

Abstract
In anaphase, the spindle dictates the site of contractile ring assembly. Assembly and ingression of the contractile ring involves activation of myosin-II and actin polymerization, which are triggered by the GTPase RhoA. In many cells, the central spindle affects division plane positioning via unknown molecular mechanisms. Here, we dissect furrow formation in human cells and show that the RhoGEF ECT2 is required for cortical localization of RhoA and contractile ring assembly. ECT2 concentrates on the central spindle by binding to centralspindlin. Depletion of the centralspindlin component MKLP1 prevents central spindle localization of ECT2; however, RhoA, F-actin, and myosin still accumulate on the equatorial cell cortex. Depletion of the other centralspindlin component, CYK-4/MgcRacGAP, prevents cortical accumulation of RhoA, F-actin, and myosin. CYK-4 and ECT2 interact, and this interaction is cell cycle regulated via ECT2 phosphorylation. Thus, central spindle localization of ECT2 assists division plane positioning and the CYK-4 subunit of centralspindlin acts upstream of RhoA to promote furrow assembly.
Introduction
Shortly after the metaphase–anaphase transition, cells must ensure that the spindle poles are on opposite sides of the division plane so that the segregated sets of chromosomes are partitioned into separate cells. In some cells, such as budding and fission yeast, division plane positioning precedes anaphase onset, in which case the cell must coordinate the position of the spindle with the preexisting division plane. In contrast, in animal cells, the division plane is determined at the metaphase to anaphase transition and its position is dictated by the spindle. In animal cells, the spindle can regulate division plane establishment by at least two distinct pathways (for review see Glotzer, 2004). One pathway involves the central spindle, a set of antiparallel microtubule bundles that assembles during anaphase. A second pathway involves astral microtubules. The relative importance of these two pathways differs among organisms. Despite the fundamental importance of the process, no molecular pathway has yet been defined that explains how the anaphase spindle positions the division plane.
In animal cells, the GTPase RhoA has a central role in furrow formation. When RhoA is depleted genetically or inactivated by the bacterial enzyme C3, furrow formation is abrogated (Kishi et al., 1993; Jantsch-Plunger et al., 2000). The GTPase RhoA is involved in activating actin polymerization by regulating formin proteins (Alberts, 2001), which have a critical role in actin filament nucleation and elongation (Kovar et al., 2003; Romero et al., 2004). In addition, RhoA can activate myosin by promoting phosphorylation of myosin light chain (Kimura et al., 1996; Kosako et al., 2000). Cleavage furrow formation might result from the local activation of RhoA, which could promote key aspects of contractile ring assembly.
RhoA is activated by guanine nucleotide exchange factors (GEFs) that stabilize the nucleotide-free, transition state of small GTPases. There are large numbers of proteins containing RhoGEF domains; these proteins have diverse biological functions. One such RhoGEF, ECT2, originally identified as a protooncogene (Miki et al., 1993), is a critical RhoGEF for cytokinesis in animal cells. Overexpression of the NH2 terminus of ECT2, lacking the GEF domain, inhibits completion of cytokinesis in mammalian cells (Tatsumoto et al., 1999). Loss-of-function studies confirm the requirement for ECT2. The Drosophila melanogaster orthologue of ECT2, Pebble, is required for cytokinesis (Lehner, 1992; Prokopenko et al., 1999), the Caenorhabditis elegans orthologue, LET-21, is likewise required for formation of a cleavage furrow (Dechant and Glotzer, 2003), and depletion of ECT2 prevents cytokinesis in human cells (Kim et al., 2005).
ECT2 is an excellent candidate molecule to integrate the spatial and temporal information that directs establishment of the contractile ring. In vertebrate cells, ECT2 localizes to the central spindle (Tatsumoto et al., 1999), which is implicated in the spatial control of cytokinesis. Central spindle assembly and completion of cytokinesis requires the centralspindlin complex that consists of a Rho GTPase activating protein (GAP), CYK-4/MgcRacGAP, and a kinesin-6, MKLP1 (Mishima et al., 2002). In Drosophila, the orthologue of ECT2, Pebble, binds to the orthologue of CYK-4, RacGAP50C (Somers and Saint, 2003). This interaction was suggested to recruit Pebble/ECT2 to the vicinity of the central spindle where it could locally control formation of the contractile ring (Saint and Somers, 2003). However, whether an ECT2–CYK-4 complex assembles in other organisms is not known and, more importantly, the functional significance of this complex has not been addressed.
In this study, we have developed a fluorescently tagged RhoA protein whose cortical localization correlates with RhoA activation. Using this probe and other tools, we have molecularly dissected the requirements for RhoA localization to the cell cortex during cytokinesis. We have found that the central spindle restricts RhoA localization to a well defined region of the equatorial cell cortex. However, the central spindle and the localization of ECT2 to the central spindle are not strictly essential for cortical RhoA localization. In contrast, the centralspindlin component CYK-4 and the RhoGEF ECT2 are essential for RhoA localization and contractile ring assembly. Furthermore, ECT2 associates with CYK-4 in a cell cycle–regulated manner. The interaction is weak during metaphase due to mitotic phosphorylation of ECT2 and increases during anaphase when ECT2 is dephosphorylated. We propose that the centralspindlin component CYK-4 can act as an essential activator of ECT2 that couples cytokinesis to mitotic exit.
Results
RhoA localizes in early anaphase
Genetic studies indicate that RhoA regulates cytokinesis by controlling contractile ring formation. Biochemical pathways are known through which RhoA promotes actin polymerization and myosin activation. Because contractile ring formation occurs at a discrete site, we investigated whether RhoA is spatially regulated during cytokinesis. Under certain fixation conditions, RhoA is highly concentrated at the site of contractile ring formation (Yonemura et al., 2004). We confirmed that RhoA is discretely localized when cells are fixed with TCA (Fig. 1 A), though not with formaldehyde or methanol (not depicted). The cortical domain of RhoA localization overlaps extensively with the region where nonmuscle myosin IIB accumulates (Fig. 1 A). The cortical pool of RhoA appears to be selectively retained during TCA fixation (unpublished data), and this correlates well with the regions where its effectors are active. This domain is positioned between the separating chromosomes and is closely apposed to the central spindle during early anaphase, as revealed by CYK-4 localization. Aurora B kinase, which exhibits both central spindle and cortical localization during early anaphase, is directly adjacent to the cortical domain where myosin II concentrates (Fig. 1 A).
Figure 1.
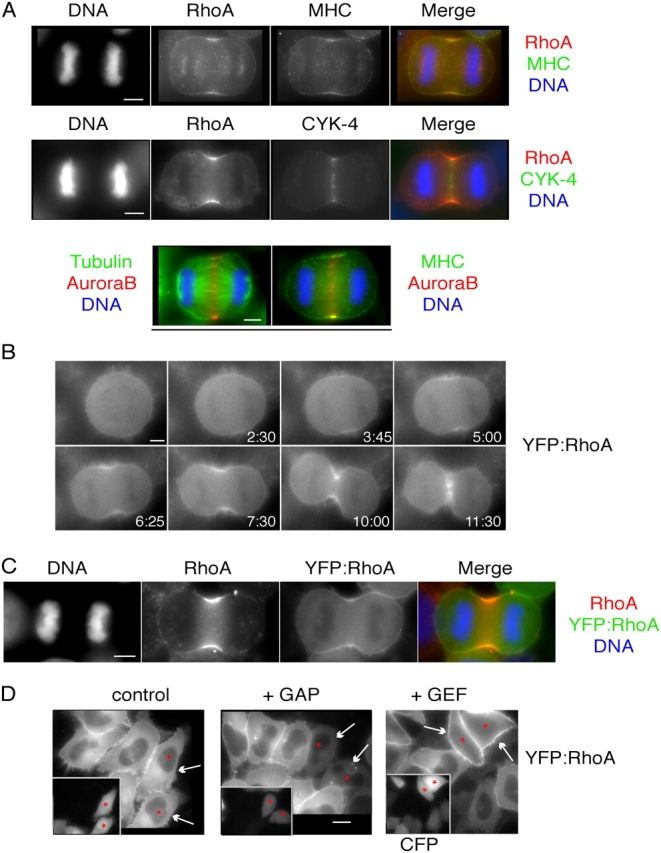
RhoA localizes to the equatorial cell cortex in early anaphase. (A) HeLa cells were fixed with TCA and stained with antibodies directed against RhoA (red) and either myosin IIB heavy chain or CYK-4 (green). HeLa cells fixed with methanol were stained with antibodies directed against aurora B (red) and tubulin or myosin II (green). (B) YFP:RhoA concentrates at the equatorial cortex before furrow formation. A HeLa cell stably expressing YFP:RhoA is shown at successive time points during cytokinesis (Video 2). This cell was treated with lamin B siRNA. (C) YFP:RhoA colocalizes with endogenous RhoA. Cells stably expressing YFP:RhoA were fixed with TCA and stained with antibodies directed against RhoA (red) and YFP (green). The anti-RhoA antibody does not cross react with YFP:RhoA. (D) Cortical localization of YFP:RhoA increases upon RhoA activation and decreases upon RhoA inhibition (arrows). The GAP domain of RhoGAP1 or the GEF domain of ECT2 were coexpressed with CFP in YFP:RhoA cells. CFP-positive cells (inset) are indicated (asterisks). Images obtained from the same cell are underlined. Bars: (A–C) 5 μm; (D) 10 μm.
