm in diameter were irreversibly photobleached for 2.5 s each in heterochromatic as well as in euchromatic regions. Fluorescence recovery in the same areas was imaged at regular time points in a time series (Fig. 3 A). The corresponding fluorescence intensity values that were collected from 37 cells were plotted in a FRAP curve (after correction for photobleaching). The t1/2 of fluorescence recovery, which is a measure for the speed by which molecules in the bleached area are replaced by molecules from the environment, was ∼19.0 s within heterochromatin domains (Fig. 3 B and Table I). Remarkably, EYFP-SUV39H1 recovery reached 70% as a maximum after 140 s, suggesting that ∼30% of SUV39H1 is stably bound within heterochromatic domains on the time scale of our experiments.
Figure 3.
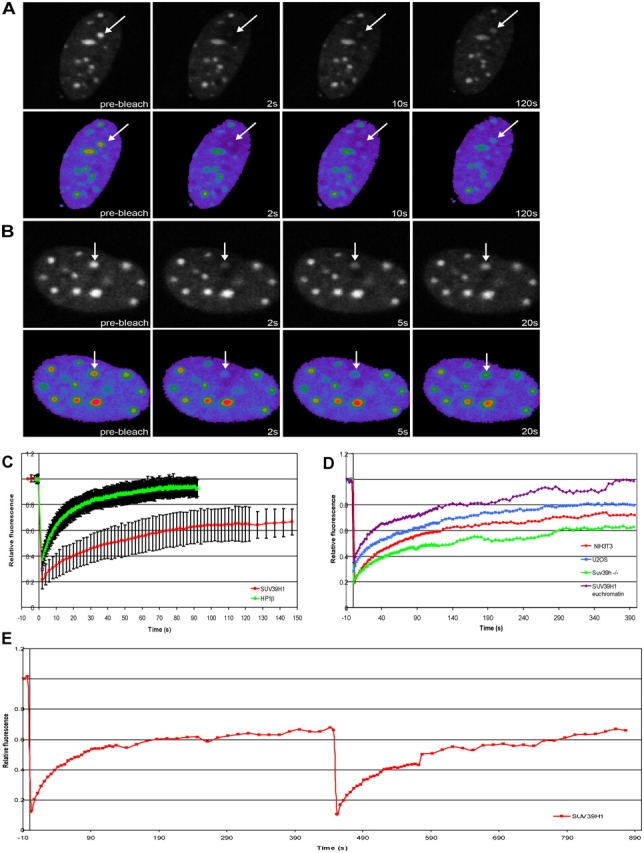
FRAP analysis of EYFP-SUV39H1 and EYFP-HP1β shows that SUV39H1 is a less dynamic component of heterochromatin. NIH3T3 cells were transfected with EYFP-SUV39H1 (A) or EYFP-HP1β (B). A heterochromatic area was selected and photobleached. Images were recorded just before bleaching and at different time intervals after bleaching. Arrows indicate the photobleached region. To illustrate the recovery of fluorescence more clearly, pseudocolor images of the bleached cells are shown. Fluorescent intensities range from blue (low) to red (high). (C) Relative fluorescence intensities are displayed in recovery curves. Fluorescence recovery of EYFP-SUV39H1 reached a plateau at ∼70% after 140 s. Fluorescence recovery of EYFP-HP1β reached a plateau at ∼95% after 60 s. The curves represent mean values from 37 and 24 cells, respectively. Error bars represent SD. (D) FRAP curves calculated after extended recovery periods of EYFP-SUV39H1 in heterochromatin of U2OS cells, of Suv39h dn PMEFs, and of NIH3T3 cells and in euchromatin of NIH3T3 cells. (E) FRAP curves obtained from two successive FRAP measurements. After fluorescence recovery after the first bleach, the same region was bleached for the second time, and the fluorescence recovery was measured. EYFP-SUV39H1 fully recovered after the second bleach to 70% of the initial amount of fluorescence measured before the first bleach.
Table I. FRAP values measured in this study.
| Construct | t 1/2 | t 1/2 TSA | t 1/2 5-aza-C |
|---|---|---|---|
| s | s | s | |
| EYFP-SUV39H1 (heterochromatin) |
19.0 | 7.7 | 14.6 |
| EYFP-SUV39H1 (euchromatin) |
11.8 | ND | ND |
| EYFP-SUV39H1 (heterochromatin U2OS) |
15.0 | ND | ND |
| EYFP-SUV39H1 (heterochromatin Suv39h−/−) |
15.0 | ND | ND |
| EYFP–SUV39H1-ΔSET | 8.6 | 6.0 | 3.0 |
| EYFP–SUV39H1-Nchromo | 3.8 | ND | ND |
| EYFP–SUV39H1-chromo | 0.6 | ND | ND |
| EYFP–SUV39H1-ΔN89 | 0.5 | ND | ND |
| EYFP-HP1β | 4.2 | 1.8 | 4.2 |
To ensure the existence of an immobile or considerably less mobile SUV39H1 fraction in heterochromatin domains, we measured the dynamics of EYFP-SUV39H1 by FRAP using extended recovery periods in NIH3T3 and U2OS cells as well as in Suv39h dn PMEFs. EYFP-SUV39H1 recovery reached a maximum of 80% after 275 s in U2OS cells (t1/2 of ∼15 s), a maximum of 70% after 140 s in NIH3T3 cells, and only a maximum of 60% after 290 s in Suv39h dn PMEFs (t1/2 of ∼15 s) in heterochromatic domains (Fig. 3 D and Table I). This suggests that 20–40% of SUV39H1 is stably bound within heterochromatic domains depending on cell type. It should be noted, however, that it proved difficult to obtain consistent FRAP data from U2OS cells because its heterochromatin is too dispersed to selectively photobleach with high accuracy. For this reason, the mobile fraction of EYFP-SUV39H1 is probably <80%. Within euchromatic regions of NIH3T3 cells, we measured a t1/2 of 11.8 s and no immobile fraction for EYFP-SUV39H1 (Fig. 3 D and Table I).
The presence of an immobile or considerably less mobile fraction of SUV39H1 in heterochromatin was further investigated by performing two successive FRAP measurements on the same heterochromatic domain in NIH3T3 cells. EYFP-SUV39H1 was photobleached in a heterochromatic domain, and, after a recovery period of 450 s, the same area was photobleached for the second time, after which the fluorescence recovery was measured again. During the second FRAP, an immobile fraction would be invisible because it was bleached during the first FRAP. As shown in Fig. 3 E, after the second bleach, a full recovery of fluorescence was measured, which was 70% of the initial amount of fluorescence that was measured before the first photobleaching. Thus, this experiment confirmed the existence of an immobile fraction, at least on the time scale of the experiment.
Because various lines of evidence suggest that SUV39H1 interacts with HP1 proteins (Aagaard et al., 1999; Melcher et al., 2000) and that HP1 proteins are very mobile in the nucleus of living cells (Cheutin et al., 2003; Festenstein et al., 2003), we sought to compare the dynamic properties of SUV39H1 with those of HP1 proteins in NIH3T3 cells. Fusion constructs of HP1α and HP1β with EYFP were made and transiently transfected into cells. Fusion proteins were shown to localize to the same nuclear sites as their endogenous counterparts, as revealed by immunocytochemistry with specific anti-HP1 antibodies. Cells showing moderate expression levels were selected, and, upon bleaching of small areas inside the nucleus that corresponded to heterochromatin and euchromatin, recovery of fluorescence was recorded. Consistent with previous data, HP1 proteins revealed a very dynamic behavior in NIH3T3 cells (Fig. 3 B). For EYFP-HP1β, we measured a t1/2 of ∼4.2 s and a maximum fluorescence recovery of ∼95%, which was reached after 60 s from bleaching (Fig. 3 C and Table I). Similar kinetics of EYFP-HP1β was measured when CFP-SUV39H1 and EYFP-HP1β were coexpressed in the same cells (unpublished data). Together, these results suggest that SUV39H1 is more stably bound to chromatin than HP1 proteins and that a significant fraction of SUV39H1 is recruited to chromatin in an independent fashion or at least is not in a stable complex together with HP1 proteins.
