

Abstract
The origin recognition complex (ORC) ensures exactly one round of genome replication per cell cycle through acting as a molecular switch that precisely controls the assembly, firing, and inactivation of the replication initiation machinery. Recent data indicate that it may also coordinate the processes of mitosis and cytokinesis and ensure the proper distribution of replicated genome to daughter cells. We have found that the ORC core subunits are highly expressed in the nervous system. They are selectively localized to the neuronal somatodendritic compartment and enriched in the membrane fraction. siRNA knockdown of ORC subunits dramatically reduced dendritic branch formation and severely impeded dendritic spine emergence. Expression of ORC ATPase motif mutants enhanced the branching of dendritic arbors. The ORC core complex thus appears to have a novel role in regulating dendrite and dendritic spine development in postmitotic neurons.
Introduction
Dendrites and dendritic spines are the major sites of information processing and storage in the nervous system. They divide neurons into dynamic electrochemical compartments that serve integral roles in neuronal computation (Hausser et al., 2000). The function of these compartments depends greatly on the morphology of the dendritic tree that is sculpted during neural development. In the past decades, major insights have been gained into how intrinsic factors and extrinsic signals control and guide the development of dendrites and dendritic spines and how patterned neural activity shapes this process (Hering and Sheng, 2001; Whitford et al., 2002; Wong and Ghosh, 2002; Jan and Jan, 2003; Van Aelst and Cline, 2004). Nonetheless, large gaps still exist in our knowledge about how all these pathways integrate and execute their function at the molecular level and orchestrate the development of dendritic morphology. In particular, little is known about the molecular circuit neurons use to make the binary decision on initiating the formation of a new dendritic branch or spine.
The origin recognition complex (ORC; Bell and Stillman, 1992) is a hexameric protein complex key to initiating DNA replication during the cell cycle (Kelly and Brown, 2000; Bell, 2002; Bell and Dutta, 2002). It is part of the protein machinery responsible for one of the central processes of life, genome replication (Baker and Bell, 1998). Structurally the mammalian ORC is composed of four core (Orc2–5) and two peripheral subunits (Orc1 and Orc6; Dhar et al., 2001; Vashee et al., 2001), among which three (Orc1, Orc4, and Orc5) belong to the AAA+ family of ATPases (Neuwald et al., 1999). During early interphase when the Cdk activity level is low, the ORC initiates the assembly of a prereplication complex, which triggers DNA replication when the Cdk activity level rises as the cells undergo G1–S transition. At the same time it prevents replication reinitiation through multiple mechanisms including an ATPase-dependent conformational change of the large subunit Orc1 and modification of several other subunits, which result in the inactivation of the complex (Lee and Bell, 2000; Nguyen et al., 2001; Li and DePamphilis, 2002; Mendez et al., 2002). Thus, the ORC acts as a Cdk-regulated ATPase-dependent molecular switch for initiating DNA replication during the cell cycle, ensuring that each wave of Cdk activation is translated into one and only one round of genome replication.
The extraordinary properties of the ORC and its associated signaling circuits make it an ideal protein machine for coupling Cdk activation and genome replication. Recent evidence suggests that the ORC may be reused later in the cell cycle to couple Cdk activation with cytokinesis, the process where the cytoplasm of a cell is divided into two parts so that each would inherit one copy of the genome replicated earlier in the cell cycle. Orc6 has been found to localize to the spindle midzone of mitotic cells and its loss of function leads to accumulation of multinucleate cells (Prasanth et al., 2002; Chesnokov et al., 2003). Orc2 has also been found to associate with centrosomes and centromeres, where it is required for proper segregation of replicated chromosomes (Prasanth et al., 2004). Thus, the ORC can orchestrate not only the nuclear event of DNA replication but also cellular morphogenetic processes such as cytokinesis where the exact division of one cell into two, once per cell cycle, is equally critical for safeguarding genome integrity. In this report, we describe a novel role for the ORC in initiating dendritic branch and spine formation in postmitotic neurons and discuss the implication of its unexpected function in the nervous system.
Results
Expression of ORC subunits in the nervous system
Our initial encounter with the ORC genes was during our search of the RIKEN EST database for the adult mouse brain. As brain tissues mostly consist of postmitotic cells, it was a surprise to us that several ORC subunits appeared so frequently in the database. However, we noticed that one ORC subunit, Orc3, had been found to localize to the Drosophila neuromuscular junction (NMJ) and is required for its normal development and function (Pinto et al., 1999; Rohrbough et al., 1999). To further explore our finding, we did a series of Northern analyses on various adult mouse brain tissues (Fig. 1 and Fig. S1 available at http://www.jcb.org/cgi/content/full/jcb.200505075/DC1). We found that orc2–5 are expressed at high levels (Fig. 1 A), whereas orc6 is expressed at a moderate level in adult brain tissues including cerebral cortex, hippocampus, and cerebellum (Fig. 1 B). By contrast, orc1 is not expressed at a detectable level (Fig. 1 B). This expression pattern is consistent with the structural relationship between the ORC subunits in that Orc2–5 subunits are known to form the core of the ORC and thus seem likely to be coregulated at the transcriptional level. As all the ORC subunits are required for DNA replication (Kelly and Brown, 2000; Bell, 2002; Bell and Dutta, 2002), the absence of orc1 strongly suggests that the ORC may have a different role in the brain from regulating DNA replication. Consistent with this, we found that several components downstream of the ORC in DNA replication, including mcm2, mcm4, mcm6, and cdc6, are barely detectable in the adult brain (Fig. S1).
Figure 1.
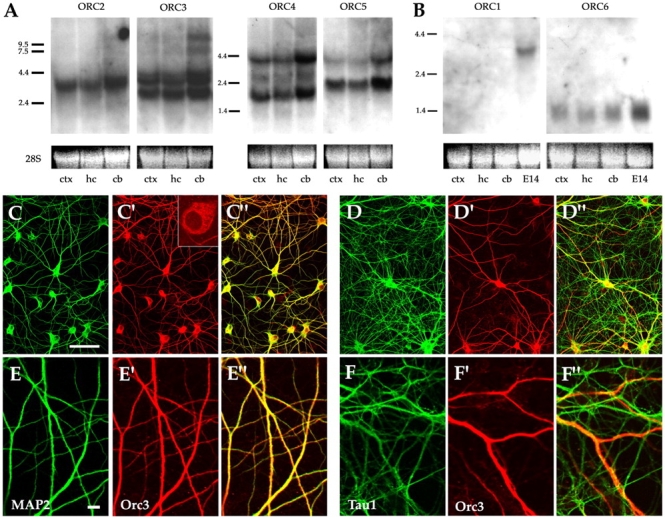
ORC core subunits are expressed in the brain and localized to dendrites. (A and B) Northern analysis of ORC subunit expression in adult mouse brain tissues: cerebral cortex (ctx), hippocampus (hc), and cerebellum (cb). E14 denotes RNA from day 14 embryos. 28S denotes 28S ribosomal RNA. The multiple transcripts for orc3–5 are likely due to alternative initiation. (C–C″ and E–E″) Confocal micrographs of hippocampal neurons stained for Orc3 (red) and the dendritic marker MAP2 (green). (C′, inset) Close-up view of neuronal soma. (D–D″ and F–F″) Confocal micrographs of neurons stained for Orc3 (red) and the axonal marker Tau1 (green). Sometimes the Orc3 staining appears yellow due to axons fasciculating along dendrites. Bars, 50 μm.
