

Abstract
Macroautophagy is a key pathway for the clearance of aggregate-prone cytosolic proteins. Currently, the only suitable pharmacologic strategy for up-regulating autophagy in mammalian cells is to use rapamycin, which inhibits the mammalian target of rapamycin (mTOR), a negative regulator of autophagy. Here we describe a novel mTOR-independent pathway that regulates autophagy. We show that lithium induces autophagy, and thereby, enhances the clearance of autophagy substrates, like mutant huntingtin and α-synucleins. This effect is not mediated by glycogen synthase kinase 3β inhibition. The autophagy-enhancing properties of lithium were mediated by inhibition of inositol monophosphatase and led to free inositol depletion. This, in turn, decreased myo-inositol-1,4,5-triphosphate (IP3) levels. Our data suggest that the autophagy effect is mediated at the level of (or downstream of) lowered IP3, because it was abrogated by pharmacologic treatments that increased IP3. This novel pharmacologic strategy for autophagy induction is independent of mTOR, and may help treatment of neurodegenerative diseases, like Huntington's disease, where the toxic protein is an autophagy substrate.
Introduction
The ubiquitin–proteasome and autophagy–lysosomal pathways are the two major routes for protein and organelle clearance in eukaryotic cells. Proteasomes predominantly degrade short-lived nuclear and cytosolic proteins. The bulk degradation of cytoplasmic proteins or organelles is mediated largely by macroautophagy, generally referred to as autophagy. It involves the formation of double-membrane structures, called autophagosomes, which fuse with lysosomes to form autolysosomes. Autophagy substrates generally have long half-lives (Klionsky and Emr, 2000).
Autophagy also can help cells clear the toxic, long-lived, aggregate-prone proteins, like mutant huntingtin and A53T, and A30P mutants of α-synuclein (Ravikumar et al., 2002; Webb et al., 2003), which cause neurodegenerative disorders, such as Huntington's disease (HD) (Rubinsztein, 2002) and forms of Parkinson's disease (PD) (Polymeropoulos et al., 1997; Kruger et al., 1998), respectively. Induction of autophagy reduced the levels of mutant huntingtin and protected against its toxicity in cells (Ravikumar et al., 2002), and in transgenic Drosophila and mouse models of HD (Ravikumar et al., 2004). The only suitable pharmacologic strategy for up-regulating autophagy in mammalian brains is to use rapamycin, or its analogs, that inhibit the mammalian target of rapamycin (mTOR), a negative regulator of autophagy.
HD is an autosomal-dominant neurodegenerative disorder that is caused by a CAG trinucleotide repeat expansion in the huntingtin gene (Huntington's Disease Collaborative Research Group, 1993), which results in an expansion of the polyglutamine tract in the NH2 terminus of the huntingtin protein. Asymptomatic individuals have ≤35 CAG repeats, whereas HD is caused by ≥36 repeats (Rubinsztein et al., 1996). Mutant huntingtin accumulates in intraneuronal aggregates (also called inclusions). Huntingtin is cleaved to form NH2-terminal fragments that consist of the first 100–150 residues containing the expanded polyglutamine tract, which are believed to be the toxic species found in the aggregates. Thus, HD pathogenesis frequently is modeled with exon 1 fragments that contain expanded polyglutamine repeats, which cause aggregate formation and toxicity in cell models and in vivo (Rubinsztein, 2002).
PD is another condition that is associated with aggregate formation. The intraneuronal Lewy body aggregates that characterize PD have the protein α-synuclein as a major component. The A53T and A30P point mutations in α-synuclein cause autosomal dominant forms of Parkinson's disease (Polymeropoulos et al., 1997; Kruger et al., 1998). Unlike wild-type α-synuclein, the clearance of these mutant forms is retarded when autophagy is inhibited (Webb et al., 2003; Cuervo et al., 2004). Although these forms of α-synuclein aggregate in vivo, we do not observe overt aggregation in our cell lines (Webb et al., 2003). Furthermore, unlike wild-type α-synuclein, these mutant forms are not cleared by the chaperone-mediated autophagy pathway (Cuervo et al., 2004), which is distinct from macroautophagy (called “autophagy” in this paper). Hence, we have used these mutations as model autophagy substrates.
We showed previously that lithium is protective in HD cell models, because it reduced mutant huntingtin aggregates and cell death (Carmichael et al., 2002). Lithium is used as a mood-stabilizing treatment of bipolar disorder, which is characterized by recurrent fluctuations in mood (Manji and Lenox, 1998). Although lithium has been used for decades for bipolar and unipolar affective disorders, the mechanism that underlies its therapeutic action is not understood fully. Here we describe a novel role for lithium as an inducer of autophagy. Lithium facilitated the clearance of known autophagy substrates, like mutant huntingtin and A53T, and A30P mutants of α-synuclein. Lithium induced autophagy by inhibiting inositol monophosphatase (IMPase), which led to reduced free inositol and myo-inositol-1,4,5-triphosphate (IP3) levels. This represents a novel pathway for regulation of mammalian autophagy.
Results
Lithium facilitates clearance of mutant huntingtin and α-synuclein
The possibility that lithium regulates the clearance of certain proteins was suggested by our observations that it significantly reduced aggregation and cell death in COS-7 (nonneuronal) and SK-N-SH (neural precursor) cells (Carmichael et al., 2002) (Fig. 1, A and B), that were caused by truncated forms of mutant huntingtin. In our earlier experiments, we showed protective effects with concentrations from 2.5–5 mM (Carmichael et al., 2002). Here we used 10 mM, because this concentration was used in the seminal paper from Harwood and colleagues that demonstrated that lithium, carbamazepine, and valproate decreased inositol levels (Williams et al., 2002). Expression levels of EGFP-tagged huntingtin exon 1 with 74 polyglutamine repeats (EGFP-HDQ74) correlate with aggregate formation (the proportion of transfected cells with aggregates) in such cell models (Narain et al., 1999; Ravikumar et al., 2002). We previously showed that EGFP-HDQ74 is cleared by autophagy. This is not the result of protein overexpression or the EGFP tag, because blocking autophagy had no obvious effect on the levels of isolated EGFP, but increased levels of HA-tagged HDQ74 as well as EGFP-HDQ74 (Ravikumar et al., 2002).
Figure 1.

Lithium facilitates clearance of mutant huntingtin fragment. COS-7 (A) and SK-N-SH (B) cells transfected with pEGFP-HDQ74 were treated with or without 10 mM LiCl or 10 mM NaCl for 48 h. The effects of treatment on the percentage of EGFP-HDQ74-positive cells with aggregates or apoptotic morphology (cell death) were expressed as odds ratios. Stable inducible PC12 cells expressing EGFP-HDQ74 were induced with doxycycline for 8 h, then transgene expression was switched off for 120 h (by removing doxycycline), with (+) or without (−) 10 mM LiCl (C) or 10 mM NaCl (D). Clearance of soluble EGFP-HDQ74 was analyzed by immunoblotting with antibody against EGFP (i). Densitometry analysis of soluble EGFP-HDQ74 relative to actin (ii) was done from three independent experiments. Untreated cells were termed “120 h off”. (E) The percentage of EGFP-positive cells with aggregates in stable PC12 cells expressing EGFP-HDQ74, induced and treated for 120 h as in Fig. 1, C and D with 10 mM LiCl or 10 mM NaCl, were assessed and expressed as odds ratio compared with control condition (120 h off). (F) Clearance of aggregated and soluble EGFP-HDQ74 in stable PC12 cells as in Fig. 1 C, treated with (+) or without (−) 10 mM LiCl for 120 h, was analyzed by immunoblotting with antibody against EGFP. The aggregated EGFP-HDQ74 is seen in the stacking gel. ***, P < 0.001; NS, non-significant.
To investigate if the reduced aggregation of the huntingtin construct was due to enhanced clearance, we used a stable doxycycline-inducible PC12 cell line expressing EGFP-HDQ74, where transgene expression is first induced by adding doxycycline and then switched off by removing doxycycline from the medium. If one follows transgene expression levels at various times after switching off expression after an initial induction period, one can assess if specific agents alter the clearance of the transgene product, as the amount of transgene product decays when synthesis is stopped (Ravikumar et al., 2002). Using this paradigm, we observed that lithium (but not NaCl) significantly enhanced clearance of soluble EGFP-HDQ74 (Fig. 1, C and D) and reduced aggregates at 120 h (Fig. 1 E; Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200504035/DC1) and 170 h (Fig. S1, B and C). Furthermore, at 120 h, lithium enhanced the clearance of the insoluble mutant huntingtin that gets retarded in the stacking gel (Fig. 1 F). However, because this is probably less reliable quantified than scoring aggregates, we have used aggregate counts (e.g., Fig. 1 E) as a measure of insoluble huntingtin clearance. The clearance of aggregates is likely to be a consequence of removal of aggregate precursors (soluble and oligomeric species), rather than big aggregates that are much larger than typical autophagosomes, and will also not be able to enter the proteasome barrel (Ravikumar et al., 2004).
