

Abstract
Mutations in GDAP1 lead to severe forms of the peripheral motor and sensory neuropathy, Charcot-Marie-Tooth disease (CMT), which is characterized by heterogeneous phenotypes, including pronounced axonal damage and demyelination. We show that neurons and Schwann cells express ganglioside-induced differentiation associated protein 1 (GDAP1), which suggest that both cell types may contribute to the mixed features of the disease. GDAP1 is located in the mitochondrial outer membrane and regulates the mitochondrial network. Overexpression of GDAP1 induces fragmentation of mitochondria without inducing apoptosis, affecting overall mitochondrial activity, or interfering with mitochondrial fusion. The mitochondrial fusion proteins, mitofusin 1 and 2 and Drp1(K38A), can counterbalance the GDAP1-dependent fission. GDAP1-specific knockdown by RNA interference results in a tubular mitochondrial morphology. GDAP1 truncations that are found in patients who have CMT are not targeted to mitochondria and have lost mitochondrial fragmentation activity. The latter activity also is reduced strongly for disease-associated GDAP1 point mutations. Our data indicate that an exquisitely tight control of mitochondrial dynamics, regulated by GDAP1, is crucial for the proper function of myelinated peripheral nerves.
Introduction
Charcot-Marie-Tooth disease (CMT) is the most frequent inherited peripheral neuropathy in humans; it affects 1 in 2,500 people (Berger et al., 2002). CMT is grouped into demyelinating and axonal forms according to electrophysiologic and pathologic findings. The defects in demyelinating types are believed to originate in myelinating Schwann cells, whereas neurons are first affected in axonal forms. However, recent studies on the molecular and cellular basis of CMT suggest that this distinction might not be absolute (Berger et al., 2002; Suter and Scherer, 2003). Demyelinating forms are associated with reduced nerve conduction velocity, segmental de- and remyelination, and “onion bulb” formations (CMT1, CMT3, and CMT4). The axonal forms (CMT2) show axonal degeneration without overt demyelinating lesions (Berger et al., 2002; Suter and Scherer, 2003). Among the recessive forms, the subtype CMT4A is the most frequent (Nelis et al., 2002), and disease-associated mutations in GDAP1 (ganglioside-induced differentiation associated protein 1) have been identified. Initially, Baxter and colleagues (2002) reported patients with an early onset of the disease and severe symmetric weakness and atrophy of the feet and hands. Motor and sensory nerve conduction velocities were reduced which was consistent with a demyelinating form of CMT (Baxter et al., 2002). Cuesta et al. (2002) described a classic axonal phenotype, without apparent demyelination, with hoarse voice and vocal cord paresis as additional symptoms that was associated with other mutations in GDAP1. Later, even patients with an identical mutation (homozygous GDAP1 S194X) were depicted with demyelinating or axonal phenotypes (Baxter et al., 2002; Nelis et al., 2002). Today, approximately 20 CMT-associated GDAP1 alterations, including point mutations and changes leading to truncated proteins (see Online Mendelian Inheritance in Man [OMIM] number *606598), have been described, causing axonal, demyelinating, or intermediate forms (Di Maria et al., 2004). No obvious correlation between the position and nature of the mutations and the electrophysiologic or neuropathologic phenotype could be discerned. Thus, the issue of axonal versus demyelinating phenotypes and the cellular origin and basis of the disease remain open.
The available information about the biology of GDAP1 is scarce. GDAP1 was identified as a transcript that was up-regulated after ganglioside-induced cholinergic differentiation of the mouse neuroblastoma cell line Neuro2a (Liu et al., 1999). Furthermore, an increase of GDAP1 mRNA expression also was found in neural-differentiated P19 cells and during development of the mouse brain (Liu et al., 1999).
Some hints about the molecular function of GDAP1 have been provided by bioinformatic analyses (Marco et al., 2004) confirming and extending previous reports (Baxter et al., 2002; Cuesta et al., 2002; Nelis et al., 2002). These studies demonstrated that GDAP1 and related proteins in invertebrates and vertebrates contain the characteristic domains of GSTs (GST-N, GST-C). Based on its particular domain features, GDAP1 was proposed as the founder of a new GST family. Members of this family are characterized by an enlarged interdomain between the GST-N and GST-C domains, and contain COOH-terminal hydrophobic stretches with potential transmembrane features (Marco et al., 2004).
Here we show that Schwann cells and neurons of myelinated peripheral nerves express GDAP1. Thus, both cell types might be affected directly by the consequences of the disease-causing mutations in GDAP1. Within the cell, GDAP1 functions as a tail-anchored protein of the mitochondrial outer membrane and promotes fragmentation of mitochondria. On this basis, we define GDAP1 as a novel regulator of the mitochondrial network. This ability is lost or reduced in CMT-associated mutated forms of GDAP1 and is very likely to contribute to the axonal and/or demyelinating defects that are seen in patients. The precise regulation of fusion and fission of mitochondria seems to be crucial for the integrity of the peripheral nervous system (PNS).
Results
GDAP1 is expressed by myelinating Schwann cells and neurons of the PNS
To examine the function of GDAP1 in peripheral nerves and to determine its role in neuropathies, we developed rabbit antisera against two GDAP1-specific peptides (Fig. S1; available at http://www.jcb.org/cgi/content/full/jcb.200507087/DC1). Western blot analysis of extracts that were derived from various neural tissues revealed that GDAP1 is expressed most prominently in different regions of the central nervous system, including the cortex, cerebellum, thalamus, olfactory bulb, and spinal cord (Fig. 1 A). Consistent with the consequences of GDAP1 mutations leading to motor and sensory neuropathies, we also found GDAP1 expression in sciatic nerves and in dorsal root ganglia (DRG) (Fig. 1 A). The results were identical with antisera against each of the two GDAP1 peptides (unpublished data). Furthermore, the levels of GDAP1 protein correlated well with parallel measurements of GDAP1 transcripts that were determined by quantitative RT-PCR (unpublished data) and with previously published data (Cuesta et al., 2002).
Figure 1.
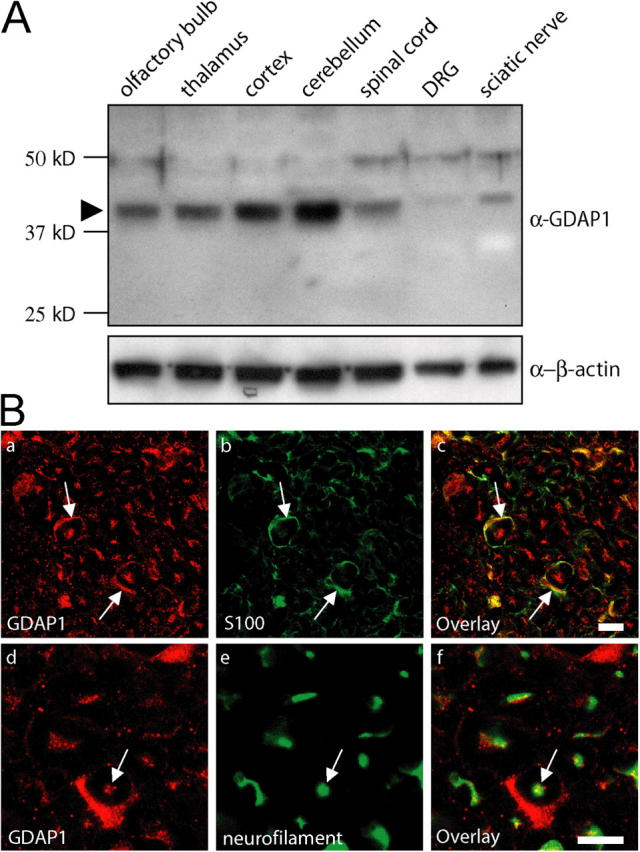
GDAP1 protein is expressed in Schwann cells and neurons. (A) 15 μg of protein lysate from the indicated neuronal tissues was analyzed by Western blot. In all tissues tested, GDAP1 was detected at the predicted size of 41.5 kD (arrowhead). The expression was highest in central nervous system tissues. The additional weak bands at ∼48 kD and ∼75 kD were not detected with another GDAP1 antiserum, and thus, were considered to be unspecific. (B) GDAP1 expression on cryosections of rat sciatic nerve. The signal colocalizes on single-plane confocal pictures (0.5-μm sections) with the Schwann cell marker S100 (1B, a–c) and the neuronal marker neurofilament (1B, d–f). Bars, 10 μm.