To investigate the role of ORC in neurons, we used antibodies against Orc3 to stain cultured hippocampal neurons. Interestingly, we found punctate Orc3 staining not in the nucleus, the usual location for ORC, but in the cytoplasm of neurons (Fig. 1 C′, inset, and Fig. S1). In neurons at the early stage of differentiation before axonal polarization, Orc3 appears to localize to all neuronal processes. After the emergence of axons, however, Orc3 seems to be excluded from the axons but persists in dendrites (Fig. 1, C–F). Double staining with the dendritic marker MAP2 revealed extensive overlap between Orc3 and MAP2 immunoreactivity (Fig. 1, C–C″ and E–E″), whereas very little overlap was observed between Orc3 and Tau1, the axonal microtubule binding protein (Fig. 1, D–D″ and F–F″). When EGFP fusion reporters of Orc2, Orc3, and Orc5 were expressed in neurons, they also seemed to colocalize with endogenous Orc3 in dendrites (Fig. S2 available at http://www.jcb.org/cgi/content/full/jcb.200505075/DC1). These results thus further argue that the ORC subunits may have a novel function in neurons.
Association of ORC with neuronal membranes
The coexpression of the ORC core subunits in the brain and their colocalization in neuronal processes suggest that, as in DNA replication, these subunits (Orc2–5) may also exist and function as a complex in neurons. To determine if this is the case, we tried various immunoprecipitation approaches but without success. We then used the approach of brain homogenate fractionation. First we fractionated brains of early postnatal rat pups into cytoplasmic and membrane fractions. We found that, interestingly, all three subunits we examined, Orc3–5, were detected in the membrane fraction except for Orc3, which was also detected in the cytosol (Fig. 2 A). This suggests that the majority of the ORC subunits are associated with neuronal membrane while a fraction of Orc3 may exist in a second cytoplasmic pool. To determine the nature of neuronal membrane with which ORC is associated, we further separated it into microsome, synaptic vesicle, and lysed synaptosome fractions. We found that most of Orc3–5 proteins were associated with the microsome and synaptosome fractions and very little of them, particularly for Orc4 and Orc5, were detected in the synaptic vesicle fraction (Fig. 2 B). Similar patterns of subunit distribution were also observed in adult brain fractions (Fig. S2). In addition, consistent with the fact that none of the ORC subunits possess transmembrane motifs, we found that either high salt or chaotropic conditions would disrupt their association with the membrane. These results are thus consistent with the idea that the ORC subunits may exist primarily as a complex in the nervous system.
Figure 2.

ORC core subunits are associated with neuronal membranes. Western analysis of P10 rat brain fractions. (A) Immunoblot of Orc3, Orc4, and Orc5 in crude brain fractions: low-speed centrifugation pellet (P1), cytosol (C), and membrane fraction (M, duplicates). (B) Immunoblot of Orc3, Orc4, and Orc5 in rat brain fractions: postnuclear supernatant (S1), medium-speed centrifugation supernatant (S2), crude synaptosomal fraction (CS), cytosol (C), microsomal fraction (Mi), synaptic vesicle-enriched fraction (SV), and lysed synaptosomal fraction (LS).
ORC is required for dendritic arbor development
To determine ORC function in neurons, we used a vector-based siRNA approach (Brummelkamp et al., 2002). Eight nonoverlapping siRNA constructs were designed, each derived from a 19- to 21-mer oligonucleotide in the coding region of the murine orc3 gene. We found that four of them, siRNA1, 2, 4, and 7, caused nearly complete degradation of the mouse orc3 mRNA expressed in COS cells (Fig. 3 A). We also confirmed that they led to severe reduction of the mouse Orc3 protein expressed in COS cells (Fig. 3 B). In addition, when transfected into hippocampal neurons, they also caused a significant loss of endogenous Orc3 protein (Fig. 3, C and C′). As all four constructs caused similar phenotypes, we focused our analysis on two of them, siRNA1 and 4.
Figure 3.
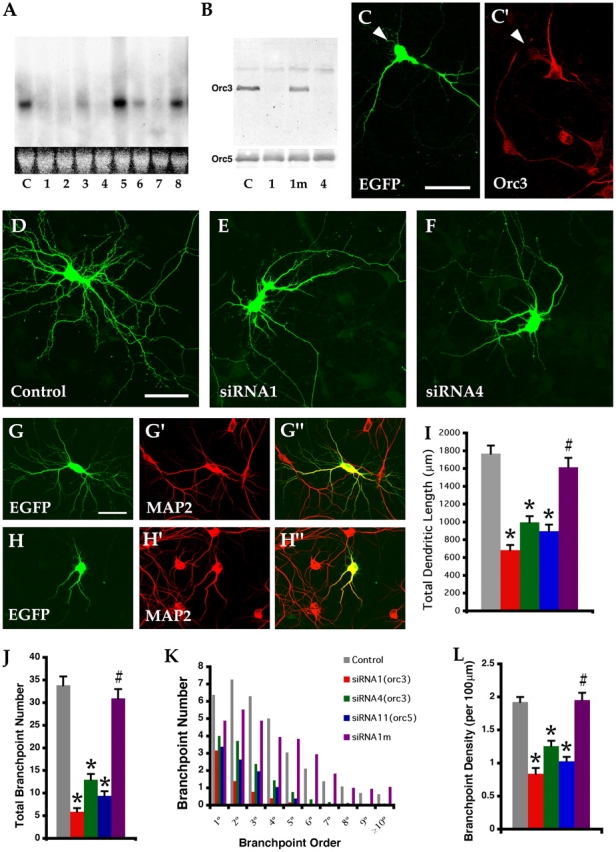
orc3 is required for dendritic growth and branching. (A) Northern analysis of mouse orc3 expression in COS cells transfected with control (lane C) or orc3 siRNA constructs (lanes 1–8). (B) Western analysis of mouse Orc3 protein expression in COS cells transfected with control (C) or orc3 siRNA constructs (1, 1m, and 4). (C–C′) Confocal micrographs of hippocampal neurons stained for Orc3 (B′, red). A siRNA1-transfected neuron was labeled by EGFP (B, green). Notice the loss of Orc3 staining from the transfected cell (arrowhead). (D–F) Confocal micrographs of 10 DIV neurons transfected on 7 DIV with control (C), siRNA1 (D), or siRNA4 (E) constructs and EGFP (green). (G–G″ and H–H″) Confocal micrographs of neurons transfected with control (G–G″) or siRNA1 constructs (H–H″) and stained for EGFP (green) and MAP2 (red). (I–L) Quantitative analysis of dendritic morphology. Total dendritic length of neurons transfected with orc3 (siRNA1, n = 26, and siRNA4, n = 24) or orc5 (siRNA11, n = 24) siRNA or mutated siRNA1 (siRNA1m, n = 17) is compared with control (n = 27, all ≥3 experiments) neurons (I). Total branchpoint numbers (J), branchpoint distribution (K), and density (L) are also compared. Error bars reflect SEM. *, P < 1.0 × 10−6 (all compared with control). #, P > 0.28 (compared with control); P < 1.0 × 10−9 (compared with siRNA1). Bars, 50 μm.