The SET domain with adjacent regions mediates stable binding of SUV39H1 to heterochromatin
Three distinct protein domains have been identified in SUV39H1, of which aa 3–44 at the NH2 terminus form the HP1β interaction surface that, together with the adjacent chromodomain (aa 44–88), direct accumulation at heterochromatic regions (Melcher et al., 2000). The COOH-terminal SET domain (aa 249–412) has been shown to be responsible for H3K9 methylation but has also been suggested to modulate heterochromatin association of SUV39H1 (Melcher et al., 2000; Lachner et al., 2001). To investigate how the different domains contribute to the kinetic behavior of SUV39H1 at chromatin in vivo, we generated various deletion mutants of SUV39H1 according to Melcher et al. (2000), fused them to EYFP, and transiently expressed them in NIH3T3 cells. A cartoon depicting the deletion mutants is shown in Fig. 4. First, we analyzed the spatial distribution of fusion proteins in the cell nucleus. Cells moderately expressing EYFP–SUV39H1-ΔSET (aa 3–249 of Suv39H1 fused to EYFP) or EYFP–SUV39H1-Nchromo (aa 3–118 of Suv39H1 fused to EYFP) revealed the characteristic heterochromatic localization pattern that has also been observed for the full-length protein and after staining with DAPI or HP1 antibodies (Fig. 5 A, top). However, when expressed in U2OS cells, slightly different localization patterns were observed. The patterns seemed more dotlike when compared with localization of the full-length fusion protein (Fig. 5, C and D, top). Nevertheless, the expressed EYFP–SUV39H1-ΔSET and EYFP–SUV39H1-Nchromo fusion proteins were of the correct size, as determined by Western blotting (Fig. 5 B). Confocal analysis of U2OS cells expressing the fusion proteins that were stained with an anticentromere antibody showed EYFP–SUV39H1-ΔSET (Fig. 5 C, bottom) or EYFP–SUV39H1-Nchromo (Fig. 5 D, bottom) localization only at the centromeres. This localization in interphase nuclei is consistent with the finding that SUV39H1-ΔSET and SUV39H1-Nchromo localize more exclusively to the centromeres of metaphase chromosomes (Melcher et al., 2000).
Figure 4.

Mutant SUV39H1 constructs. Overview of the SUV39H1 deletion mutants fused to EYFP. The chromodomain is shown as a gray shaded box, and the SET domain is in black. The numbers refer to amino acid positions in the SUV39H1 protein.
Figure 5.
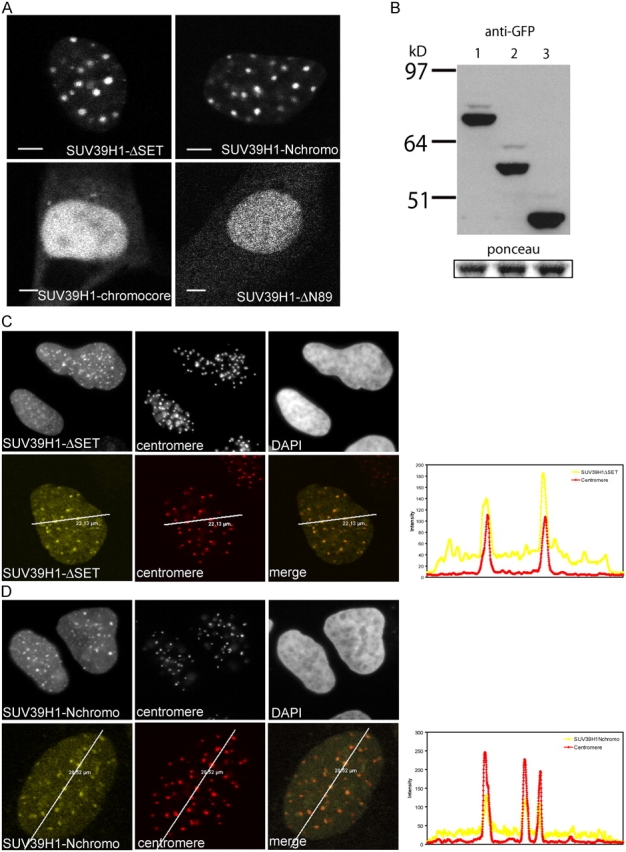
SUV39H1-ΔSET and SUV39H1-Nchromo are more concentrated at centromeres than SUV39H1. (A) Localization of the various SUV39H1 mutants in NIH3T3 cells. Bar, 10 μm. (B) Western blot showing expression of EYFP-SUV39H1 (lane 1), EYFP–SUV39H1-ΔSET (lane 2), and EYFP–SUV39H1-Nchromo (lane 3). Ponceau staining shows equal loading of the gel. (C) The top panel shows the distribution of EYFP–SUV39H1-ΔSET, centromeres, and DNA in a U2OS cell. The bottom panel shows a single optical section of a U2OS cell expressing EYFP–SUV39H1-ΔSET (left) and stained for centromeres (middle). Colocalization of the two is shown in the right image. The line scan (diagonal lines through the images) shows the local intensity distribution of EYFP–SUV39H1-ΔSET in yellow and centromere labeling in red. (D) Simultaneous detection of EYFP–SUV39H1-Nchromo (left) and centromeres (middle) in a U2OS cell. DNA is stained by DAPI (top right). The bottom panel shows a single optical section and a line scan (diagonal lines through the images) giving the local intensity distribution of EYFP–SUV39H1-Nchromo (yellow) and centromere staining (red).
Next, we performed FRAP analysis to determine the kinetic properties of SUV39H1-ΔSET and SUV39H1-Nchromo mutants in living cells at heterochromatic regions (Fig. 6, A and B). In NIH3T3 cells, we measured a t1/2 of recovery of ∼8.6 s for EYFP–SUV39H1-ΔSET and of ∼3.8 s for EYFP–SUV39H1-Nchromo. Notably, these proteins reached ∼90% recovery at 120 s and ∼95% at 40 s, respectively (Fig. 6 C and Table I). These results indicate that both SUV39H1 mutants are more dynamic than the full-length protein and that there is no significant stably bound fraction. Furthermore, the t1/2 of fluorescence recovery and the mobile fraction of EYFP–SUV39H1-Nchromo are comparable with that of HP1β. Coexpression experiments in NIH3T3 cells revealed that neither the dynamics nor the localization of HP1 significantly changed as a result of EYFP–SUV39H1-ΔSET or EYFP–SUV39H1-Nchromo expression (unpublished data).
Figure 6.