In humans, the most prominent result of mutations in GDAP1 is a pronounced motor and sensory neuropathy with a remarkable variability in phenotypic expression that ranges from primarily demyelinating to strong axonal involvement, and mixed forms with both alterations. Unraveling this complex situation, in efforts to clarify the underlying disease mechanisms, requires the determination of whether GDAP1 is expressed exclusively by only one of the two cell types involved—Schwann cells or neurons—or both. Such an analysis recently elucidated that mutations in the N-myc downstream-regulated gene 1 (NDRG1) primarily target myelinating Schwann cells in the hereditary motor and sensory neuropathy-Lom (Berger et al., 2004). Thus, we have analyzed the expression of GDAP1 in peripheral nerves in detail. GDAP1 was found in cross sections of rat sciatic nerves and colocalized with the Schwann cell marker S100 (Fig. 1 B, a–c) and the neuronal marker neurofilament (Fig. 1 B, d–f; neurofilament-positive axons stained for GDAP1 with punctuated patterns). Consistent with these observations, the cell bodies of motoneurons in the spinal cord (not depicted) and sensory neurons in DRG (Fig. S2; available at http://www.jcb.org/cgi/content/full/jcb.200507087/DC1) were labeled with anti-GDAP1 antibodies. The specificity was confirmed by successful blocking of the staining by preincubation of the serum with the antigenic peptide (Fig. S2), and concordant results for GDAP1 mRNA expression were obtained by in situ hybridization on spinal cord and DRG sections (not depicted). Teased sciatic nerve fiber preparations revealed that GDAP1 is present in the cytoplasm of myelinating Schwann cells, with particularly strong staining in the perinuclear region and Schmidt-Lanterman incisures (unpublished data). We conclude that PNS neurons and Schwann cells, both commonly involved in the etiology of motor and sensory neuropathies, express GDAP1 with potential functional implications.
GDAP1 is a mitochondrial protein
We next determined the precise subcellular localization of GDAP1. In COS-7 cells that were transfected with an untagged GDAP1 full-length expression construct, we found GDAP1 in distinct structures with tubular and vesicular appearances throughout the cytoplasm at the onset of expression (Fig. 2 A). These GDAP1-positive formations were identified as mitochondria by congruent costaining with the mitochondrial markers MitoTracker (Fig. 2 A, a–d), cytochrome c (Fig. 4 B), and porin (not depicted). No appreciable overlap was found with other membranous compartments, including the Golgi complex (marker giantin; Fig. 2 A, e–h) or the ER (marker protein disulfide isomerase [PDI]; Fig. 2 A, i–l).
Figure 2.
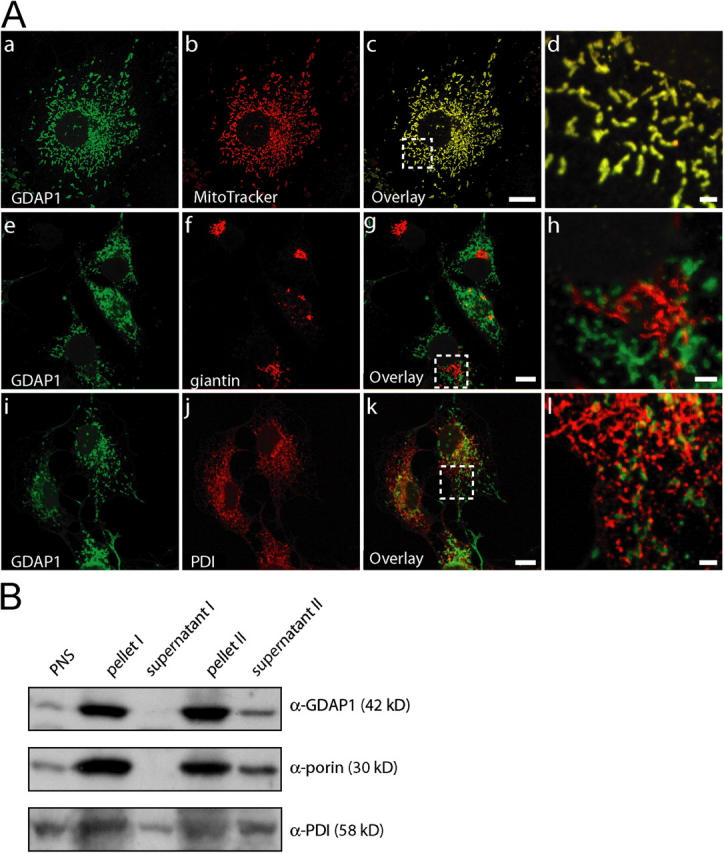
GDAP1 is a mitochondrial protein. (A) Transfected COS-7 cells, 12 h after transfection with GDAP1 expression constructs. GDAP1 colocalizes with the mitochondrial marker MitoTracker Red (a–d), but not with the Golgi complex marker giantin (e–h) or the ER marker PDI (i–l) on single-plane confocal pictures (0.4-μm sections). Bars in c, g, and k, 10 μm; bars in d, h, and l, 2 μm. Boxes in c, g, and k indicate the magnified areas in d, h, and l. (B) Mitochondria from endogenously GDAP1-expressing N1E-115 cells were enriched in a differential centrifugation approach. 20 μg of protein yielded by the centrifugation steps were loaded per lane. The mitochondrial protein porin and GDAP1, but not the ER protein PDI, showed increased levels in the pellets.
Figure 4.
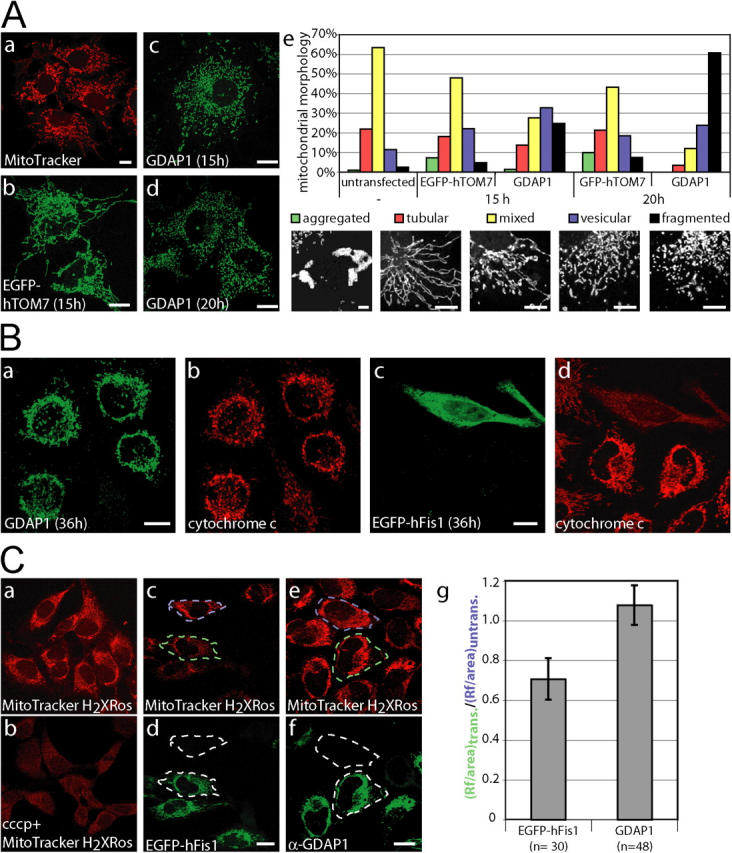
GDAP1 promotes mitochondrial fission. (A, a) Untransfected COS-7 cells display vesicular and tubular mitochondria, labeled with MitoTracker Red. (b) Transient transfection with the mitochondrial EGFP-fusion protein hTOM7 does not change this appearance. (c) The mitochondrial architecture is altered to a partially fragmented appearance 15 h after transfection with GDAP1. (d) After 20 h, most mitochondria are fragmented. (e) The morphologic results were quantified by classifying the appearance of mitochondria in GDAP1-expressing cells. Representative images for aggregated, tubular, mixed, vesicular, and fragmented mitochondrial appearance are shown. Bars: (a–d) 10 μm, (e) 5 μm. ∼500 transfected cells per condition from three independent experiments were counted. All changes are highly significant (probability > chi square > 0.001, comparing all conditions versus control transfected cells in a contingency analysis using JMP 5 [JMP Discovery]). (B) HeLa cells were transfected transiently with GDAP1 (a, b), or 0.5 μg plasmid DNA coding for the fission factor EGFP-hFis1 (c, d) and fixed 36 h after transfection. The expression of EGFP-Fis1 leads to a release of cytochrome c into the cytoplasm (c, d). Exclusive mitochondrial localization of cytochrome c was observed in cells expressing GDAP1 (a, b). (C) The mitochondrial uptake of the dye MitoTrackerH2XRos into HeLa cells is dependent on the activity of the mitochondria (a) and can be blocked with the protonophore cccp (b; 10 μM, 45 min) (b). HeLa cells were transfected with expression constructs for EGFP-hFis1 (0.25 μg; c, d) and GDAP1 (0.5 μg; e, f) and labeled with MitoTrackerH2Xros, 24 h after transfection. HeLa cells expressing EGFP-hFis1 display a weaker mitochondrial staining with MitoTrackerH2Xros (dashed green area) compared with untransfected cells (dashed blue box) (c, d). No difference between cells expressing GDAP1 and untransfected cells was found (compare dashed green and blue area) (e, f). The observation shown on the representative confocal images (a–f) was quantified using ImageJ (NIH) on 15 independent confocal pictures from three independent experiments (g; n, number of cells). Bars, (B and C) 10 μm.
To exclude potential localization artifacts that are due to overexpression, we also examined the intracellular confinement of endogenous GDAP1. In agreement with previous results, we found GDAP1 staining of mitochondria in Neuro2a (Liu et al., 1999), N1E-115 cells, C2C12 cells (not depicted), and primary Schwann cells in culture (Fig. S2). Finally, GDAP1 and porin colocalized in sections of rat DRG (Fig. S2).