Because TCA fixation could induce the redistribution of RhoA, we used another independent method to study RhoA localization. We examined the localization of a YFP-tagged variant of human RhoA. In cleaving HeLa cells, YFP:HsRhoA did not localize distinctly during cytokinesis (Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200501097/DC1). However, YFP-tagged C. elegans RhoA (YFP:RhoA hereafter) localized discretely in dividing HeLa cells and this pattern was highly similar to the localization of endogenous RhoA in TCA-fixed cells (Video 2, available at http://www.jcb.org/cgi/content/full/jcb.200501097/DC1; and Fig. 1, B and C). Human and C. elegans RhoA are 96% similar (88% identical) and all but two of the substitutions are in the COOH-terminal third of the protein. YFP:RhoA is functional in HeLa cells as indicated by its ability to recruit myosin (Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200501097/DC1).
Cortical localization of YFP:RhoA correlates with RhoA activation
To test whether cortical YFP:RhoA reflects active RhoA, we overexpressed either the GAP domain from mouse RhoGAP1 or the COOH-terminal GEF domain from ECT2 and observed the effects on YFP:RhoA localization in interphase HeLa cells (Fig. 1 D and Fig. S1 B). The signal from YFP:RhoA was diminished and less cortical in cells that overexpress the GAP domain; these cells also lack stress fibers (Fig. 1 D and Fig. S1 B). Conversely, overexpression of the GEF domain induced enrichment of YFP:RhoA at the cortex and an increase in stress fibers. (Fig. 1 D and Fig. S1 B). Thus, the amount of cortically localized YFP:RhoA correlates well with the activation level of RhoA.
RhoA is required for furrow formation and myosin recruitment
During cytokinesis, cortically localized YFP:RhoA colocalizes with its downstream effectors, which is consistent with prior genetic studies suggesting that RhoA is a critical regulator of cytokinesis. Therefore, we addressed whether RhoA is functionally required for the localization of actin and myosin II. HeLa cells were treated with siRNAs directed against RhoA. RhoA-depleted cells were cytokinesis defective (36% binucleate, n = 260 fixed cells). In anaphase cells lacking detectable RhoA, actin (n = 13/14 cells) and myosin II (n = 22/24 cells) failed to localize at the presumptive furrow (Fig. 2 A).
Figure 2.

RhoA regulates myosin II accumulation and localizes in the absence of myosin II or F-actin. (A) RhoA is required for accumulation of myosin II and actin. HeLa cells depleted of RhoA were fixed and stained with the indicated antibodies and rhodamine phalloidin. (B) Neither myosin activity nor myosin is required for RhoA localization. HeLa cells were treated with 100 μM Blebbistatin (−, active) or Blebbistatin (+, inactive) for 45 min or treated with siRNAs to myosin heavy chain before fixation and immunofluorescence. (C) F-actin is not required for RhoA localization. HeLa cells treated with 5 μM LatA for 45 min were fixed and stained with the indicated antibodies and rhodamine phalloidin. Images obtained from the same cell are underlined. Bars, 5 μm.
We next evaluated whether RhoA localization requires myosin or actin. Blebbistatin, a small molecule inhibitor of myosin activity, causes a penetrant cytokinesis defect, as shown previously (Straight et al., 2003). However, RhoA localized normally in the presence of Blebbistatin (n = 43) as did myosin (n = 14; Fig. 2 B). Similarly, depletion of the total pool of myosin II using siRNAs also caused early cytokinesis defects and prevented cell flattening during ana/telophase. RhoA accumulation at the presumptive furrow was not affected by depletion of myosin II (n = 31; Fig. 2 B). Thus RhoA is required for myosin II localization, but not vice versa. Next, we used Latrunculin A (LatA), which causes actin filaments to disassemble by sequestering actin monomers, to assess whether F-actin is required for RhoA localization. Cells treated with LatA displayed early cytokinesis defects and loss of myosin localization (n = 20; Fig. 2 C). However, RhoA accumulated at the site of the presumptive furrow as in nontreated cells (n = 27; Fig. 2 C). Thus, RhoA accumulates at the equatorial cortex independently of actin and myosin. These data suggest that local accumulation of RhoA may promote the assembly of actin filaments, which are required in turn for myosin recruitment that is independent of its motor activity.
ECT2 localizes to the central spindle and is required for RhoA localization and furrow formation
Because RhoA is an important early regulator of cytokinesis and its localization, as measured by YFP:RhoA and by immunofluorescence, correlates with active RhoA, we used RhoA localization as an assay to investigate the mechanism that initiates cytokinesis. In HeLa cells, during interphase, ECT2 and the centralspindlin component CYK-4 are nuclear (Fig. 3 A). At anaphase onset, ECT2 colocalizes with CYK-4 in the central spindle and remains associated with the central spindle through furrow ingression until the midbody stage (Fig. 3 A; Tatsumoto et al., 1999).
Figure 3.

ECT2 localizes to the central spindle and regulates RhoA localization and cytokinesis. (A) ECT2 localizes to the central spindle and colocalizes with the centralspindlin complex. (B) ECT2 is required for furrow formation. ECT2 depleted or control cells undergoing cytokinesis were imaged by DIC microscopy (Videos 3 and 4). (C) Neither RhoA, myosin, nor actin localize to the cell cortex in ECT2-depleted cells, but the central spindle assembles normally. ECT2-depleted or control cells were fixed and stained with the indicated antibodies or rhodamine phalloidin. Images obtained from the same cell are underlined. Bars: (A, top) 10 μm; (A–C) 5 μm.
To confirm that human ECT2 is required for cytokinesis, like its orthologues in Drosophila and C. elegans, we used RNAi to deplete endogenous ECT2 from HeLa cells. Penetrant cytokinesis defects were observed as expected for an upstream regulator of RhoA (92% binucleate, n = 200 cells by fixed analysis). Time-lapse imaging of ECT2-depleted cells revealed that the most severely affected cells remained round during anaphase and did not exhibit furrow ingression (Fig. 3 B and Videos 3 and 4, available at http://www.jcb.org/cgi/content/full/jcb.200501097/DC1).