Cultured hippocampal pyramidal neurons undergo several stages of development including initial determination and extension of axons followed by elaboration of dendrites. The selective localization of the Orc3 protein to the dendrites and the association of ORC subunits with neuronal membranes of early postnatal brains suggest that the ORC may be involved in dendritic development. We therefore transfected the siRNA constructs together with EGFP into 7 d in vitro (DIV) neurons and examined them on 10 DIV. We found that orc3 knockdown severely impaired dendritic growth and branching of hippocampal neurons. When compared with neurons transfected with a control construct that has no homology to any known mouse or rat genes (Fig. 3 D), siRNA1- or 4-transfected neurons showed greatly shortened dendrites with severely reduced branching (Fig. 3, E and F). Quantification revealed a 61% reduction of total dendritic length caused by siRNA1 and a 44% reduction by siRNA4 (Fig. 3 I). Moreover, these neurons suffered an 83% drop in the total number of branchpoints in case of siRNA1 and a 62% drop in case of siRNA4, with branchpoints higher than the order of six or seven near completely eliminated (Fig. 3, J and K). These phenotypes do not seem to be due to nonspecific effects of siRNA as the control construct did not produce such a phenotype nor did we observe abnormalities in the nuclear morphology of these neurons. Furthermore, the siRNA constructs that failed to effectively degrade the orc3 mRNA did not produce such phenotypes. Mutations of two nucleotides in siRNA1 (siRNA1m) that abolished its capacity to interfere with Orc3 expression (Fig. 3 B) also eliminated its effects on dendritic development (Fig. 3, I–L). Moreover, overexpression of a truncated version of Orc3 that interferes with ORC complex assembly (Dhar et al., 2001) also impaired dendritic growth and branching of transfected neurons (Fig. S3 available at http://www.jcb.org/cgi/content/full/jcb.200505075/DC1). These results thus strongly argue that the observed effects were due to specific knockdown of endogenous Orc3 protein in transfected neurons.
To determine other potential effects, we next examined expression of the dendritic marker MAP2. We found that, despite their simplified dendritic trees, orc3 knockdown neurons showed relatively normal MAP2 staining (Fig. 3, G–G″ and H–H″), suggesting that the identity of these processes was not affected by loss of Orc3. Consistent with the selective dendritic localization of Orc3, we also observed relatively normal growth and morphology of axons and staining of the axonal marker Tau1 (unpublished data) as well as normal size of neuronal somas (Fig. S4 available at http://www.jcb.org/cgi/content/full/jcb.200505075/DC1). In addition, we also observed relatively normal expression of the neuron-specific tubulin isoform TubβIII (unpublished data). Thus, orc3 seems to be quite specifically involved in regulating dendritic growth and branching of hippocampal neurons at this stage of differentiation. However, we did frequently observe an additional effect in siRNA1- or 4-transfected neurons in that their dendrites seemed to be somewhat thicker and they appeared to have a looser organization of microtubules (Fig. S4). Because we did not observe this with an orc5 siRNA construct (see Fig. 4), it suggests that orc3 may have an additional function separate from the core complex in neurons. This is consistent with our finding that, unlike other subunits, Orc3 was also found in the cytoplasmic fraction of brain homogenates (Fig. 2).
Figure 4.

orc5 is required for dendritic growth and branching. (A and B) Confocal micrographs of 10 DIV neurons transfected on 7 DIV with control (A) or orc5 siRNA (siRNA11) (B) constructs and EGFP (green). Quantitative analysis of dendritic morphology of siRNA11-transfected neurons is shown in Fig. 3 (I–L). Bar, 50 μm.
orc3 knockdown severely impeded both dendritic growth and branching of transfected neurons. To determine what may be the primary cause of the phenotype, we then compared the dendritic branchpoint density of transfected neurons with control neurons. We found that both siRNA1 and siRNA4 caused a significant decrease in branchpoint density defined as the number of branchpoints per 100 μm dendritic length (Fig. 3 L). Although the density for control neurons on 10 DIV is ∼1.9, it is only 0.8 for siRNA1-treated neurons, ∼57% lower, and around 1.3 for siRNA4-treated neurons, ∼35% lower. Thus, a primary effect of orc3 knockdown seems to be a reduction in dendritic branchpoint formation, which may lead to retarded dendritic growth as a consequence. Consistent with this interpretation, we found that siRNA1- or 4-transfected neurons exhibited many fewer dendritic filopodia at later stages (see next section and Fig. 5), an effect that may explain the reduction in dendritic branchpoint formation, as imaging data have shown that dendritic branches seem to develop from stabilized filopodia both in vitro and in vivo (Dailey and Smith, 1996; Niell et al., 2004).
Figure 5.
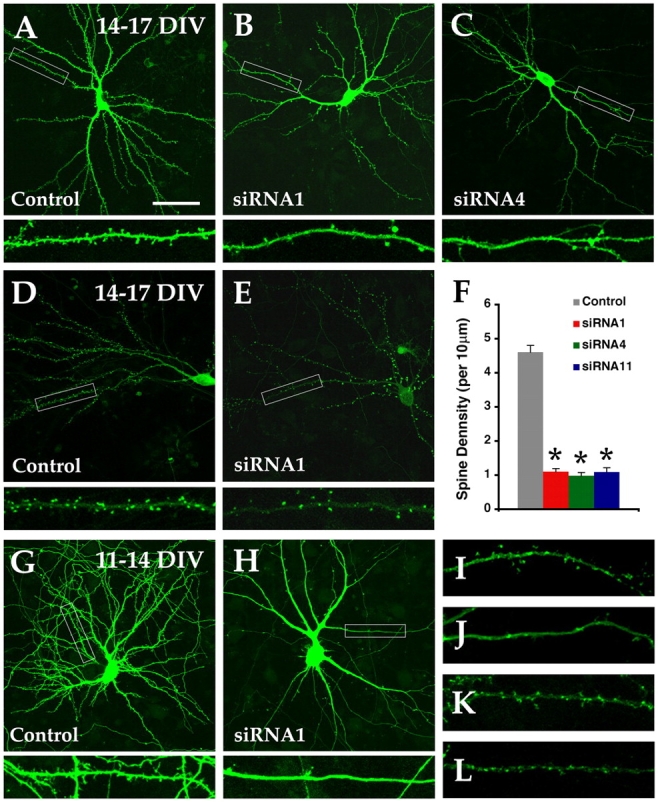
orc3 is required for dendritic spine development and filopodial formation. (A–C) Confocal micrographs of 17 DIV neurons transfected on 14 DIV with control (A) or orc3 siRNA (B and C) constructs and EGFP–actin (green). Boxed areas are shown enlarged at the bottom of each panel. (D and E) Confocal micrographs of 17 DIV neurons transfected with control (D) or siRNA1 (E) and PSD95–EGFP (green). (F) Quantitative analysis of dendritic spine density of 17 DIV neurons transfected with control (n = 28), orc3 (siRNA1, n = 23, and siRNA4, n = 22), or orc5 (siRNA11 n = 14, all ≥3 experiments) siRNA constructs. Error bars reflect SEM. *, P < 1.0 × 10−13 (all compared with control). (G and H) Confocal micrographs of 14 DIV neurons transfected on 11 DIV with control (G) or siRNA1 (H) and EGFP (green). (I and J) Confocal micrographs of EGFP–Mena in control (I) and siRNA1-treated (J) neurons. (K and L) Confocal micrographs of EGFP–mCDC10 in control (K) and siRNA1-treated (L) neurons. Bar, 50 μm.