Furthermore, we assessed clearance of A53T and A30P α-synuclein mutants. After induction of stable inducible PC12 cell lines (Webb et al., 2003) and subsequent removal of doxycycline, lithium treatment for 24 h significantly enhanced the clearance of A53T (Fig. 2 A) and A30P (Fig. 2 B) α-synuclein mutants; no effect was observed with NaCl (Fig. 2 C).
Figure 2.
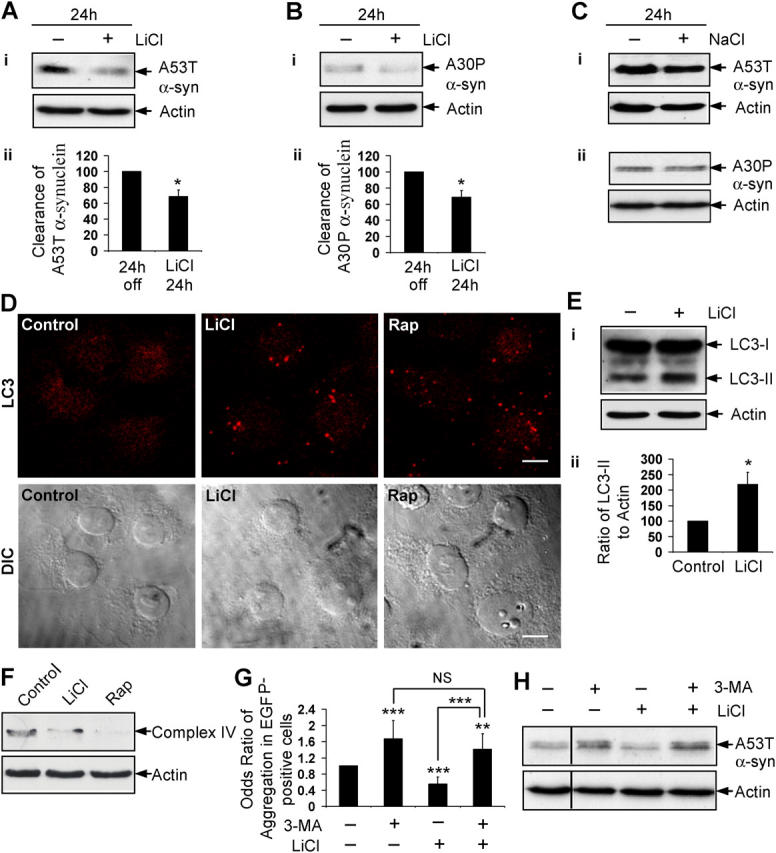
Lithium induces autophagy. Stable inducible PC12 cell lines expressing A53T (A) or A30P (B) α-synuclein mutants were induced with doxycycline for 48 h; expression of transgene was switched off for 24 h, with (+) or without (−) 10 mM LiCl. (i) Clearance of mutant α-synucleins was analyzed by immunoblotting with antibody against HA. (ii) Densitometry analysis of mutant α-synuclein relative to actin was done from three independent experiments. The untreated cells were termed “24 h off.” (C) Clearance of A53T (i) and A30P (ii) α-synuclein mutants in stable PC12 cells as in Fig. 2, A and B, treated with (+) or without (−) 10 mM NaCl for 24 h, was analyzed by immunoblotting with antibody against HA. (D) COS-7 cells treated with or without 10 mM LiCl or 0.2 μM rapamycin (Rap) for 24 h, were analyzed by immunofluorescence with antibody against LC3 using a confocal microscope. Bar, 20 μm. (E) COS-7 cells, treated as in Fig. 2 D, were analyzed by immunoblotting (i) with antibody against LC3. Densitometry analysis of LC3-II levels relative to actin (ii) was done from three independent experiments. (F) COS-7 cells were treated with or without 10 mM LiCl or 0.2 μM rapamycin (Rap) for 120 h, and mitochondrial load was assessed by immunoblotting with antibody to complex IV. (G) The percentage of EGFP-HDQ74–positive cells with aggregates in COS-7 cells treated with (+) or without (−) 10 mM 3-MA, 10 mM LiCl, or both for 48 h as in Fig. 1 A, were expressed as odds ratio. (H) Clearance of A53T α-synuclein in stable inducible PC12 cells as in Fig. 2 A, treated with (+) or without (−) 10 mM 3-MA, 10 mM LiCl, or both for 24 h, was analyzed by immunoblotting with antibody against HA. *, P < 0.05; **, P < 0.01; ***, P < 0.001; NS, non-significant.
Lithium induces autophagy to enhance clearance of mutant proteins
We assessed if lithium induced autophagy, because the clearance of EGFP-HDQ74 and the A53T and A30P α-synuclein mutants are strongly dependent on this pathway (Ravikumar et al., 2002, 2004; Webb et al., 2003). The microtubule-associated protein 1 light chain 3 (LC3), a homologue of Apg8p that is essential for autophagy in yeast, is processed posttranslationally into LC3-I, which is cytosolic. LC3-I is converted to LC3-II, which associates with autophagosome membranes (Kabeya et al., 2000). Because LC3 specifically associates with autophagosomes, the numbers of LC3-positive vesicles reflect autophagosome number (Kabeya et al., 2000), which is a specific and sensitive assay that has been used widely (for review see Mizushima, 2004). Lithium increased the number of LC3-positive autophagic vesicles in COS-7 cells, like rapamycin (Fig. 2 D), and increased the levels of LC3-II (Fig. 2 E). The modest increase in LC3-II is similar to that observed when autophagy was induced (Kabeya et al., 2000). We next transfected COS-7 or HeLa cells with LC3 fused to EGFP (EGFP-LC3). EGFP-LC3 localizes only to autophagic membranes but not other membrane structures (Kabeya et al., 2000). Because EGFP-LC3 overexpression does not affect autophagic activity, numbers of EGFP-LC3 vesicles have been used frequently to assess autophagic activity (Mizushima et al., 2004). A significantly greater proportion of lithium-treated cells expressing EGFP-LC3 had overt vesicle formation compared with untreated cells, even at a therapeutically-relevant dose (1 mM) (Fig. S2, A and B; available at http://www.jcb.org/cgi/content/full/jcb.200504035/DC1). Furthermore, we assessed the mitochondrial load in cells that were treated with lithium by immunoblotting for mitochondrial complex IV, because mitochondria are endogenous autophagy substrates (Klionsky and Emr, 2000). After 120 h of treatment with lithium, complex IV levels were reduced in COS-7 cells (Fig. 2 F). Autophagy induction with rapamycin also reduced levels of complex IV (Fig. 2 F), and complex IV accumulates when autophagy was inhibited (e.g., with 3-methyl adenine) (unpublished data).
If lithium facilitates clearance of EGFP-HDQ74 and mutant α-synucleins through autophagy, then it should be blocked by the autophagy inhibitor, 3-methyladenine (3-MA). 3-MA increased EGFP-HDQ74 aggregate formation in COS-7 cells (Ravikumar et al., 2002) (Fig. 2 G; Fig. S2 C), and delayed clearance of A53T α-synuclein (Webb et al., 2003) (Fig. 2 H). Furthermore, 3-MA completely abolished the effect of lithium in reducing EGFP-HDQ74 aggregation (Fig. 2 G; Fig. S2 C), or promoting A53T α-synuclein clearance (Fig. 2 H). Thus, lithium induces autophagy and enhances the clearance of known autophagy substrates, such as EGFP-HDQ74 and mutant α-synucleins.
Lithium enhances clearance of mutant proteins by inositol monophosphatase inhibition
Lithium inhibits a number of enzymes, including glycogen synthase kinase-3β (GSK-3β) and IMPase (Gould et al., 2002; Coyle and Duman, 2003). To test if either of these enzymes was involved in autophagy regulation, we used specific inhibitors of GSK-3β (SB216763) (Coghlan et al., 2000) and IMPase (L-690,330) (Atack et al., 1993) (Fig. S3; available at http://www.jcb.org/cgi/content/full/jcb.200504035/DC1). L-690,330 reduced aggregation and cell death that were caused by EGFP-HDQ74 in COS-7 and SK-N-SH cells, whereas SB216763 increased EGFP-HDQ74 aggregates but reduced cell death, which was consistent with our earlier findings (Carmichael et al., 2002) (Fig. 3, A and B). The idea that IMPase was involved in autophagy regulation was supported further because L-690,330 facilitated clearance of soluble EGFP-HDQ74 (Fig. 3 C) and the mutant α-synucleins (Fig. 3 D), whereas SB216763 had no effect on clearance of soluble EGFP-HDQ74 (Fig. 3 E) and seemed to slow clearance of mutant α-synucleins in stable PC12 cell lines (Fig. 3 F). Because SB216763, if anything, slows clearance of these substrates, the enhanced clearance of mutant huntingtin and α-synucleins that is mediated by lithium cannot be due to the drug inhibiting GSK-3β.
Figure 3.