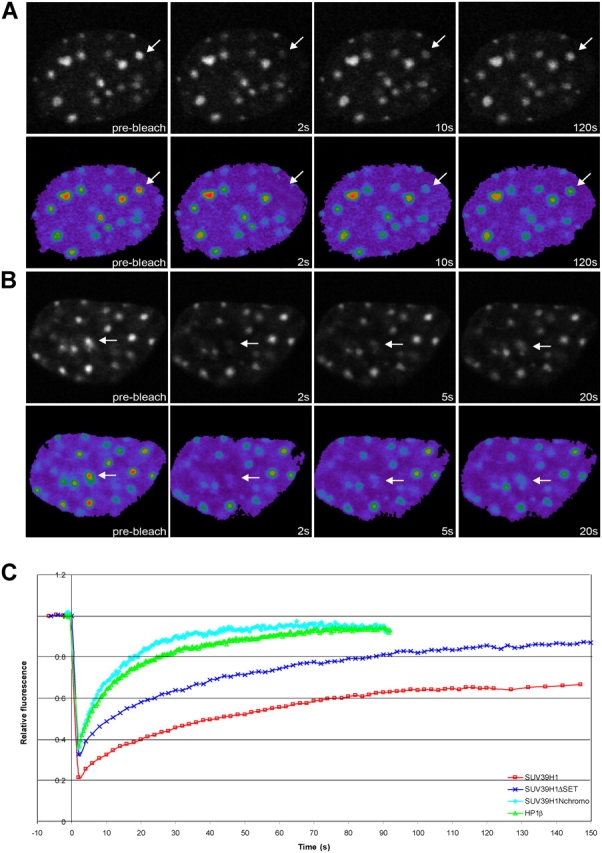
SUV39H1-ΔSET and SUV39H1-Nchromo have a higher mobility rate than full-length SUV39H1. After transfection of NIH3T3 cells with EYFP–SUV39H1-ΔSET (A) or EYFP–SUV39H1-Nchromo (B), a heterochromatic area was selected and photobleached. Images were recorded just before and at different time intervals after bleaching. Arrows indicate the photobleached areas. (C) The corresponding FRAP curves are plotted together with the FRAP curves for EYFP-SUV39H1 and EYFP-HP1β. These curves indicate that EYFP–SUV39H1-ΔSET and EYFP–SUV39H1-Nchromo are more dynamic than the full-length protein. The FRAP curves for the two mutant proteins represent means from 30 and 14 cells, respectively.
The mutants EYPF–SUV39H11-chromocore (aa 37–118 of Suv39H1 fused to EYFP) and EYPF–SUV39H1-ΔN89 (aa 89–412 of Suv39H1 fused to EYFP) localized diffusely throughout the nucleus in NIH3T3 cells (Fig. 5 A, bottom) as well as in human U2OS cells (not depicted). FRAP analysis revealed that EYPF–SUV39H1-chromocore and EYPF–SUV39H1-ΔN89 gave very rapid recovery times (∼0.6 and ∼0.5 s respectively; Table I). Expression of both fusion proteins was checked by Western blotting and showed their expected sizes (not depicted). These data indicate that EYPF–SUV39H1-chromocore and EYPF–SUV39H1-ΔN89 mutants diffuse freely throughout the nucleus, which is consistent with immunocytochemical data (Melcher et al., 2000), and do not (or to a minor extent) interact with chromatin.
SUV39H1 dynamics at centromeric heterochromatin is independent of its methyltransferase activity
Our data and that of others suggest that targeting of SUV39H1 to centromeric heterochromatin is mediated by the HP1-binding domain together with the chromodomain and that this targeting is even more profound in absence of the SET domain (Melcher et al., 2000). Meanwhile, a SUV39H1 mutant lacking the SET domain becomes more mobile at centromeric heterochromatin. This suggests that the SET domain is not essential for recruitment, but it still may play a role in stabilizing the interaction of full-length SUV39H1 with centromeric heterochromatin. To investigate whether it is the methyltransferase enzymatic activity of the SET domain that mediates SUV39H1 binding to heterochromatin, we analyzed the mobility of two point mutants; one with impaired methyltransferase activity (SUV39H1-H324L) and one with enhanced methyltransferase activity (SUV39H1-H320R). Both mutants were fused to EYFP and revealed the same localization as EYFP-SUV39H1 when transiently transfected into NIH3T3 cells. FRAP analysis of both point mutants revealed that the fluorescence recovery times and the mobile fractions at heterochromatin did not significantly deviate from that of wild-type SUV39H1 (Fig. 7), indicating that the binding of SUV39H1 is not mediated by its methyltransferase enzymatic activity.
Figure 7.

Methyltranferase activity has no influence on SUV39H1 dynamics. NIH3T3 cells were transfected with EYFP–SUV39H1-H324L or with EYFP–SUV39H1-H320R. Heterochromatic areas were selected and photobleached. Images were recorded just before and at different time intervals after photobleaching. The corresponding FRAP curves are plotted together with the FRAP curve for EYFP-SUV39H1. The curves represent mean values from 28 and 25 cells, respectively.
SUV39H1 interacts with HP1 on chromatin, as measured by FRET
FRAP analysis suggests that the binding of SUV39H1 to heterochromatin is strengthened by the SET domain (Fig. 6). Although previous work suggested that the binding of SUV39H1 to chromatin was not governed by an interaction with HP1 (Melcher et al., 2000), this interaction may still play a role in stabilizing the binding of SUV39H1 to pericentric heterochromatin in vivo. Thus, to examine the binding characteristics of EYFP-SUV39H1 to chromatin in more detail, we analyzed the interaction of full-length and mutant EYFP-SUV39H1 with HP1 in vivo by using FRET. FRET only occurs when two fluorescent proteins are at very close proximity (2–7 nm), meaning that they have to be in direct interaction. At present, there are various ways of measuring FRET in cells, each having their specific requirements. We measured FRET by two independent methods using the same experimental conditions and microscope setup. The three-filter method measures the sensitized emission of the acceptor molecule (Xia and Liu, 2001), and the fluorescence lifetime imaging microscopy (FLIM) method measures the decrease in donor fluorescence lifetime (Gadella, 1999), which occurs as a consequence of FRET.
U2OS cells were transiently cotransfected with EYFP-HP1β and ECFP-SUV39H1, EYFP-HP1β and ECFP–SUV39H1-ΔSET, or with EYFP-HP1β and ECFP–SUV39H1-ΔN89. As a negative control, cells were transfected with ECFP-SUV39H1 only. For each combination, 16–30 cells were analyzed using the three-filter method as well as the FLIM method. Cells expressing EYFP-HP1β together with ECFP-SUV39H1 or ECFP–SUV39H1-ΔSET revealed sensitized emission (Fig. 8, A and B), a significant decrease in mean donor lifetime, respectively, and 2.2 and 2.1 against 2.5 ns measured in control cells (Table II). Spatial analysis revealed most prominent FRET at heterochromatic areas (Fig. 8, A and B). Cells that were cotransfected with EYFP-HP1β and ECFP–SUV39H1-ΔN89 did not show sensitized emission (Fig. 8 C) or a decrease in donor lifetime (Table II). These results indicate that SUV39H1 and SUV39H1-ΔSET, but not SUV39H1-ΔN89, interact with HP1β in living cells. Moreover, the data suggest that the potential role of the SET domain in binding of SUV39H1 to heterochromatin is not mediated by an interaction with HP1.
Figure 8.

FRET measurements confirm the interaction between different autofluorescent fusion proteins. U2OS cells were cotransfected with ECFP-SUV39H1 and EYFP-HP1β (A), with ECFP–SUV39H1-ΔSET and EYFP-HP1β (B), or with ECFP–SUV39H1-ΔN89 and EYFP-HP1β (C). A CFP, YFP, and FRET image was acquired from all cells for spectral measurements, and a FLIM stack was collected to calculate fluorescent lifetimes. The CFP, YFP, and FRET images were first corrected for pixel shift and background. Then, the NFRET values were calculated and displayed in pseudocolors (Nfret). Absence of FRET is indicated by blue as depicted in the scale. The fluorescent lifetime values are also displayed in pseudocolors (FLIM). Blue (as depicted in the scale) indicates a lifetime of 2.5 ns and an absence of FRET.
Table II. Mean τφ and τ mod.
| Construct | τφ | SD | τ Mod | SD |
|---|---|---|---|---|
| ns | ns | |||
| ECFP-SUV39H1 | 2.5 | 0.1 | 3.1 | 0.1 |
| ECFP-SUV39H1/EYFP-HP1β | 2.2 | 0.1 | 2.9 | 0.1 |
| ECFP–SUV39-ΔSET/EYFP-HP1β | 2.1 | 0.1 | 2.7 | 0.1 |
| ECFP–SUV39H1-Nchromo/EYFP-HP1β | 2.2 | 0.2 | 2.8 | 0.1 |
| ECFP–SUV39H1-chromocore/EYFP-HP1β | 2.6 | 0.1 | 3.1 | 0.1 |
| ECFP–SUV39-ΔN89/EYFP-HP1β | 2.5 | 0.1 | 3.0 | 0.2 |
Values are means from 16 to 30 cells.