To support further the localization of GDAP1 to mitochondria, we used a cell fractionation approach. Mitochondria were enriched by differential centrifugation (Graham, 2004), and the various steps were analyzed by Western blotting. Cell fractionations were performed with transfected Cos-7 cells (not depicted) and with endogenously GDAP1-expressing N1E-115 cells (Fig. 2 B). As expected, GDAP1 levels were enhanced specifically by this procedure in parallel with porin, an abundant transmembrane protein of the outer mitochondrial membrane, and in contrast to proteins of the ER, like PDI. Thus, GDAP1 is located in mitochondria.
GDAP1 is an integral membrane protein of the outer mitochondrial membrane
To elucidate the localization of GDAP1 within the mitochondria, we purified mitochondria from COS-7 cells, 24 h after transfection with a GDAP1 expression construct. Mitochondria were resuspended and digested with an excess of proteinase K in the presence or absence of detergent (Olichon et al., 2002), and analyzed by Western blot. The GDAP1 signal was lost upon proteinase incubation, whereas cytochrome c, located in the intermembrane space, was protected and disappeared only if increasing amounts of digitonin or SDS were added (Fig. 3 A). Because the antisera used in this analysis recognize the NH2-terminal proximal segment of GDAP1 (Fig. S1), these data indicate that the NH2 terminus of GDAP1, and thereby, the GST domains, are exposed to the cellular cytoplasm. In support of this interpretation, we found GDAP1 in the outer mitochondrial membrane by ultrastructural analysis using immunogold labeling (unpublished data).
Figure 3.

GDAP1 is a transmembrane protein of the outer mitochondrial membrane. (A) A crude mitochondrial extract of transfected COS-7 cells was digested with proteinase K in the presence of increasing concentrations of digitonin. The digest was stopped after 30 min on ice with an excess of PMSF, and was analyzed by Western blot. The signal for GDAP1 was lost in the absence of digitonin. Cytochrome c was protected against the digest until the mitochondrial outer membrane was permeabilized with high concentrations of digitonin, or with SDS (bottom lane). (B) The mitochondrial pellet of transfected COS-7 cells was resuspended in buffer (control), in 1 M NaCl, in 0.1 M carbonate (pH 11), or in buffer with 0.1% Triton X-100, and centrifuged to separate the soluble protein supernatants from membranous pellets. GDAP1 and the known transmembrane protein porin are found in the supernatant only in the presence of detergent.
We next asked whether GDAP1 is associated peripherally with mitochondrial membranes or whether it is an integral membrane protein as proposed (Marco et al., 2004). Isolated mitochondria were resuspended in various buffers and, after centrifugation, supernatants and membranous pellets were analyzed by Western blotting (Sogo and Yaffe, 1994; Olichon et al., 2002; Stojanovski et al., 2004) (Fig. 3 B). GDAP1 remained associated with the mitochondrial pellet after treatment with 1 M sodium chloride or 0.1 M carbonate buffer (pH 11). Detergent treatment released GDAP1 to the supernatant, comparable to the integral membrane protein porin. Together, these experiments demonstrate that GDAP1 is an integral membrane protein of the outer mitochondrial membrane.
GDAP1 regulates the architecture of mitochondria
In various experiments, we observed time-dependent changes in the mitochondrial morphology upon GDAP1 transfection. With prolonged expression of GDAP1, the mitochondria in COS-7 cells became more fragmented, and tubular forms were less prominent (Fig. 4 A, c and d). This effect was not seen in mock-transfected cells (Fig. 4 A, a). We reasoned that overexpression of any protein of the outer mitochondrial membrane could cause this effect. However, mitochondrial morphology was not appreciably altered upon transient expression of the GFP-tagged human transporter of the outer membrane subunit 7 (EGFP-hTOM7), in agreement with previous results (Stojanovski et al., 2004) (Fig. 4 A, b). This indicates a specific effect of GDAP1 in our experimental settings. Thus, we quantified the morphologic alterations caused by GDAP1 overexpression. To this end, we defined five distinct mitochondrial architectures within cells (for examples, see Fig. 4 A, e, bottom panel): perinuclear aggregated (“aggregated”), predominantly tubular (“tubular”), tubular and vesicular (“mixed”), predominantly vesicular but with some tubular structures (“vesicular”), and completely vesicular or fragmented mitochondria (“fragmented”). This analysis revealed that 15 h after transfection with GDAP1, fragmented mitochondria increase significantly at the expense of tubular and mixed mitochondrial formations compared with control cells (Fig. 4 A, e). As suspected from our previous impressions, this shift toward mitochondrial fragmentation was even more pronounced 20 h after transfection.
GDAP1-induced fragmentation is not associated with defective overall mitochondrial physiology
Fragmentation of mitochondria can occur in response to apoptotic stimuli (Lee et al., 2004). Thus, we tested whether GDAP1 overexpression induces apoptosis. Initial qualitative observations of transiently transfected HeLa, Neuro2a, or COS-7 cells did not suggest massive induction of apoptosis. To analyze this issue more precisely, we transfected HeLa cells with GDAP1 or EGFP-hFis1, a protein that is known to induce mitochondrial fission and apoptosis (James et al., 2003); fixed the cells after 36 h; and stained for cytochrome c and GDAP1. As anticipated, EGFP-hFis1 promoted the release of cytochrome c (note the diffuse cytochrome c staining exclusively in the transfected cell, Fig. 4 B, d), whereas in cells expressing GDAP1, cytochrome c was restricted to mitochondria (Fig. 4 B, a and b).
Loss of the mitochondrial transmembrane potential (ΔΨm) also triggers the fragmentation of mitochondria (Legros et al., 2002). Thus, we asked whether the fragmentation that is induced by overexpression of GDAP1 might be secondary to an effect on ΔΨm. The uptake of the dye, MitoTrackerH2XRos, into mitochondria is dependent on mitochondrial activity (Chen and Cushion, 1994), which provides a mean to test this possibility (Fig. 4 C, a and b). Transient transfection with low amounts of an EGFP-hFis1 expression construct served as positive control (Yoon, 2004). Dye uptake was unchanged in GDAP1-expressing cells compared with nontransfected HeLa cells, whereas it was reduced to 70% in EGFP-hFis1–positive HeLa cells. In addition, we generated stably transfected Neuro2a cells that inducibly overexpress GDAP1. Using 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide in flow cytometry (Cossarizza et al., 1993), we confirmed that overexpression of GDAP1 does not induce apoptosis or influence ΔΨm in these cells (unpublished data).
GDAP1-induced mitochondrial fragmentation is blocked by dynamin-related protein 1 (K38A) coexpression and counterbalanced by mitofusin 1 and 2
The shape of mitochondria depends on the balance of fusion and fission. Thus, we tested whether the induction of fragmentation by GDAP1 can be counterbalanced by proteins that are known to induce fusion or block fission. The dynamin-related protein 1 (Drp1) plays a central role in the fission of mitochondria; the dominant-negative Drp1(K38A) form blocks mitochondrial fission (Smirnova et al., 1998). We coexpressed GDAP1 with Drp1 or Drp1(K38A) in COS-7 cells, and analyzed the mitochondrial morphology of cells that express both proteins 17 h after transfection. Western blotting confirmed comparable expression levels of the transfected constructs (unpublished data). Coexpression of GDAP1 and Drp1 did not influence GDAP1-induced mitochondrial fragmentation considerably (Fig. 5 A, a–f). This outcome was expected, because overexpression of Drp1 per se does not change the mitochondrial architecture; there is an abundant pool of endogenous Drp1 in the cell (Yoon, 2004). In contrast, if coexpressed with GDAP1, Drp1(K38A) partially inhibited the fragmentation of mitochondria (Fig. 5 A, g–i). An increased number of GDAP1-Drp1(K38A) doubly expressing cells still had fragmented mitochondria compared with EGFP-hTOM7 control-transfected cells; however, this number was reduced significantly compared with GDAP1-only expressing cells (Fig. 5 A, j). Also, more cells than controls showed extended tubular mitochondrial morphology, the most prominent phenotype observed when Drp1(K38A) is expressed alone (85%; unpublished data). In summary, coexpression of Drp1(K38A) can block GDAP1-induced fragmentation partially, but without restoring the normal pattern completely.
Figure 5.

GDAP1-induced fragmentation is counterbalanced by the activity of fusion-promoting factors. (A) COS-7 cells were cotransfected with 0.25 μg of a GDAP1 expression vector and 0.25 μg of an empty vector (a–c), a vector coding for Drp1 (d–f), or dominant-negative Drp1(K38A) (g–i). 17 h after transfection, cells were counterstained with MitoTracker Red. Fixed cells were probed with anti-GDAP1 and anti-myc antibodies. (B) In analogy to the fission-blocking approach with Drp1(K38A), cotransfections of GDAP1 were performed with the fusion-inducing Mfn1 (a) or Mfn2 (c), or with the activity-deficient mutants Mfn1(K88T) or Mfn2(K109A) (b, d). Representative confocal images (0.3-μm sections) from three independent experiments are shown. The results were quantified (A, j and B, e) to show the percentage of transfected cells that express both proteins and belong to different mitochondrial classifications (n > 500 cells per condition). Bars, 10 μm.