Lithium accelerates clearance of mutant huntingtin fragment by inositol monophosphatase inhibition. The percentage of EGFP-HDQ74–positive cells with aggregates and cell death in COS-7 (A) and SK-N-SH (B) cells as in Fig. 1, A and B, treated with or without 10 μM SB216763 or 100 μM L-690,330 for 48 h, were expressed as odds ratios. (C) Clearance of soluble EGFP-HDQ74 in stable inducible PC12 cell lines as in Fig. 1 C, treated with (+) or without (−) 100 μM L-690,330 for 170 h, was analyzed by immunoblotting with antibody against EGFP (i) and densitometry (ii). (D) Clearance of A53T (i) and A30P (ii) α-synucleins in stable inducible PC12 cells as in Fig. 2, A and B, treated with (+) or without (−) 100 μM L-690,330 for 24 h, was analyzed by immunoblotting with antibody against HA. (E) Clearance of soluble EGFP-HDQ74 in stable inducible PC12 cell lines as in Fig. 1 C, treated with (+) or without (−) 10 μM SB216763 for 170 h, was analyzed by immunoblotting with antibody against EGFP (i) and densitometry (ii). (F) Clearance of A53T (i) and A30P (ii) α-synucleins in stable inducible PC12 cells as in Fig. 2, A and B, treated with (+) or without (−) 10 μM SB216763 for 24 h, was analyzed by immunoblotting with antibody against HA. ***, P < 0.001; NS, non-significant.
Lithium induces autophagy by inhibiting IMPase
We tested if the enhanced clearance of these proteins mediated by L-690,330 was by autophagy or the proteasomal route, using inhibitors of autophagy (3-MA) and the proteasome (lactacystin). Both of these inhibitors reduced clearance of A53T α-synuclein in the PC12 stable line (Fig. 4, A and B), and increased EGFP-HDQ74 aggregates in COS-7 cells (Fig. 4, C and D); this was consistent with our previous observations that these proteins are cleared by autophagy and the proteasome (Ravikumar et al., 2002; Webb et al., 2003). When autophagy was inhibited by 3-MA, L-690,330 could not facilitate clearance of A53T α-synuclein or reduce EGFP-HDQ74 aggregates (Fig. 4, A and C). However, cells that were treated with the proteasome inhibitor, lactacystin, and L-690,330 had enhanced clearance of A53T α-synuclein and significantly reduced EGFP-HDQ74 aggregates, compared with cells that were treated with lactacystin alone (Fig. 4, B and D). This suggested that the enhanced clearance of these proteins was mediated by IMPase inhibition by way of a nonproteasomal route. We confirmed that L-690,330 enhanced clearance of these proteins through the autophagic route by showing more LC3-stained autophagic vesicles in COS-7 cells after L-690,330 treatment (Fig. 4 E). Furthermore, L-690,330 also decreased mitochondrial load as judged by complex IV (Fig. 4 F) and cytochrome c (not depicted) levels in COS-7 cells. Accordingly, the IMPase inhibitory activity of lithium can account for its induction of autophagy.
Figure 4.
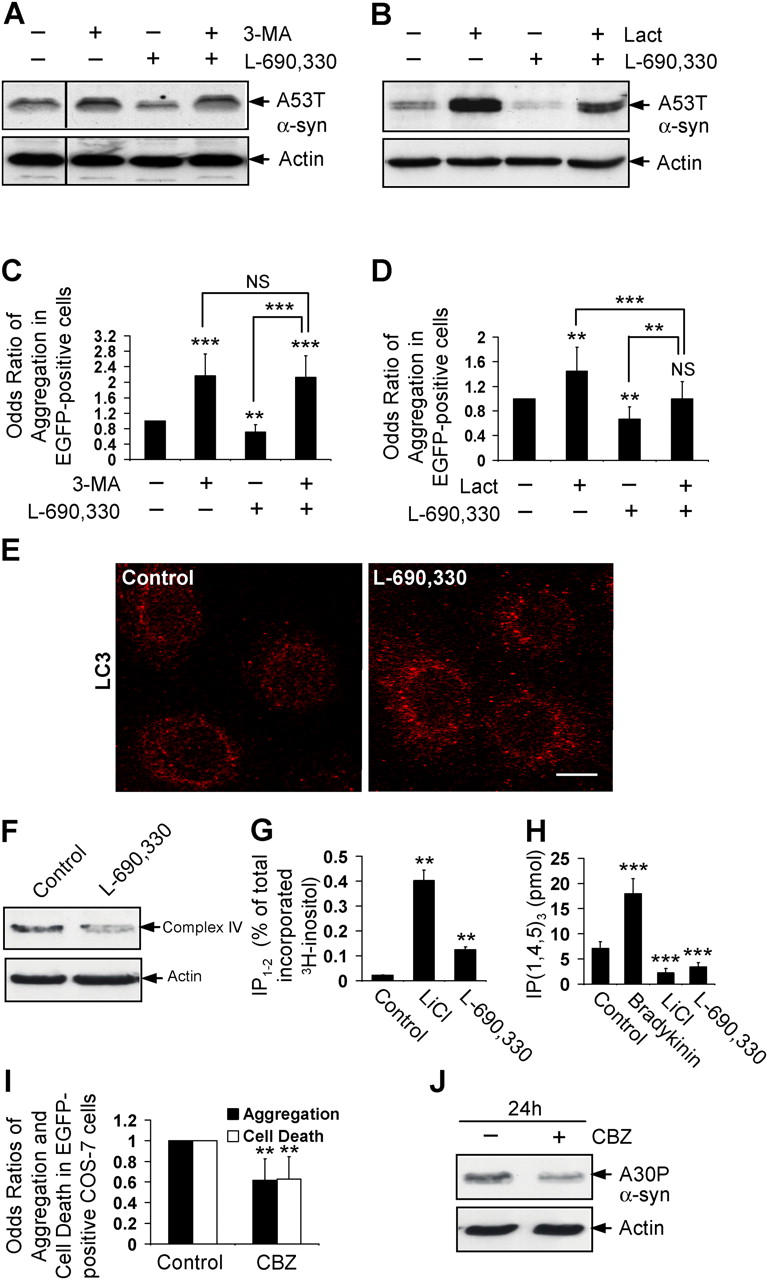
Lithium induces autophagy by inositol monophosphatase inhibition. Clearance of A53T α-synuclein in stable inducible PC12 cells as in Fig. 2 A, treated with (+) or without (−) 10 mM 3-MA, 100 μM L-690,330, or both (A), or 10 μM lactacystin (Lact), 100 μM L-690,330, or both (B) for 24 h, was analyzed by immunoblotting with antibody against HA. The percentage of EGFP-HDQ74–positive cells with aggregates in COS-7 cells as in Fig. 1 A, treated with (+) or without (−) 10 mM 3-MA, 100 μM L-690,330, or both (C), or 10 μM lactacystin (Lact), 100 μM L-690,330, or both (D) for 48 h, were expressed as odds ratio. (E) COS-7 cells treated with (+) or without (−) 100 μM L-690,330 for 24 h were analyzed by immunofluorescence with antibody against LC3 using a confocal microscope. Bar, 20 μm. (F) COS-7 cells were treated with or without 100 μM L-690,330 for 170 h, and mitochondrial load was assessed by immunoblotting with antibody to complex IV. (G) Levels of IP1-2 were measured in COS-7 cells treated with 10 mM LiCl or 100 μM L-690,330 for 24 h. (H) Levels of IP3 were measured in COS-7 cells treated with 2 μM bradykinin, 10 mM LiCl, or 100 μM L-690,330 for 5 min. (I) The percentage of EGFP-HDQ74–positive cells with aggregates and cell death in COS-7 cells as in Fig. 1 A, treated with or without 50 μM CBZ for 48 h, were expressed as odds ratios. (J) Clearance of A30P α-synuclein in stable inducible PC12 cells as in Fig. 2 B, treated with (+) or without (−) 50 μM CBZ for 24 h, was analyzed by immunoblotting with antibody against HA. **, P < 0.01; ***, P < 0.001; NS, non-significant.
We confirmed that lithium and L-690,330 were inhibiting IMPase in our experimental conditions, because they increased levels of inositol mono- and bis-phosphate (IP1-2) (Fig. 4 G) and decreased IP3 levels (Fig. 4 H), a consequence of reduced free inositol (Berridge et al., 1989). Lithium significantly reduced IP3 levels at time points from 1 min to 24 h (Fig. S4 A; available at http://www.jcb.org/cgi/content/full/jcb.200504035/DC1).
Mood-stabilizing drugs facilitate clearance of mutant proteins
Inositol depletion is a common mechanism for mood stabilizing drugs like lithium, carbamazepine (CBZ), and valproic acid (VPA) (Williams et al., 2002). Consistent with a role for inositol depletion in autophagy regulation, CBZ significantly reduced EGFP-HDQ74 aggregates and attenuated polyglutamine toxicity in COS-7 cells (Fig. 4 I), and enhanced clearance of A30P α-synuclein (Fig. 4 J). Similar results were seen with VPA (unpublished data). Thus, drugs that can deplete intracellular inositol may be of therapeutic value in HD and related neurodegenerative diseases.