Previous in vitro studies have shown that the first 89 amino acids are indeed responsible for the interaction of SUV39H1 with HP1β (Melcher et al., 2000). To confirm that this interaction also occurs in living cells, we cotransfected EYFP-HP1β together with ECFP–SUV39H1-Nchromo or ECFP–SUV39H1-chromo. Again, cells were analyzed by the three-filter as well as by the FLIM method. Cells cotransfected with EYFP-HP1β and ECFP–SUV39H1-Nchromo showed a clear, sensitized emission and a decrease in lifetime (2.2 ns, Table II), whereas cells cotransfected with EYFP-HP1β and ECFP–SUV39H1-chromocore did not show sensitized emission or a decrease in lifetime (2.6 ns, Table II). This confirms the fact that it is also the most NH2-terminal part of SUV39H1 that interacts with HP1 in living cells.
DNA demethylation increases SUV39H1 mobility
Evidence is accumulating that a tight interplay exists between DNA methylation and histone modification. Interactions of SUV39H1 with different DNA methyltransferases (Fuks et al., 2003) as well as with methyl–cytosine guanine dinucleotide–binding domain proteins (Fujita et al., 2003) have been described previously. To explore the role of DNA methylation in strengthening the interaction between SUV39H1 and heterochromatin, we treated cells with the DNA demethylating agent 5-aza-2′deoxycytidine (5-aza-C). Cell sorting analysis revealed that this treatment resulted in some decrease of cells that are in S or G2 phase of the cell cycle (9 vs. 20% in untreated cells), with a vast majority of cells in G1 (90 vs. 78%). Furthermore, we showed by Western blotting that this treatment had no significant influence on the trimethylation of H3K9 or acyetylation of histones (see Fig. 10 D). Next, we analyzed the mobility of EYFP-SUV39H1 by FRAP, and the resulting curves show that DNA demethylation increases the mobility of SUV39H1 (t1/2 of ∼14.6 s compared with ∼19.0 s in untreated cells; Fig. 9). This suggests that the interaction of SUV39H1 with components of the DNA methylation machinery or with the unmethylated DNA itself has changed. However, the mobility of SUV39H1 in 5-aza-C–treated cells was still less than determined for SUV39H1-ΔSET in untreated cells, indicating that these interactions are not the sole mechanisms by which SUV39H1 binds to pericentromeric heterochromatin. Furthermore, the observation that EYFP–SUV39H1-ΔSET becomes more mobile in 5-aza-C–treated cells compared with nontreated cells (Fig. 9) suggests that it is not the SET domain of SUV39H1 that mediates the interaction with components of the DNA methylation machinery or with DNA itself.
Figure 10.
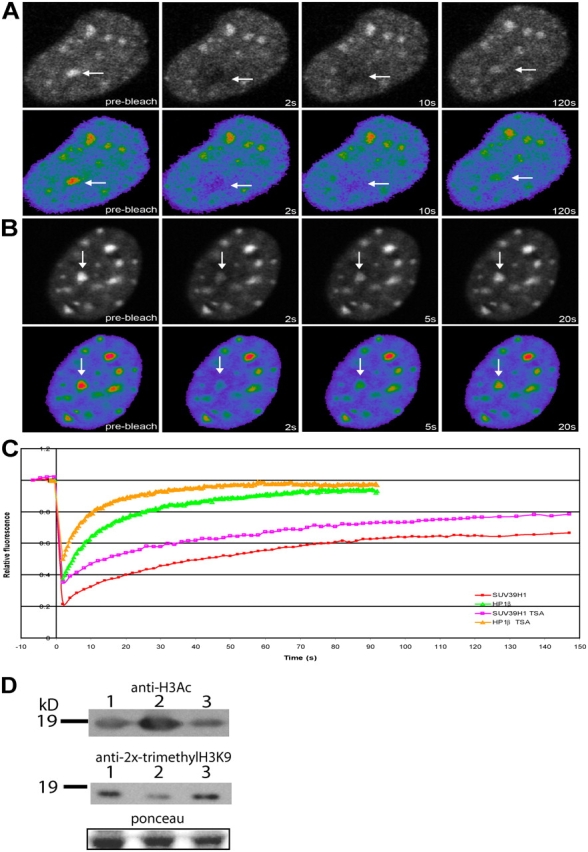
Inhibition of histone deacetylase activity increases the dynamics of both SUV39H1 and HP1β. NIH3T3 cells were transfected with EYFP-SUV39H1 (A) or EYFP-HP1β (B) and treated with 50 ng/ml TSA. After 18–22 h of TSA treatment, a heterochromatic area was selected and photobleached. Images were recorded just before and at different time intervals after photobleaching. Arrows indicate the photobleached areas. (C) The corresponding FRAP curves are plotted together with the FRAP curves for EYFP-SUV39H1 and EYFP-HP1β obtained from nontreated cells. Curves represent means from 22 and 8 cells, respectively. (D) Western blots showing the levels of acetylation and trimethylation in nontreated (lane 1), TSA-treated (lane 2), and 5-aza-C–treated (lane 3) NIH3T3 cells. Ponceau staining shows equal protein loading.
Figure 9.

DNA demethylation increases the dynamics of both SUV39H1 and SUV39H1-ΔSET but not that of HP1β. NIH3T3 cells were transfected with EYFP-SUV39H1, EYFP–SUV39H1-ΔSET, or with EYFP-HP1β and were treated with 5 μM 5-aza-C. After 48–52 h of treatment, a heterochromatic area was selected and photobleached. Images were recorded just before and at different time intervals after photobleaching. The corresponding FRAP curves are plotted together with the FRAP curves for EYFP-SUV39H1, EYFP–SUV39H1-ΔSET, and EYFP-HP1β obtained from nontreated cells. Curves represent the means from 18, 12, and 16 cells, respectively, and show that both EYFP-SUV39H1 and EYFP–SUV39H1-ΔSET become more dynamic after 5-aza-C treatment.
FRAP analysis of HP1β mobility in EYFP-HP1β–expressing cells treated with 5-aza-C showed that HP1β mobility is not changed compared with nontreated cells (Fig. 9).
Trichostatin A (TSA) treatment affects the binding of SUV39H1 but not SUV39H1-ΔSET to chromatin
Heterochromatin is not only characterized by hypermethylation but also by hypoacetylation of histones. The hyperacetylation of histones is a hallmark of transcriptionally active genes and has been suggested to prevent methylation of histone H3K9 and, thus, heterochromatin formation (Jenuwein and Allis, 2001). Therefore, histone deacetylases might be active at heterochromatin to maintain a hypoacetylated state. Treatment of cells with the histone deacetylase inhibitor TSA has been shown to result in the dissociation of HP1 from pericentromeric heterochromatin domains, probably by preventing methylation of H3 by SUV39H1 (Taddei et al., 2001). To test whether the in vivo kinetics of SUV39H1 binding to chromatin alters as a consequence of hyperacetylation, NIH3T3 cells expressing EYFP-SUV39H1 were treated with TSA. As shown by Western blotting, this treatment indeed resulted in an increased amount of acetylated histones and in a decrease in trimethylated H3K9 (Fig. 10 D). Cell sorting analysis revealed that this treatment resulted in some decrease of cells that were in S or G2 phase of the cell cycle (12 vs. 20% in untreated cells), with a vast majority of cells in G1 (87 vs. 78%). TSA-treated cells revealed a less pronounced localization of EYFP-SUV39H1 at heterochromatic regions, which is consistent with previous data (Maison et al., 2002; Cheutin et al., 2003). Furthermore, FRAP analysis revealed a faster fluorescence recovery of EYFP-SUV39H1 in TSA-treated cells (t1/2 of ∼7.7 s) compared with untreated cells (t1/2 of ∼19.0 s). Also, the total mobile fraction of EYFP-SUV39H1 at chromatin increased from ∼70 to ∼90% (Fig. 10, A and C, and Table I). TSA treatment had no significant effect on the kinetics of SUV39H1-ΔSET (t1/2 of ∼8.6 s). However, consistent with previous data (Cheutin et al., 2003), EYFP-HP1β mobility increased after TSA treatment (Fig. 10 B) to a t1/2 of ∼1.8 s compared with 4.2 s in untreated cells (Fig. 10 C). These data show that hyperacetylation of histones reduces the binding of EYFP-SUV39H1 and EYFP-HP1β to heterochromatin but does not prevent a transient SET-independent interaction of EYFP-SUV39H1 with chromatin.