Mitofusin (Mfn) 1 and 2 are essential for mitochondrial fusion and maintenance of mitochondrial morphology (Chen et al., 2003; Rojo et al., 2002; Santel and Fuller, 2001; Santel et al., 2003);. We found that coexpression of GDAP1 and Mfn1 or Mfn2 led to a balanced mitochondrial architecture, similar to the morphology observed in untransfected or EGFP-hTOM7 control-transfected cells (Fig. 5 B, a and c; compare with Fig. 4 A, a and b). Quantification confirmed that the coexpression of GDAP1 with Mfn1 or Mfn2 rescues GDAP1-induced mitochondrial fragmentation (Fig. 5 B, e). This effect is specific and depends on the activity of Mfn's because altered forms of Mfn1 and Mfn2 that carry mutations in their GTPase G1 motives (Mfn1[K88T] and Mfn2[K109A]) (Chen et al., 2003), were unable to prevent GDAP1-induced fragmentation (Fig. 5 B, b, d, e). We conclude that GDAP1 and Mfn's can counterbalance their opposite effects on structural mitochondria dynamics.
GDAP1 does not reduce mitochondrial fusion
Our previous experiments have shown that the overexpression of GDAP1 promotes mitochondrial fragmentation. This mitochondrial morphology might result from increased mitochondrial fission, reduced mitochondrial fusion, or a combination of both effects. Using polyethylene glycol (PEG)-based cell fusion technique, we examined the effect of GDAP1 on mitochondrial fusion (Mattenberger et al., 2003). HeLa cells that were transfected transiently with mitochondrial targeted DsRed (mtDsRed) were coplated with HeLa cells that were transfected transiently with mitochondrial targeted GFP (mtGFP; see Fig. 7, a–c) or GDAP1 (see Fig. 7, d–f). The cells were fused with PEG and cultivated in the presence of cycloheximide. As reported previously, we found mitochondrial fusion in hybrids of cells transfected with mtDsRed fused to cells transfected with mtGFP 3–4 h after cell fusion. This process was almost complete 6 h after cell fusion (Fig. 6, a–c, g; Mattenberger et al., 2003). An identical timing and extent of mitochondrial fusion was observed in cell hybrids of cells that were transfected with mtDsRed fused with cells transfected with GDAP1 (Fig. 6, d–g). Thus, GDAP1 overexpression does not interfere significantly with mitochondrial fusion, and indicates that GDAP1 is a fission-inducing factor.
Figure 7.
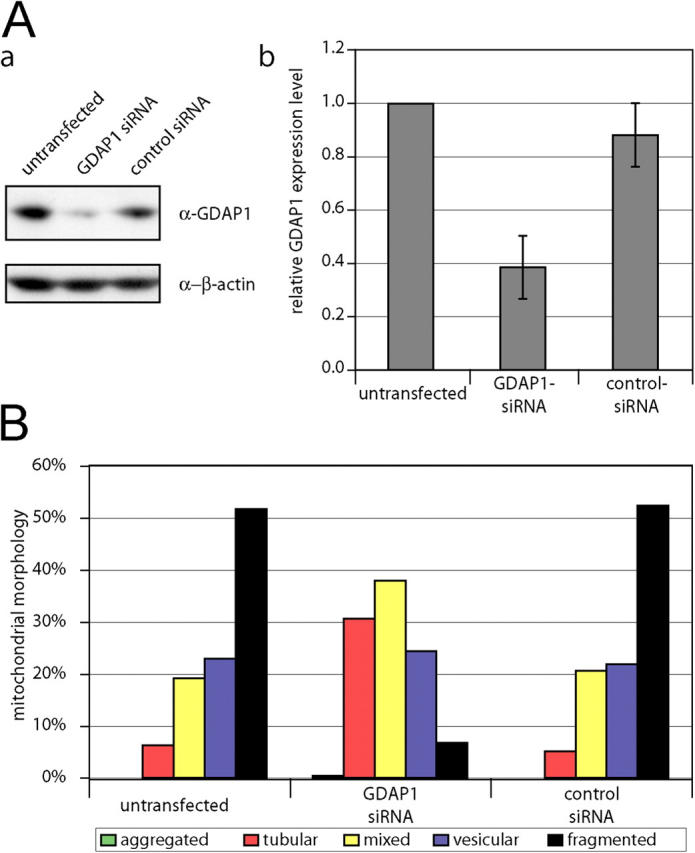
GDAP1 knock-down leads to elongation of mitochondria. Neuroblastoma N1E-115 cells were transfected with double-stranded small interfering RNA against GDAP1, with the scrambled control, or were left untransfected. (A, a) Equal amounts of cell lysates were separated and immunoblotted against GDAP1 and β-actin as loading control. (b) The relative expression levels of GDAP1 in correlation to the β-actin levels are represented as the mean of three independent transfections. (B) The influence of GDAP1 knock-down on the mitochondrial architecture was quantified by counting in blinded experiments. At least 750 cells per condition were counted from three independent experiments. Transfection with GDAP1-specific small interfering RNA leads to a significant increase in tubular mitochondria (probability > chi square > 0.0001), whereas the control transfection did not change the mitochondrial architecture (probability > chi square < 0.5).
Figure 6.
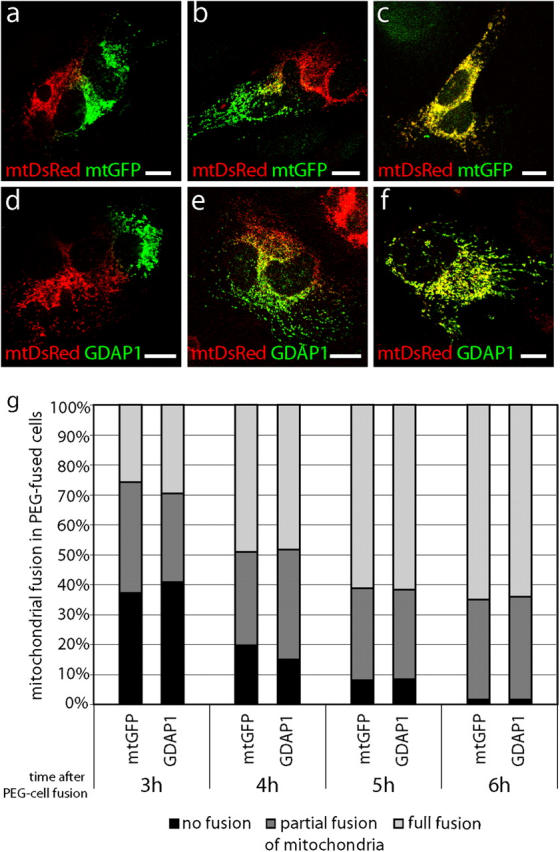
GDAP1 expression does not block mitochondrial fusion. HeLa cells that were transfected transiently with mtDsRed were coplated with HeLa cells, which were transfected transiently with mtGFP (a–c) or GDAP1 (d–f). Cells were fused with PEG and cultivated in the presence of cycloheximide. 3–6 h after PEG fusion, the cell hybrids were analyzed for fusion of the mitochondrial markers. Cell hybrids with no mitochondrial fusion (a, d), with partial mitochondrial fusion (b, e), and with full mitochondrial fusion (c, f) are depicted in single-plane confocal pictures. Cell hybrids were counted from three independent experiments (n > 120 hybrids per time point). The percentage of hybrids with the different degrees of mitochondrial fusion at the time points indicated is shown (g). Bars, 10 μm.
GDAP1 knock-down causes elongated mitochondria
To confirm that GDAP1 is regulating the mitochondrial morphology, we performed RNA interference (RNAi) knock-down experiments in N1E-115 neuroblastoma cells that endogenously express GDAP1. We were able to reduce GDAP1 expression levels by 60% (Fig. 7 A, a and b). This reduction could not be determined reliably at the single-cell level by immunocytochemistry. Thus, we counted at least 750 randomly chosen cells per condition in blinded experiments. Transfections with GDAP1-specific RNAi led to an increase of cells with a tubular mitochondrial morphology compared with untransfected or control transfected cells (Fig.7 B); this further substantiated our conclusion that GDAP1 promotes mitochondrial fission.
Disease-associated truncated GDAP1 proteins fail to reach the mitochondria
We next asked whether mutations of GDAP1 found in patients who have CMT have lost their mitochondrial targeting and/or the ability to induce fission of mitochondria. To answer this question, we generated a set of GDAP1 expression constructs, that represents three different stop mutations and five different point mutations (Fig. 9) that were identified previously in patients who had CMT (Baxter et al., 2002; Cuesta et al., 2002; Nelis et al., 2002; Ammar et al., 2003; Azzedine et al., 2003; Boerkoel et al., 2003; Senderek et al., 2003; Di Maria et al., 2004). Analysis in transfected COS-7 cells revealed that the truncated proteins GDAP1 T288fs290X and GDAP1 S194X were not targeted efficiently to mitochondria, but were localized diffusely throughout the cell (Fig. 8 A, a–f). No mitochondrial fragmentation-inducing activity was associated with these mutants (Fig. 8 A, b, e, h). The product of the shortest construct, GDAP1(Q163X), was not detectable with our antisera after transfection, although the epitope was present in the sequence (see Fig. S1; Fig. 8 A, g). We favor the interpretation that the GDAP1(Q163X) mutation generates a highly unstable protein, which suggests that the corresponding disease allele behaves like a null mutation.
Figure 9.