Our results have so far suggested that the ORC subunits may function primarily as a complex in neurons. If this is the case, one would predict that knockdown of other subunits should give phenotypes similar to orc3. To test this, we screened for siRNA constructs that would interfere with Orc5 subunit function. We found that one of the five designed constructs, siRNA11, led to nearly complete degradation of mouse orc5 mRNA expression in COS cells (Fig. S5 available at http://www.jcb.org/cgi/content/full/jcb.200505075/DC1). When transfected into neurons, siRNA11 resulted in a dramatic simplification of the dendritic trees (Fig. 4 B), whereas the other orc5 constructs had no obvious effects. The total dendritic length of siRNA11-treated neurons was reduced by 49% and the total number of branchpoints dropped by 72% (Fig. 3, I–K). Most importantly, the branchpoint density of siRNA11-treated neurons was reduced to about 1.0, a 47% drop from the normal value of 1.9 (Fig. 3 L), indicating that reduced branchpoint formation is a primary effect of orc5 knockdown. Thus, orc5 loss of function yielded a phenotype closely resembling that of orc3 loss of function, supporting the idea that they indeed function as a complex. Consistent with this, a truncated Orc3 protein that interferes with ORC complex assembly also impairs dendritic growth and branching when overexpressed in hippocampal neurons (Fig. S3). Based on these results, we conclude that the ORC core subunits function primarily as a complex and regulate dendrite development through controlling branchpoint formation in hippocampal neurons.
ORC is required for initiating dendritic spine formation
Dendritic arbor development of hippocampal neurons is followed by formation of dendritic spines, tiny protrusions on dendrites where the majority of excitatory synapses in the brain are located (Yuste and Bonhoeffer, 2004). To determine if the ORC is involved in spine development, we transfected 14 DIV neurons with siRNA1 or 4 and examined them on 17 DIV. We found that orc3 knockdown severely impeded the development of dendritic spines (Fig. 5, A–C). Normally dendrites of 17 DIV neurons are covered by a large number of spines that are spaced more or less evenly along the dendrites; in the orc3 knockdown neurons, however, very few spines were found along the dendrites and they appeared irregularly. For control neurons, the density of spines along dendrites is ∼4.6 per 10 μm. For siRNA1-treated neurons, however, the density dropped to ∼1.1, a 76% decrease; for siRNA4-treated neurons, it dropped to ∼1.0, a 78% decrease (Fig. 5 F). These results suggest that orc3 is required for the development of dendritic spines on the dendrites of hippocampal neurons. Moreover, consistent with the results on dendritic development, siRNA11, which targets orc5, also caused a 76% decrease in spine density from ∼4.6 to 1.1 per 10 μm in transfected neurons (Fig. 5 F and Fig. S5). Thus, the ORC is required for spine development along neuronal dendrites.
Dendritic spines normally undergo a gradual process of maturation from thin, long filopodia to mushroom-shaped protrusions (Hering and Sheng, 2001; Yuste and Bonhoeffer, 2004). In control neurons, the majority of dendritic spines on 17 DIV assume a shape with enlarged heads (Fig. 5 A), consistent with a relatively high degree of maturation. In siRNA1-, 4-, or 11-transfected neurons, we noticed that the few spines that developed also had a similar morphology (Fig. 5, B and C, and Fig. S6 B available at http://www.jcb.org/cgi/content/full/jcb.200505075/DC1), suggesting that they might have matured properly. To test this, we examined the expression of a number of postsynaptic markers in these spines. We found that, as in spines of control neurons on 17 DIV (Fig. 5 D), nearly all the spines of siRNA1- or 4-transfected neurons exhibited strong signals for the major postsynaptic scaffold protein PSD95, as revealed by a PSD95–EGFP reporter (Fig. 5 E). This pattern was further confirmed by immunostaining for endogenous PSD95 protein with an antibody. Similarly, the vast majority of these spines were also found to be positive for the AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptor subunit GluR1 by immunostaining (unpublished data). These results indicate that, despite the dramatic reduction in the number of developing spines, the absence of ORC function does not seem to affect their maturation, suggesting that the ORC might be required in an early step of spine development.
Recent in vivo imaging data indicate that dendritic branching seems to depend on the formation of synapses on newly extended filopodia, which subsequently stabilize a subset of the filopodia and allow them to mature into branches (Niell et al., 2004). It has been suggested that dendritic spine development may also take place in a similar synaptotropic fashion. To determine if the ORC is required for filopodial stabilization, we next examined neurons of earlier stages when most of the dendritic protrusions are still in the form of filopodia. We transfected siRNA constructs into 11 DIV neurons and examined them on 14 DIV. We found that on 14 DIV, orc3 knockdown resulted in a severe loss of dendritic filopodia on transfected neurons. We normally observed large numbers of filopodia on the dendrites of control neurons (Fig. 5 G). However, very few such protrusions were found on siRNA1-treated neurons (Fig. 5 H). These results thus suggest that the ORC may be required in the very early step of spine development, the initiation of dendritic filopodia, although we cannot exclude the possibility that it may also have a role in their stabilization. To determine how the ORC may regulate filopodial formation, we then probed the dendritic cytoskeleton using a number of EGFP reporters. We found that in control neurons, EGFP–Mena, a marker that labels filopodia before their emergence (Svitkina et al., 2003; Mejillano et al., 2004), displayed periodic clusters along dendrites and decorated tips of protruding filopodia (Fig. 5 I). By contrast, very few such clusters were observed along the dendrites of siRNA1-treated neurons (Fig. 5 J). Similar results were observed using an EGFP–VASP reporter (unpublished data). As Orc6 has been found to interact with the septin proteins, we also used a mCDC10 reporter to determine its localization but found relatively normal clusters of EGFP–mCDC10 along dendrites in orc3 knockdown neurons (Fig. 5, K and L). These results thus indicate that the ORC might regulate the organization of the actin cytoskeleton in emerging dendritic filopodia. As its role in controlling dendritic branching also seems to depend on filopodial formation (Dailey and Smith, 1996; Niell et al., 2004), our data thus indicate that the ORC may regulate both dendrite and spine development through controlling the key common step of dendritic filopodial initiation. Depending on the stage of neuronal differentiation, the failure in filopodial initiation may either dramatically reduce dendritic branching or severely impair spine development.
Orc4 ATPase motif mutants and dendritic branching
One of the critical properties of the ORC in regulating DNA replication is its ability to act as a molecular switch that not only precisely couples origin firing with Cdk activation but also triggers complex inactivation once replication is initiated. Among the mechanisms that contribute to ORC inactivation is the intrinsic ATPase activity of the ORC that induces a conformational change upon ATP hydrolysis (Lee and Bell, 2000). During DNA replication, Orc1 seems to be the main subunit responsible for this function and mutations in its ATPase motifs have been found to interfere with DNA replication (Chesnokov et al., 2001; Klemm and Bell, 2001). Among the core subunits expressed in the nervous system, Orc4 and Orc5 also belong to the AAA+ family of ATPases, which typically contain two well-conserved motifs termed Walker A and B (Neuwald et al., 1999). However, only Orc4 has both of the ATPase motifs conserved (Chesnokov et al., 2001), raising the possibility that these motifs may play a role in regulating ORC activity in neurons.