Discussion
SUV39H1, the human homologue of the D. melanogaster position effect variegation modifier Su(var)3-9, catalyzes the methylation of histone H3K9, thereby creating binding sites for HP1. In particular, SUV39H1 catalyzes trimethylation events, which are characteristic of constitutive pericentromeric heterochromatin formation. Despite the fact that much is known about the catalytic activity of SUV39H1, little is known about how this protein is recruited to and interacts with chromatin. In this study, we report that SUV39H1 that is localized at pericentromeric heterochromatin consists of two populations: a mobile and an immobile one. Furthermore, the mobile population of SUV39H1 appears to be significantly less mobile than HP1. These findings suggest that SUV39H1 may, in addition to its catalytic activity, also serve a structural role in chromatin. In addition, our data support the idea that HP1 is not necessarily recruited in a complex with SUV39H1 to pericentromeric chromatin.
Initially, HP1 had been considered a stable component of heterochromatin in order to maintain a compact, inaccessible chromatin conformation that would exclude transcriptional activity. This concept has changed because HP1 proteins were shown by FRAP analysis to be highly mobile in living cells (Cheutin et al., 2003; Festenstein et al., 2003). Recently, various other structural chromatin-binding proteins were shown to interact transiently, indicating that transient interactions are a common feature of chromatin proteins (Christensen et al., 2002; Harrer et al., 2004; Phair et al., 2004). These findings led to the suggestion that the dynamic nature of architectural chromatin-associated proteins would be essential to regulate gene-related processes (Harrer et al., 2004; Phair et al., 2004). This idea is consistent with recent findings showing that condensed chromatin domains are accessible for high molecular weight dextrans (Verschure et al., 2003) as well as for gene regulatory and chromatin proteins (Chen et al., 2005). Our observation that a substantial fraction of SUV39H1 is stably associated with chromatin during an extended period of time suggests, however, that not all chromatin-associated proteins are necessarily in continuous flux with the nucleoplasm. The significant immobile fraction of SUV39H1 might be indicative of chromatin conformations that are not prone to processes regulating transcriptional activity but that play a role in the structural organization of the cell nucleus. This might be particularly true for centromeric and telomeric regions, which have been implicated to play roles in spatial nuclear organization and generally show constrained movements in live cells (Molenaar et al., 2003). Consistent with this idea is the observation that changes in DNA methylation and histone acetylation have significant effects on the architecture of interphase chromosome arms but not on the positioning of centromeres and telomeres in wheat cell nuclei (Santos et al., 2002).
SUV39H1, however, is not only found at constitutive silent regions but also at genomic regions displaying transcriptional activity (Greil et al., 2003). Furthermore, transcriptionally inactive genes are found in open chromatin structures, whereas transcriptionally active genes are found in domains of compact chromatin (Gilbert et al., 2004). Hence, the classical strict distinction between heterochromatin (that it is supposed to be transcriptionally inactive) and euchromatin (that it is supposed to be transcriptionally active) does not exist. Because it is not possible to spatially define and distinguish small transcriptionally active and inactive regions in living cells expressing EYFP-SUV39H1, it was not possible to reproducibly analyze and compare the dynamic properties of SUV39H1 association at sites that were distinct from the relatively large heterochromatin domains. Therefore, the FRAP data obtained from nuclear regions that were adjacent to the pericentromeric heterochromatin in NIH3T3 cells is likely a reflection of SUV39H1 mobility rates at various levels of chromatin compaction. Nevertheless, the fact that we, on average, measured higher mobility rates in regions that we defined as euchromatic suggests that SUV39H1 is more dynamic at less condensed chromatin than at pericentromeric heterochromatin.
Recently, a slow moving fraction of HP1 has been identified at pericentromeric heterochromatin by using fluorescence correlation microscopy and FRAP (Schmiedeberg et al., 2004). However, this fraction appears to be relatively small (7% in NIH3T3 cells) when compared with the relatively large immobile fraction of SUV39H1 (∼30% in NIH3T3 cells) that we measured. Furthermore, the fast fluorescence recovery rate of HP1 (t1/2 of ∼4 s) compared with the slow recovery rate of SUV39H1 (t1/2 of ∼19 s) support the idea that both proteins are not necessarily recruited as a complex to chromatin, which is in contradiction with current models (Lachner et al., 2001; Maison and Almouzni, 2004). Our findings and the data of others are consistent with a model that SUV39H1 is a structural component of heterochromatin and is able to recruit HP1 by direct protein–protein interaction (Stewart et al., 2005). Interestingly, another SET domain–containing protein, multiple myeloma SET II, has recently been shown to bind to a nucleoplasmic structure (possibly chromatin) with even higher affinity than SUV39H1 (Keats et al., 2005). This finding suggests that the tight association of SUV30H1 with chromatin is a more common feature that is shared by other SET proteins. However, multiple myeloma SET II belongs to another subfamily than does SUV39, whose function is unknown and may not even possess histone methyltransferase activity.
To identify the protein domain that is responsible for stable binding of the immobile population of SUV39H1 to pericentromeric heterochromatin, we characterized the exchange rate of various deletion mutants of SUV39H1 at pericentromeric heterochromatin in living cells by FRAP analysis. This analysis revealed that in addition to the NH2 terminus and the adjacent chromodomain of SUV39H1, the SET domain also plays a role in chromatin binding. Consistent with previous data (Melcher et al., 2000), we observed that SUV39H1-ΔSET localization was mainly restricted to pericentromeric heterochromatin regions compared with wide-spread chromatin associations of full-length SUV39H1. This suggests that the SET domain plays a role in chromatin association. Our finding that SUV39H1-ΔSET is highly mobile at the pericentromeric heterochromatin regions in living cells reinforces this suggestion and indicates that the SET domain mediates the stable interaction of SUV39H1 with pericentromeric chromatin, whereas the other domains are responsible for the recruitment of SUV39H1 to chromatin. Furthermore, FRAP analysis of the point mutants SUV39H1-H324L and SUV39H1-H320R revealed that it is not the histone methyltransferase activity of the SET domain that stabilizes binding of SUV39H1. This is consistent with previous localization data showing that heterochromatin association of SUV39H1 can be uncoupled from its intrinsic histone methyltransferase activity (Lachner et al., 2001).