We next characterized the localization of RhoA and centralspindlin in cells depleted of ECT2. Time-lapse analysis of YFP:RhoA revealed that this reporter failed to localize in ECT2-depleted cells (Video 5, available at http://www.jcb.org/cgi/content/full/jcb.200501097/DC1; n = 11 noningressing cells). Immunofluorescence analysis of anaphase cells revealed that all cells fully depleted of ECT2 failed to ingress (n = 24). Cortical localization of RhoA was abolished in all noningressing cells (n = 24; incompatible fixation precluded costaining of ECT2 and RhoA; Fig. 3 C). Myosin recruitment (n = 14) and actin accumulation (n = 17) were also abolished in cells lacking ECT2 (Fig. 3 C). However, centralspindlin localization was not affected in these cells (Fig. 3 C). Similarly, centralspindlin and ECT2 localized in cells depleted of RhoA (Fig. 2 A). These results demonstrate that ECT2 is required for RhoA activation and contractile ring formation but central spindle assembly is independent of ECT2.
MKLP1 is required for central spindle assembly and ECT2 localization but not RhoA recruitment
Localization of ECT2 to the central spindle could regulate cleavage furrow formation by creating a localized pool of active RhoA, as previously proposed (Somers and Saint, 2003). To test this possibility, we depleted MKLP1 from HeLa cells using RNAi. MKLP1 forms a heterotetramer with CYK-4 and is required for central spindle formation (Mishima et al., 2002). Other investigators have demonstrated that cells depleted of MKLP1 do not form a well organized central spindle, yet are able to form cleavage furrows that ingress extensively and regress (Matuliene and Kuriyama, 2002). As expected, loss of MKLP1 caused abnormal central spindle formation and both CYK-4 (n = 25) and ECT2 (n = 43/47) failed to localize (Fig. 4 A). Interestingly, two classes of cytokinesis phenotypes were observed in time-lapse recordings of MKLP1-depleted cells. About 1/3 of cells (9/28) displayed a late cytokinesis defect, as observed previously. However, in the remaining cells, weak or no furrow formation was observed. Importantly, RhoA was cortically localized in all cells (Fig. 4 B). However, the domain of cortically localized RhoA was expanded as compared with control cells (Fig. 4 B). To quantify this difference, we measured the angle subtended by the cortical region where RhoA concentrated in fixed cells. MKLP1 depletion affected cell shape, therefore the cell dimensions were measured and these parameters were plotted for each cell (Fig. 5 A). Control cells were elliptical and the zone of RhoA was quite restricted (Fig. 4 B and 5 A). MKLP1-depleted cells that formed furrows were elliptical like the control cells, but the RhoA zone was somewhat expanded (Fig. 4 B and 5 A). In contrast, cells without furrows were less elliptical and had the greatest expansion of the RhoA zone (Fig. 4 B and 5 A).
Figure 4.

MKLP1 is required for ECT2 localization. (A) Cells depleted of MKLP1 were fixed and stained with the indicated antibodies. (B) MKLP1 restricts the localization of RhoA, but it does not block cortical localization of RhoA. MKLP-depleted cells were fixed and stained with the indicated antibodies or rhodamine phalloidin. Cells that exhibit the two classes of cytokinesis phenotypes are shown. Bars, 5 μm.
Figure 5.

Expansion of cortically localized RhoA upon MKLP1 depletion and aurora inhibition. (A) Quantitation of RhoA localization in MKLP1-depleted cells and control cells. Cell morphology and the extent of RhoA localization in fixed and live cells were measured as indicated. Fixed cells were scored for the presence of a furrow and live cells were scored for furrow formation during the course of the recording. Fixed cells with partly condensed, and segregated DNA were measured. Live cells were measured when RhoA localization was most restricted. (B) YFP:RhoA-expressing cells were depleted of MKLP1 by RNAi. YFP:RhoA was imaged and shown at successive time points during cytokinesis. Two phenotypes were observed. Either RhoA remained somewhat restricted and these cells formed furrows that regress; otherwise RhoA spread out along the cell cortex and furrows did not form (Videos 6 and 7). (C) HeLa cells stably expressing YFP:RhoA were treated with 100 nM Hesperadin and YFP:RhoA was imaged during cytokinesis (Videos 8 and 9). Bars, 5 μm.
To better correlate the extent of the zone of RhoA with the phenotype of the cell, we performed time-lapse analysis of MKLP1-depleted cells expressing YFP:RhoA and measured the zone of RhoA during anaphase (Fig. 5 B and Videos 6 and 7, available at http://www.jcb.org/cgi/content/full/jcb.200501097/DC1). In control cells, YFP:RhoA localization was quantitatively similar to that of endogenous RhoA in fixed cells. Depletion of MKLP1 caused an increase in the size of the YFP:RhoA zone. The cells that formed furrows were more elliptical and contained a more restricted zone of RhoA than cells that did not furrow (Fig. 5, A and B). Immunofluorescence analysis of fixed cells showed that myosin II (n = 11) and actin (n = 13) localization were also expanded, implying that the extended pool of localized RhoA is active (Fig. 4 B). These data suggest that the central spindle is not required for cortical localization of active RhoA, but, rather, it functions to restrict the zone of active RhoA to a narrow region. Thus, failure to localize ECT2 to the midzone results in a variable broadening of the RhoA zone that can interfere with cleavage furrow formation.
Aurora kinase is required for central spindle assembly and furrow formation but not RhoA recruitment
To perturb the central spindle by an independent means to evaluate its involvement in restricting accumulation of active RhoA, we depleted aurora B kinase by siRNAs or inhibited its activity by treatment with the aurora inhibitor Hesperadin (Hauf et al., 2003). Cells lacking aurora B activity displayed variable cytokinesis defects, similar to cells lacking MKLP1. In both live and fixed cells, aurora B inhibition induced expansion of the region of cortical RhoA localization (Fig. 5 C; Videos 8 and 9, available at http://www.jcb.org/cgi/content/full/jcb.200501097/DC1 [n = 12 live cells]; and not depicted). Furthermore, the same expansion of the zone of RhoA was observed in time-lapse analysis of cells simultaneously depleted of MKLP1 and treated with the aurora inhibitor (n = 9 cells; unpublished data). These data provide independent evidence that the central spindle plays a role in refining the localization of active RhoA to the site of the cleavage furrow.
CYK-4 is required for central spindle assembly, ECT2 localization, furrow formation, and RhoA recruitment
We next examined the consequences of CYK-4 depletion on cytokinesis. In C. elegans, CYK-4 is required for central spindle formation and CYK-4–depleted embryos form ingressing cleavage furrows that regress late in cytokinesis (Jantsch-Plunger et al., 2000). HeLa cells depleted of detectable CYK-4 by RNAi failed to form a central spindle, and both ECT2 (n = 15) and MKLP1 (n = 15) failed to localize. Strikingly, these cells showed an early cytokinesis phenotype: the cells were round and furrow formation did not occur. Importantly, RhoA failed to accumulate at the cortex in both live and fixed cells lacking CYK-4 (n = 19 noningressing live cells; n = 45 fixed cells; Fig. 6 and Video 10, available at http://www.jcb.org/cgi/content/full/jcb.200501097/DC1). Actin (n = 14) and myosin (n = 11) also failed to localize in CYK-4–depleted cells (Fig. 6). The cytokinesis phenotype caused by CYK-4 depletion closely parallels that caused by loss of ECT2, suggesting that CYK-4 may regulate RhoA activation and thereby contractile ring assembly. This function of CYK-4 does not appear to be shared by its partner protein in the centralspindlin complex, MKLP1.
Figure 6.

CYK-4 is required for central spindle formation and is essential for active RhoA localization. Cells depleted of CYK-4 were fixed and stained with the indicated antibodies or rhodamine phalloidin. YFP:RhoA localization was also abolished in CYK-4–depleted cells (Video 10). Images obtained from the same cell are underlined. Bars, 5 μm.
CYK-4 binds to ECT2 in a phosphoregulated manner
As mentioned previously, the Drosophila orthologues of ECT2 and CYK-4 interact in a yeast two-hybrid assay as well as in embryo extracts (Somers and Saint, 2003). These observations prompted us to determine if endogenous ECT2 and CYK-4 interact in human cells. Indeed, CYK-4 coimmunoprecipitated with ECT2 from mitotic HeLa cell lysates (Fig. 7 A). Significantly more CYK-4 coprecipitated with MKLP1, suggesting that only a fraction of the centralspindlin complex associates with ECT2.