The effect of lithium on clearance of mutant proteins is regulated by IP3 levels
To determine the importance of inositol levels in autophagy regulation, we tested if the effect of lithium on autophagy could be overcome by the addition of extracellular inositol in the form of myo-inositol (Williams et al., 2002). We also used an inhibitor of prolyl oligopeptidase activity, called prolyl endopeptidase inhibitor 2 (PEI), which elevates intracellular IP3 and abolishes some other effects of lithium (Williams et al., 1999) (Fig. S4 B). Lithium may deplete inositol by inhibiting IMPase and by decreasing the transport of myo-inositol into cells (Lubrich and van Calker, 1999). Accordingly, we pretreated cells with myo-inositol and PEI before adding lithium. As predicted, myo-inositol and PEI pretreatment raised IP3 levels in COS-7 cells that were treated with lithium, compared with lithium treatment alone (Fig. 5 A). Myo-inositol and PEI significantly reversed the protective effect of lithium on EGFP-HDQ74–induced aggregation and cell death in COS-7 cells (Fig. 5 B). (The failure of myo-inositol and PEI to reverse completely the lithium effect on EGFP-HDQ74 aggregation at 48 h [Fig. 5 B]—despite their effects on IP3 levels at 5 min [Fig. 5 A]—can be explained by the different timings of these observations.) These compounds also inhibited the effect of lithium on the clearance of soluble EGFP-HDQ74 (Fig. 5 C) and A53T α-synuclein (Fig. 5 D) in stable PC12 cell lines.
Figure 5.
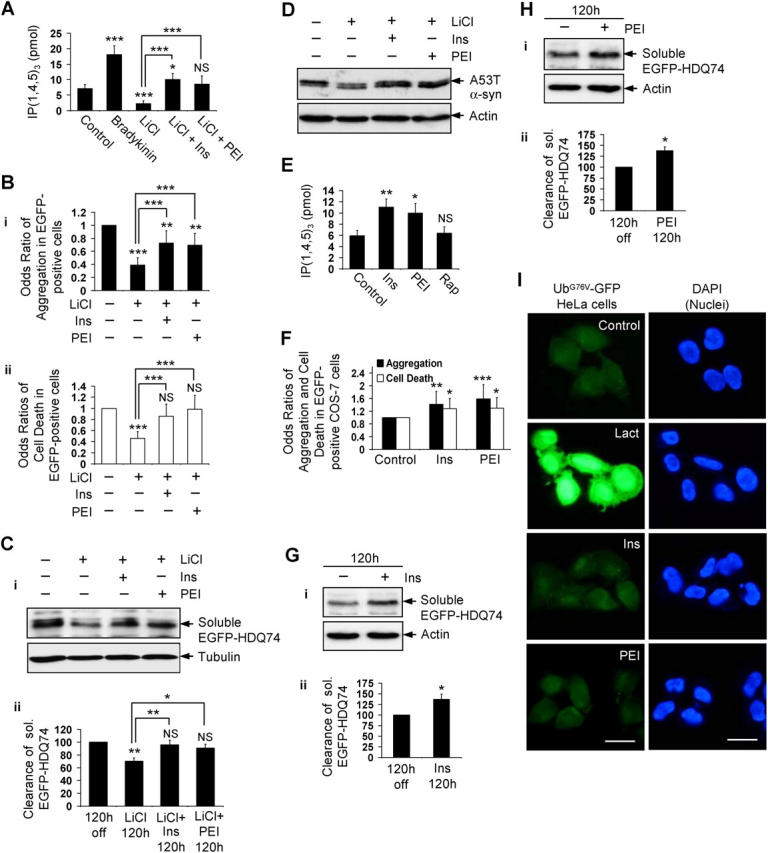
Lithium accelerates clearance of mutant huntingtin fragment by reducing free inositol and IP 3 levels. (A) IP3 levels were measured in COS-7 cells treated for 5 min with or without 2 μM bradykinin, 10 mM LiCl, or 10 mM LiCl pretreated for 5 min with 1 mM myo-inositol (Ins) or 24 μM prolyl endopeptidase inhibitor 2 (PEI). (B) The percentage of EGFP-HDQ74–positive cells with aggregates (i) and cell death (ii) in COS-7 cells as in Fig. 1 A, either left untreated or treated with 10 mM LiCl with (+) or without (−) 1mM myo-inositol or 24 μM PEI for 48 h, were expressed as odds ratios. (C) Clearance of soluble EGFP-HDQ74 in stable PC12 cells as in Fig. 1 C, either left untreated or treated with 10 mM LiCl with (+) or without (−) 1 mM myo-inositol or 24 μM PEI for 120 h, was analyzed by immunoblotting with antibody against EGFP (i) and densitometry (ii). (D) Clearance of A53T α-synuclein in stable PC12 cells as in Fig. 2 A, either left untreated or treated with 10 mM LiCl with (+) or without (−) 1 mM myo-inositol or 24 μM PEI for 24 h, was analyzed by immunoblotting with antibody against HA. (E) IP3 levels were measured in COS-7 cells treated with or without 1 mM myo-inositol, 24 μM PEI or 0.2 μM rapamycin (Rap) for 5 min. (F) The percentage of EGFP-HDQ74–positive cells with aggregates and cell death in COS-7 cells as in Fig. 1 A, treated with or without 1 mM myo-inositol or 24 μM PEI for 48 h, were expressed as odds ratios. Clearance of soluble EGFP-HDQ74 in stable PC12 cells as in Fig. 1 C, treated with (+) or without (−) 1 mM myo-inositol (G) or 24 μM PEI (H) for 120 h, was analyzed by immunoblotting with antibody against EGFP (i) and densitometry (ii). (I) HeLa cells expressing UbG76V-GFP reporter, treated with or without 10 μM lactacystin (Lact), 1 mM myo-inositol, or 24 μM PEI for 24 h, were analyzed by fluorescence microscopy. Bar, 20 μm. *, P < 0.05; **, P < 0.01; ***, P < 0.001; NS, non-significant; Ins, myo-inositol.
Single treatments of myo-inositol or PEI, which each increased intracellular IP3 (Fig. 5 E), increased EGFP-HDQ74 aggregates and cell death in COS-7 cells (Fig. 5 F) and significantly inhibited the clearance of soluble EGFP-HDQ74 (Fig. 5, G and H). We next tested whether myo-inositol or PEI inhibited proteasome activity using HeLa cells stably expressing the UbG76V-GFP reporter (Dantuma et al., 2000), a fluorescent-specific proteasome substrate. Treatment with the proteasome inhibitor, lactacystin, led to massive accumulation of the fluorescent substrate, whereas myo-inositol or PEI had no effect on its levels, compared with the untreated (control) cells (Fig. 5 I). We showed above that lithium (which has IMPase inhibitory activity) and an IMPase inhibitor reduced intracellular IP3 and induced autophagy (Fig. 2, D–F; Fig. 4, E, F, and H). Because increasing intracellular IP3 inhibited the clearance of mutant huntingtin, these data suggest that lithium is inducing autophagy by virtue of its IMPase inhibitory activity, acting at the level of (or downstream of) lowered IP3.
Lithium and IMPase inhibitor induce autophagy independently of mTOR inhibition
We were interested if this novel means of pharmacologic regulation of autophagy was independent of the known pathway that is regulated negatively by mTOR. The activity of mTOR, a protein kinase, can be inferred by the levels of phosphorylation of its substrates, ribosomal S6 protein kinase (S6K1, also known as p70S6K) and eukaryotic initiation factor 4E-binding protein 1 (4E-BP1) at Thr389 and Thr37/46, respectively (Fig. S5 A; available at http://www.jcb.org/cgi/content/full/jcb.200504035/DC1), and the phosphorylation of the ribosomal S6 protein (S6P), a substrate of S6K1 (Schmelzle and Hall, 2000). Whereas rapamycin (a specific mTOR inhibitor) reduced phosphorylation of S6K1, S6P, and 4E-BP1 in COS-7 cells as expected, lithium or L-690,330 did not reduce their phosphorylation (Fig. 6, A–D; Fig. S5 B), which suggests that their effects are independent of mTOR inhibition.
Figure 6.