The mechanism that mediates the stable interaction of SUV39H1 via the SET domain with pericentromeric chromatin remains elusive. It has been described previously that SUV39H1 not only interacts with HP1 (Aagaard et al., 1999) but also interacts with components of the DNA methylation pathway (Fujita et al., 2003; Fuks et al., 2003) and with histone deacetylases (Vaute et al., 2002). Our FRET analysis reveals that SUV39H1 and HP1 indeed interact at pericentromeric chromatin in live cells but that this interaction does not mediate the stable binding of SUV39H1 with heterochromatin. Furthermore, we could find no indication that binding of the immobile SUV39H1 population to pericentromeric chromatin is mediated by DNA methylation. Interestingly, treatment of cells with the deacetylation inhibitor TSA resulted in a higher mobility of SUV39H1 and in a significant reduction of the immobile SUV39H1 fraction at pericentromeric heterochromatin, whereas the kinetic behavior of SUV39H1-ΔSET did not change. This suggests that histone deacetylase activity is required for the stable interaction of SUV39H1 to pericentromeric chromatin via the SET domain. Remarkably, exposure of HeLa cells to TSA was previously reported to relocate centromeres from a more internal position to the nuclear periphery (Taddei et al., 2001; Maison et al., 2002), supporting the idea that the immobile population of SUV39H1 may contribute to maintaining nuclear architecture. By immunoprecipitation and activity assays, it has been demonstrated that the NH2-terminal part of SUV39H1 can interact with histone deacetylase 1 and 2 (Vaute et al., 2002). This interaction, which might be direct, may help to recruit SUV39H1 to heterochromatin but does not, however, explain the role of the SET domain in maintaining an immobile population of SUV39H1. Recent studies indicate that a yet unidentified RNA may contribute to HP1 and may also contribute to SUV39H1 binding to pericentromeric heterochromatin (Maison et al., 2002; Muchardt et al., 2002). We are currently investigating the interesting possibility that an RNA component contributes to the establishment of an immobile population of SUV39H1 at pericentromeric heterochromatin.
In summary, our data have important implications for understanding the mechanisms through which SUV39H1 exerts its biological functions. It is intriguing that the SET domain not only displays histone methyltransferase activity but also plays a role in the binding of SUV39H1 to heterochromatin. In particular, the SET domain is likely to be involved in stabilizing the association of a population of SUV39H1 proteins to heterochromatin. We speculate that this stable, immobile population of SUV39H1 plays an essential role in the maintenance of a functional nuclear architecture and may also act as an interaction platform for other heterochromatin proteins. Future studies will address how the stable association of SUV39H1 at heterochromatin is maintained. This is particularly relevant because the SET domain of SUV39H1 has also been identified as being a protein–protein interaction domain with possible implications in cancer development (Schneider et al., 2002). For example, the SET domain was shown to interact with Sbf-1, a protein with the ability to transform fibroblasts (Cui et al., 1998). It can be speculated that this interaction may interfere with the functioning of SUV39H1 either by deregulating methyltransferase activity or possibly by direct destabilization of the chromatin conformation by weakening the binding of SUV39H1 to chromatin.
Materials and methods
Construction of autofluorescent fusion proteins
SUV39H1 (nt 46–1284 of NM_003173) was amplified from cDNA that was generated from RNA isolated from human osteosarcoma cells (U2OS). This was done by using the forward primer 5′-GCGCGCGAATTCTATGGCGGAAAATTTAAAAGG-3′ containing the EcoRI site and the reverse primer 5′-GCGCGCGGTACCCTAGAAGAGGTATTTGCGGC-3′ containing the KpnI site. SUV39H1 deletion mutants were generated according to Melcher et al. (2000). SUV39H11-ΔSET was amplified from pEYFP-SUV39H1 with the full-length forward primer and the reverse primer 5′-GCGCGCGGTACCCCGGAAGATGCAGAGGTCAT-3′ containing the KpnI site. SUV39H1-Nchromo was made by using the full-length forward primer and the reverse primer 5′-GCGCGCGGTACCCAGGTAGTTGGCCAAGCTTG-3′ containing the KpnI site. SUV39H1-chromocore was made by using the forward primer 5′-GCGCGCGAATTCTAGGAACCTCTATGACTTTGA-3′ containing the EcoRI site and the Nchromo reverse primer. SUV39H11-ΔN89 was made by using forward primer 5′-GCGCGCGAATTCTCACAAGGACTTAGAAAGGGA-3′ containing the EcoRI site and the full-length reverse primer for. Purified PCR fragments were inserted in frame into the EcoRI-KpnI fragment of pECFP-C1 and pEYFP-C1 (CLONTECH Laboratories, Inc.).
HP1β was amplified with the forward primer 5′-GCGCGGTACCCATGGGGAAAAAACAAAACAAG-3′ containing the KpnI site and the reverse primer 5′-GCGCCCCGGGCGTTCTTGTCATCTTTTT-3′ containing the XmaI site and was cloned in the KpnI-XmaI fragment of pECFP-N1 and pEYFP-N1 (CLONTECH Laboratories, Inc.). Two SUV39H1 point mutants were made using the QuikChange II Site-Directed Mutagenesis Kit (Stratagene). For generating point mutant SUV39H11-H324L, we used the sense primer 5′-CTATGGCAACATCTCCCGCTTTGTCAACCACAGTTG-3′ and the antisense primer 5′-CAACTGTGGTTGACAAAGCGGGAGATGTTGCCATAG-3′. For generating SUV39H1-H320R, the sense primer was 5′-CATCTCCCACTTTGTCAACCTCAGTTGTGACCCCAAC-3′ and the antisense primer was 5′-GTTGGGGTCACAACTGAGGTTGACAAAGTGGGAGATG-3′. All constructs were verified by sequencing.
Cell culture and transfection
U2OS cells and NIH3T3 mouse fibroblast cells were cultured on 3.5-cm glass bottom petri dishes (MatTek) in DME without phenol red and containing 1.0 mg/ml glucose, 4% FBS, 2 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin buffered with 25 mM Hepes, pH 7.2 (all from Invitrogen). Wild-type and Suv39h dn PMEFs (provided by T. Jenuwein, Research Institute of Molecular Pathology, Vienna, Austria) were cultured as described previously (Lehnertz et al., 2003). Transient transfections were performed at ∼70–80% confluence using 1 μl LipofectAMINE 2000 (Invitrogen) and 0.75–1.5 μg DNA. For TSA treatment, cells were incubated with 50 ng/ml TSA (Sigma-Aldrich) for 18–22 h. For 5-aza-C treatment, cells were incubated with 5 μM 5-aza-C (Sigma-Aldrich) for 48–54 h.
Immunocytochemistry
Antibodies that were used in this study are listed as follows: human anticentromere (Antibodies, Inc.), mouse antiheterochromatin protein 1α and β (both from Euromedex), rabbit anti–2x-trimethylated H3K9 (provided by T. Jenuwein), and the appropriate secondary antibodies. These antibodies were diluted 1:100, 1:500, 1:500, and 1:250, respectively, in TBS containing 0.5% (wt/vol) blocking reagent (Roche) and 0.1% Tween 20.
Cells that were grown on microscopic glass slides were washed in PBS and fixed in 2% formaldehyde in PBS for 5 min for incubation with the anticentromere antibody or with 4% formaldehyde and 0.5% Triton X-100 (Sigma-Aldrich) in 0.1× PBS for 5 min. Subsequently, cells were permeabilized in PBS containing 1% Triton X-100 for 15 min, washed three times in PBS, and washed once in TBS containing 0.1% Tween 20. Then, cells were incubated with the first antibody for 45 min for the anticentromere or for 3 h at 37°C followed by three washes in TBS/0.1% Tween 20. Finally, cells were incubated with the secondary antibody for 45 min at 37°C, washed in TBS/0.1% Tween 20, and mounted in Citifluor (Agar Scientific, Ltd.) containing 400 μg/ml DAPI (Sigma-Aldrich).
Cell sorting
Cells were stained with propidium iodide according to the DNAcon3 kit (DakoCytomation). Cells were sorted in FACS (Becton Dickinson) and analyzed with Cell Quest software (BD Biosciences).