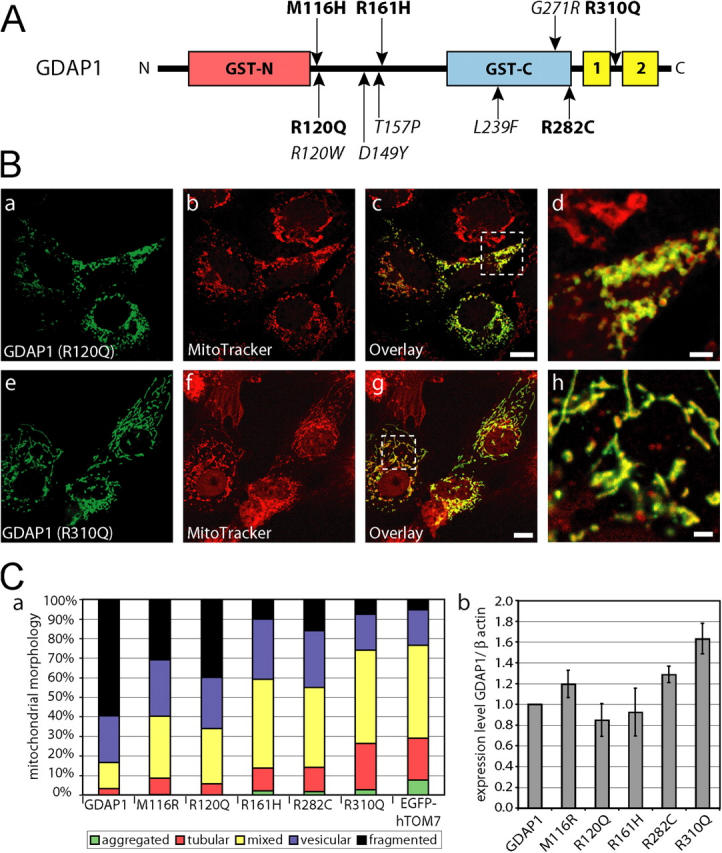
Disease-related GDAP1 point mutations are impaired in fission activity. (A) Schematic representation of the GDAP1 protein with known CMT-associated point mutations indicated. Mutations examined are in bold. (B) Transfected COS-7 cells were counterstained with MitoTracker Red before fixation and were stained with GDAP1 antibodies. All tested point mutations colocalized with mitochondria. Representative confocal pictures of GDAP1(R120Q) (a–d) and GDAP1(R310Q) (e–h) are shown. Bars in c, g, 10 μm; bars in d, h, 2 μm. (C, a) The influence on the mitochondrial architecture was quantified for all point mutations. We counted at least 500 cells per point mutation that expressed GDAP1 or one of the point mutations tested. The cells were grouped into five mitochondrial classifications. All changes are highly significant (probability > chi square > 0.001, comparing all conditions versus GDAP1 [wild-type] transfected cells in a contingency analysis). The expression levels of the tested GDAP1 point mutations were comparable to the GDAP1 wild-type protein in transfected COS-7 cells (quantified as ratio of the anti-GDAP1/anti-β-actin signals in cell lysates of sister plates [b]).
Figure 8.
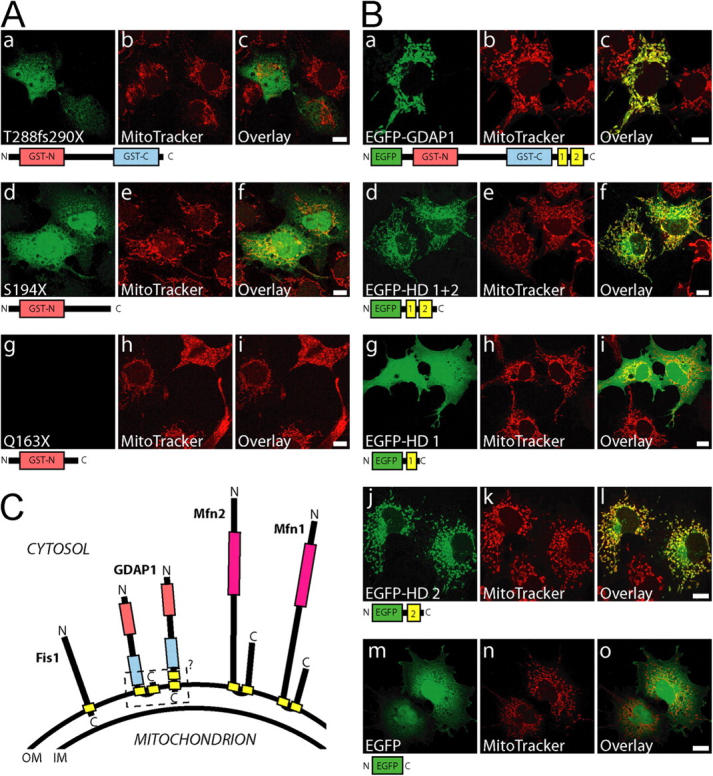
The COOH terminus of GDAP1 bears the mitochondrial-targeting signal. (A) Transiently transfected COS-7 cells were counterstained with MitoTracker Red before fixation; this was followed by GDAP1-antibody staining. The frame shift mutation GDAP1(T288fs290X) (a–c) and the stop mutation GDAP1(S194X) (d–f) have lost mitochondrial targeting. The shortest truncation tested, GDAP1(Q163X), was not detectable (g–i). (B) EGFP was fused to the NH2 terminus of the GDAP1 full-length protein (a–c), or the COOH-terminal parts of GDAP1 coding for the hydrophobic domains (HD) (d–f), for the first hydrophobic domain (g–i) or the second hydrophobic domain only (j–l). EGFP alone is not appreciably found in mitochondria (m–o). All images represent single 0.3-μm sections. All bars, 10 μm. (C) Schematic representation of known mammalian tail-anchored proteins that regulate the mitochondrial morphology. All proteins expose the NH2 terminus with the predicted catalytic domains (pink box, GTPase domain; red box, GST-N; blue box, GST-C) toward the cytosol. Hydrophobic domains are in yellow. For GDAP1, the exact localization of the hydrophobic domain 1 and of the COOH terminus remains unsolved (dashed box). IM, inner membrane; OM, outer membrane.
The COOH-terminal transmembrane domain of GDAP1 contains the mitochondrial-targeting information
Computer analysis revealed that GDAP1 carries no canonical mitochondrial targeting signal (unpublished data). Because the GDAP1 truncation T288fsX290 apparently has lost the mitochondrial-targeting information, the deleted 68 COOH-terminal amino acids are likely to harbor such signals. To assess this issue in more detail, we generated fusion proteins in which portions of GDAP1 were linked to the COOH terminus of EGFP. A full-length EGFP-GDAP1 fusion protein, colocalized with MitoTracker, demonstrated that the EGFP tagging allows efficient mitochondrial targeting in transfected COS-7 cells (Fig. 8 B, a–c). As expected, the last 78 amino acids of GDAP1 (EGFP-GDAP1280–358) were sufficient for mitochondrial localization (Fig. 8 B, d–f), and confirmed that the signal is close to the COOH terminus. Our further experimental strategy was based on the knowledge that mitochondrial proteins with a COOH-terminal hydrophobic domain, surrounded by basic amino acids, are targeted to the outer mitochondrial membrane (Kutay et al., 1993). Mfn2 belongs to this class of proteins, called “tail-anchored proteins.” For Mfn2, proper targeting requires the two transmembrane domains surrounded by basic amino acids, and coiled-coiled domains (Rojo et al., 2002). For GDAP1, the situation seems to be related, but different. An EGFP–fusion protein with the first hydrophobic domain, including its flanking regions (EGFP-GDAP1280–316), is not targeted to mitochondria (Fig. 8 B, g–i). However, the second hydrophobic domain and flanking regions are sufficient to direct an EGFP–fusion protein (EGFP-GDAP1308–358) to mitochondria (Fig. 8 B, j–l). Notably, cells with a particularly high expression of fusion protein exhibited an additional perinuclear staining (Fig. 8 B, f and l), which likely was an artifact of overexpression.
Disease-associated point mutations affect the ability of GDAP1 to induce fragmentation of mitochondria
Mutations leading to motor and sensory neuropathies are likely to yield important information about particular GDAP1 domains that are required for the proper function of the protein. Thus, we tested various GDAP1 point mutations (Fig. 9 A; M116H, R120Q, R161H, R282C, R310Q), in addition to the truncation mutants that were described above. Each of the selected point mutant forms of GDAP1 protein was targeted to mitochondria in transfected COS-7 cells (for examples, see Fig. 9 B). However, most interestingly, the influence on the mitochondrial architecture differed among the point mutants tested. Each mutant showed some impairment in the ability to induce fragmentation compared with wild-type GDAP1 protein, as indicated by the variable reduction of the fraction of cells with only fragmented mitochondria (Fig. 9 C, a). The mutants R161H and R310Q were completely inactive with respect to the promotion of mitochondrial fragmentation, and showed morphologies that were comparable to those seen in control transfections. We conclude that GDAP1 mutations that are found in patients who have CMT are defective in the ability of GDAP1 to induce mitochondrial fragmentation, albeit to different degrees.
Discussion
We have identified GDAP1 as a novel regulator of mitochondria. GDAP1 is a component of the outer mitochondrial membrane and, in concert with other proteins, influences the balance between fused and fragmented mitochondria. The mitochondrial localization of GDAP1 is required for this activity; the necessary information for proper organelle targeting is contained within the COOH-terminal part of the polypeptide. Analysis of GDAP1 mutations associated with hereditary motor and sensory neuropathies revealed that both features of GDAP1—proper localization and the modulating activity on mitochondria—are likely to be crucial for the functional integrity of myelinated peripheral nerves.