To test this, we generated a point mutation in the Walker B motif of Orc4 where the glutamate residue (Glu 157) known to be essential for ATP binding/hydrolysis in other ATPase family members is replaced by glutamine (O4EQ). We first tried to determine the effects of overexpressing this construct on dendritic spine development. However, possibly due to the relatively low level of O4EQ protein expression and/or its inability to incorporate itself into the ORC complex in well-differentiated neurons, we were unable to observe consistent and convincing effects. We next turned to examine its effects on dendrite development of earlier stage neurons. We found that, interestingly, when transfected into neurons at the time of plating, O4EQ significantly increased the elaboration of dendritic branches of hippocampal neurons. In 7 DIV neurons transfected with wild-type orc4 (O4WT), the dendritic branches were mostly confined near the cell body and were mostly of low branchpoint orders (Fig. 6, A and A′). In neurons overexpressing O4EQ, however, dendrites of many cells frequently extended away from the cell body and had higher order branches (Fig. 6, B and B′). Quantification revealed that the total branchpoint number increased more than 60%, from ∼24 for O4WT cells to over 39 for O4EQ cells (Fig. 6, D and E). Moreover, the branchpoint density showed an average increase of 32% from ∼2.1 for O4WT cells to over 2.7 for O4EQ cells (Fig. 6 F). Thus, O4EQ, behaving as an apparent gain of function mutant, seemed to affect dendritic development mainly through promoting branchpoint formation, suggesting that the Walker B motif of Orc4 may be normally involved in down-regulating ORC activity.
Figure 6.
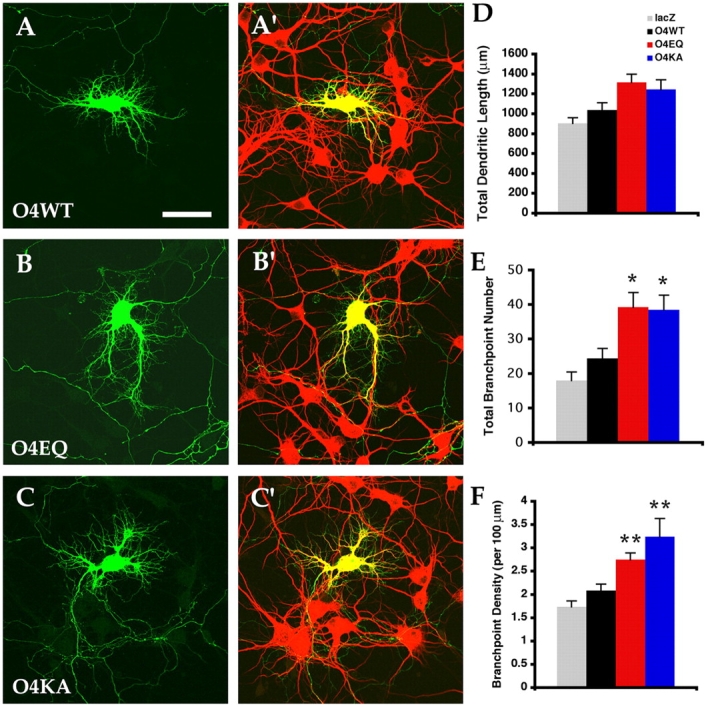
orc4 ATPase motif mutants promote dendritic branching. (A–C and A′–C′) Confocal micrographs of 7 DIV neurons transfected on 0 DIV with wild-type (O4WT, A and A′) or mutant (O4EQ, B and B′; O4KA, C and C′) orc4 constructs and EGFP (green). Neurons were also stained for MAP2 (red). (D–F) Quantitative analysis of dendritic morphology. Differences are not observed between O4WT (n = 49) and O4EQ (n = 43) or O4KA (n = 27) in total dendritic length (D), but found in total branchpoint numbers (E) and branchpoint density (F). Error bars reflect SEM. *, P < 0.008; **, P < 0.002 (all compared with O4WT). Bar, 50 μm.
To further test Orc4 function, we generated another mutated form of Orc4 where the conserved lysine residue (Lys 71) in its Walker A motif is replaced by alanine (O4KA). We found that, similar to O4EQ, O4KA expression also significantly enhanced the branching of dendritic arbors (Fig. 6, C and C′) and increased the total branchpoint number by ∼58% (Fig. 6, D and E). The branchpoint density for O4KA cells reached over 3.2, 55% higher than O4WT cells (Fig. 6 F). These results thus support the interpretation that the ATPase motifs of Orc4 may normally act to down-regulate ORC function in neurons.
Discussion
The formation of dendrites and dendritic spines, the major sites of information processing and storage in the nervous system, is tightly regulated during neural development and plasticity. We have found that the ORC, a protein complex originally identified for its key role in regulating genome replication, has a surprising and novel function in postmitotic neurons by regulating dendrite and spine development. We found that the ORC core subunits are highly expressed in the adult brain, selectively localized to neuronal dendrites, and preferentially associated with neuronal membranes. Our analyses of orc3 and orc5 loss of function phenotypes in hippocampal pyramidal neurons revealed that the ORC core subunits function primarily as a complex to regulate dendrite and spine development through controlling the initiation of dendritic filopodia. Our mutational studies of the Orc4 subunit indicated that its ATPase motifs might be involved in down-regulating ORC activity in neurons. Thus, the ORC core complex, similar to its role as a molecular switch in controlling the initiation of DNA replication in dividing cells, appears to play an active regulatory role in dendritic branch and spine formation of postmitotic neurons.
The ORC, acting as a switch in initiating DNA replication, plays a key role in maintaining genomic integrity that ensures exactly one round of genome replication per cell cycle. As it is one of the key components of the DNA replication machinery, this raises the question how it may regulate the process of neuronal morphogenesis. Recent findings of ORC function in the later events of the cell cycle including cytokinesis and mitosis have provided some clues. Here the Orc6 and Orc2 subunits are associated with the spindle midzone, cleavage furrow, and centrosomes and are required to coordinate these processes of major cytoskeletal reorganization for the proper division of the cytoplasm and the accurate transmission of the genome to daughter cells (Prasanth et al., 2002, 2004; Chesnokov et al., 2003). Although the mechanistic details of ORC function in cytokinesis and mitosis are still unclear, these findings nonetheless demonstrate that the ORC is not limited in its capacity to regulating DNA replication but can also interact with components of the actin–microtubule cytoskeleton and the cell membrane and participate in processes of cellular morphogenesis outside the nucleus.
Dendrite branching and spine formation, like cytokinesis, are also very different from the process of DNA replication. On the other hand, in vivo imaging studies have found that synaptic growth at the Drosophila NMJ, at least morphologically, is surprisingly similar to yeast budding, the unique cytokinetic process of budding yeast (Zito et al., 1999). This suggests that there may be commonalities underlying neuronal morphogenesis and cytokinesis. Indeed, the Orc3 protein has been found to localize to the Drosophila NMJ and orc3 mutants display impaired NMJ development and function (Rohrbough et al., 1999). Orc6 function during cytokinesis has also been found to depend on its interaction with a septin protein (Chesnokov et al., 2003), a component well known for its role in organizing the actin cytoskeleton at yeast budding sites. Our findings that the ORC may regulate dendrite and spine development through controlling the organization of the actin cytoskeleton are therefore consistent with these observations.