Protein blot analysis
Cells were lysed in NuPAGE LDS Sample Preparation Buffer (Invitrogen). Protein samples were then size fractionated on Novex 4–12% BisTris gradient gels using MOPS buffer (Invitrogen) and were subsequently transferred onto Hybond-C extra membranes (GE Healthcare) using a submarine system (Invitrogen). Blots were stained for total protein using Ponceau S (Sigma-Aldrich). After blocking with PBS containing 0.1% Tween 20 and 5% milk powder, the membranes were incubated with anti-GFP antibody (1:500; Roche), anti-H3Ac antibody (1:1,000; Upstate Biotechnology), and anti–2x-trimethylated H3K9 (1:500). The secondary antibodies that were used were anti–rabbit (1:2,000) and anti–mouse (1:5,000) HRP-conjugated antibodies (Pierce Chemical Co.). Bound antibodies were detected by chemiluminescence using ECL Plus (GE Healthcare).
Microscopy
Wide-field microscopy was performed on a microscope (model DMRXA; Leica) equipped with a 100-W mercury arc lamp and a 100× NA 1.3 plan Apo objective. Colocalization studies were performed on a confocal microscope system (model TCS/SP2; Leica). Image stacks were acquired with a 100× NA 1.4 plan Apo objective and were analyzed with Leica confocal software.
FRAP
Photobleaching experiments were performed on a confocal microscope. The temperature of the cells was maintained at 37°C by a heating ring surrounding the culture chamber (Harvard App., Inc.) and a microscope objective heater (Bioptechs). A 100× NA 1.4 plan Apo lens and a 514-nm laser line was used for photobleaching. Selected areas were subjected to five excitation pulses of 500 ms each at high laser power. Images were collected at time intervals of 450 ms from 5 s for 1.5–10 min after bleaching. The first postbleach image was acquired 2 s after photobleaching. To better quantify the recovery of fast moving SUV39H1 deletion mutants, selected areas were bleached once for 500 ms, and the first postbleach image was acquired after 500 ms. Quantitation of the fluorescent recovery was performed using Leica confocal software and Microsoft Excel. The recovery curves were corrected for background, fluorescence fading, and decrease in fluorescence during photobleaching. The t1/2 value was defined as the time required for reaching half-maximum recovery and was calculated from the corrected recovery curves.
FRET imaging
FRET was measured using a microscope (Axiovert 135 TV; Carl Zeiss MicroImaging, Inc.) equipped with a 100-W mercury arc lamp, a 100× plan Neofluar NA 1.3 phase objective, and a CCD camera (Micromax; Princeton Instruments). The spectral FRET measurements using three-filter sets were performed essentially as described by Xia and Liu (2001). CFP images were obtained with a filter set containing a 436/20-nm bandpass (BP) excitation filter, a 510-nm dichroic mirror, and a 480/200-nm BP emission filter. YFP images were obtained with a filter set containing a 500/20-nm BP filter, a 520-nm dichroic mirror, and a 535/30-nm BP emission filter (all from Chroma Technology Corp.). FRET images were acquired with a filter set containing a 436/20-nm BP excitation filter, a 510-nm dichroic mirror, and a 535/30-nm BP emission filter. The set of three images was corrected for background and pixel shift. Images were further processed, and the relative FRET efficiencies were calculated on a pixel-by-pixel basis. For the calculation of relative FRET values, the formula as described previously by Xia and Liu (2001) was used:
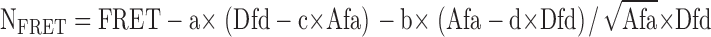 |
(1) |
where NFRET is the normalized FRET value and FRET, Dfd, and Afa are intensities measured through FRET, CFP, and YFP filter sets. a–d are bleed-through percentages measured in cells expressing CFP or YFP only. a and b are the percentages of YFP and CFP, respectively, that bleed through the FRET filter set. c is the percentage of CFP that bleeds through the YFP filter set, and d is the percentage of YFP that bleeds through the CFP filter set. Values for a–d were 22.6, 55.4, 0.4, and 0.6%, respectively. Calculated FRET values were displayed in pseudocolors.
FLIM was performed essentially as described previously (Gadella, 1999) using a microscope (Axiovert 135 TV; Carl Zeiss MicroImaging, Inc.) equipped with a 100-W mercury arc lamp, a 100× plan Neofluar NA 1.3 phase objective, and an image intensifier (model C5825; Hamamatsu) modulated at 40,000 MHz. ECFP was excited with a 458-nm argon/krypton laser line (Innova 70C; Coherent) modulated at 80,000 MHz using an acousto-optic modulator and a filter set containing a 458/10-mn BP excitation filter, a 470-nm dichroic mirror, and a 480/20-nm BP emission filter. 20 phase images (one every 36°C; 10 forward and 10 back to correct for bleaching) were acquired using a CCD camera (CoolSNAP fx; Photometrics). Reference phase settings and modulation were set every 15 min using a freshly prepared solution of 100 nM rhodamine 6G in 100% ethanol with a single component lifetime of 4.0 ns. Fluorescence lifetime values τϕ and τmod were calculated with the equations τϕ = (1/ω) tanΔϕ and τM = (1/ω)√(1/M2 − 1). Lifetime values were calculated for each pixel and were displayed in pseudocolors.
Acknowledgments
We are particularly grateful to Thomas Jenuwein for supplying reagents and advice and to Prim Singh for critical reading of the manuscript. We thank Dorus Gadella for his help building the FLIM system and Shosh Knaan for help with the cell sorting experiments.
This work was partially supported by Cyttron (grant BSIK03036).
C. Molenaar's present address is Developmental Genetics Laboratory, Cancer Research UK, London WC2A 3PX, England, UK.
Abbreviations used in this paper: 5-aza-C, 5-aza-2′deoxycytidine; BP, bandpass; dn, double null; FLIM, fluorescence lifetime imaging microscopy; FRET, fluorescence resonance energy transfer; HP1, heterochromatin protein 1; PMEF, primary mouse embryonic fibroblast; TSA, trichostatin A.
References
- Aagaard, L., G. Laible, P. Selenko, M. Schmid, R. Dorn, G. Schotta, S. Kuhfittig, A. Wolf, A. Lebersorger, and P.B. Singh, et al. 1999. Functional mammalian homologues of the Drosophila PEV-modifier Su(var)3-9 encode centromere-associated proteins which complex with the heterochromatin component M31. EMBO J. 18:1923–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister, A.J., P. Zegerman, J.F. Partridge, E.A. Miska, J.O. Thomas, R.C. Allshire, and T. Kouzarides. 2001. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 410:120–124. [DOI] [PubMed] [Google Scholar]
- Chen, D., M. Dundr, C. Wang, A. Leung, A. Lamond, T. Misteli, and S. Huang. 2005. Condensed mitotic chromatin is accessible to transcription factors and chromatin structural proteins. J. Cell Biol. 168:41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheutin, T., A.J. McNairn, T. Jenuwein, D.M. Gilbert, P.B. Singh, and T. Misteli. 2003. Maintenance of stable heterochromatin domains by dynamic HP1 binding. Science. 299:721–725. [DOI] [PubMed] [Google Scholar]
- Christensen, M.O., M.K. Larsen, H.U. Barthelmes, R. Hock, C.L. Andersen, E. Kjeldsen, B.R. Knudsen, O. Westergaard, F. Boege, and C. Mielke. 2002. Dynamics of human DNA topoisomerases IIα and IIβ in living cells. J. Cell Biol. 157:31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, X., I. De Vivo, R. Slany, A. Miyamoto, R. Firestein, and M.L. Cleary. 1998. Association of SET domain and myotubularin-related proteins modulates growth control. Nat. Genet. 18:331–337. [DOI] [PubMed] [Google Scholar]
- Festenstein, R., S.N. Pagakis, K. Hiragami, D. Lyon, A. Verreault, B. Sekkali, and D. Kioussis. 2003. Modulation of heterochromatin protein 1 dynamics in primary mammalian cells. Science. 299:719–721. [DOI] [PubMed] [Google Scholar]
- Fujita, N., S. Watanabe, T. Ichimura, S. Tsuruzoe, Y. Shinkai, M. Tachibana, T. Chiba, and M. Nakao. 2003. Methyl-CpG binding domain 1 (MBD1) interacts with the Suv39h1-HP1 heterochromatic complex for DNA methylation-based transcriptional repression. J. Biol. Chem. 278:24132–24138. [DOI] [PubMed] [Google Scholar]
- Fuks, F., P.J. Hurd, R. Deplus, and T. Kouzarides. 2003. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 31:2305–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadella, T.W.J. 1999. Fluorescence lifetime imaging microscopy (FLIM):instrumentation and applications. In Fluorescent and Luminescent Probes for Biological Activity. A Practical Guide for Technology for Quantitative Real-Time Analysis. W.T. Mason, editor. Academic Press, New York. 467–489.