Figure 7.

CYK-4 and ECT2 directly interact and associate in vivo in a phosphorylation-dependent manner. (A) ECT2, CYK-4, and MKLP1 were precipitated and the immunoprecipitates were subjected to Western blotting with anti–CYK-4 antibodies. (B) Recombinant, soluble ECT2 E5 was incubated with CBD beads or CBD beads bound to CBD-CYK-4-N or CBD-CSC-1. Bound proteins were analyzed by Coomassie staining. (C) Schematic showing the boundaries of deletion constructs. (D) The tandem BRCT domains are sufficient for efficient binding of CYK-4. E. coli–expressed derivatives bound to chitin beads were used to pull-down proteins from a mitotic cell lysate. Bound proteins were analyzed by Western blotting with anti–CYK-4 antibodies. (E) The ECT-2 truncation derivative E5 colocalizes with CYK-4. Myc-tagged ECT2 E5 construct was transfected into HeLa cells, fixed, and stained for myc and CYK-4. (F) The association between CYK-4 and the ECT2 BRCT domains is phosphodependent. Mitotic cell lysates were treated with λ phosphatase before binding to chitin beads loaded with the indicated proteins. Bound proteins were analyzed by Western blotting with anti–CYK-4 antibodies.
In addition to the GEF domain, ECT2 also contains two BRCA1 COOH terminus (BRCT) domains in its NH2-terminal region, which are predicted to mediate protein–protein interactions. Therefore, we determined if the ECT2 BRCT domains are sufficient to bind to CYK-4. Using recombinant fragments, we found that ECT2 could efficiently bind to immobilized CYK-4 NH2 terminus, whereas a control fusion protein and empty beads bound only trace amounts of ECT2 (Fig. 7 B).
To more precisely define the region of ECT2 that binds to CYK-4, we prepared a set of truncation derivatives and produced the fusion proteins in E. coli (Fig. 7 C). These derivatives were used as affinity reagents to bind factors in mitotic lysates from HeLa cells. The NH2-terminal region of ECT2 was sufficient to pull-down CYK-4 (Fig. 7 D). ECT2 fragments containing both BRCT domains (E3, E4, and E5) retrieved significantly more CYK-4 in comparison to fragments containing only the first BRCT domain (E1); the second BRCT domain (E2) did not retrieve detectable CYK-4. Moreover, we observed that, like endogenous ECT2, truncation derivative E5 localized to the central spindle during anaphase (Fig. 7 E).
Recent studies suggest that tandem BRCT domains constitute a phosphopeptide binding domain (Manke et al., 2003). Therefore, we determined if the ability of recombinant ECT2 to associate with CYK-4 from mitotic lysates required phosphorylation of proteins in the lysate. Proteins in the lysate were dephosphorylated before the pull-down with recombinant ECT2 fragment E3. Fragment E2 was used as a negative control. Phosphatase treatment significantly reduced the ability of fragment E3 to bind to CYK-4 (Fig. 7 F). Thus, the association between ECT2 and CYK-4 is enhanced by phosphorylation. Because the ECT2 fragments used in this assay were bacterially expressed and are unlikely to be phosphorylated, a protein in the lysate must be phosphorylated for efficient binding of CYK-4 to ECT2. Because CYK-4 binds directly to ECT2, this protein could be CYK-4 itself.
CYK-4–ECT2 binding is cell cycle regulated
Next, we investigated whether the abundance of the ECT2–CYK-4 complex is regulated during mitosis. Lysates were prepared from metaphase-arrested cells and at intervals after release from metaphase. ECT2 was immunoprecipitated and the associated proteins were analyzed by Western blotting. The association between CYK-4 and ECT2 was not detectable in metaphase extracts. However, 60 min after release from metaphase, when most cells are in early anaphase, the abundance of the ECT2–CYK-4 complex increased dramatically (Fig. 8 A). Using an anti–phospho-TP antibody, we found that ECT2 was phosphorylated in metaphase. The phospho-TP signal on ECT2 decreased concomitantly with an increase in abundance of the ECT2–CYK-4 complex (Fig. 8 A). To determine whether CDK1 activity acutely inhibits the ECT2–CYK-4 interaction during metaphase, we treated nocodazole-arrested cells with Roscovitine, an inhibitor of CDK1. Roscovitine treatment significantly increased the association between ECT2 and CYK-4 (Fig. 8 B). These data indicate that ECT2–CYK-4 complex formation is inhibited during metaphase and this inhibition is relieved in anaphase. The inhibitor studies implicate CDK1 as a negative regulator of this interaction.
Figure 8.

The association between CYK-4 and ECT2 is cell cycle regulated. (A) Time course analysis of the association of CYK-4 and ECT2 during mitotic exit. HeLa cells were arrested in metaphase with nocodazole and released. Lysates were prepared at the indicated times and ECT2 was immunoprecipitated. Immunoprecipitates were Western blotted with anti–CYK-4 and anti–phospho-Thr-Pro antibodies to detect phosphorylation of ECT2. (B) CDK1 activity antagonizes the interaction between CYK-4 and ECT2. Metaphase-arrested HeLa cells were treated with Roscovitine for 30 min, ECT2 was immunoprecipitated, and the immunoprecipitates were Western blotted with anti–CYK-4 antibodies. (C) Recombinant ECT2 pulls down CYK-4 equally efficiently in metaphase and anaphase. HeLa cells were arrested in metaphase with nocodazole and released from the arrest. Lysates were prepared at the indicated times and ECT2 binding proteins were recovered with chitin beads loaded with the E3 fusion protein. Bound proteins analyzed by Western blotting with anti–CYK-4 antibodies.
Metaphase phosphorylation of ECT2 inhibits CYK-4 binding
We sought to understand the molecular mechanism that regulates the abundance of the ECT2–CYK-4 complex. Perhaps the phosphorylated residue recognized by the BRCT domains of ECT2 is absent in metaphase and accumulates in anaphase. Alternatively, CYK-4 may be phosphorylated but contains an additional modification during metaphase that inhibits its association with ECT2. To test these two possibilities, we used bacterially expressed ECT2 fragments to pull-down proteins from lysates prepared at metaphase and at various times during anaphase and analyzed the associated proteins by Western blotting. Recombinant ECT2 NH2 terminus (E3) pulled down equivalent amounts of CYK-4 from metaphase and anaphase extracts (Fig. 8 C), suggesting that in metaphase CYK-4 has the modifications necessary to bind to ECT2 and does not contain modifications that prevent this interaction.
A third possibility is that ECT2–CYK-4 complex formation may be inhibited during metaphase via regulation of ECT2. Because the phospho-TP signal on ECT2 inversely correlates with CYK-4 binding, we prepared myc-tagged ECT2 fragments (Fig. 9 A) to identify the metaphase-specific phospho-TP site to test whether this phosphorylation regulates complex formation. We transfected cells with myc-tagged versions of the NH2- and COOH-terminal halves of ECT2. The cells were arrested in metaphase and lysates were collected at metaphase and at various times after release. The expressed proteins were immunoprecipitated and analyzed by Western blotting with the anti–phospho-TP antibody. The NH2-terminal half (E5) was highly phosphorylated in metaphase and was dephosphorylated as the cells entered anaphase (Fig. 9 B). The COOH-terminal half did not react strongly with the antibody. To narrow down the region, we prepared additional deletion constructs (Fig. 9 A). These constructs were transfected into HeLa cells that were subsequently arrested in metaphase and the tagged proteins were immunoprecipitated. In metaphase cells, fragment E5 was reactive with the phospho-TP antibody and did not coprecipitate significant amounts of CYK-4 (Fig. 9 C). In contrast, the slightly shorter fragment, E4, did not react with the phospho-TP antibody and precipitated significantly more CYK-4 (Fig. 9 C). Thus, the association between ECT2 and CYK-4 inversely correlates with mitotic phosphorylation of ECT2.