It is well-established that the nervous system is especially sensitive to mitochondrial dysfunction (Bouillot et al., 2002; Bossy-Wetzel et al., 2003; Finsterer, 2004). Furthermore, a critical role for mitochondria is discussed in the context of diseases and syndromes with liability to neuropathies, including diabetes (Vincent et al., 2004) or degeneration related to ageing (Bossy-Wetzel et al., 2003). We have found that GDAP1, the product of the disease-causing gene in particular forms of the neurodegenerative disorder CMT, is a key regulator of mitochondrial fragmentation. The observed fragmentation effect most likely is linked closely to mitochondrial fission. This important specific aspect of the biology of mitochondria has been appreciated fully only recently (Mozdy and Shaw, 2003; Chen and Chan, 2004; Rube and van der Bliek, 2004). In relation to diseases, two proteins that are involved in mitochondrial fusion are involved in neuropathies (Bossy-Wetzel et al., 2003). Mutations in the dynamin-related GTPase, OPA-1, lead to optic atrophy (Delettre et al., 2000); mutations in MFN2 were linked recently to CMT2A (Zuchner et al., 2004). We describe that GDAP1 also takes part in the regulation of mitochondrial dynamics by promoting mitochondrial fission. The fission effect is not secondary as a result of the induction of apoptosis or the reduction of the ΔΨm. GDAP1-induced mitochondrial fragmentation was counteracted by the fusion inducing factors, Mfn1 and Mfn2 and the dominant negative fission-blocking Drp1(K38A). PEG-mediated cell fusions further demonstrated that GDAP1 expression does not interfere with mitochondrial fusion. These findings suggest that common elements in the disease mechanisms of CMT are caused by mutations in GDAP1 and MFN2, although GDAP1 and Mfn2 have opposite effects on the mitochondrial morphology. The relation between GDAP1 and Mfn2 is intriguing if the two phenotypes of the associated diseases are compared. Mfn2 has been described exclusively in the context of axonal neuropathies in CMT2A, which indicates a neuronal basis of the disease mechanism. GDAP1 has been associated with axonal, demyelinating, and mixed phenotypes in CMT. Thus, a neuronal disease origin also might be favored in CMT caused by mutations in GDAP1 if an altered mitochondrial regulation by mutations in GDAP1 and MFN2 is put forward as the hypothetic basis of both genetically distinct disorders. However, we found that GDAP1 is expressed by myelinating Schwann cells as well as motor and sensory neurons; it is possible that the disease primarily affects Schwann cells, neurons, or both. Cell-type specific elimination of GDAP1 individually from neurons or Schwann cells will be required to resolve this issue.
Truncations and point mutations in GDAP1 are associated with CMT. Our results indicate that the COOH-terminal truncations that were tested have lost mitochondrial localization, whereas the examined GDAP1 proteins carrying missense mutations remained correctly targeted to mitochondria. These data are consistent with a recent report on the subcellular localization and effects of some GDAP1 mutations (Pedrola et al., 2005). We further demonstrate that GDAP1 proteins carrying disease-related mutations have lost the ability to induce mitochondrial fragmentation to various degrees. However, we have been unable to correlate the activity differences to disease onset, severity, and/or pathology. The variability of the phenotypes seems to be too high, even between relatives with the identical mutation (Azzedine et al., 2003).
The mitochondrial network is dynamic, and shifting the balance of fusion and fission is a major regulatory mechanism. Although the functional consequences of these events are understood poorly, it is obvious that complex and divergent stimuli influence the mitochondrial appearance (Goglia et al., 1999; Bach et al., 2003; Bossy-Wetzel et al., 2003; Honda and Hirose, 2003). The finding that mutations in genes affect this process in hereditary motor and sensory neuropathies suggests that myelinated peripheral nerves are particularly dependent on the proper function and control of this system. Whether this vulnerability is the consequence of a deregulation of the mitochondrial architecture or an impaired mitochondrial transport (Shy, 2004) remains to be determined.
Recently, evidence for mitochondrial fusion intermediates and an Mfn complex mediating mitochondrial tethering was provided (Koshiba et al., 2004; Meeusen et al., 2004). How the potential GST protein GDAP1, might, on a molecular level, be involved in the regulation of the mitochondrial architecture remains elusive. On a highly speculative note, the mitochondrial morphology is dependent on glutathione (GSH). Depletion of GSH leads to mitochondrial fusion, and results in a tubular mitochondrial network (Soltys and Gupta, 1994), although this effect may not be based solely on the GSH concentration (Bowes and Gupta, 2005). It remains unknown whether these are genuine hints that GDAP1 is a mitochondrial fission-inducing factor that is dependent on GSH.
In summary, we introduce GDAP1 as a key regulator of mitochondrial dynamics. If this activity is lost, the integrity of peripheral nerves is disturbed and leads to myelin and axonal defects. We anticipate that further elucidation of the cellular and molecular mechanisms that underlie the pathology of GDAP1-based CMT will help to uncover the function of GDAP1 and mitochondrial dynamics in peripheral nerves. Conversely, further examination of the molecular function of GDAP1 in the biology of mitochondria will contribute significantly to our basic understanding of CMT.
Materials and methods
RNA and protein isolation from mouse tissue
Tissues of C57BL/6 mice were isolated at postnatal day 68, flash frozen in liquid nitrogen, and stored at −80°C. Total RNA and protein was extracted with Trizol reagent using a Polytron PT homogenizer, according to the manufacturer's instructions (Invitrogen). RNA was quantified by photometry, and protein concentrations were determined using the Dc-Protein Assay (Bio-Rad Laboratories).
Constructs
GDAP1 cDNA was amplified by RT-PCR from adult mouse brain using the following primers: 5′-CGGATCCATGGCTCGGAGGCAGGAC-3′, 5′-CGTCGACTAGAAATAATTTGGTCTGG-3′. GDAP1-like-1 cDNA was cloned using 5′-CGGATCCATGGCGACCCCCAAC-3′ and 5′-CCCAGCCAGCTCTAGATGT-3′. Point mutations and truncations were generated by PCR with appropriate primer pairs. All cDNAs were cloned into the pGEM-T vector and verified by sequencing. The cDNAs were subcloned into pcDNA3.1 (Invitrogen) or EGFP-C1 (CLONTECH Laboratories, Inc.). Alternatively, the coding region for EGFP was replaced by the HA epitope (HA-C1 vector, provided by A. Hirschy, Swiss Federal Institute of Technology, Zürich, Switzerland). EGFP-hTOM7 and EGFP-hFis1 expression constructs were provided M. Ryan (La Trobe University, Melbourne, Australia). Mfn1-10xMyc, Mfn1(K88T)-10xMyc, Mfn2-16xMyc, Mfn2(K109A)-16xMyc, Drp1-6xMyc, and Drp(K38A)6xMyc were provided by D. Chan (California Institute of Technology, Pasadena, California). MtDsRed2 and mtGFP were from CLONTECH Laboratories, Inc.
Cell culture
Rat Schwann cells were prepared essentially as described by Brockes et al. (1979). Cells were grown in Schwann cell medium (DME; Invitrogen), containing 10% FCS, 4 μg/ml glial growth factor (crude pituitary extract (Sigma-Aldrich), and 4 μM forskolin (Sigma-Aldrich). For growth arrest, cells were incubated for 3 d in low serum medium (DME/F12 medium (Invitrogen), 0.5% FCS, 4 μM forskolin, 100 μg/ml human apo-transferrin, 60 ng/ml progesterone, 16 μg/ml putrescine, 10 μg/ml insulin, 400 ng/ml L-thyroxin, 160 ng/ml selenium, 0.02 ng/ml triiodothyronine, and 300 μg/ml BSA (Fluka). Supplements were from Sigma-Aldrich, unless stated otherwise. COS-7 cells, HeLa cells, and N1E-115 cells were maintained in DME containing 10% FCS, 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). COS-7 and HeLa cells were transfected using Fugene 6, according to the recommendations of the manufacturer (Roche). RNAi transfections on N1E-115 cells were done with 1.7 μg/ml lipofectamine 2000 (Invitrogen) and 55 nM Stealth RNAi (Invitrogen; GDAP1-specific: GGCCA CUCAG AUCAU UGAUU AUCUU; control: GGCAC UCCUA GGUUA AUUAU ACCUU). The cells were fixed or harvested 48 h after the start of transfection.
Cell fractionation
The differential centrifugation protocol to enrich for mitochondria was adopted from Graham (2004). Proteinase digestion was performed as described elsewhere (Olichon et al., 2002). Proteinase K (50 μg/ml) was added to the cell fractionation buffer with increasing concentrations of digitonin, or SDS. After 30 min on ice, the digest was stopped with 0.2 mM PMSF. To test for membrane integration of GDAP1, the first mitochondrial pellet from the differential centrifugation procedure was split and resuspended with a vortex mixer for 30 s at full speed in four different buffers: the homogenization buffer with 0.25 M sucrose (control), the control buffer plus 1 M sodium chloride (1 M NaCl), the control buffer plus 0.1% Triton X-100, or in 0.1 M sodium carbonate pH 11.0 (carbonate). The samples were centrifuged again for 20 min at 8000 g, 4°C. Equal volumes of the supernatants and pellets were analyzed by Western blot (Olichon et al., 2002; Stojanovski et al., 2004).