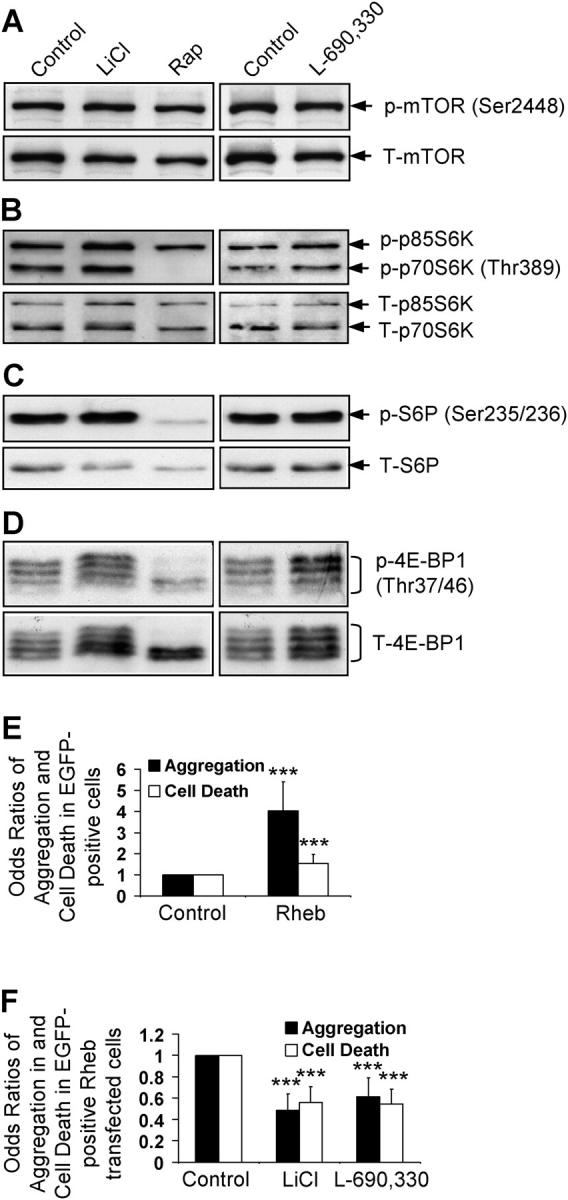
Lithium and IMPase inhibitor do not impair mTOR signaling. COS-7 cells treated with or without 10 mM LiCl, 0.2 μM rapamycin (Rap), or 100 μM L-690,330 for 24 h, were analyzed for mTOR activity by immunoblotting for levels of phospho and total mTOR (A), ribosomal S6 protein kinase (B), S6P (C), and 4E-BP1 (D). (E and F) COS-7 cells transfected with pEGFP-HDQ74 along with empty vector (pCDNA3.1) or pRheb at 1:3 ratio (E). The rheb-transfected cells were treated with or without 10 mM LiCl or 100 μM L-690,330; the control represents untreated rheb-transfected cells (F). The proportion of GFP-positive cells with aggregates or cell death were assessed after 48 h and expressed as odds ratios. ***, P < 0.001.
Overexpression of the small G-protein rheb, which greatly enhances mTOR signaling (Manning and Cantley, 2003), markedly increased EGFP-HDQ74 aggregates and cell death in COS-7 cells (Ravikumar et al., 2004) (Fig. 6 E). However, lithium or L-690,330 reduced EGFP-HDQ74 aggregates and cell death in rheb-transfected cells (Fig. 6 F); this suggested that induction of autophagy by IMPase inhibition occurs even when mTOR is activated.
The effect of IP3 on autophagy is unlikely to be a downstream consequence of mTOR inhibition, because rapamycin had no effect on IP3 levels (Fig. 5 E). Furthermore, neither myo-inositol nor PEI abolished the protective effect of rapamycin on polyglutamine toxicity in COS-7 cells (Fig. 7 A), and the increased clearance of soluble EGFP-HDQ74 and A53T α-synuclein in stable PC12 cell lines (Fig. 7, B and C), in contrast to what we observed in the context of lithium. Therefore, rapamycin and intracellular inositol seem to regulate autophagy independently.
Figure 7.
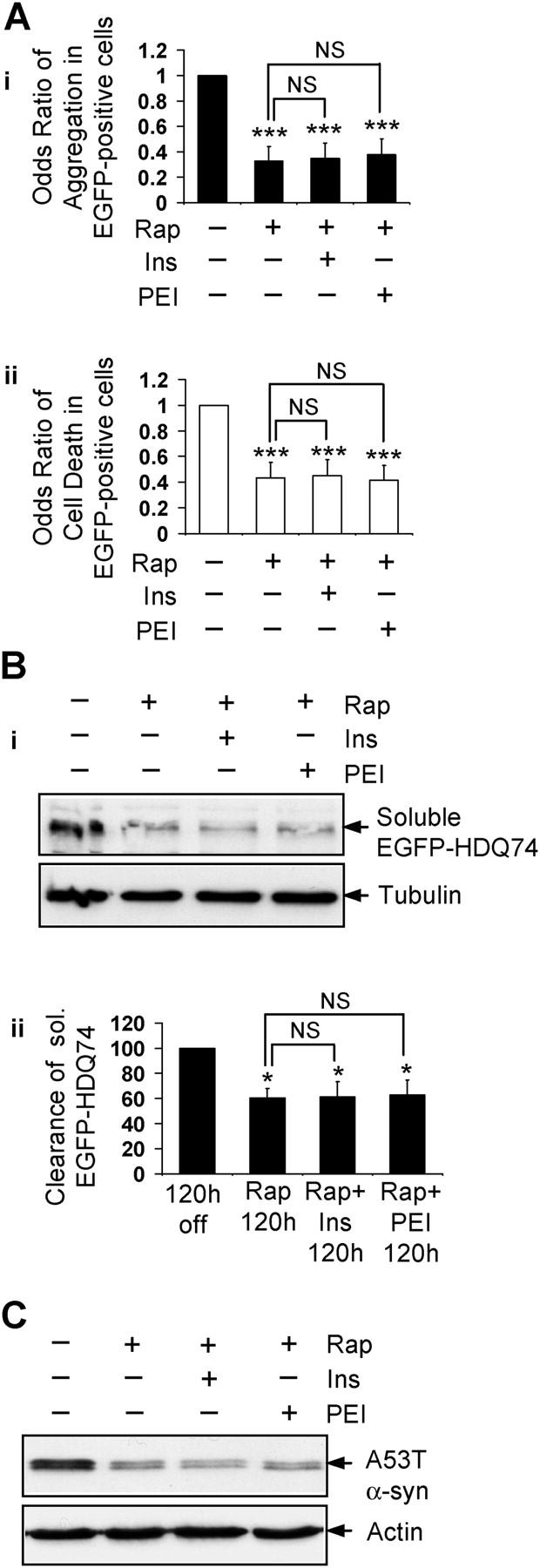
The ability of rapamycin to enhance clearance of mutant huntingtin and α-synuclein is not impaired by increasing intracellular IP 3 . (A) The percentage of EGFP-HDQ74-positive cells with aggregates (i) and cell death (ii) in COS-7 cells as in Fig. 1 A, either left untreated or treated with 0.2 μM rapamycin with (+) or without (−) 1 mM myo-inositol (Ins) or 24 μM PEI for 48 h, were expressed as odds ratios. (B) Clearance of soluble EGFP-HDQ74 in stable PC12 cells as in Fig. 1 C, either left untreated or treated with 0.2 μM rapamycin with (+) or without (−) 1 mM myo-inositol or 24 μM PEI for 120 h, was analyzed by immunoblotting with antibody against EGFP (i) and densitometry (ii). (C) Clearance of A53T α-synuclein in stable PC12 cells as in Fig. 2 A, either left untreated or treated with 0.2 μM rapamycin (Rap) with (+) or without (−) 1 mM myo-inositol or 24 μM PEI for 24 h, was analyzed by immunoblotting with antibody against HA. *, P < 0.05; ***, P < 0.001; NS, non-significant; Ins, myo-inositol.
Inhibition of IMPase and mTOR additively enhance the clearance of mutant huntingtin and α-synuclein
We confirmed the prediction that lithium and rapamycin should have additive effects in reducing EGFP-HDQ74 aggregates and cell death in COS-7 cells (Fig. 8 A), compared with the single treatments of lithium or rapamycin. Furthermore, lithium and rapamycin together facilitated greater clearance of soluble EGFP-HDQ74 at 72 h (Fig. 8 B) and A53T α-synuclein at 8 h (Fig. 8 C), compared with either compound alone. To demonstrate this effect clearly we chose early time points at which we do not observe obvious reductions of the levels of these proteins when the cells are treated with either of the compounds alone. We have confirmed that the rapamycin concentration that was used in these experiments is close to saturating, because a further increase in the rapamycin dose did not lead to more clearance of soluble EGFP-HDQ74 (Fig. 8 D).
Figure 8.