Figure 9.

Identification of a phosphorylation site that regulates the CYK-4–ECT-2 interaction. (A) Schematic of the ECT2 constructs used. (B) The NH2- and COOH-terminal halves (E5 and C-term) of ECT2 were transfected into HeLa cells, which were then arrested in metaphase with nocodazole. Cells were released from the block and collected at the indicated times. The myc-tagged ECT2 constructs were immunoprecipitated and Western blotted with anti-myc and anti–phospho-Thr-Pro antibodies. (C) ECT2 fragments are differentially competent to bind CYK-4 during metaphase. The differential association inversely correlates with reactivity with anti–phospho-Thr-Pro antibody. ECT2 constructs were transfected into HeLa cells, which were then arrested in metaphase with nocodazole. Lysates were prepared and the myc-tagged ECT2 constructs were immunoprecipitated and Western blotted with anti-CYK-4, anti-myc, and anti–phospho-Thr-Pro antibodies. (D) Identification of threonine 342, which, when mutated to alanine, prevents TP phosphorylation and allows ECT2 to bind CYK-4 during metaphase. This experiment was performed as described in C.
To obtain direct evidence that mitotic phosphorylation of ECT2 inhibits its association with CYK-4, we mutated threonine 342, which is one of the TP sites in the region that distinguishes fragments E4 and E5, to alanine in fragment E6, which comprises residues 1–352. E6, E6* (T342A), and fragment E4 were transfected into HeLa cells. The cells were arrested in metaphase and the tagged proteins were immunoprecipitated. Fragment E6 was reactive with the phospho-TP antibody and did not precipitate significant amounts of CYK-4 (Fig. 9 C). However, E6*(T342A) did not react with the phospho-TP antibody and, importantly, this mutant precipitated significantly more CYK-4 than E6. These data indicate that T342 is phosphorylated during metaphase and this phosphorylation inhibits ECT2 from binding to CYK-4. To summarize, the ability of the NH2 terminus of ECT2 to bind a phosphoprotein, probably CYK-4, inhibited phosphorylation of ECT2 in metaphase.
Discussion
We have investigated the molecular requirements for cleavage furrow formation in cultured human cells. We report that the central spindle–associated RhoGEF ECT2 is required for cortical localization of the GTPase RhoA and for accumulation of two major constituents of the contractile ring, myosin II and F-actin. Depletion of the centralspindlin component MKLP1 causes delocalization of ECT2, and yet RhoA, F-actin, and myosin accumulate on the cell cortex and, when the cortical zone of RhoA localization is sufficiently narrow, cleavage furrow formation occurs. Thus, central spindle localization of ECT2 is not required for furrow formation. In contrast, depletion of the centralspindlin component CYK-4 prevents cortical localization of RhoA, actin, and myosin and prevents cleavage furrow formation. Therefore, in human cells, CYK-4 acts upstream of RhoA. CYK-4 binds directly to ECT2 and this interaction is cell cycle regulated. We propose that CYK-4 activates the RhoGEF activity of ECT2, which promotes activation of RhoA. ECT2 localization to the central spindle helps restrict RhoA activity to the equatorial cell cortex at anaphase. However, ECT2 can activate RhoA globally and additional mechanisms contribute to equatorial localization of RhoA.
Spatial regulation of RhoA
Cytokinesis begins at the metaphase to anaphase transition. At this time, components of the central spindle are activated by dephosphorylation. Non-kinetochore, antiparallel microtubules are bundled by kinesin motors including the centralspindlin complex and several microtubule-associated proteins, in particular PRC1 (for review see Glotzer, 2005). In HeLa cells, these bundles, and therefore centralspindlin and ECT2, are most abundant in the center of the cell. However, these bundles are also present in the subcortical cytoplasm underlying the region where RhoA concentrates. Cortical RhoA recruitment requires ECT2, and this pool of RhoA colocalizes with myosin II and actin, suggesting that it is active. The actin and myosin that are recruited by RhoA then participate in contractile ring assembly leading to cleavage furrow formation.
Due to their overlapping localization patterns, centralspindlin was proposed to recruit Pebble/ECT2 to the cell cortex overlying the central spindle where the exchange factor could activate RhoA (Somers and Saint, 2003). We tested this appealing hypothesis by depleting MKLP1 and by inhibiting aurora B kinase, two treatments that result in delocalization of CYK-4 and ECT2. A significant fraction of such cells form cleavage furrows that later regress, whereas other cells do not form furrows. In all cells, RhoA is cortically localized and actin and myosin accumulate. We observed that cells in which RhoA was more restricted (though slightly broader than control cells) form furrows, whereas cells in which the RhoA zone was greatly expanded do not form furrows. These data suggest that furrow formation will not occur if the actively constricting zone exceeds a certain size. Because cortical localization of RhoA requires ECT2, we conclude that delocalized ECT2 is active. These results are consistent with previous observations in which disruption of the central spindle by depletion of either MKLP1 or PRC1 did not abrogate cleavage furrow formation (Matuliene and Kuriyama, 2002; Mollinari et al., 2004). The ability to follow cortical RhoA localization in living cells has enabled us to extend these observations. In particular, we have shown that localization of ECT2 to the central spindle contributes to spatial regulation of RhoA, but it is not the sole means by which RhoA activity is localized (Fig. 10).
Figure 10.

The phenotypes of depleted cells and the regulated association of CYK-4 and ECT2. (A) A schematic model summarizing the phenotypes of cells depleted of MKLP1, CYK-4, and ECT2. (B) A model for the regulated association of CYK-4 and ECT2. See text for details.
How does RhoA become cortically localized when ECT2 is not concentrated on the central spindle, as in MKLP1-depleted cells? In C. elegans, we have demonstrated that two pathways redundantly specify furrow formation (Dechant and Glotzer, 2003). One pathway relies on the central spindle and the second involves astral microtubules. In several biological contexts, microtubules inhibit cortical contractility (Danowski, 1989; Ren et al., 1999; Pletjushkina et al., 2001), and because the density of astral microtubules is higher at the polar cortex than at the equatorial cortex, this would result in increased equatorial contractility and allow formation of an ingressing cleavage furrow. Importantly, cytokinesis is delayed when only one pathway is operable, indicating that both pathways are active in wild-type embryos. By analogy, we suspect that, in MKLP1-depleted human cells, as in C. elegans embryos lacking ZEN-4/MKLP1, astral microtubules regulate cortical contractility.
Mode of RhoA activation
Both ECT2 and CYK-4 are required for cortical localization of RhoA, accumulation of F-actin and myosin, and furrow formation. The biochemical mechanism by which ECT2 activates RhoA is well defined: it induces exchange of GDP for GTP on RhoA. How does CYK-4 activate RhoA? This may involve intramolecular autoinhibition of ECT2, as has been seen previously in other GEFs (Aghazadeh et al., 2000). Consistent with this idea, the NH2 terminus of ECT2 binds to and inhibits the COOH-terminal GEF domain (Saito et al., 2004; Kim et al., 2005). We have shown that the NH2 termini of ECT2 and CYK-4 interact directly in vitro. Based on the precedence for intramolecular inhibition of RhoGEFs, it is tempting to speculate that CYK-4 activates ECT2 by relieving its autoinhibition (Fig. 10).
It is remarkable that CYK-4, which contains a RhoGAP domain, appears to activate a RhoGEF. If the GEF activity of ECT2 activates RhoA, what would prevent the RhoGAP domain from CYK-4 from immediately inactivating RhoA, leading to a futile cycle? The solution to this apparent paradox could come from the fact that CYK-4 has a low GAP activity toward RhoA. Thus, in the ECT2–CYK-4 complex, the balance of activities may favor RhoA activation. The GAP domain of CYK-4 may act on another GTPase, such as Rac or Cdc42 (D'Avino et al., 2004; Oceguera-Yanez et al., 2005), or it may not be of great importance to cytokinesis (Goldstein et al., 2005), or it may function late in division when the nature of the ECT2–CYK-4 complex changes. The potential of a futile cycle is more acute for Rac and Cdc42 because, in vitro, ECT2 activates all three GTPases to a similar extent (Tatsumoto et al., 1999) and CYK-4 is ∼30-fold more active against Rac and Cdc42 than RhoA (Toure et al., 1998). Further analysis of the role of the CYK-4 GAP domain will be important to fully understand this interesting question.