The ORC plays a key role not only in the interphase of the cell cycle by regulating genome replication but also in the later events of the cell cycle where it is involved in coordinating the processes of cytoplasmic division and chromosome segregation. As it is one of the cornerstones of the cell cycle machinery, our observation on ORC function in neuronal dendrite development suggests that there might be wider commonality between the cell cycle and neuronal morphogenesis than previously recognized. Indeed, many key components of the cell cycle machinery have been recently implicated in the differentiation of postmitotic neurons. For example, the kinesin-like protein mKLP-1, which plays an essential role in cytokinesis (Glotzer, 2001), has been found to localize to the dendrites and is required for dendritic differentiation (Yu et al., 1997, 2000). Family members of the cytoplasmic polyadenylation element binding (CPEB) protein, which coordinates mitotic progression through controlling local protein translation around the mitotic spindle (Groisman et al., 2002), also promote protein translation in neuronal dendrites (Huang et al., 2003) and regulate synapse-specific long-term facilitation (Si et al., 2003). The Polo family protein kinase SNK, whose family member Plk1 plays prominent roles in the cell cycle (Barr et al., 2004), has also been found to regulate activity-dependent spine synapse remodeling (Pak and Sheng, 2003). The anaphase promoting complex, well known for its role in cell cycle progression (Harper et al., 2002), plays key roles in the development of the Drosophila NMJ (van Roessel et al., 2004) and Caenorhabditis elegans synapses (Juo and Kaplan, 2004). The parallel functions of these key cell cycle components therefore suggest that the function of the ORC in dendrite development is not an exception, but may reflect an emerging underlying commonality between these two biological processes. They suggest that a large part of the cell cycle apparatus may have been coopted and tinkered with during evolution to fulfill new functions in postmitotic neurons.
The function of the ORC during the cell cycle is to faithfully translate each wave of Cdk activation into exactly another copy of the genome and, in most cases, another copy of the cell. Viewed from another angle, it may also be described as to translate the waves of Cdk activation a cell experiences into the copy number of the genome or the number of cells. From this perspective, it is interesting to note that the formation of new dendritic branches and spines in neurons also seems to involve an all or none decision that depends on ORC function. It is tempting to speculate that there might also be waves of Cdk-like activity in neurons that may be responsible for triggering the activity of the ORC and regulating dendritic branch and spine formation. Most interestingly, recent data have shown that dendritic spines continue to appear and disappear in the adult cortex and experience-driven plasticity is accompanied by increased spine synapse turnover (Grutzendler et al., 2002; Trachtenberg et al., 2002). As the ORC is continuously expressed in many areas of the adult nervous system, this raises the possibility that it may be a key part of the molecular machinery that translates patterns of neural activity into patterns of neuronal connectivity and that is believed to underlie long-term memory of sensory experience.
Materials and methods
Molecular biology
The siRNA constructs for orc3 and orc5 knockdown were generated either by cloning annealed oligonucleotides into the pSilencer plasmid (Ambion) or by cloning PCR-produced fragments into the pCR4–TOPO vector (Invitrogen). The orc3 gene sequences based on which the siRNA constructs were designed are: gactgcttcctcattcagt (siRNA1), aagcaacagttgtgacagctg (siRNA2), agcccctaagtgttctgtgc (siRNA4), and aagaagccaaccaagtttgaa (siRNA7). The control siRNA (Ambion) has with no homology to any known mouse or rat genes. The sequence for siRNA1m is gactTcttcctcattAagt. The orc5 gene sequence based on which siRNA11 was designed is aagagacatggaagcaaatct. The wild-type orc4 expression vector was generated by cloning a PCR fragment of orc4 cDNA into the pcDNA3.1/V5–His TOPO vector (Invitrogen). The mutant orc4 expression constructs were generated using a PCR-based mutagenesis strategy. The bacterial expression vectors for producing recombinant mOrc3 and mOrc5 proteins were made by cloning PCR fragments of murine orc3 and orc5 cDNAs into the pET-21a(+) vector (Novagen) and transformed into BL21-CodonPlus (DE3)-RIL–competent cells (Stratagene). RNA was prepared from brain tissues or transfected COS cells (72 h after transfection) using the RNAzol B reagent (Tel-Test) for Northern analysis and probed with 32P-labeled cDNA fragments.
Neuronal culture and transfection
Primary hippocampal neurons were prepared from E18 rat embryos and cultured in high density on poly-l-lysine–coated coverslips in Neurobasal medium supplemented with B27 (Invitrogen). Animal use was in accordance with institutional guidelines. Neurons were transfected either with Effectene (Qiagen), Lipofectamine 2000 (Invitrogen) reagents, or using the calcium phosphate method. A ratio of 2.5:1 or higher between siRNA or cDNA expression constructs and reporter plasmids was maintained to ensure that labeled cells were transfected with the intended constructs. For siRNA constructs, neurons were transfected on 7, 11, or 14 DIV as specified in the text. For orc4 expression constructs, neurons were transfected on the day of plating and examined on 7 DIV.
Immunochemistry
Hippocampal neurons were fixed in a 1:1 mixture of culture medium and 8% paraformaldehyde/8% sucrose/PBS for 10 min at room temperature, except for PSD95 staining, when it was followed by a 5-min fixation in −20°C methanol. Primary antibodies used for immunofluorescent staining included: affinity-purified rabbit anti-Xorc3 (0.6 μg/ml; gift of P. Carpenter, University of Texas, Houston, TX), custom rabbit anti-mOrc3 serum (1:1,000), mouse monoclonal anti-MAP2 clone AP-20 (1:250; Sigma-Aldrich), monoclonal anti–Tau-1 (1:400; Chemicon), monoclonal anti–β-tubulin isotype III (1:100; Sigma-Aldrich), monoclonal anti-PSD95 (1:200; Affinity Bioreagents), rabbit anti-GluR1 (1:200; Upstate Biotechnology), and Alexa488-conjugated rabbit anti-GFP (1:500; Molecular Probes). Secondary antibodies used for immunostaining included: Cy3-conjugated goat anti–mouse IgG (1:500; Jackson ImmunoResearch Laboratories) and Cy3-conjugated goat anti–rabbit IgG (1:500; Jackson ImmunoResearch Laboratories). Rabbit anti-mOrc3 and chicken anti-mOrc5 antibodies were custom raised against bacterially expressed mOrc3 and mOrc5 protein inclusion bodies at Covance Research Products, Inc. and affinity purified with recombinant proteins bound to nitrocellulose filters. Goat anti-ORC4L antibody for Western blot analysis was purchased from Abcam, Inc.