Cell fusions
For cell fusion, HeLa cells were transfected transiently with mtDsRed, mtGFP, or GDAP1. 8 h after the start of transfection, the cells were coplated on coverslips and cultivated overnight. The next morning, cycloheximide was added to the medium (1 mM). After 30 min, the cells were fused on a drop of prewarmed 50% w/v PEG 6000 (Sigma-Aldrich) in PBS/glucose (1 g/l) for 2 min. The cells were washed intensively with PBS/glucose and were kept in cultivation medium plus cycloheximide.
Antibodies
Polyclonal rabbit anti-GDAP1 antisera were generated by Pineda. Two different peptides were used as antigen (peptide 1: MARRQDEARAGVPL; peptide 2: FLDERTPRLMPDEGS). The antisera were used in a 1:7,500 dilution for Western blotting and 1:1,000 for immunofluorescence. Unless indicated otherwise, the sera against peptide 1 (serum 1) and peptide 2 (serum 2) were used and gave identical results. For immunohistochemistry of paraffin sections, we used serum 1, which was purified with the Montage antibody purification Kit (Millipore). The monoclonal mouse antibodies against β actin (clone AC-74), neurofilament 160 (clone NN18), and S-100 β-subunit (clone SH-B1) were from Sigma-Aldrich. Monoclonal anti-cytochrome c antibody (clone 7H8.2C12 and clone 6H2.B4) and the polyclonal rabbit anti-caspase3 antibody were obtained from BD Biosciences. The monoclonal rat anti-HA (clone 3F10) was purchased from Roche. The polyclonal rabbit anti-PDI antisera were provided by Dr. A. Helenius (Swiss Federal Institute of Technology, Zürich, Switzerland).
Immunohistochemistry
Cells were fixed with 2.5% PFA/PBS for 30 min at room temperature. Tissue was fixed overnight with 3.7% formalin, dehydrated, and embedded in paraffin in a Tissue Processor TP1010 (Leica). 6-μm sections were prepared using a Rotary Microtome HM 330 (Microm). For immunohistochemistry, fixed cells and rehydrated sections were incubated with 0.1% Triton X-100/PBS for 10 min. After washing with PBS, the specimens were incubated with 10% goat serum in PBS for 1 h at room temperature, followed by incubation with primary antibodies in 10% goat serum/PBS for 90 min at room temperature or overnight at 4°C. After washing, samples were exposed to appropriate secondary antibodies. Cy3- and Cy5-conjugated antibodies were from the Jackson Laboratory, secondary antibodies with Alexa-dyes from Molecular Probes. Samples were incubated with DAPI (Roche) and mounted in Immu-Mount (ThermoShandon). Specimens were analyzed with a Zeiss Axiophot microscope (Carl Zeiss Microimaging, Inc.). Alternatively, we used confocal microscopy on an inverted microscope DM IRB/E equipped with a true confocal scanner TCS SP1, a PL APO 63×/1.32 oil immersion objective (Leica) as well as argon, helium–neon lasers. Image processing was done on a Silicon Graphics workstation using Imaris (Bitplane AG). For labeling of mitochondria, we used MitoTracker Red (Molecular Probes) following the manufacturer's recommendations. MitoTracker Red CM-H2XRos was added to the medium in a final concentration of 0.5 μM for 30 min. After this pulse, the cells were cultivated in growth medium without dye for 10 min before fixation. To determine the relative uptake of MitoTrackerH2XRos into transfected cells, we analyzed single-plane confocal images. Using ImageJ, we measured the relative fluorescence (RF) intensity of MitoTrackerH2XRos and the area. We determined the RF/area in transfected cells and in untransfected cells on the same picture. For each picture, the RF/area of transfected cells was divided by the RF/area of untransfected cells. The average and the standard deviation of several pictures (n ≥ 6) were determined using two-tailed unpaired t test.
Western blotting
After SDS-PAGE, proteins were transferred onto polyvinylidene difluoride membrane (Millipore). Blots were blocked with 10% nonfat dry milk powder in TBS. The primary antibodies were diluted according to the manufacturer's recommendation or as described above. Blots were washed with 0.05% Tween 20/TBS. Secondary antibodies were from Santa Cruz Biotechnology, Inc., DakoCytomation, and Southern Biotechnology Associates, Inc. Immunoreactive bands were visualized by Western Lightning (PerkinElmer) or CDP-Star (Roche).
Online supplemental material
Fig. S1 shows validation of GDAP1-specific antisera. Fig. S2 shows that endogenous GDAP1 is found in mitochondria of Schwann cells and PNS neurons.
Acknowledgments
We gratefully acknowledge the gift of reagents by Drs. A. Hirschy, M. Wegner (University of Erlangen-Nürnberg, Erlangen, Germany), M. Ryan, D. Chan, and A. Helenius and thank Dr. S. Atanasoski for support with the Western blot analysis.
This work was supported by the Swiss National Science Foundation and the National Center of Competence in Research “Neural Plasticity and Repair” to U.S. and FIRB-RBAU01KJE4_002 to A. Schenone.
Abbreviations used in this paper: CMT, Charcot-Marie-Tooth disease; ΔΨm, mitochondrial transmembrane potential; DRG, dorsal root ganglia; Drp1, dynamin-related protein 1; GDAP1 ganglioside-induced differentiation associated protein 1; Mfn, mitofusin; mtDsRed, mitochondrial targeted DsRed; mtGFP, mitochondrial targeted GFP; PDI, protein disulfide isomerase; PEG, polyethylene glycol; PNS, peripheral nervous system; hTOM7, human transporter of the outer membrane subunit 7; RF, relative fluorescence; RNAi, RNA interference.
References
- Ammar, N., E. Nelis, L. Merlini, N. Barisic, R. Amouri, C. Ceuterick, J.J. Martin, V. Timmerman, F. Hentati, and P. De Jonghe. 2003. Identification of novel GDAP1 mutations causing autosomal recessive Charcot-Marie-Tooth disease. Neuromuscul. Disord. 13:720–728. [DOI] [PubMed] [Google Scholar]
- Azzedine, H., M. Ruberg, D. Ente, C. Gilardeau, S. Perie, B. Wechsler, A. Brice, E. LeGuern, and O. Dubourg. 2003. Variability of disease progression in a family with autosomal recessive CMT associated with a S194X and new R310Q mutation in the GDAP1 gene. Neuromuscul. Disord. 13:341–346. [PubMed] [Google Scholar]
- Bach, D., S. Pich, F.X. Soriano, N. Vega, B. Baumgartner, J. Oriola, J.R. Daugaard, J. Lloberas, M. Camps, J.R. Zierath, et al. 2003. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J. Biol. Chem. 278:17190–17197. [DOI] [PubMed] [Google Scholar]
- Baxter, R.V., K. Ben Othmane, J.M. Rochelle, J.E. Stajich, C. Hulette, S. Dew-Knight, F. Hentati, M. Ben Hamida, S. Bel, J.E. Stenger, et al. 2002. Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat. Genet. 30:21–22. [DOI] [PubMed] [Google Scholar]
- Berger, P., E.E. Sirkowski, S.S. Scherer, and U. Suter. 2004. Expression analysis of the N-Myc downstream-regulated gene 1 indicates that myelinating Schwann cells are the primary disease target in hereditary motor and sensory neuropathy-Lom. Neurobiol. Dis. 17:290–299. [DOI] [PubMed] [Google Scholar]
- Berger, P., P. Young, and U. Suter. 2002. Molecular cell biology of Charcot-Marie-Tooth disease. Neurogenetics. 4:1–15. [DOI] [PubMed] [Google Scholar]
- Boerkoel, C.F., H. Takashima, M. Nakagawa, S. Izumo, D. Armstrong, I. Butler, P. Mancias, S.C. Papasozomenos, L.Z. Stern, and J.R. Lupski. 2003. CMT4A: identification of a Hispanic GDAP1 founder mutation. Ann. Neurol. 53:400–405. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel, E., M.J. Barsoum, A. Godzik, R. Schwarzenbacher, and S.A. Lipton. 2003. Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr. Opin. Cell Biol. 15:706–716. [DOI] [PubMed] [Google Scholar]
- Bouillot, S., M.L. Martin-Negrier, A. Vital, X. Ferrer, A. Lagueny, D. Vincent, M. Coquet, J.M. Orgogozo, B. Bloch, and C. Vita. 2002. Peripheral neuropathy associated with mitochondrial disorders: 8 cases and review of the literature. J. Peripher. Nerv. Syst. 7:213–220. [DOI] [PubMed] [Google Scholar]
- Bowes, T.J., and R.S. Gupta. 2005. Induction of mitochondrial fusion by cysteine-alkylators ethacrynic acid and N-ethylmaleimide. J. Cell Physiol. 202:796–804. [DOI] [PubMed] [Google Scholar]
- Brockes, J.P., K.L. Fields, and M.C. Raff. 1979. Studies on cultured rat Schwann cells. I. Establishment of purified populations from cultures of peripheral nerve. Brain Res. 165:105–118. [DOI] [PubMed] [Google Scholar]