Temporal regulation of RhoA
The interaction between ECT2 and CYK-4 is regulated by at least two phosphorylation events. First, phosphorylation of a factor, likely CYK-4, enhances the binding of CYK-4 to the tandem BRCT domains of ECT2. Recombinant ECT2 can pull-down CYK-4 from lysates at all stages of mitosis so this phosphorylation is not highly regulated. In contrast, the interaction between endogenous CYK-4 and ECT2 is cell cycle regulated; it is reduced in metaphase as compared with anaphase. This regulation is due, at least in part, to phosphorylation of ECT2 at threonine 342. Phosphorylation of ECT2 T342, adjacent to the second BRCT domain, inhibits its binding to CYK-4 during metaphase (Fig. 10). The kinase responsible has not been unambiguously identified. However, we favor the interpretation that CDK1 is responsible because T342 is strongly phosphorylated in metaphase and is dephosphorylated upon anaphase onset, and a CDK1 inhibitor enhances ECT2–CYK-4 complex formation. Although this site (SLMTPNSNK) does not have a basic residue in the +2 or +3 position as is commonly found in CDK1 sites, other CDK1 sites are known that lack the basic residue (Yamashiro et al., 1995; Esashi et al., 2005). Negative regulation of ECT2 by CDK1 is consistent with several previous results. In Drosophila, nondegradable cyclin derivatives, which prolong CDK1 activity, antagonize mutations in the Pebble gene (Echard and O'Farrell, 2003). Furthermore, high CDK1 levels have been shown to inhibit cortical recruitment of myosin II (Royou et al., 2002). Finally, measurements of RhoA•GTP levels in human cells indicate that the levels of active RhoA increase from metaphase to anaphase (Kimura et al., 2000). These earlier findings are all consistent with CDK1-mediated inhibition of ECT2 activation by CYK-4.
Applicability to other systems
The regulatory mechanisms described here may apply in other animal cells, though perhaps not in all details. The cortical localization of RhoA to the nascent furrow appears widely conserved. The distribution of YFP:RhoA that we have observed in living cells resembles the localization of RhoA in fixed vertebrate cells (Yonemura et al., 2004) and of a GFP:RBD fusion protein that binds specifically to RhoA•GTP (Bement et al., 2005). Given the well documented requirement for RhoA in cytokinesis and its ability to regulate actin polymerization and myosin activity, local accumulation of active RhoA may be a general mechanism for furrow formation.
However, the regulatory mechanisms responsible for the spatial and temporal regulation of RhoA may differ somewhat in other systems. For example, in C. elegans embryos, furrow formation occurs when CYK-4 is depleted, indicating that CYK-4 is not an essential activator of the ECT2 orthologue LET-21. If the role of CYK-4 in human cells is to engage the tandem BRCT domains of ECT2 and relieve autoinhibition, perhaps in C. elegans other factors serve this function.
Drosophila differs from mammalian cells in that the ECT2 orthologue Pebble does not localize to the central spindle. Rather, this critical GEF localizes to a ring in the cell cortex (Prokopenko et al., 1999). Because Pebble and RacGAP50C/CYK-4 interact in Drosophila (Somers and Saint, 2003), it is not clear why Pebble does not accumulate on the central spindle. Most importantly, it is not yet known if Pebble localization requires either component of the centralspindlin complex, Pav/MKLP1 or RacGAP50C. Drosophila also differs from human cells and C. elegans embryos in that the assembly of a contractile ring requires the MKLP1 orthologue Pavarotti (Adams et al., 1998). The basis for this requirement is not known. The central spindle–independent pathway observed in human cells and C. elegans embryos may be absent in Drosophila, which would predict global RhoA recruitment. Alternatively, if loss of Pavarotti destabilized the CYK-4 orthologue, then the Pavarotti mutants would fail to recruit cortical RhoA and resemble the CYK-4–depleted cells shown here.
Although the basic machinery for cytokinesis is conserved, differences do exist in different cell types. In addition, cytokinesis is affected by the degree to which cells are attached to the substrate. Further analysis at the biochemical and in vivo at the molecular level using probes like the one presented here will help to assess the generality of this mechanism.
Materials and methods
Cell culture
HeLa cells were grown in DME supplemented with 10% FCS, 2 mM l-glutamine, 100 U penicillin, and 0.1 mg/ml streptomycin. LipofectAmine PLUS reagent (Invitrogen) was used for DNA transfections.
Antibodies
The following commercial antibodies were used. For immunostaining, primary antibodies were a follows: mouse anti-RhoA and rabbit anti-RhoA (Santa Cruz Biotechnology, Inc.) and rabbit anti-MHC (nonmuscle IIB; Sigma-Aldrich) were used at 1:100; aurora B (Transduction labs), rhodamine phalloidin (Molecular Probes), DM 1α monoclonal anti–α-tubulin (Sigma-Aldrich), and monoclonal anti-GFP mixture (clone 7.1 and 13.1; Roche) were used at 1:300, rat anti-tubulin YOL/34 was used at 1:50. Alexa-labeled secondary antibodies (Molecular Probes) were used at 1:500. For Western blotting, anti-phospho-Thr-Pro (Cell Signaling Technology) and rabbit anti-Myc (CM-100; Gramsch Labs) were used at 1:500 and 1:1,000, respectively.
An ECT2 antibody was raised in rabbits immunized with recombinant ECT2 (1–421) and affinity purified. Rabbit and mouse anti–HsCYK-4 and anti–HsMKLP-1 were described previously (Mishima et al., 2002).
RNA interference and drug treatments
We used siRNA duplexes targeting the following sequences for RNA-mediated inhibition: RhoA, GCCGGUGAAACCUGAAGAA; MHC, CUGUAUCCUGCUUCAGACU; ECT2, GGCGGAAUGAACAGGAUUU; CYK-4, CCUCUUCUGACCUUUCGCC; MKLP1, CGACAUAACUUACGACAAAUU; aurora B, GUCCCAGAUAGAGAAGGAG; lamin, ACCAGGUGGAGCAGUAUAA (Dharmacon). Oligos were designed using the siRNA Design program (Dharmacon) in combination with BLAST to ensure specificity.
For optimal RNAi, HeLa cells (∼45% confluence) were grown in the presence of 2.5 mM thymidine for 18–24 h, and then released for 8 h after which the cells were transfected with siRNAs using Oligofectamine (Invitrogen) as described by Dharmacon. After 4–5 h, siRNAs were removed and cells were grown in the presence of 2.5 mM thymidine for 12–13 h. Finally, cells were released from the second thymidine block and fixed 10–11 h later when the majority of cells were in anaphase. Control cells were treated with Oligofectamine alone and different oligos were used in parallel, and distinct phenotypes were observed. The efficiency of protein depletion was assessed by Western blotting (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200501097/DC1).
Actin filaments were disrupted with 5 μM LatA (Molecular Probes). Myosin activity was inhibited using 100 μM Blebbistatin (Toronto Research). Aurora kinase activity was inhibited with100 nM Hesperadin (Boehringer). Microtubules were depolymerized using 300 nM nocodazole (Sigma-Aldrich). For inhibitor experiments, drugs were added for 45 min 9–10 h after release from the second thymidine block, and the cells were collected for staining. Control treatments with solvent (DMSO or PBS) were performed in parallel.
YFP-RhoA constructs
Full-length cDNA fragments for human and C. elegans RhoA were cloned in frame into pEYFP C1 (BD Biosciences) to generate the YFP:HsRhoA and YFP:CeRhoA (YFP:RhoA) fusions. A stable line expressing YFP:RhoA was generated by G418 selection followed by cloning by limiting dilution. Clones were screened by fluorescence microscopy and one line was obtained that stably expressed the fusion protein.