Brain fractionation
Rat brain homogenate was fractionated after standard protocols (De Camilli et al., 1983; Cho et al., 1992; Schilling et al., 1999; Lee et al., 2001; Ehlers, 2003). In brief, P10 rat or adult mouse brain homogenate was centrifuged at 1,000 g to remove nuclei and large debris (P1), and the postnuclear supernatant (S1) was centrifuged at 100,000 g to obtain crude cytosol (C) and membrane (M) fractions. To subfractionate the membrane fraction, supernatant S1 was centrifuged at 10,000 g to obtain a crude synaptosomal pellet fraction (CS). The supernatant (S2) was further centrifuged at 140,000 g to obtain cytosol (C) and microsomal (Mi) fractions. In parallel, the crude synaptosomal fraction (CS) was lysed by hypoosmotic shock and centrifuged at 25,000 g to obtain a synaptic vesicle–enriched fraction (SV) and a lysed synaptosomal fraction (LS). The membrane fractions were solubilized in nuclear lysis buffer (50 mM Tris, pH 7.5, 0.5 M NaCl, 1% NP40, 1% deoxycholate, 0.1% SDS, 2 mM EDTA plus protease inhibitors) and all fractions quantified using the Bio-Rad Dc protein assay. Samples were separated on SDS-PAGE gradient gels (Bio-Rad) and transferred to nitrocellulose membrane for Western analysis.
Microscopy and quantification
Glass coverslips with cultured neurons were mounted with ProLong antifade medium (Molecular Probes) after the appropriate immunochemical procedure. Digital images of neurons were captured using a LSM 5 PASCAL confocal microscope (Carl Zeiss MicroImaging, Inc.) using a Plan-Neofluar 40× (NA = 1.30; Carl Zeiss MicroImaging, Inc.) oil objective at room temperature. Neuronal dendritic trees were traced manually using NIH Image software (version 1.62). Data were collected from at least three duplicate experiments for each construct. For analysis of dendritic morphology, branches shorter than 3.3 μm (15 pixels) were ignored in the tracing process. For spine density analysis, all protrusions on dendrites were counted whether or not they had enlarged heads. Statistical analysis was done using the Student's t test and P values smaller than 0.01 (P < 0.01) were considered significant.
Online supplemental material
Fig. S1 shows that the DNA replication components downstream of ORC are not expressed but Orc3 and Orc5 proteins are detected in the adult mouse brain. Fig. S2 shows that the ORC subunits colocalize with endogenous Orc3 as EGFP fusion proteins and cofractionate in adult mouse brain homogenate. Fig. S3 shows that overexpression of a truncated form of Orc3 impaired dendritic growth and branching. Fig. S4 shows that soma size is unaffected but dendritic microtubule organization is altered in orc3 knockdown neurons. Fig. S5 shows that orc5 is required for dendritic spine formation. Online supplemental materials are available at http://www.jcb.org/cgi/content/full/jcb.200505075/DC1.
Acknowledgments
We are indebted to Phil Carpenter for the gift of Xenopus Orc3 antibody, Song Hu for the EGFP–Mena/VASP reporters, Mike Stryker for critical reading of an earlier version of the manuscript, and members of the Reichardt Lab for helpful discussion.
Z. Huang was supported by a Helen Hay Whitney fellowship. L.F. Reichardt is an investigator of the Howard Hughes Medical Institute.
Abbreviations used in this paper: DIV, days in vitro; NMJ, neuromuscular junction; ORC, origin recognition complex.
This article contains online supplemental material.
References
- Baker, T.A., and S.P. Bell. 1998. Polymerases and the replisome: machines within machines. Cell. 92:295–305. [DOI] [PubMed] [Google Scholar]
- Barr, F.A., H.H. Sillje, and E.A. Nigg. 2004. Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol. 5:429–440. [DOI] [PubMed] [Google Scholar]
- Bell, S.P. 2002. The origin recognition complex: from simple origins to complex functions. Genes Dev. 16:659–672. [DOI] [PubMed] [Google Scholar]
- Bell, S.P., and A. Dutta. 2002. DNA replication in eukaryotic cells. Annu. Rev. Biochem. 71:333–374. [DOI] [PubMed] [Google Scholar]
- Bell, S.P., and B. Stillman. 1992. ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature. 357:128–134. [DOI] [PubMed] [Google Scholar]
- Brummelkamp, T.R., R. Bernards, and R. Agami. 2002. A system for stable expression of short interfering RNAs in mammalian cells. Science. 296:550–553. [DOI] [PubMed] [Google Scholar]
- Chesnokov, I., D. Remus, and M. Botchan. 2001. Functional analysis of mutant and wild-type Drosophila origin recognition complex. Proc. Natl. Acad. Sci. USA. 98:11997–12002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesnokov, I.N., O.N. Chesnokova, and M. Botchan. 2003. A cytokinetic function of Drosophila ORC6 protein resides in a domain distinct from its replication activity. Proc. Natl. Acad. Sci. USA. 100:9150–9155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, K.O., C.A. Hunt, and M.B. Kennedy. 1992. The rat brain postsynaptic density fraction contains a homolog of the Drosophila discs-large tumor suppressor protein. Neuron. 9:929–942. [DOI] [PubMed] [Google Scholar]
- Dailey, M.E., and S.J. Smith. 1996. The dynamics of dendritic structure in developing hippocampal slices. J. Neurosci. 16:2983–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Camilli, P., S.M. Harris Jr., W.B. Huttner, and P. Greengard. 1983. Synapsin I (Protein I), a nerve terminal-specific phosphoprotein. II. Its specific association with synaptic vesicles demonstrated by immunocytochemistry in agarose-embedded synaptosomes. J. Cell Biol. 96:1355–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar, S.K., L. Delmolino, and A. Dutta. 2001. Architecture of the human origin recognition complex. J. Biol. Chem. 276:29067–29071. [DOI] [PubMed] [Google Scholar]
- Ehlers, M.D. 2003. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat. Neurosci. 6:231–242. [DOI] [PubMed] [Google Scholar]
- Glotzer, M. 2001. Animal cell cytokinesis. Annu. Rev. Cell Dev. Biol. 17:351–386. [DOI] [PubMed] [Google Scholar]
- Groisman, I., M.Y. Jung, M. Sarkissian, Q. Cao, and J.D. Richter. 2002. Translational control of the embryonic cell cycle. Cell. 109:473–483. [DOI] [PubMed] [Google Scholar]
- Grutzendler, J., N. Kasthuri, and W.B. Gan. 2002. Long-term dendritic spine stability in the adult cortex. Nature. 420:812–816. [DOI] [PubMed] [Google Scholar]
- Harper, J.W., J.L. Burton, and M.J. Solomon. 2002. The anaphase-promoting complex: it's not just for mitosis any more. Genes Dev. 16:2179–2206. [DOI] [PubMed] [Google Scholar]
- Hausser, M., N. Spruston, and G.J. Stuart. 2000. Diversity and dynamics of dendritic signaling. Science. 290:739–744. [DOI] [PubMed] [Google Scholar]