Induction of autophagy by two independent pathways has an additive effect on the clearance of mutant proteins. (A) The percentage of EGFP-HDQ74–positive cells with aggregates (i) and cell death (ii) in COS-7 cells as in Fig. 1 A, treated with (+) or without (−) 10 mM LiCl, 0.2 μM rapamycin (Rap), or both for 48 h, were expressed as odds ratios. (B) Clearance of soluble EGFP-HDQ74 in stable PC12 cells as in Fig. 1 C, treated with (+) or without (−) 10 mM LiCl, 0.2 μM rapamycin (Rap), or both for 72 h, was analyzed by immunoblotting with antibody against EGFP (i) and densitometry (ii). (C) Clearance of A53T α-synuclein in stable PC12 cells as in Fig. 2 A, treated with (+) or without (−) 10 mM LiCl, 0.2 μM rapamycin (Rap), or both for 8 h, was analyzed by immunoblotting with antibody against HA. (D) Clearance of soluble EGFP-HDQ74 in stable PC12 cells as in Fig. 1 C, treated with (+) or without (−) 0.1, 0.2, or 0.4 μM rapamycin (Rap) for 72 h, was analyzed by immunoblotting with antibody against EGFP. (E) The percentage of EGFP-HDQ74–positive cells with aggregates (i) and cell death (ii) in COS-7 cells as in Fig. 1 A, treated with (+) or without (−) 100 μM L-690,330, 0.2 μM rapamycin (Rap), or both for 48 h, were expressed as odds ratios. (F) Clearance of soluble EGFP-HDQ74 in stable PC12 cells as in Fig. 1 C, treated with (+) or without (−) 100 μM L-690,330, 0.2 μM rapamycin (Rap), or both for 72 h, was analyzed by immunoblotting with antibody against EGFP (i) and densitometry (ii). (G) Clearance of A53T α-synuclein in stable PC12 cells as in Fig. 2 A, treated with (+) or without (−) 100 μM L-690,330, 0.2 μM rapamycin (Rap), or both for 8 h, was analyzed by immunoblotting with antibody against HA. *, P < 0.05; **, P < 0.01; ***, P < 0.001; NS, non-significant.
Consistent with the above observations, the combination of L-690,330 and rapamycin had an enhanced protective effect on EGFP-HDQ74–mediated toxicity in COS-7 cells (Fig. 8 E), and facilitated greater clearance of soluble EGFP-HDQ74 and A53T α-synuclein at early time points in stable PC12 cell lines, compared with the individual treatments (Fig. 8, F and G). Thus, two independent pathways of inducing autophagy in mammalian cells have an additive effect, which is reflected by the greater clearance of mutant proteins because of simultaneous inhibition of mTOR and IMPase. This provides a new direction for combinatorial treatment of neurodegenerative disorders (e.g., HD) by enhancing autophagy by two different routes.
Discussion
Lithium induces autophagy and enhances clearance of aggregate-prone proteins, like mutant huntingtin and A53T, and A30P mutants of α-synuclein in PC12, SK-N-SH (both neuronal precursor cell lines), and COS-7 (nonneuronal) cells at different time points (Fig. 1, A–C, E, and F; Fig. S1, A–C; Fig. 2, A, B, and D–F). These aggregate-prone proteins are distinct confirmed autophagy substrates (Ravikumar et al., 2002; Webb et al., 2003). This effect is not mediated by GSK-3β inhibition (Fig. 3, A, B, E, and F), but is a consequence of IMPase inhibition, because an IMPase inhibitor, L-690,330, mediates similar effects (Fig. 3, A–D; Fig. 4, E and F). This assertion was confirmed further by showing that the effects of lithium on the clearance of EGFP-HDQ74 and mutant α-synuclein were abrogated with myo-inositol (Fig. 5, A–D), which increases intracellular IP3 (Fig. 5 E). Furthermore, increasing the levels of IP3 by using PEI (Fig. 5 E) also abolished the effect of lithium on the clearance of these mutant proteins (Fig. 5, A–D). Moreover, free inositol (myo-inositol) inhibited clearance of autophagy substrates (Fig. 5, F and G), which provided further support for the role of IMPase as a regulator of autophagy, because this enzyme decreases the free inositol pool. Decreased free inositol leads to decreased IP3 levels (Berridge et al., 1989). Our data suggest that the autophagy effect is mediated at the level of (or downstream of) lowered IP3, because it was abrogated by PEI that increased intracellular levels of IP3 (Fig. 5, E, F, and H). This represents a novel pathway of mammalian autophagy regulation. It will be extremely challenging to characterize the molecular mechanism whereby IP3 regulates autophagy, because the machinery that regulates the rate of mammalian autophagy under physiologic conditions is poorly understood. It is still not clear how mTOR regulates mammalian autophagy.
Lithium is a major therapy for affective disorders (Manji and Lenox, 1998). It takes 3–4 wk to stabilize mood, although brain inositol levels in patients who have bipolar disorder are reduced by lithium much sooner after treatment is initiated (Moore et al., 1999). Because autophagy modulates the half-lives of many long-lived proteins (Klionsky and Emr, 2000; Ravikumar et al., 2002, 2004; Webb et al., 2003), it is tempting to speculate that part of the therapeutic efficacy of lithium and related drugs (e.g., CBZ and valproate) may be mediated by clearance of very long half-life autophagy substrates that may contribute to the disease. Steady-state levels of some long half-life proteins could take weeks (many multiples of the half-life) to change in a biologically significant way after the half-life is shortened. This is a plausible explanation for why mood-stabilizing drugs have acute effects on free inositol levels long before they begin to mediate clinical effects. The different lag times that are seen with various mood-stabilizing drugs may be related to their pharmacokinetics—the fact that these drugs may deplete intracellular inositol by different mechanisms (Gould et al., 2002; Ju et al., 2004; Shaltiel et al., 2004) and/or effects on other target enzymes that are unrelated to autophagy. This speculative model does not mean necessarily that abnormal protein degradation is a feature of affective disorders. It is tempting to speculate whether trials of rapamycin or related autophagy-inducing drugs may be worthwhile for mood disorders, given the lack of understanding of their biology and the enormous patient need.
Although the inositol depletion hypothesis may be one of the most widely accepted hypotheses for lithium's actions in bipolar affective disorder (Silverstone et al., 2005), it remains controversial. In rodents, lithium (and valproate) reduces myo-inositol levels and increases IP1 concentrations in the brain (Silverstone et al., 2005). Although the studies in humans have been more difficult to interpret, this may be due, in part, to technical limitations of magnetic resonance spectroscopy methods in humans, and sample size and clinical heterogeneity issues (for review see Silverstone et al., 2005). Nevertheless, the meta-analysis of data from manic and depressed studies generally is consistent with the inositol depletion hypothesis of lithium action in these disorders (Silverstone et al., 2005). It is clearly outside the realm of this study to resolve the controversy of the inositol depletion hypothesis for lithium. Note also that lithium can reduce inositol levels by two mechanisms. The most commonly considered is by inhibition on inositol monophosphatase. However, lithium also inhibits inositol uptake into cells (Lubrich and van Calker, 1999). We performed most studies using 10 mM lithium, following the precedent of Williams et al. (1999). Although this is higher than the desired concentration in humans, we were able to show that the autophagy effects at this dose were mediated specifically by inositol depletion, because they were attenuated by myo-inositol and prolyl endopeptidase inhibition. We previously showed that 2.5 mM lithium reduced polyglutamine aggregation and toxicity in cell models (Carmichael et al., 2002). Furthermore, 1 mM lithium (approximating the therapeutic levels in humans [Camus et al., 2003]) induces autophagy (Fig. S2, A and B). Even if lithium turns out to be not very effective at inducing autophagy in humans, similar effects may be achieved with other drugs that reduce inositol levels, like valproate or CBZ (Williams et al., 2002) (Fig. 4, I and J; not depicted).
These issues do not detract from our fundamental cell biologic elucidation of a novel mTOR-independent autophagy regulatory pathway. This may be of potential value in neurodegenerative diseases that are caused by aggregate-prone proteins, such as HD (Ravikumar et al., 2004). Some benefit was reported with lithium in a mouse model of HD (Wood and Morton, 2003). Combination therapy with more moderate IMPase and mTOR inhibition may be safer for long-term treatment than using doses of either inhibitor that result in more severe perturbation of a single pathway.
Materials and methods
Plasmids
HD gene exon 1 fragment with 74 polyglutamine repeats (Q74) in pEGFP-C1 (CLONTECH Laboratories, Inc.) was described and characterized previously (Narain et al., 1999). EGFP-LC3 (gift from T. Yoshimori, National Institute of Genetics, Mishima, Shizouka, Japan) and rheb (gift from K.L. Guan, University of Michigan, Ann Arbor, MI) constructs were obtained.
Mammalian cell culture and transfection
African green monkey kidney cells (COS-7) and human neuroblastoma cells (SK-N-SH) were maintained in DME (Sigma-Aldrich) supplemented with 10% FBS (Sigma-Aldrich), 100 U/ml penicillin/streptomycin, and 2 mM l-glutamine (Sigma-Aldrich) at 37°C, 5% CO2. Cells were plated in six-well dishes at a density of 105 cells per well for 24 h, and transfected with pEGFP-HDQ74 using LipofectAMINE reagent for COS-7 cells and LipofectAMINE PLUS reagent for SK-N-SH according to the manufacturer's protocol (Invitrogen). Transfection mixture was replaced after 4 h incubation at 37°C by various compounds, such as 10 mM LiCl, 0.2 μM rapamycin, 10 mM 3-MA, 10 μM lactacystin, 50 μM CBZ, 1 mM myo-inositol (all from Sigma-Aldrich), 10 mM NaCl (BDH), 10 μM SB216763 (Tocris), 100 μM L-690,330 (Tocris), or 24 μM prolyl endopeptidase inhibitor 2 (Z-PP-CHO, Calbiochem). Transfected cells were fixed with 4% paraformaldehyde (Sigma-Aldrich) after 48 h and mounted in DAPI (3 mg/ml; Sigma-Aldrich) over coverslips on glass slides and analyzed for aggregation and cell death. For immunoblotting, cells were plated at a density of 3 × 105 cells per well. HeLa cells stably expressing UbG76V-GFP reporter (gift from N.P. Dantuma, Karolinska Institutet, Stockholm, Sweden) were grown in similar media used for growing COS-7 cells, which is supplemented with 0.5 mg/ml G418.