- Garcia-Cao, M., R. O'Sullivan, A.H. Peters, T. Jenuwein, and M.A. Blasco. 2004. Epigenetic regulation of telomere length in mammalian cells by the Suv39h1 and Suv39h2 histone methyltransferases. Nat. Genet. 36:94–99. [DOI] [PubMed] [Google Scholar]
- Gilbert, N., S. Boyle, H. Fiegler, K. Woodfine, N.P. Carter, and W.A. Bickmore. 2004. Chromatin architecture of the human genome: gene-rich domains are enriched in open chromatin fibers. Cell. 118:555–566. [DOI] [PubMed] [Google Scholar]
- Greil, F., I. van der Kraan, J. Delrow, J.F. Smothers, E. de Wit, H.J. Bussemaker, R. van Driel, S. Henikoff, and B. van Steensel. 2003. Distinct HP1 and Su(var)3-9 complexes bind to sets of developmentally coexpressed genes depending on chromosomal location. Genes Dev. 17:2825–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrer, M., H. Luhrs, M. Bustin, U. Scheer, and R. Hock. 2004. Dynamic interaction of HMGA1a proteins with chromatin. J. Cell Sci. 117:3459–3471. [DOI] [PubMed] [Google Scholar]
- Jenuwein, T., and C.D. Allis. 2001. Translating the histone code. Science. 293:1074–1080. [DOI] [PubMed] [Google Scholar]
- Keats, J.J., C.A. Maxwell, B.J. Taylor, M.J. Hendzel, M. Chesi, P.L. Bergsagel, L.M. Larratt, M.J. Mant, T. Reiman, A.R. Belch, and L.M. Pilarski. 2005. Overexpression of transcripts originating from the MMSET locus characterizes all t(4;14)(p16;q32)-positive multiple myeloma patients. Blood. 105:4060–4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachner, M., N. O'Carroll, S. Rea, K. Mechtler, and T. Jenuwein. 2001. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 410:116–120. [DOI] [PubMed] [Google Scholar]
- Lehnertz, B., Y. Ueda, A.A. Derijck, U. Braunschweig, L. Perez-Burgos, S. Kubicek, T. Chen, E. Li, T. Jenuwein, and A.H. Peters. 2003. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 13:1192–1200. [DOI] [PubMed] [Google Scholar]
- Maison, C., and G. Almouzni. 2004. HP1 and the dynamics of heterochromatin maintenance. Nat. Rev. Mol. Cell Biol. 5:296–304. [DOI] [PubMed] [Google Scholar]
- Maison, C., D. Bailly, A.H. Peters, J.P. Quivy, D. Roche, A. Taddei, M. Lachner, T. Jenuwein, and G. Almouzni. 2002. Higher-order structure in pericentric heterochromatin involves a distinct pattern of histone modification and an RNA component. Nat. Genet. 30:329–334. [DOI] [PubMed] [Google Scholar]
- Melcher, M., M. Schmid, L. Aagaard, P. Selenko, G. Laible, and T. Jenuwein. 2000. Structure-function analysis of SUV39H1 reveals a dominant role in heterochromatin organization, chromosome segregation, and mitotic progression. Mol. Cell. Biol. 20:3728–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar, C., K. Wiesmeijer, N.P. Verwoerd, S. Khazen, R. Eils, H.J. Tanke, and R.W. Dirks. 2003. Visualizing telomere dynamics in living mammalian cells using PNA probes. EMBO J. 22:6631–6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchardt, C., M. Guilleme, J.S. Seeler, D. Trouche, A. Dejean, and M. Yaniv. 2002. Coordinated methyl and RNA binding is required for heterochromatin localization of mammalian HP1alpha. EMBO Rep. 3:975–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, A.H., D. O'Carroll, H. Scherthan, K. Mechtler, S. Sauer, C. Schofer, K. Weipoltshammer, M. Pagani, M. Lachner, and A. Kohlmaier, et al. 2001. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell. 107:323–337. [DOI] [PubMed] [Google Scholar]
- Peters, A.H., S. Kubicek, K. Mechtler, R.J. O'Sullivan, A.A. Derijck, L. Perez-Burgos, A. Kohlmaier, S. Opravil, M. Tachibana, and Y. Shinkai, et al. 2003. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol. Cell. 12:1577–1589. [DOI] [PubMed] [Google Scholar]
- Phair, R.D., P. Scaffidi, C. Elbi, J. Vecerova, A. Dey, K. Ozato, D.T. Brown, G. Hager, M. Bustin, and T. Misteli. 2004. Global nature of dynamic protein-chromatin interactions in vivo: three-dimensional genome scanning and dynamic interaction networks of chromatin proteins. Mol. Cell. Biol. 24:6393–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos, A.P., R. Abranches, E. Stoger, A. Beven, W. Viegas, and P.J. Shaw. 2002. The architecture of interphase chromosomes and gene positioning are altered by changes in DNA methylation and histone acetylation. J. Cell Sci. 115:4597–4605. [DOI] [PubMed] [Google Scholar]
- Schmiedeberg, L., K. Weisshart, S. Diekmann, G. Meyer Zu Hoerste, and P. Hemmerich. 2004. High- and low-mobility populations of HP1 in heterochromatin of mammalian cells. Mol. Biol. Cell. 15:2819–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, R., A.J. Bannister, and T. Kouzarides. 2002. Unsafe SETs: histone lysine methyltransferases and cancer. Trends Biochem. Sci. 27:396–402. [DOI] [PubMed] [Google Scholar]
- Schotta, G., A. Ebert, V. Krauss, A. Fischer, J. Hoffmann, S. Rea, T. Jenuwein, R. Dorn, and G. Reuter. 2002. Central role of Drosophila SU(VAR)3-9 in histone H3-K9 methylation and heterochromatic gene silencing. EMBO J. 21:1121–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart, M.D., J. Li, and J. Wong. 2005. Relationship between histone H3 lysine 9 methylation, transcription repression, and heterochromatin protein 1 recruitment. Mol. Cell. Biol. 25:2525–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei, A., C. Maison, D. Roche, and G. Almouzni. 2001. Reversible disruption of pericentric heterochromatin and centromere function by inhibiting deacetylases. Nat. Cell Biol. 3:114–120. [DOI] [PubMed] [Google Scholar]
- Vaute, O., E. Nicolas, L. Vandel, and D. Trouche. 2002. Functional and physical interaction between the histone methyl transferase Suv39H1 and histone deacetylases. Nucleic Acids Res. 30:475–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verschure, P.J., I. van der Kraan, E.M. Manders, D. Hoogstraten, A.B. Houtsmuller, and R. van Driel. 2003. Condensed chromatin domains in the mammalian nucleus are accessible to large macromolecules. EMBO Rep. 4:861–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wreggett, K.A., F. Hill, P.S. James, A. Hutchings, G.W. Butcher, and P.B. Singh. 1994. A mammalian homologue of Drosophila heterochromatin protein 1 (HP1) is a component of constitutive heterochromatin. Cytogenet. Cell Genet. 66:99–103. [DOI] [PubMed] [Google Scholar]
- Xia, Z., and Y. Liu. 2001. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys. J. 81:2395–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]