Expression constructs
ECT2 and CYK-4 constructs were prepared in pCMV-(myc)3 and/or pET–chitin binding domain (CBD) for transfection experiments and bacterial expression, respectively. The precise fragments are described in Figs. 7 and 9. The GAP domain (201–439) of mouse RhoGAP1 was cloned into pcDNA3.1+. Bacterial proteins were expressed in BL21(DE3)-RIL. Expression was induced with IPTG for 4 h at 37°C. Bacteria were collected and lysed by sonication in 50 mM Tris, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 1% Triton X-100, 1 mM DTT, 10 μg/ml leupeptin, 10 μg/ml pepstatin, and 1 mM PMSF. The lysates were centrifuged for 30 min at 4°C, and the recombinant proteins were affinity purified with chitin beads (New England Biolabs, Inc.), washed extensively, and stored in the lysis buffer supplemented with 10% glycerol at −80°C.
To assess the regulation of YFP:RhoA by GAP and GEF activity, constructs were cotransfected with pECFP C1 (BD Biosciences) into YFP:RhoA cells at a ratio of 5:1.
Immunolocalization
Coverslips containing cells were collected and washed in warm PBS or cytoskeletal buffer. The cells were fixed in −20°C MeOH (to stain ECT2, tubulin, MKLP1, CYK-4, and myosin II), 4% PFA (to label actin), or 4°C 10% TCA (to stain RhoA, MKLP1, CYK-4, and myosin II [Yonemura et al., 2004]) for 15 min. Immunostaining was performed according to standard procedures using Alexa 488, Alexa 568, and Cy5-labeled secondary antibodies. Actin was labeled with rhodamine phalloidin and DNA was labeled with Hoechst 33342. Slides were mounted in 50% glycerol, 0.1 M Tris, pH 8.8 and 4% n-propyl gallate. Slides were observed on microscopes (Axioplan; Carl Zeiss MicroImaging, Inc.) with 40×/1.3 or 63×/1.4 objectives and appropriate filters. Images were obtained with a CoolSnap FX camera. Image processing (scaling from 12 to 8 bit, cropping, rotating, brightness and contrast adjustment, and color combining) was performed with MetaMorph, ImageJ, (rsb.info.nih.gov/ij/), and Adobe PhotoShop.
Live imaging
Cells were imaged with an imaging system (DeltaVision) controlled by Softworx. HeLa cells were transfected with appropriate siRNAs for 4–5 h, and then released into siRNA-free media and filmed with DIC optics with a 40×/1.35 objective beginning 24 h after RNAi treatment. Cells were filmed every 15 s from metaphase or early anaphase until after cell separation (between 40 to 100 images collected per cell). To image the YFP:RhoA cell line, typically 300-ms exposures were collected every 15 s.
Immunoprecipitation and pull-downs
Mitotic HeLa cells were collected 10 h after release from a double thymidine block. To make metaphase lysates, cells were released from the thymidine block for 7.5 h, at which time 40 ng/ml nocodazole was added for a further 3.5 h. To collect cells in anaphase, the cells were released from nocodazole and collected at the indicated times. Where applicable, metaphase-arrested cells were treated with 100 μM Roscovitine for 30 min. Cells were lysed in lysis buffer (20 mM Hepes, pH 7.2, 0.1% Triton X-100, 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, 10 μg/ml leupeptin, 10 μg/ml pepstatin, 1 mM PMSF, 20 mM NaF, and 200 μM microcysteine). The lysates were centrifuged 2 × 10 min at 14 krpm at 4°C. Antibodies were added for 40 min, after which the immune complexes were collected with protein A–Sepharose for 1 h at 4°C. The beads were washed extensively and run on SDS-PAGE followed by Western blotting. Phosphatase treatment was performed with λ phosphatase (New England Biolabs, Inc.) for 60 min at 30°C.
For direct binding assays, the bacterially expressed ECT2 NH2 terminus was isolated in soluble form by TEV cleavage of CBD-ECT2-E5. This fraction was incubated with CBD beads or bead-bound CBD-CSC-1 (Romano et al., 2003) or CBD-CYK-4 in 20 mM Hepes, pH 7.2, 50 mM NaCl, 5 mM MgCl2, 0.1% Triton X-100, 1 mM DTT, 10 μg/ml leupeptin, 10 μg/ml pepstatin, and 1 mM PMSF for 2 h at 4°C. Beads were washed four times in binding buffer and analyzed by SDS-PAGE and Coomassie staining.
Online supplemental material
Online supplemental material shows the regulation of stress fibers by overexpressed GEF and GAP domains, the rescue of myosin treatment by YFP:RhoA in RhoA-depleted cells, and the extent of protein depletion by RNAi and videos showing phenotypes observed by time-lapse recordings. The online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200501097/DC1.
Acknowledgments
The authors would like to thank the Institute of Molecular Pathology and Boehringer Ingelheim for financial support.
Ö. Yüce and A. Piekny contributed equally to this paper.
A. Piekny and M. Glotzer's present address is Dept. of Molecular Genetics and Cell Biology, University of Chicago, Chicago, IL 60637.
Abbreviations used in this paper: BRCT, BRCA1 COOH terminus; CBD, chitin binding domain; GAP, GTPase activating protein; GEF, guanine nucleotide exchange factor; LatA, Latrunculin A.
References
- Adams, R.R., A.A. Tavares, A. Salzberg, H.J. Bellen, and D.M. Glover. 1998. pavarotti encodes a kinesin-like protein required to organize the central spindle and contractile ring for cytokinesis. Genes Dev. 12:1483–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghazadeh, B., W.E. Lowry, X.Y. Huang, and M.K. Rosen. 2000. Structural basis for relief of autoinhibition of the Dbl homology domain of proto-oncogene Vav by tyrosine phosphorylation. Cell. 102:625–633. [DOI] [PubMed] [Google Scholar]
- Alberts, A.S. 2001. Identification of a carboxyl-terminal diaphanous-related formin homology protein autoregulatory domain. J. Biol. Chem. 276:2824–2830. [DOI] [PubMed] [Google Scholar]
- Bement, W.M., H.A. Benink, and G. von Dassow. 2005. A microtubule-dependent zone of active RhoA during cleavage plane specification. J. Cell Biol. 170:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Avino, P.P., M.S. Savoian, and D.M. Glover. 2004. Mutations in sticky lead to defective organization of the contractile ring during cytokinesis and are enhanced by Rho and suppressed by Rac. J. Cell Biol. 166:61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danowski, B.A. 1989. Fibroblast contractility and actin organization are stimulated by microtubule inhibitors. J. Cell Sci. 93:255–266. [DOI] [PubMed] [Google Scholar]
- Dechant, R., and M. Glotzer. 2003. Centrosome separation and central spindle assembly act in redundant pathways that regulate microtubule density and trigger cleavage furrow formation. Dev. Cell. 4:333–344. [DOI] [PubMed] [Google Scholar]
- Echard, A., and P.H. O'Farrell. 2003. The degradation of two mitotic cyclins contributes to the timing of cytokinesis. Curr. Biol. 13:373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esashi, F., N. Christ, J. Gannon, Y. Liu, T. Hunt, M. Jasin, and S.C. West. 2005. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature. 434:598–604. [DOI] [PubMed] [Google Scholar]
- Glotzer, M. 2004. Cleavage furrow positioning. J. Cell Biol. 164:347–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glotzer, M. 2005. The molecular requirements for cytokinesis. Science. 307:1735–1739. [DOI] [PubMed] [Google Scholar]