Inducible PC12 stable cell lines expressing EGFP-tagged exon 1 of HD gene (EGFP-HDQ74) (Wyttenbach et al., 2001) and HA-tagged A53T and α-synuclein mutants (Webb et al., 2003), were maintained at 75 μg/ml hygromycin B (Calbiochem) in standard DME with 10% horse serum (Sigma-Aldrich), 5% FBS, 100 U/ml penicillin/streptomycin, 2 mM l-glutamine, and 100 μg/ml G418 (GIBCO BRL) at 37°C, 10% CO2.
Quantification of aggregate formation and cell death
∼200 EGFP-positive cells were counted for proportion of cells with aggregates, as described previously (Narain et al., 1999). Nuclei were stained with DAPI; those that showed apoptotic morphology (fragmentation or pyknosis) were considered abnormal. These criteria are specific for cell death, and correlate highly with propidium iodide staining in live cells (Wyttenbach et al., 2002). Analysis was performed using a Nikon Eclipse E600 fluorescence microscope (plan-apo 60×/1.4 oil immersion lens at room temperature); the observer was blinded to identity of slides. Slides were coded and the code was broken after completion of the experiment. All experiments were done in triplicate at least twice. Images were acquired with a Nikon Digital Camera DXM1200 using Nikon Eclipse E600 fluorescence microscope (40× plan-fluor 40x/0.75 lens or plan-apo 60×/1.4 oil immersion lens at room temperature). Acquisition software was Nikon ACT-1 version 2.12; Adobe Photoshop 6.0 (Adobe Systems, Inc.) was used for subsequent image processing.
Clearance of mutant huntingtin and α-synucleins
Stable inducible PC12 cell lines expressing EGFP-HDQ74, or A53T or A30P α-synuclein mutants, were plated at 3 × 105 per well in six-well dishes and induced with 1 μg/ml doxycycline (Sigma-Aldrich) for 8 h and 48 h, respectively. Expression of transgenes was switched off by removing doxycycline from the medium (Ravikumar et al., 2002; Webb et al., 2003); cells were treated with or without various compounds for 120 h for EGFP-HDQ74 clearance and 24 h for mutant α-synuclein clearance. For additive effect of LiCl or L-690,330 with rapamycin, treatment was done for 72 h for EGFP-HDQ74 clearance and 8 h for mutant α-synuclein clearance. The medium with various compounds was changed every 24 h. Cells were fixed and mounted in DAPI, or processed for immunoblotting analysis with EGFP for soluble EGFP-HDQ74 clearance or HA for mutant α-synuclein clearance.
Western blot analysis
Cell pellets were lysed on ice in Laemmli buffer (62.5 mM Tris-HCl pH 6.8, 5% β-mercaptoethanol, 10% glycerol, and 0.01% bromophenol blue) for 30 min in the presence of protease inhibitors (Roche Diagnostics). Samples were subjected to SDS-PAGE, and proteins were transferred to nitrocellulose membrane (GE Healthcare). Primary antibodies used include anti-EGFP (8362–1, CLONTECH Laboratories, Inc.), anti-HA (12CA5, Covance), anti-mTOR (2972), anti–phospho-mTOR (Ser2448) (2971), anti-p70 S6 kinase (9202), anti–phospho-p70 S6 kinase (Thr389) (9206), anti–4E-BP1 (9452), anti–phospho-4E-BP1 (Thr37/46) (9459), anti-S6 ribosomal protein (2212), and anti–phospho-S6 ribosomal protein (Ser235/236) (2211), all from Cell Signaling Technology; anti-complex IV subunit IV (A-21348, Molecular Probes), anti-LC3 (gift from T. Yoshimori), anti-actin (A2066, Sigma-Aldrich), and anti-tubulin (clone DM 1A, Sigma-Aldrich). Blots were probed with anti–mouse or anti–rabbit IgG-HRP (GE Healthcare) and visualized using ECL or ECL Plus detection kit (GE Healthcare).
Immunocytochemistry
COS-7 cells were fixed with 4% paraformaldehyde. Primary antibodies included anti-LC3 (gift from T. Yoshimori, National Institute of Genetics, Japan) and anti–phospho-S6 ribosomal protein (Ser235/236) (2211, Cell Signaling Technology). Standard fluorescence method was used for detection; secondary antibodies used were goat anti–rabbit Alexa 488 Green and Alexa 594 Red (Cambridge Biosciences). Images were acquired on a Zeiss LSM510 META confocal microscope (63× 1.4NA plan-apochromat oil immersion) at room temperature using Zeiss LSM510 v3.2 software; Adobe Photoshop 6.0 (Adobe Systems, Inc.) was used for subsequent image processing.
Inositol phosphate measurements
IP1-2 were assayed as described previously (Harnett and Harnett, 1993). COS-7 cells were labeled with myo-[3H]inositol (1 μCi/106 cells) for 24 h, stimulated, and then subjected to chloroform/methanol (1:2) extraction followed by Bligh-Dyer phase separation. Levels of IP1-2 were determined by liquid scintillation counting of fractions eluted following Dowex (formate form) ion exchange chromatography of aliquots of the aqueous phase. Results were calculated as a percentage of total incorporated radioactivity. Levels of inositol-1,4,5-trisphosphate [IP3(1,4,5)] were measured in perchloric acid extracted samples (107 cells/aliquot) using the [3H] Biotrak Assay System (GE Healthcare) according to the manufacturer's instructions. Data are presented as mean ± SD of triplicate measurements and are representative of at least three separate experiments.
Statistical analysis
Pooled estimates for the changes in aggregate formation or cell death, resulting from perturbations assessed in multiple experiments, were calculated as odds ratios with 95% confidence intervals. We have used this method frequently in the past to allow analysis of data from multiple independent experiments (Wyttenbach et al., 2001, 2002; Carmichael et al., 2002). Odds ratios and P values were determined by unconditional logistical regression analysis, using the general log-linear analysis option of SPSS 9 software. Densitometry analysis was done by Scion Image Beta 4.02 software on immunoblots from three independent experiments. Significance for clearance of mutant proteins was determined by factorial ANOVA test using STATVIEW software, version 4.53 (Abacus Concepts) and the error bar denotes standard error of the mean.
Online supplemental material
Fig. S1 shows lithium facilitated clearance of mutant huntingtin aggregates, an effect not seen with NaCl. Fig. S2 shows lithium-induced autophagy to facilitate clearance of mutant proteins. Fig. S3 shows a schematic diagram of some molecular targets of lithium. Fig. S4 shows aspects of the inositol cycle pertinent to lithium. Fig. S5 shows the mTOR pathway in relation to autophagy. Online supplemental material available at http://www.jcb.org/cgi/content/full/jcb.200504035/DC1.
Acknowledgments
We thank T. Yoshimori for the antibody to LC3 and EGFP-LC3 construct, K.L. Guan for rheb construct, N.P. Dantuma for UbG76V-GFP HeLa cells, and C. O'Kane for valuable input. We would like to thank B. Ravikumar and members of D.C. Rubinsztein's laboratory for technical assistance and helpful discussions.
We are grateful for Gates Cambridge Scholarship (S. Sarkar), Wellcome Trust Senior Fellowship in Clinical Science (D.C. Rubinsztein), Academy of Medical Sciences (A.M.S.)/Medical Research Council (M.R.C.) Clinical Scientist Fellowship (R.A. Floto), a Prize Studentship (Z. Berger), and Overseas Research Students Award (Z. Berger). This work was supported by an M.R.C. program grant and by E.U. Framework VI (EUROSCA).
Abbreviations used in this paper: 3-MA, 3-methyladenine; 4E-BP1, eukaryotic initiation factor 4E-binding protein 1; CBZ, carbamazepine; EGFP-HDQ74, EGFP-tagged huntingtin exon 1 with 74 polyglutamine repeats; GSK-3β, glycogen synthase kinase 3β; HD, Huntington's disease; IMPase, inositol monophosphatase; IP1-2, inositol mono- and bis-phosphate; IP3, myo-inositol-1,4,5-triphosphate; LC3, microtubule-associated protein 1 light chain 3; mTOR, mammalian target of rapamycin; PD, Parkinson's disease; PEI, prolyl endopeptidase inhibitor; S6P, ribosomal S6 protein.