- Hering, H., and M. Sheng. 2001. Dendritic spines: structure, dynamics and regulation. Nat. Rev. Neurosci. 2:880–888. [DOI] [PubMed] [Google Scholar]
- Huang, Y.S., J.H. Carson, E. Barbarese, and J.D. Richter. 2003. Facilitation of dendritic mRNA transport by CPEB. Genes Dev. 17:638–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan, Y.N., and L.Y. Jan. 2003. The control of dendrite development. Neuron. 40:229–242. [DOI] [PubMed] [Google Scholar]
- Juo, P., and J.M. Kaplan. 2004. The anaphase-promoting complex regulates the abundance of GLR-1 glutamate receptors in the ventral nerve cord of C. elegans. Curr. Biol. 14:2057–2062. [DOI] [PubMed] [Google Scholar]
- Kelly, T.J., and G.W. Brown. 2000. Regulation of chromosome replication. Annu. Rev. Biochem. 69:829–880. [DOI] [PubMed] [Google Scholar]
- Klemm, R.D., and S.P. Bell. 2001. ATP bound to the origin recognition complex is important for preRC formation. Proc. Natl. Acad. Sci. USA. 98:8361–8367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, D.G., and S.P. Bell. 2000. ATPase switches controlling DNA replication initiation. Curr. Opin. Cell Biol. 12:280–285. [DOI] [PubMed] [Google Scholar]
- Lee, S.H., J.G. Valtschanoff, V.N. Kharazia, R. Weinberg, and M. Sheng. 2001. Biochemical and morphological characterization of an intracellular membrane compartment containing AMPA receptors. Neuropharmacology. 41:680–692. [DOI] [PubMed] [Google Scholar]
- Li, C.J., and M.L. DePamphilis. 2002. Mammalian Orc1 protein is selectively released from chromatin and ubiquitinated during the S-to-M transition in the cell division cycle. Mol. Cell. Biol. 22:105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejillano, M.R., S. Kojima, D.A. Applewhite, F.B. Gertler, T.M. Svitkina, and G.G. Borisy. 2004. Lamellipodial versus filopodial mode of the actin nanomachinery: pivotal role of the filament barbed end. Cell. 118:363–373. [DOI] [PubMed] [Google Scholar]
- Mendez, J., X.H. Zou-Yang, S.Y. Kim, M. Hidaka, W.P. Tansey, and B. Stillman. 2002. Human origin recognition complex large subunit is degraded by ubiquitin-mediated proteolysis after initiation of DNA replication. Mol. Cell. 9:481–491. [DOI] [PubMed] [Google Scholar]
- Neuwald, A.F., L. Aravind, J.L. Spouge, and E.V. Koonin. 1999. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 9:27–43. [PubMed] [Google Scholar]
- Nguyen, V.Q., C. Co, and J.J. Li. 2001. Cyclin-dependent kinases prevent DNA re-replication through multiple mechanisms. Nature. 411:1068–1073. [DOI] [PubMed] [Google Scholar]
- Niell, C.M., M.P. Meyer, and S.J. Smith. 2004. In vivo imaging of synapse formation on a growing dendritic arbor. Nat. Neurosci. 7:254–260. [DOI] [PubMed] [Google Scholar]
- Pak, D.T., and M. Sheng. 2003. Targeted protein degradation and synapse remodeling by an inducible protein kinase. Science. 302:1368–1373. [DOI] [PubMed] [Google Scholar]
- Pinto, S., D.G. Quintana, P. Smith, R.M. Mihalek, Z.H. Hou, S. Boynton, C.J. Jones, M. Hendricks, K. Velinzon, J.A. Wohlschlegel, et al. 1999. latheo encodes a subunit of the origin recognition complex and disrupts neuronal proliferation and adult olfactory memory when mutant. Neuron. 23:45–54. [DOI] [PubMed] [Google Scholar]
- Prasanth, S.G., K.V. Prasanth, K. Siddiqui, D.L. Spector, and B. Stillman. 2004. Human Orc2 localizes to centrosomes, centromeres and heterochromatin during chromosome inheritance. EMBO J. 23:2651–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanth, S.G., K.V. Prasanth, and B. Stillman. 2002. Orc6 involved in DNA replication, chromosome segregation, and cytokinesis. Science. 297:1026–1031. [DOI] [PubMed] [Google Scholar]
- Rohrbough, J., S. Pinto, R.M. Mihalek, T. Tully, and K. Broadie. 1999. latheo, a Drosophila gene involved in learning, regulates functional synaptic plasticity. Neuron. 23:55–70. [DOI] [PubMed] [Google Scholar]
- Schilling, G., J.D. Wood, K. Duan, H.H. Slunt, V. Gonzales, M. Yamada, J.K. Cooper, R.L. Margolis, N.A. Jenkins, N.G. Copeland, et al. 1999. Nuclear accumulation of truncated atrophin-1 fragments in a transgenic mouse model of DRPLA. Neuron. 24:275–286. [DOI] [PubMed] [Google Scholar]
- Si, K., M. Giustetto, A. Etkin, R. Hsu, A.M. Janisiewicz, M.C. Miniaci, J.H. Kim, H. Zhu, and E.R. Kandel. 2003. A neuronal isoform of CPEB regulates local protein synthesis and stabilizes synapse-specific long-term facilitation in aplysia. Cell. 115:893–904. [DOI] [PubMed] [Google Scholar]
- Svitkina, T.M., E.A. Bulanova, O.Y. Chaga, D.M. Vignjevic, S. Kojima, J.M. Vasiliev, and G.G. Borisy. 2003. Mechanism of filopodia initiation by reorganization of a dendritic network. J. Cell Biol. 160:409–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachtenberg, J.T., B.E. Chen, G.W. Knott, G. Feng, J.R. Sanes, E. Welker, and K. Svoboda. 2002. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 420:788–794. [DOI] [PubMed] [Google Scholar]
- Van Aelst, L., and H.T. Cline. 2004. Rho GTPases and activity-dependent dendrite development. Curr. Opin. Neurobiol. 14:297–304. [DOI] [PubMed] [Google Scholar]
- van Roessel, P., D.A. Elliott, I.M. Robinson, A. Prokop, and A.H. Brand. 2004. Independent regulation of synaptic size and activity by the anaphase-promoting complex. Cell. 119:707–718. [DOI] [PubMed] [Google Scholar]
- Vashee, S., P. Simancek, M.D. Challberg, and T.J. Kelly. 2001. Assembly of the human origin recognition complex. J. Biol. Chem. 276:26666–26673. [DOI] [PubMed] [Google Scholar]
- Whitford, K.L., P. Dijkhuizen, F. Polleux, and A. Ghosh. 2002. Molecular control of cortical dendrite development. Annu. Rev. Neurosci. 25:127–149. [DOI] [PubMed] [Google Scholar]
- Wong, R.O., and A. Ghosh. 2002. Activity-dependent regulation of dendritic growth and patterning. Nat. Rev. Neurosci. 3:803–812. [DOI] [PubMed] [Google Scholar]
- Yu, W., C. Cook, C. Sauter, R. Kuriyama, P.L. Kaplan, and P.W. Baas. 2000. Depletion of a microtubule-associated motor protein induces the loss of dendritic identity. J. Neurosci. 20:5782–5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, W., D.J. Sharp, R. Kuriyama, P. Mallik, and P.W. Baas. 1997. Inhibition of a mitotic motor compromises the formation of dendrite-like processes from neuroblastoma cells. J. Cell Biol. 136:659–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste, R., and T. Bonhoeffer. 2004. Genesis of dendritic spines: insights from ultrastructural and imaging studies. Nat. Rev. Neurosci. 5:24–34. [DOI] [PubMed] [Google Scholar]
- Zito, K., D. Parnas, R.D. Fetter, E.Y. Isacoff, and C.S. Goodman. 1999. Watching a synapse grow: noninvasive confocal imaging of synaptic growth in Drosophila. Neuron. 22:719–729. [DOI] [PubMed] [Google Scholar]