- Goldstein, A.Y., Y.N. Jan, and L. Luo. 2005. Function and regulation of Tumbleweed (RacGAP50C) in neuroblast proliferation and neuronal morphogenesis. Proc. Natl. Acad. Sci. USA. 102:3834–3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauf, S., R.W. Cole, S. LaTerra, C. Zimmer, G. Schnapp, R. Walter, A. Heckel, J. Van Meel, C.L. Rieder, and J.M. Peters. 2003. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore–microtubule attachment and in maintaining the spindle assembly checkpoint. J. Cell Biol. 161:281–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jantsch-Plunger, V., P. Gönczy, A. Romano, H. Schnabel, D. Hamill, R. Schnabel, A.A. Hyman, and M. Glotzer. 2000. CYK-4: a Rho family GTPase activating protein (GAP) required for central spindle formation and cytokinesis. J. Cell Biol. 149:1391–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J.E., D.D. Billadeau, and J. Chen. 2005. The tandem BRCT domains of Ect2 are required for both negative and positive regulation of Ect2 in cytokinesis. J. Biol. Chem. 280:5733–5739. [DOI] [PubMed] [Google Scholar]
- Kimura, K., M. Ito, M. Amano, K. Chihara, Y. Fukata, M. Nakafuku, B. Yamamori, J. Feng, T. Nakano, K. Okawa, et al. 1996. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science. 273:245–248. [DOI] [PubMed] [Google Scholar]
- Kimura, K., T. Tsuji, Y. Takada, T. Miki, and S. Narumiya. 2000. Accumulation of GTP-bound RhoA during cytokinesis and a critical role of ECT2 in this accumulation. J. Biol. Chem. 275:17233–17236. [DOI] [PubMed] [Google Scholar]
- Kishi, K., T. Sasaki, S. Kuroda, T. Itoh, and Y. Takai. 1993. Regulation of cytoplasmic division of Xenopus embryo by rho p21 and its inhibitory GDP/GTP exchange protein (rho GDI). J. Cell Biol. 120:1187–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosako, H., T. Yoshida, F. Matsumura, T. Ishizaki, S. Narumiya, and M. Inagaki. 2000. Rho-kinase/ROCK is involved in cytokinesis through the phosphorylation of myosin light chain and not ezrin/radixin/moesin proteins at the cleavage furrow. Oncogene. 19:6059–6064. [DOI] [PubMed] [Google Scholar]
- Kovar, D.R., J.R. Kuhn, A.L. Tichy, and T.D. Pollard. 2003. The fission yeast cytokinesis formin Cdc12p is a barbed end actin filament capping protein gated by profilin. J. Cell Biol. 161:875–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner, C.F. 1992. The pebble gene is required for cytokinesis in Drosophila. J. Cell Sci. 103:1021–1030. [DOI] [PubMed] [Google Scholar]
- Manke, I.A., D.M. Lowery, A. Nguyen, and M.B. Yaffe. 2003. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science. 302:636–639. [DOI] [PubMed] [Google Scholar]
- Matuliene, J., and R. Kuriyama. 2002. Kinesin-like protein CHO1 is required for the formation of midbody matrix and the completion of cytokinesis in mammalian cells. Mol. Biol. Cell. 13:1832–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki, T., C.L. Smith, J.E. Long, A. Eva, and T.P. Fleming. 1993. Oncogene ect2 is related to regulators of small GTP-binding proteins. Nature. 362:462–465. [DOI] [PubMed] [Google Scholar]
- Mishima, M., S. Kaitna, and M. Glotzer. 2002. Central spindle assembly and cytokinesis require a kinesin-like protein/RhoGAP complex with microtubule bundling activity. Dev. Cell. 2:41–54. [DOI] [PubMed] [Google Scholar]
- Mollinari, C., J.P. Kleman, Y. Saoudi, S.A. Jablonski, J. Perard, T.J. Yen, and R.L. Margolis. 2004. Ablation of PRC1 by small interfering RNA demonstrates that cytokinetic abscission requires a central spindle bundle in mammalian cells, whereas completion of furrowing does not. Mol. Biol. Cell. 16:1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oceguera-Yanez, F., K. Kimura, S. Yasuda, C. Higashida, T. Kitamura, Y. Hiraoka, T. Haraguchi, and S. Narumiya. 2005. Ect2 and MgcRacGAP regulate the activation and function of Cdc42 in mitosis. J. Cell Biol. 168:221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletjushkina, O.J., Z. Rajfur, P. Pomorski, T.N. Oliver, J.M. Vasiliev, and K.A. Jacobson. 2001. Induction of cortical oscillations in spreading cells by depolymerization of microtubules. Cell Motil. Cytoskeleton. 48:235–244. [DOI] [PubMed] [Google Scholar]
- Prokopenko, S.N., A. Brumby, L. O'Keefe, L. Prior, Y. He, R. Saint, and H.J. Bellen. 1999. A putative exchange factor for Rho1 GTPase is required for initiation of cytokinesis in Drosophila. Genes Dev. 13:2301–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, X.D., W.B. Kiosses, and M.A. Schwartz. 1999. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 18:578–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano, A., A. Guse, I. Krascenicova, H. Schnabel, R. Schnabel, and M. Glotzer. 2003. CSC-1: a subunit of the Aurora B kinase complex that binds to the survivin-like protein BIR-1 and the incenp-like protein ICP-1. J. Cell Biol. 161:229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero, S., C. Le Clainche, D. Didry, C. Egile, D. Pantaloni, and M.F. Carlier. 2004. Formin is a processive motor that requires profilin to accelerate actin assembly and associated ATP hydrolysis. Cell. 119:419–429. [DOI] [PubMed] [Google Scholar]
- Royou, A., W. Sullivan, and R. Karess. 2002. Cortical recruitment of nonmuscle myosin II in early syncytial Drosophila embryos: its role in nuclear axial expansion and its regulation by Cdc2 activity. J. Cell Biol. 158:127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint, R., and W.G. Somers. 2003. Animal cell division: a fellowship of the double ring? J. Cell Sci. 116:4277–4281. [DOI] [PubMed] [Google Scholar]
- Saito, S., X.F. Liu, K. Kamijo, R. Raziuddin, T. Tatsumoto, I. Okamoto, X. Chen, C.C. Lee, M.V. Lorenzi, N. Ohara, and T. Miki. 2004. Deregulation and mislocalization of the cytokinesis regulator ECT2 activate the Rho signaling pathways leading to malignant transformation. J. Biol. Chem. 279:7169–7179. [DOI] [PubMed] [Google Scholar]
- Somers, W.G., and R. Saint. 2003. A RhoGEF and Rho family GTPase-activating protein complex links the contractile ring to cortical microtubules at the onset of cytokinesis. Dev. Cell. 4:29–39. [DOI] [PubMed] [Google Scholar]
- Straight, A.F., A. Cheung, J. Limouze, I. Chen, N.J. Westwood, J.R. Sellers, and T.J. Mitchison. 2003. Dissecting temporal and spatial control of cytokinesis with a myosin II Inhibitor. Science. 299:1743–1747. [DOI] [PubMed] [Google Scholar]
- Tatsumoto, T., X. Xie, R. Blumenthal, I. Okamoto, and T. Miki. 1999. Human ECT2 is an exchange factor for rho GTPases, phosphorylated in G2/M phases, and involved in cytokinesis. J. Cell Biol. 147:921–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toure, A., O. Dorseuil, L. Morin, P. Timmons, B. Jegou, L. Reibel, and G. Gacon. 1998. MgcRacGAP, a new human GTPase-activating protein for Rac and Cdc42 similar to Drosophila rotundRacGAP gene product, is expressed in male germ cells. J. Biol. Chem. 273:6019–6023. [DOI] [PubMed] [Google Scholar]
- Yamashiro, S., Y. Yamakita, K. Yoshida, K. Takiguchi, and F. Matsumura. 1995. Characterization of the COOH terminus of non-muscle caldesmon mutants lacking mitosis-specific phosphorylation sites. J. Biol. Chem. 270:4023–4030. [DOI] [PubMed] [Google Scholar]
- Yonemura, S., K. Hirao-Minakuchi, and Y. Nishimura. 2004. Rho localization in cells and tissues. Exp. Cell Res. 295:300–314. [DOI] [PubMed] [Google Scholar]