References
- Atack, J.R., S.M. Cook, A.P. Watt, S.R. Fletcher, and C.I. Ragan. 1993. In vitro and in vivo inhibition of inositol monophosphatase by the bisphosphonate L-690,330. J. Neurochem. 60:652–658. [DOI] [PubMed] [Google Scholar]
- Berridge, M.J., C.P. Downes, and M.R. Hanley. 1989. Neural and developmental actions of lithium: a unifying hypothesis. Cell. 59:411–419. [DOI] [PubMed] [Google Scholar]
- Camus, M., G. Hennere, G. Baron, G. Peytavin, L. Massias, F. Mentre, and R. Farinotti. 2003. Comparison of lithium concentrations in red blood cells and plasma in samples collected for TDM, acute toxicity, or acute-on-chronic toxicity. Eur. J. Clin. Pharmacol. 59:583–587. [DOI] [PubMed] [Google Scholar]
- Carmichael, J., K.L. Sugars, Y.P. Bao, and D.C. Rubinsztein. 2002. Glycogen synthase kinase-3β inhibitors prevent cellular polyglutamine toxicity caused by the Huntington's disease mutation. J. Biol. Chem. 277:33791–33798. [DOI] [PubMed] [Google Scholar]
- Coghlan, M.P., A.A. Culbert, D.A. Cross, S.L. Corcoran, J.W. Yates, N.J. Pearce, O.L. Rausch, G.J. Murphy, P.S. Carter, L. Roxbee Cox, et al. 2000. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem. Biol. 7:793–803. [DOI] [PubMed] [Google Scholar]
- Coyle, J.T., and R.S. Duman. 2003. Finding the intracellular signaling pathways affected by mood disorder treatments. Neuron. 38:157–160. [DOI] [PubMed] [Google Scholar]
- Cuervo, A.M., L. Stefanis, R. Fredenburg, P.T. Lansbury, and D. Sulzer. 2004. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 305:1292–1295. [DOI] [PubMed] [Google Scholar]
- Dantuma, N.P., K. Lindsten, R. Glas, M. Jellne, and M.G. Masucci. 2000. Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat. Biotechnol. 18:538–543. [DOI] [PubMed] [Google Scholar]
- Gould, T.D., G. Chen, and H.K. Manji. 2002. Mood stabilizer psychopharmacology. Clin. Neurosci. Res. 2:193–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harnett, W., and M.M. Harnett. 1993. Inhibition of murine B cell proliferation and down-regulation of protein kinase C levels by a phosphorylcholine-containing filarial excretory-secretory product. J. Immunol. 151:4829–4837. [PubMed] [Google Scholar]
- Huntington's Disease Collaborative Research Group. 1993. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosome. Cell. 72:971–983. [DOI] [PubMed]
- Ju, S., G. Shaltiel, A. Shamir, G. Agam, and M.L. Greenberg. 2004. Human 1-D-myo-inositol-3-phosphate synthase is functional in yeast. J. Biol. Chem. 279:21759–21765. [DOI] [PubMed] [Google Scholar]
- Kabeya, Y., N. Mizushima, T. Ueno, A.Yamamoto, T. Kirisako, T. Noda, E. Kominami, Y. Ohsumi, and T. Yoshimori. 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19:5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky, D.J., and S.D. Emr. 2000. Autophagy as a regulated pathway of cellular degradation. Science. 290:1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger, R., W. Kuhn, T. Muller, D. Woitalla, M. Graeber, S. Kosel, H. Przuntek, J.T. Epplen, L. Schols, and O. Riess. 1998. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat. Genet. 18:106–108. [DOI] [PubMed] [Google Scholar]
- Lubrich, B., and D. van Calker. 1999. Inhibition of the high affinity myo-inositol transport system: a common mechanism of action of antibipolar drugs? Neuropsychopharmacology. 21:519–529. [DOI] [PubMed] [Google Scholar]
- Manji, H.K., and R.H. Lenox. 1998. Lithium: a molecular transducer of mood-stabilization in the treatment of bipolar disorder. Neuropsychopharmacology. 19:161–166. [DOI] [PubMed] [Google Scholar]
- Manning, B.D., and L.C. Cantley. 2003. Rheb fills a GAP between TSC and TOR. Trends Biochem. Sci. 28:573–576. [DOI] [PubMed] [Google Scholar]
- Mizushima, N. 2004. Methods for monitoring autophagy. Int. J. Biochem. Cell Biol. 36:2491–2502. [DOI] [PubMed] [Google Scholar]
- Mizushima, N., A. Yamamoto, M. Matsui, T. Yoshimori, and Y. Ohsumi. 2004. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell. 15:1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, G.J., J.M. Bebchuk, J.K. Parrish, M.W. Faulk, C.L. Arfken, J. Strahl-Bevacqua, and H.K. Manji. 1999. Temporal dissociation between lithium-induced changes in frontal lobe myo-inositol and clinical response in manic-depressive illness. Am. J. Psychiatry. 156:1902–1908. [DOI] [PubMed] [Google Scholar]
- Narain, Y., A. Wyttenbach, J. Rankin, R.A. Furlong, and D.C. Rubinsztein. 1999. A molecular investigation of true dominance in Huntington's disease. J. Med. Genet. 36:739–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos, M.H., C. Lavedan, E. Leroy, S.E. Ide, A. Dehejia, A. Dutra, B. Pike, H. Root, J. Rubenstein, R. Boyer, et al. 1997. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 276:2045–2047. [DOI] [PubMed] [Google Scholar]
- Ravikumar, B., R. Duden, and D.C. Rubinsztein. 2002. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 11:1107–1117. [DOI] [PubMed] [Google Scholar]
- Ravikumar, B., C. Vacher, Z. Berger, J.E. Davies, S. Luo, L.G. Oroz, F. Scaravilli, D.F. Easton, R. Duden, C.J. O'Kane, and D.C. Rubinsztein. 2004. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36:585–595. [DOI] [PubMed] [Google Scholar]
- Rubinsztein, D.C. 2002. Lessons from animal models of Huntington's disease. Trends Genet. 18:202–209. [DOI] [PubMed] [Google Scholar]
- Rubinsztein, D.C., J. Leggo, R. Coles, E. Almqvist, V. Biancalana, J.-J. Cassiman, K. Chotai, M. Connarty, D. Craufurd, A. Curtis, et al. 1996. Phenotypic characterisation of individuals with 30-40 CAG repeats in Huntington's disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36-39 repeats. Am. J. Hum. Genet. 59:16–22. [PMC free article] [PubMed] [Google Scholar]
- Schmelzle, T., and M.N. Hall. 2000. TOR, a central controller of cell growth. Cell. 103:253–262. [DOI] [PubMed] [Google Scholar]
- Shaltiel, G., A. Shamir, J. Shapiro, D. Ding, E. Dalton, M. Bialer, A.J. Harwood, R.H. Belmaker, M.L. Greenberg, and G. Agam. 2004. Valproate decreases inositol biosynthesis. Biol. Psychol. 56:868–874. [DOI] [PubMed] [Google Scholar]
- Silverstone, P.H., B.M. McGrath, and H. Kim. 2005. Bipolar disorder and myo-inositol: a review of magnetic resonance spectroscopy findings. Bipolar Disord. 7:1–10. [DOI] [PubMed] [Google Scholar]
- Webb, J.L., B. Ravikumar, J. Atkins, J.N. Skepper, and D.C. Rubinsztein. 2003. Alpha-synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 278:25009–25013. [DOI] [PubMed] [Google Scholar]
- Williams, R.S., M. Eames, W.J. Ryves, J. Viggars, and A.J. Harwood. 1999. Loss of a prolyl oligopeptidase confers resistance to lithium by elevation of inositol (1,4,5) trisphosphate. EMBO J. 18:2734–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, R.S., L. Cheng, A.W. Mudge, and A.J. Harwood. 2002. A common mechanism of action for three mood-stabilizing drugs. Nature. 417:292–295. [DOI] [PubMed] [Google Scholar]
- Wood, N.I., and A.J. Morton. 2003. Chronic lithium chloride treatment has variable effects on motor behaviour and survival of mice transgenic for the Huntington's disease mutation. Brain Res. Bull. 61:375–383. [DOI] [PubMed] [Google Scholar]
- Wyttenbach, A., J. Swartz, H. Kita, T. Thykjaer, J. Carmichael, J. Bradley, R. Brown, M. Maxwell, A. Schapira, T.F. Orntoft, et al. 2001. Polyglutamine expansions cause decreased CRE-mediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntington's disease. Hum. Mol. Genet. 10:1829–1845. [DOI] [PubMed] [Google Scholar]
- Wyttenbach, A., O. Sauvageot, J. Carmichael, C. Diaz-Latoud, A.P. Arrigo, and D.C. Rubinsztein. 2002. Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum. Mol. Genet. 11:1137–1151. [DOI] [PubMed] [Google Scholar]