- Chen, F., and M.T. Cushion. 1994. Use of fluorescent probes to investigate the metabolic state of Pneumocystis carinii mitochondria. J. Eukaryot. Microbiol. 41:79S. [PubMed] [Google Scholar]
- Chen, H., and D.C. Chan. 2004. Mitochondrial dynamics in mammals. Curr. Top. Dev. Biol. 59:119–144. [DOI] [PubMed] [Google Scholar]
- Chen, H., S.A. Detmer, A.J. Ewald, E.E. Griffin, S.E. Fraser, and D.C. Chan. 2003. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 160:189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossarizza, A., M. Baccarani-Contri, G. Kalashnikova, and C. Franceschi. 1993. A new method for the cytofluorimetric analysis of mitochondrial membrane potential using the J-aggregate forming lipophilic cation 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (JC-1). Biochem. Biophys. Res. Commun. 197:40–45. [DOI] [PubMed] [Google Scholar]
- Cuesta, A., L. Pedrola, T. Sevilla, J. Garcia-Planells, M.J. Chumillas, F. Mayordomo, E. LeGuern, I. Marin, J.J. Vilchez, and F. Palau. 2002. The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat. Genet. 30:22–25. [DOI] [PubMed] [Google Scholar]
- Delettre, C., G. Lenaers, J.M. Griffoin, N. Gigarel, C. Lorenzo, P. Belenguer, L. Pelloquin, J. Grosgeorge, C. Turc-Carel, E. Perret, et al. 2000. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 26:207–210. [DOI] [PubMed] [Google Scholar]
- Di Maria, E., R. Gulli, P. Balestra, D. Cassandrini, S. Pigullo, L. Doria-Lamba, M. Bado, A. Schenone, F. Ajmar, P. Mandich, and E. Bellone. 2004. A novel mutation of GDAP1 associated with Charcot-Marie-Tooth disease in three Italian families: evidence for a founder effect. J. Neurol. Neurosurg. Psychiatry. 75:1495–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finsterer, J. 2004. Mitochondriopathies. Eur. J. Neurol. 11:163–186. [DOI] [PubMed] [Google Scholar]
- Goglia, F., M. Moreno, and A. Lanni. 1999. Action of thyroid hormones at the cellular level: the mitochondrial target. FEBS Lett. 452:115–120. [DOI] [PubMed] [Google Scholar]
- Graham, J.M. 2004. Isolation of mitochondria from tissues and cells by differential centrifugation. Current Protocols in Cell Biology. J.S. Bonifacino, J.B. Harford, J. Lippincott-Schwartz, and K.M. Yamada, editors. Wiley InterScience, New York. 3.3.2. [DOI] [PubMed]
- Honda, S., and S. Hirose. 2003. Stage-specific enhanced expression of mitochondrial fusion and fission factors during spermatogenesis in rat testis. Biochem. Biophys. Res. Commun. 311:424–432. [DOI] [PubMed] [Google Scholar]
- James, D.I., P.A. Parone, Y. Mattenberger, and J.C. Martinou. 2003. hFis1, a novel component of the mammalian mitochondrial fission machinery. J. Biol. Chem. 278:36373–36379. [DOI] [PubMed] [Google Scholar]
- Koshiba, T., S.A. Detmer, J.T. Kaiser, H. Chen, J.M. McCaffery, and D.C. Chan. 2004. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 305:858–862. [DOI] [PubMed] [Google Scholar]
- Kutay, U., E. Hartmann, and T.A. Rapoport. 1993. A class of membrane proteins with a C-terminal anchor. Trends Cell Biol. 3:72–75. [DOI] [PubMed] [Google Scholar]
- Lee, Y.J., S.Y. Jeong, M. Karbowski, C.L. Smith, and R.J. Youle. 2004. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Biol. Cell. 15:5001–5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legros, F., A. Lombes, P. Frachon, and M. Rojo. 2002. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol. Biol. Cell. 13:4343–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, H., T. Nakagawa, T. Kanematsu, T. Uchida, and S. Tsuji. 1999. Isolation of 10 differentially expressed cDNAs in differentiated Neuro2a cells induced through controlled expression of the GD3 synthase gene. J. Neurochem. 72:1781–1790. [DOI] [PubMed] [Google Scholar]
- Marco, A., A. Cuesta, L. Pedrola, F. Palau, and I. Marin. 2004. Evolutionary and structural analyses of GDAP1, involved in Charcot-Marie-Tooth disease, characterize a novel class of glutathione transferase-related genes. Mol. Biol. Evol. 21:176–187. [DOI] [PubMed] [Google Scholar]
- Mattenberger, Y., D.I. James, and J.C. Martinou. 2003. Fusion of mitochondria in mammalian cells is dependent on the mitochondrial inner membrane potential and independent of microtubules or actin. FEBS Lett. 538:53–59. [DOI] [PubMed] [Google Scholar]
- Meeusen, S., J.M. McCaffery, and J. Nunnari. 2004. Mitochondrial fusion intermediates revealed in vitro. Science. 305:1747–1752. [DOI] [PubMed] [Google Scholar]
- Mozdy, A.D., and J.M. Shaw. 2003. A fuzzy mitochondrial fusion apparatus comes into focus. Nat. Rev. Mol. Cell Biol. 4:468–478. [DOI] [PubMed] [Google Scholar]
- Nelis, E., S. Erdem, P.Y. Van Den Bergh, M.C. Belpaire-Dethiou, C. Ceuterick, V. Van Gerwen, A. Cuesta, L. Pedrola, F. Palau, A.A. Gabreels-Festen, et al. 2002. Mutations in GDAP1: autosomal recessive CMT with demyelination and axonopathy. Neurology. 59:1865–1872. [DOI] [PubMed] [Google Scholar]
- Olichon, A., L.J. Emorine, E. Descoins, L. Pelloquin, L. Brichese, N. Gas, E. Guillou, C. Delettre, A. Valette, C.P. Hamel, et al. 2002. The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett. 523:171–176. [DOI] [PubMed] [Google Scholar]
- Pedrola, L., A. Espert, X. Wu, R. Claramunt, M.E. Shy, and F. Palau. 2005. GDAP1, the protein causing Charcot-Marie-Tooth disease type 4A, is expressed in neurons and is associated with mitochondria. Hum. Mol. Genet. 14:1087–1094. [DOI] [PubMed] [Google Scholar]
- Rojo, M., F. Legros, D. Chateau, and A. Lombes. 2002. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J. Cell Sci. 115:1663–1674. [DOI] [PubMed] [Google Scholar]
- Rube, D.A., and A.M. van der Bliek. 2004. Mitochondrial morphology is dynamic and varied. Mol. Cell. Biochem. 256-257:331–339. [DOI] [PubMed]
- Santel, A., S. Frank, B. Gaume, M. Herrler, R.J. Youle, and M.T. Fuller. 2003. Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J. Cell Sci. 116:2763–2774. [DOI] [PubMed] [Google Scholar]
- Santel, A., and M.T. Fuller. 2001. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 114:867–874. [DOI] [PubMed] [Google Scholar]
- Senderek, J., C. Bergmann, V.T. Ramaekers, E. Nelis, G. Bernert, A. Makowski, S. Zuchner, P. De Jonghe, S. Rudnik-Schoneborn, K. Zerres, and J.M. Schroder. 2003. Mutations in the ganglioside-induced differentiation-associated protein-1 (GDAP1) gene in intermediate type autosomal recessive Charcot-Marie-Tooth neuropathy. Brain. 126:642–649. [DOI] [PubMed] [Google Scholar]
- Shy, M.E. 2004. Charcot-Marie-Tooth disease: an update. Curr. Opin. Neurol. 17:579–585. [DOI] [PubMed] [Google Scholar]
- Smirnova, E., D.L. Shurland, S.N. Ryazantsev, and A.M. van der Bliek. 1998. A human dynamin-related protein controls the distribution of mitochondria. J. Cell Biol. 143:351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogo, L.F., and M.P. Yaffe. 1994. Regulation of mitochondrial morphology and inheritance by Mdm10p, a protein of the mitochondrial outer membrane. J. Cell Biol. 126:1361–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltys, B.J., and R.S. Gupta. 1994. Changes in mitochondrial shape and distribution induced by ethacrynic acid and the transient formation of a mitochondrial reticulum. J. Cell. Physiol. 159:281–294. [DOI] [PubMed] [Google Scholar]
- Stojanovski, D., O.S. Koutsopoulos, K. Okamoto, and M.T. Ryan. 2004. Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology. J. Cell Sci. 117:1201–1210. [DOI] [PubMed] [Google Scholar]
- Suter, U., and S.S. Scherer. 2003. Disease mechanisms in inherited neuropathies. Nat. Rev. Neurosci. 4:714–726. [DOI] [PubMed] [Google Scholar]
- Vincent, A.M., J.W. Russell, P. Low, and E.L. Feldman. 2004. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocr. Rev. 25:612–628. [DOI] [PubMed] [Google Scholar]
- Yoon, Y. 2004. Sharpening the scissors: mitochondrial fission with aid. Cell Biochem. Biophys. 41:193–206. [DOI] [PubMed] [Google Scholar]
- Zuchner, S., I.V. Mersiyanova, M. Muglia, N. Bissar-Tadmouri, J. Rochelle, E.L. Dadali, M. Zappia, E. Nelis, A. Patitucci, J. Senderek, et al. 2004. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 36:449–451. [DOI] [PubMed] [Google Scholar]